PIMODIVIR
VX-787, JNJ-63623872, JNJ-872, VRT-0928787, VX-787, VX 787, VX787, JNJ-872, JNJ 872, JNJ872, VRT-0928787, VRT 0928787, VRT0928787, pimodivir
(2S,3S)-3-{[5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]amino}bicyclo[2.2.2]octane-2-carboxylic acid
(2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
(2S,3S)-3-((5-Fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic Acid
MF C20H19F2N5O2, MW 399.4018
CAS 1629869-44-8
PHASE 2

| Originator |
Vertex Pharmaceuticals |
| Developer |
Janssen Pharmaceuticals |
| Mechanism Of Action |
Viral polymerase inhibitor, Viral protein inhibitor |
| Who Atc Codes |
J05A-X (Other antivirals) |
| Ephmra Codes |
J5B4 (Influenza antivirals) |
| Indication |
Influenza A |
| Paul Charifson, Michael P. Clark, Upul K. Bandarage, Randy S. Bethiel, John J. Court,Hongbo Deng, Ioana Drutu, John P. Duffy, Luc Farmer, Huai Gao, Wenxin Gu, Dylan H. Jacobs, Joseph M. Kennedy, Mark W. Ledeboer, Brian Ledford, Francois Maltais,Emanuele Perola, Tiansheng Wang, M. Woods Wannamaker, Less « |
| INNOVATOR |
Vertex Pharmaceuticals Incorporated |
Pimodivir (also known as VX-787, JNJ-872 and VRT-0928787) is a novel inhibitor of influenza virus replication that blocks the PB2 cap-snatching activity of the influenza viral polymerase complex. VX-787 binds the cap-binding domain of the PB2 subunit with a KD (dissociation constant) of 24 nM as determined by isothermal titration calorimetry (ITC).
The cell-based EC50 (the concentration of compound that ensures 50% cell viability of an uninfected control) for VX-787 is 1.6 nM in a cytopathic effect (CPE) assay, with a similar EC50 in a viral RNA replication assay. VX-787 is active against a diverse panel of influenza A virus strains, including H1N1pdm09 and H5N1 strains, as well as strains with reduced susceptibility to neuraminidase inhibitors (NAIs).

Pimodivir hydrochloride hemihydrate
RN: 1777721-70-6
UNII: A256039515, Bicyclo(2.2.2)octane-2-carboxylic acid, 3-((5-fluoro-2-(5-fluoro-1H-pyrrolo(2,3-b)pyridin-3-yl)-4-pyrimidinyl)amino)-, hydrochloride, hydrate (2:2:1), (2S,3S)-
Molecular Formula, 2C20-H19-F2-N5-O2.2Cl-H.H2-O, Molecular Weight, 889.7348
C20 H19 F2 N5 O2 . Cl H . 1/2 H2 O
Bicyclo[2.2.2]octane-2-carboxylic acid, 3-[[5-fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)-4-pyrimidinyl]amino]-, hydrochloride, hydrate (2:2:1), (2S,3S)-
Janssen Pharmaceuticals, under license from Vertex Pharmaceuticals, was developing pimodivir (first disclosed in WO2010148197), a PB2 inhibitor, for treating influenza A virus infection. In December 2016, pimodivir was reported to be in phase 2 clinical development.
Influenza spreads around the world in seasonal epidemics, resulting in the deaths of hundreds of thousands annually – millions in pandemic years. For example, three influenza pandemics occurred in the 20th century and killed tens of millions of people, with each of these pandemics being caused by the appearance of a new strain of the virus in humans. Often, these new strains result from the spread of an existing influenza virus to humans from other animal species.
Influenza is primarily transmitted from person to person via large virus-laden droplets that are generated when infected persons cough or sneeze; these large droplets can then settle on the mucosal surfaces of the upper respiratory tracts of susceptible individuals who are near (e.g. within about 6 feet) infected persons. Transmission might also occur through direct contact or indirect contact with respiratory secretions, such as touching surfaces contaminated with influenza virus and then touching the eyes, nose or mouth. Adults might be able to spread influenza to others from 1 day before getting symptoms to approximately 5 days after symptoms start. Young children and persons with weakened immune systems might be infectious for 10 or more days after onset of symptoms. [00103] Influenza viruses are RNA viruses of the family Orthomyxoviridae, which comprises five genera: Influenza virus A, Influenza virus B, Influenza virus C, Isavirus and Thogoto virus.
The Influenza virus A genus has one species, influenza A virus. Wild aquatic birds are the natural hosts for a large variety of influenza A. Occasionally, viruses are transmitted to other species and may then cause devastating outbreaks in domestic poultry or give rise to human influenza pandemics. The type A viruses are the most virulent human pathogens among the three influenza types and cause the most severe disease. The influenza A virus can be subdivided into different serotypes based on the antibody response to these viruses. The serotypes that have been confirmed in humans, ordered by the number of known human pandemic deaths, are: HlNl (which caused Spanish influenza in 1918), H2N2 (which caused Asian Influenza in 1957), H3N2 (which caused Hong Kong Flu in 1968), H5N1 (a pandemic threat in the 2007-08 influenza season), H7N7 (which has unusual zoonotic potential), H1N2 (endemic in humans and pigs), H9N2, H7N2 , H7N3 and H10N7. [00105] The Influenza virus B genus has one species, influenza B virus. Influenza B almost exclusively infects humans and is less common than influenza A. The only other animal known to be susceptible to influenza B infection is the seal. This type of influenza mutates at a rate 2-3 times slower than type A and consequently is less genetically diverse, with only one influenza B serotype. As a result of this lack of antigenic diversity, a degree of immunity to influenza B is usually acquired at an early age. However, influenza B mutates enough that lasting immunity is not possible. This reduced rate of antigenic change, combined with its limited host range (inhibiting cross species antigenic shift), ensures that pandemics of influenza B do not occur.
The Influenza virus C genus has one species, influenza C virus, which infects humans and pigs and can cause severe illness and local epidemics. However, influenza C is less common than the other types and usually seems to cause mild disease in children. [00107] Influenza A, B and C viruses are very similar in structure. The virus particle is 80-120 nanometers in diameter and usually roughly spherical, although filamentous forms can occur. Unusually for a virus, its genome is not a single piece of nucleic acid; instead, it contains seven or eight pieces of segmented negative-sense RNA. The Influenza A genome encodes 11 proteins: hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), Ml, M2, NSl, NS2(NEP), PA, PBl, PB1-F2 and PB2.
[HA and NA are large glycoproteins on the outside of the viral particles. HA is a lectin that mediates binding of the virus to target cells and entry of the viral genome into the target cell, while NA is involved in the release of progeny virus from infected cells, by cleaving sugars that bind the mature viral particles. Thus, these proteins have been targets for antiviral drugs. Furthermore, they are antigens to which antibodies can be raised. Influenza A viruses are classified into subtypes based on antibody responses to HA and NA, forming the basis of the H and N distinctions (vide supra) in, for example, H5N1. [00109] Influenza produces direct costs due to lost productivity and associated medical treatment, as well as indirect costs of preventative measures. In the United States, influenza is responsible for a total cost of over $10 billion per year, while it has been estimated that a future pandemic could cause hundreds of billions of dollars in direct and indirect costs. Preventative costs are also high. Governments worldwide have spent billions of U.S. dollars preparing and planning for a potential H5N1 avian influenza pandemic, with costs associated with purchasing drugs and vaccines as well as developing disaster drills and strategies for improved border controls.
Current treatment options for influenza include vaccination, and chemotherapy or chemoprophylaxis with anti-viral medications. Vaccination against influenza with an influenza vaccine is often recommended for high-risk groups, such as children and the elderly, or in people that have asthma, diabetes, or heart disease. However, it is possible to get vaccinated and still get influenza. The vaccine is reformulated each season for a few specific influenza strains but cannot possibly include all the strains actively infecting people in the world for that season. It takes about six months for the manufacturers to formulate and produce the millions of doses required to deal with the seasonal epidemics; occasionally, a new or overlooked strain becomes prominent during that time and infects people although they have been vaccinated (as by the H3N2 Fujian flu in the 2003-2004 influenza season). It is also possible to get infected just before vaccination and get sick with the very strain that the vaccine is supposed to prevent, as the vaccine takes about two weeks to become effective. [00111] Further, the effectiveness of these influenza vaccines is variable. Due to the high mutation rate of the virus, a particular influenza vaccine usually confers protection for no more than a few years. A vaccine formulated for one year may be ineffective in the following year, since the influenza virus changes rapidly over time, and different strains become dominant.
Also, because of the absence of RNA proofreading enzymes, the RNA- dependent RNA polymerase of influenza vRNA makes a single nucleotide insertion error roughly every 10 thousand nucleotides, which is the approximate length of the influenza vRNA. Hence, nearly every newly-manufactured influenza virus is a mutant — antigenic drift. The separation of the genome into eight separate segments of vRNA allows mixing or reassortment of vRNAs if more than one viral line has infected a single cell. The resulting rapid change in viral genetics produces antigenic shifts and allows the virus to infect new host species and quickly overcome protective immunity.
Antiviral drugs can also be used to treat influenza, with neuraminidase inhibitors being particularly effective, but viruses can develop resistance to the standard antiviral drugs.
Thus, there is still a need for drugs for treating influenza infections, such as for drugs with expanded treatment window, and/or reduced sensitivity to viral titer
U.S. Patent No. 8,829,007 discloses compounds that inhibit the replication of influenza viruses, including (2S,3S)-3-((5-fluoro-2-(5-fluoro-lH-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid (also known as VX-787). Boroylated intermediates are useful for preparing these compounds that inhibit the replication of influenza viruses. M. P. Clark et al., J. Med. Chem., 2014, 57-6668-6678. These borylated intermediates were previously prepared by incorporating a bromine at the position of the molecule to be borylated. For example, Clark reports preparing 5-chloro-3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l-tosyl-lH-pyrrolo[2,3-b]pyridine from 3-bromo-5-fluoro-lH-pyrrolo[2,3-b]pyridine.
Methods for preparing borylated compounds are described in U.S. Patent Publication Nos. 2008/0146814 and 2008/0167476.
Improved methods for preparing 3-boryl 7-azaindole compounds, such as 3-boryl-5-halo-7-azaindole compounds, in high yield and with no or few impurities are needed
Synthetic Scheme 1
(a) CHC13; (b) NaOMe, MeOH; (c) DPPA, Et3N, BnOH; (d) H2, Pd/C;
Synthetic Scheme 2
(a) Et3N, CH3CN; (b) cone. H2S04; (c) 9M H2S04; (d) Ag2C03, HOAc, DMSO, 100 °C; (e) X- phos, Pd2(dba)3, K3PO4, 2-methyl THF, H20, 120 °C (f) LiOH, THF, MeOH, 70 °C
Synthetic Scheme 3
(a) Et3N, THF; (b) chiral SFC separation; (c) 5-fluoro- l -(p-tolylsulfonyl)-3-(4,4,5,5-tetramethyl- l,3,2-dioxaborolan-
SYNTHESIS

PAPER
Journal of Medicinal Chemistry (2014), 57(15), 6668-6678
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
Michael P. Clark*†, Mark W. Ledeboer†, Ioana Davies†, Randal A. Byrn†, Steven M. Jones†, Emanuele Perola†, Alice Tsai†, Marc Jacobs†, Kwame Nti-Addae†, Upul K. Bandarage†, Michael J. Boyd†, Randy S. Bethiel†, John J. Court†, Hongbo Deng†, John P. Duffy†, Warren A. Dorsch†, Luc J. Farmer‡, Huai Gao†, Wenxin Gu†, Katrina Jackson†, Dylan H. Jacobs†, Joseph M. Kennedy†, Brian Ledford†, Jianglin Liang†, François Maltais†, Mark Murcko†, Tiansheng Wang†, M. Woods Wannamaker†, Hamilton B. Bennett†, Joshua R. Leeman†, Colleen McNeil†, William P. Taylor†, Christine Memmott†, Min Jiang†, Rene Rijnbrand†, Christopher Bral§, Ursula Germann†, Azin Nezami†, Yuegang Zhang†, Francesco G. Salituro∥, Youssef L. Bennani‡, and Paul S. Charifson†
† Vertex Pharmaceuticals Inc., 50 Northern Ave, Boston, Massachusetts 02210, United States
‡ Vertex Pharmaceuticals (Canada) Inc., 275 Armand-Frappier, Laval, Quebec H7V 4A7, Canada
§ Arrowhead Research Corporation, 465 Science Drive, Suite C, Madison, Wisconsin 53711, United States
∥ Sage Therapeutics, 215 First Street, Cambridge, Massachusetts 02141, United States
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
Publication Date (Web): July 14, 2014
Copyright © 2014 American Chemical Society
Abstract

In our effort to develop agents for the treatment of influenza, a phenotypic screening approach utilizing a cell protection assay identified a series of azaindole based inhibitors of the cap-snatching function of the PB2 subunit of the influenza A viral polymerase complex. Using a bDNA viral replication assay (Wagaman, P. C., Leong, M. A., and Simmen, K. A.Development of a novel influenza A antiviral assay. J. Virol. Methods 2002, 105, 105−114) in cells as a direct measure of antiviral activity, we discovered a set of cyclohexyl carboxylic acid analogues, highlighted by VX-787 (2). Compound 2 shows strong potency versus multiple influenza A strains, including pandemic 2009 H1N1 and avian H5N1 flu strains, and shows an efficacy profile in a mouse influenza model even when treatment was administered 48 h after infection. Compound 2represents a first-in-class, orally bioavailable, novel compound that offers potential for the treatment of both pandemic and seasonal influenza and has a distinct advantage over the current standard of care treatments including potency, efficacy, and extended treatment window.

aReagents and conditions: (a) CHCl3, 78%; (b) NaOMe, MeOH, 4 days, 85%; (c) DPPA, Et3N, BnOH, 77%; (d) H2, Pd/C, THF/MeOH, 99%; (e) 2,4-dichloro-5-fluoropyrimidine, iPr2NEt, THF, 77%; (f) SFC chiral separation; (g) 56, Pd2(dba)3, K3PO4, 2-MeTHF, water, 120 °C, 95%; (h) HCl, dioxane, MeCN, 95%; (i) NaOH, THF, MeOH, 95%.
(2S,3S)-3-((5-Fluoro-2-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic Acid
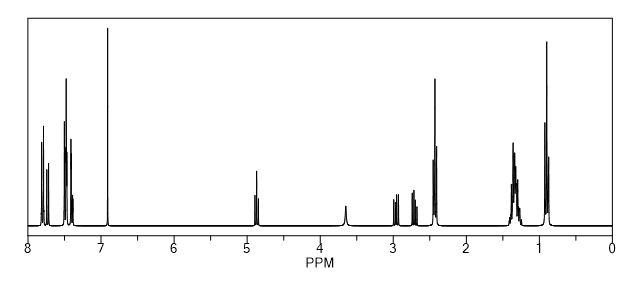
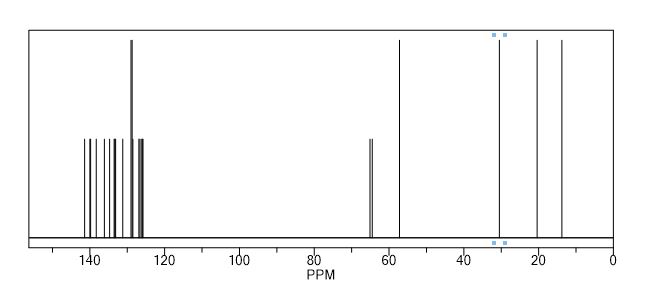
1H NMR (300 MHz, DMSO-d6) δ 12.71 (br s, 1H), 8.58 (s, 1H), 8.47 (dd, J = 9.6, 2.8 Hz, 1H), 8.41 (d, J = 4.8 Hz, 1H), 8.39–8.34 (m, 1H), 4.89–4.76 (m, 1H), 2.94 (d, J = 6.9 Hz, 1H), 2.05 (br s, 1H), 1.96 (br s, 1H), 1.68 (complex m, 7H); 13C NMR (300 MHz, DMSO-d6) δ 174.96, 157.00, 155.07, 153.34, 152.97, 145.61, 142.67, 140.65, 134.24, 133.00, 118.02, 114.71, 51.62, 46.73, 28.44, 28.00, 24.90, 23.78, 20.88, 18.98; LCMS gradient 10–90%, 0.1% formic acid, 5 min, C18/ACN, tR = 2.24 min, (M + H) 400.14; HRMS (ESI) of C20H20F2N5O2 [M + H] calcd, 400.157 95; found, 400.157 56.
PATENT
WO2010148197
(1070) (2S,3S)-3-((2-(5-fluoro-1H-pyrrolo[2,3-b]pyridm-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1070 was made in a similar fashion as described above for compounds 946 and 947.

946 (+/-) 947 (+/-)
[001117] (946) (+/-)-2,3-*r«/ts-CTt</ø-3-(2-(5-chloro-1H-pyrrolo [2,3-b] pyridin-3-yl)-5- fluoropyrimidin-4-ylamino)bicyclo[2.2.1]heptane-2-carboxylic acid & (947) (+/-)-2,3-rr««s-^xo-3-(2-(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)-5- fluoropyrimidin-4-ylamino)bicyclo[2.2.1]heptane-2-carboxylic acid
To a stirred solution of starting methyl esters, 53d, (0.076 g, 0.183 mmol) (84 : 16 = endo : exo) in THF (0.60 mL) and MeOH (0.10 mL), was added NaOH (0.10 mL of 2 M, 0.201 mmol). The reaction progress was monitored by TLC. After 30 min, additional NaOH (0.18 mL of 2 M solution, 0.37 mmol) and MeOH (0.18 mL) was added. The mixture was stirred at room temperature for a further 16 hours. The mixture was neutralized with HCl (IM) and concentrated in vacuo. Purification by preparative HPLC provided 52 mg of the major isomer (946) and 1 lmg of the minor isomer (947) as the hydrochloric acid salts.
(946) major {endo) isomer: 1H NMR (300 MHz, MeOD) δ 8.82 (d, J= 2.2 Hz, 1H), 8.48 (s, 1H), 8.39 (d, J= 2.2 Hz, 1H), 8.31 (d, J= 5.6 Hz, 1H), 5.11 (m, 1H), 2.85 (br s, 1H), 2.68 (br s, 1H), 2.62 (d, J = 4.8 Hz, 1H), 1.92 (d, J = 10.1 Hz, 1H) and 1.77 – 1.51 (m, 5H) ppm; LC/MS R, = 3.51, (M+H) 402.32.
(947) minor (exo) isomer: 1H NMR (300 MHz, MeOD) δ 8.87 (d, J = 2.1 Hz, 1H), 8.48 (s, 1H), 8.39 (d, J = 1.9 Hz, 1H), 8.30 (d, J = 5.7 Hz, 1H), 4.73 (d, J = 3.3 Hz, 1H), 3.12 (m, 1H), 2.76 (br s, 1H), 2.56 (d, J= 4.2 Hz, 1H), 1.86 (d, J= 9.5 Hz, 2H), 1.79 – 1.49 (complex m, 2H) and 1.51 (embedded d, J= 10.4 Hz, 2H) ppm; LC/MS R, = 3.42, (M+H) 402.32.
[001118] (1184) (2S,3S)-3-((2-(5-chloro-1H-pyrrolo[2,3-b]pyridin-3-yl)-5- fluoropyrimidin-4-yl)amino)bicyclo[2.2.2]octane-2-carboxylic acid
Compound 1184 was made in a similar fashion as described above for compounds 946 and 947
PATENT
WO-2016191079
EXPERIMENTAL
Example 1: Synthesis of 3-BPin-5-bromo-7-azaindole

Chemical Formula: C7H5BrN2 Chemical Formula: Ci3H16BBrN202
Molecular Weight: 197.03 Molecular Weight: 322.99
5-fluoro-7-azaindole (1 g) and THF (10 mL) were added to a small screw-top vial fitted with a septum, argon inlet and exit needle. The flask was sparged with argon for -10 minutes. The iridium catalyst [Ir(OMe)COD]2 (0.168 mg) and 2,2′-bipyridyl (0.080 mg) were added as solids and the flask was covered with the septum and sparged with argon again for -10-15 minutes. When the catalyst was added, the reaction turned a dark red/purple. The argon was then turned off, and HBPin (1.5 mL) was added via syringe. The reaction bubbled, releasing hydrogen. The hydrogen was allowed to bubble out through the bubbler outlet and once bubbling stopped, the reaction was capped and placed in an oil bath heated to 80° C.
After approx. 20 hours, the flask was allowed to cool to room temperature and a sample was pulled from the reaction flask for HPLC analysis. The reaction was then quenched with methanol (10 mL) and allowed to stir for -5 minutes before it was concentrated in vacuo to afford a dark residue (2.44 g). The residue was dissolved in methyl t-butyl ether (MTBE) (50 mL) and filtered through a silica plug (20 g, 150 mL frit). The cake was washed with MTBE (3 x 20 mL) and the filtrate was collected and concentrated in vacuo to afford 1.5 g of an off-white solid. The solid residue was taken up in 10 mL of isopropanol (IPA) and heated until it dissolved. The flask was allowed to cool to room temperature, at which point some crystals had precipitated out of solution. The flask was placed in the freezer overnight to afford white crystals.
The crystals were filtered in vacuo, washed with cold hexanes, and dried on a rotovap (yield: 0.5 g). The crystals were taken up again in hexanes (10 mL) and heated to reflux, but the crystals would not dissolve in the hexanes. Thus, the hexanes were removed on the rotovap, and the crystals were taken up in IPA (10 mL) and heated to reflux until the crystals dissolved. The flask was allowed to cool to room temperature, and then placed in the freezer overnight to crystallize, affording 300 mg (7.8 %) of product.
Example 2: Synthesis of 3-BPin-5-bromo-N-tosyl-7-azaindole

Chemical Formula: Ci4HiiBrN202S Chemical Formula: C 0H22BBrN2O4S Molecular Weight: 351.22 Molecular Weight: 477.18
A 250 mL 1-neck round bottom flask equipped with a thermocouple and argon inlet was sparged with argon for 15 minutes. 5-bromo-N-tosyl-7-azaindole (4.0 g), B2Pin2 (2.90 g), [Ir(OMe)COD]2 (0.114 g), 2,2’bipyridyl (0.054 g) and hexane (50 mL) were then added. The flask was again inerted with 3 vacuum purges. The resulting brown slurry was then heated to 60° C incrementally (setpoints: 45° C, 55° C, 58° C and 60° C) and stirred overnight.
After 15 hours at 60 ° C, HPLC of the reaction mixture indicated 3.0% starting material remaining and 94% product. After 18 hours at 60 ° C, HPLC of the reaction mixture indicated 2.7% starting material and 5% product.
The slurry was then cooled to room temperature and vacuum filtered. The solids were recombined with the mother liquor and concentrated to afford a solid. The solid was dissolved in dichloromethane (50 mL) and filtered through silica (5 g on a 60 mL frit). The plug was washed with dichloromethane (2 x 50 mL). The filtrate and washes were combined and concentrated by rotovap to approx. 50 mL. Hexane (50 mL) was added and the solution was again concentrated to approx. 50 mL. Hexane (50 mL) was again added and the solution was concentrated. Product that “bumped” was rinsed back into the flask with dichloromethane. The solution was concentrated to approx. 50 mL and hexane (35 mL) was added. The solution was then concentrated to approx. 50 mL. The slurry was vacuum filtered and the solids were washed with cold hexane, then dried by rotovap, to afford 4.7 g (88.7% of product. HPLC indicated approx. 97% purity.
Example 3: Synthesis of 3-BPin-5-fluoro-N-tosyl-7-azaindole
Chemic
Mol 
ecular Weight: 290.31
Chemical Formula: C20H22BFN2O4S
Molecular Weight: 416.27
A I L 3 -necked flask equipped with mechanical stirring, argon inlet, thermocouple, and heating mantle was sparged with argon for 15 minutes. 5-fluoro-N-tosyl-7-azaindole (50.0 g), B2Pin2 (43.7 g), [IrClCOD]2 (2.89 g), dppe (3.43 g), and heptane (500 mL) were then added to the flask. The resulting slurry was then heated to 95 C for 53 hours with stirring.
The slurry was then cooled to room temperature and the solids were collected by vacuum filtration, washed with cold hexane, and dissolved in dichloromethane (450 mL). The solution was filtered through silica (100 g on a 600 mL frit) and the plug was washed with 5 x 100 mL dichloromethane. The filtrate and washes were combined and concentrated by rotovap. Hexane (400 mL) was then added and the solution was concentrated to approx 200 mL. The slurry was then filtered and the solids washed with cold hexane, dried by rotovap to afford 56.97 g (79.6 %) of product. HPLC indicated a purity of >99%.
Example 4: Synthesis of N-Boc-3-BPin-5-fluoro-7-azaindole
Mo 204 
N-Boc-5-fluoro-7-azaindole (5.0 g), B2Pin2 (5.38 g), and hexane (50 mL) were added to a 250 mL 2-neck round bottom equipped with a condenser, magnetic stirring, heating mantle and nitrogen inlet. A colorless solution resulted with stirring. The flask was sparged with 3 nitrogen/vacuum cycles. The iridium catalyst [Ir(OMe)COD]2(0.21 g) and 2,2′-bipyridyl (0.10 g) were added as solids and another nitrogen/vacuum cycle was used to inert the flask. The resulting black solution was heated to 60° C. After 1 hour, TLC (eluting with DCM) of the reaction solution indicated that no starting material remained. The solution was cooled to room temperature and filtered through silica (10 g on a 60 mL frit). The plug was washed with dichloromethane (4 x 100 mL). The fractions were combined and concentrated by rotovap until a precipitate began to form. Dichloromethane was then added until a solution resulted and hexane (50 mL) was added. The solution was concentrated cold <25° C to approx. 50 mL. Hexane (50 mL) was added and the solution was concentrated to approx. 75 mL. The resulting white solids were collected by vacuum filtration, washed with cold hexane and dried by rotovap, to afford 4.6 g (60.5%) of product.
Example 5: Synthesis of 3-BPin-7-azaindole

Chemical Formula: C7H6N2 Chemical Formula: C13H17BN202
Molecular Weight: 118.14 Molecular Weight: 244.10
7-azaindole (5.0 g) and THF (50 mL) were added to an argon-inert small screw-top vial fitted with a septum, argon inlet and bubbler outlet. The flask was sparged with argon. The iridium catalyst [Ir(OMe)COD]2 (1.4 g) and 2,2’bipyridyl (1.4 g) were then added and the flask was again sparged with argon. The HBPin (12. 3 mL) was added by syringe and gas evolution was observed. The screwtop was sealed and the vial was placed in an oil bath and heated to 80° C for 16 hours.
The vessel was then allowed to cool to room temperature. The cap was removed and sampled while under an argon stream. The reaction appeared to stall at 50% completion. The cap was removed and 20 mL of methanol was added with visible degassing. The combined reaction solution was concentrated to an oil by rotovap (17.28 g). The crude product was dissolved in 50mL of MTBE and filtered through 50g of silica. The plug was washed with 3 x 50 mL of MTBE and the filtrate was concentrated by rotovap (13g of crude product). The crude product was dissolved in 13 mL of refluxing IPA, cooled to room temperature, and placed in the freezer. No crystals were observed.
Example 6: Synthesis of 3-BPin-5-fluoro-7-azaindole

Chemical Formula: C7H5FN2 Chemical Formula: Ci3H-ieBFN202 Molecular Weight: 136.13 Molecular Weight: 262.09
5-fluoro-7-azaindole (1.0 g) and THF (10 mL) were added to an argon-inert small screw-top vial fitted with a septum, argon inlet and bubbler outlet. The flask was sparged with argon. The iridium catalyst [Ir(OMe)COD]2 (0.24 g) and 3,4,7, 8-tetramethyl-l,10-phenanthroline (0.11 g) were then added and the flask was again sparged with argon. HBPin (2.13 mL) was added by syringe and gas evolution was observed. The screwtop was sealed and the vial was placed in an oil bath and heated to 80° C overnight.
The vial was then removed from the oil bath and allowed to cool to room temperature. The reaction was quenched by the addition of methanol (20 mL, very little gas evolution noted). The solution was then concentrated by rotovap to afford a dark oil (3.57 g). The oil was dissolved in MTBE (50 mL) and filtered though silica. The plug was washed with MTBE (4 x 25 mL) and the clear, yellow filtrate was concentrated by rotovap to an oil (2.7 g).
Upon standing overnight, solids precipitated out of the crude oil. The oil was then dissolved in refluxing hexane (3 mL, ~1 mL/g) and the solution was allowed to cool to room temperature then placed in the freezer.
The resulting white solids were collected by vacuum filtration, washed three times with cold hexane, and dried by rotovap (0.58 g crude product). HPLC indicated 92.2% purity. The crude solids were dissolved in refluxing IPA (1.2 mL, ~2 mL/g) and the resulting yellow solution was allowed to cool to room temperature (during which time crystals precipitated) then placed in the freezer. The resulting crystals were collected by vacuum filtration, washed three times with cold hexane (3x), and dried by rotovap to afford 0.33 g (17.1%) of product.
PATENT
WO2015073491
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015073491&redirectedID=true
Example 2: Preparation of Compound (l)and 2-MeTHF solvate of Compound (1)
Compound (1) can be prepared as described in WO 2010/148197. For example, an amorphous free base Compound (1) was prepared according to WO 2010/148197, followed by usual chiral separation and purification: SCF chiral chromatography with a modifier that included Et2NH (which generated Et2NH salt of Compound (1)) and then ion-exchange resin treatment. Alternatively, Compound (1) can be made by the following procedures as a 2-MeTHF solvate:
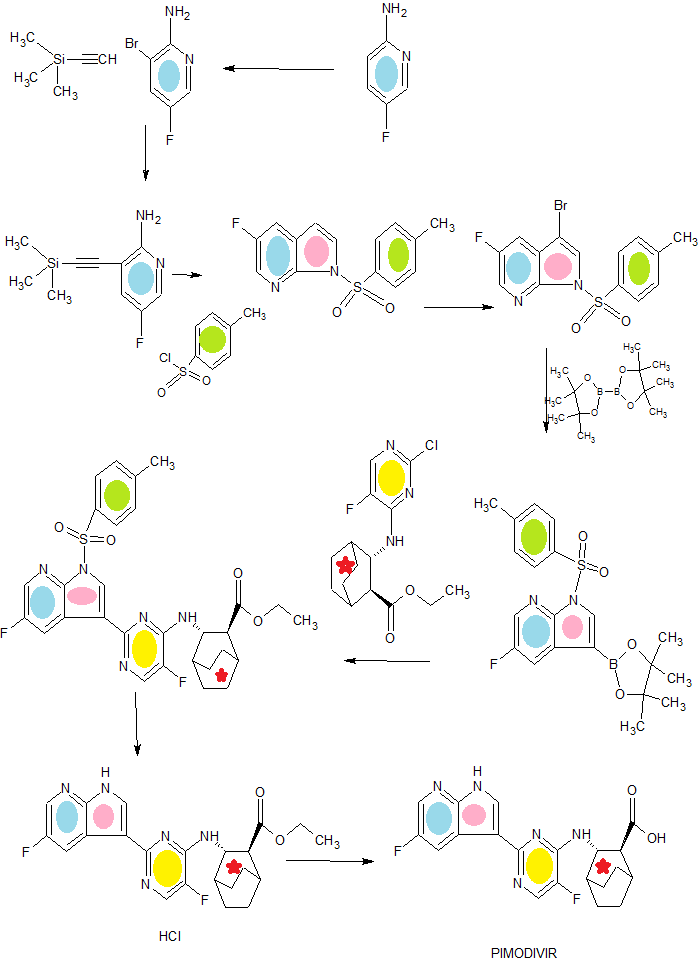
Preparation of Compound 2a (2- Amino-3-bromo-5-fluoropyridine)

1a 2a
To a slurry of 2-amino-5-fluoropyridine (6 kg, 53.6 mol) in water (24 L) at 14 °C was added over 10 minutes 48% hydrobromic acid (18.5 kg, 110 mol). The reaction was exothermic and the temperature went up to 24 °C. The mixture was re-cooled to 12 °C then bromine (9 kg, 56.3 mol) was added in nine portions over 50 minutes (exothermic, kept at 20 °C). The mixture was stirred at 22 °C overnight, and monitored by ‘HNMR of a quenched aliquot (quenched 5 drops in to mix of 1 ml 20% K2CO3, 0.3 ml 10% Na2S203 and 0.7 ml DCM. Organic layer evaporated and assayed). The mixture was cooled to 10 °C then quenched by addition of sodium bisulfite (560 g, 5.4 mol) in water (2 L), and further cooled to 0 °C. This mixture was added to a cold (-4 °C) mixture of DCM (18 L) and 5.4M sodium hydroxide (35 L, 189 mol). The bottom -35 L was filtered through a pad of Celite and then the phase break was made. The aqueous layer was re-extracted with DCM (10 L). The organics were filtered through a pad of 3 kg magnesol, washing with DCM (8 L). The filtrate was evaporated, triturated with hexane and filtered.
Despite the in-process assay indicating 97% completion, this initial product from all four runs typically contained -10% SM. These were combined and triturated in hexane (2 L per kg material) at 50 °C, then cooled to 15 °C and filtered to afford Compound 2a (30.0 kg, -95% purity, 149 mol, 67%). Mother liquors from the initial trituration and the re-purification were chromatographed (20 kg silica, eluent 25-50% EtOAc in hexane) to afford additional Compound 2a (4.7 kg, -99% purity, 24.4 mol, 11%).
Preparation of Compound 3a

To an inert 400-L reactor was charged 2a (27.5 kg, 96% purity, 138 mol), Pd(PPh3)4 (1044 g, 0.90 mol) and Cul (165 g, 0.87 mol), followed by toluene (90 kg). The mixture was de-oxygenated with three vacuum-nitrogen cycles, then triethylamine (19.0 kg, 188 mol) was added. The mixture was de-oxygenated with one more vacuum-nitrogen cycle, then
TMS-acetylene (16.5 kg, 168 mol) was added. The mixture was heated to 48 °C for 23 hours (the initial exotherm took the temperature to 53 °C maximum), then cooled to 18 °C. The slurry was filtered through a pad of Celite and washed with toluene (80 kg). The filtrate was washed with 12% Na2HP04 (75 L), then filtered through a pad of silica (25 kg), washing with 1 :1 hexane:MTBE (120 L). This filtrate was evaporated to a brown oil and then dissolved in NMP for the next step. Weight of a solution of Compound 3a – 58 kg, ~50wt%, 138 mol,
100%. 1H NMR (CDCI3, 300 MHz): δ 7.90 (s, 1H); 7.33-7.27 (m, 1H); 4.92 (s, NH2), 0.28 (s, 9H) ppm.
Preparation o Compound 4a

3a 4a
To an inert 400-L reactor was charged potassium t-butoxide (17.5 kg, 156 mol) and NMP (45 kg). The mixture was heated to 54 °C then a solution of Compound 3a (29 kg, 138 mol) in NMP (38 kg) was added over 2.75 hours and rinsed in with NMP (6 kg)
(exothermic, maintained at 70-77 °C) . The reaction was stirred at 74 °C for 2 hours then cooled to 30 °C and a solution of tosyl chloride (28.5 kg, 150 mol) in NMP (30 kg) added over 1.5 hours and rinsed in with NMP (4 kg). The reaction was exothermic and maintained at 30-43 °C. The reaction was stirred for 1 hour while cooling to 20 °C then water (220 L) was added over 35 minutes (exothermic, maintained at 18-23 °C). The mixture was stirred at 20 °C for 30 minutes then filtered and washed with water (100 L). The solids were dissolved off the filter with DCM (250 kg), separated from residual water and the organics filtered through a pad of magnesol (15 kg, top) and silica (15 kg, bottom), washing with extra DCM (280 kg). The filtrate was concentrated to a thick slurry (-50 L volume) then MTBE (30 kg) was added while continuing the distillation at constant volume (final distillate temperature of 51 °C). Additional MTBE (10 kg) was added and the slurry cooled to 15 °C, filtered and washed with MTBE (40 L) to afford Compound 4a (19.13 kg, 95% purity, 62.6 mol, 45%). Partial concentration of the filtrate afforded a second crop (2.55 kg, 91% purity, 8.0 mol, 6%). 1H NMR (CDCI3, 300 MHz): δ 8.28-8.27 (m, 1H); 8.06-8.02 (m, 2H); 7.77 (d, J= 4.0 Hz, 1H); 7.54-7.50 (m, 1H); 7.28-7.26 (m, 2H); 6.56 (d, J= 4.0 Hz, 1H); 2.37 (s, 3H) ppm.
Preparation of Compound 5a

4a 5a
To a slurry of N-bromosuccinimide (14.16 kg, 79.6 mol) in DCM (30 kg) at 15 °C was charged a solution of Compound 4a (19.13 kg, 95% purity, and 2.86 kg, 91% purity, 71.6 mol) in DCM (115 kg), rinsing in with DCM (20 kg). The mixture was stirred at 25 °C for 18 hours, and then cooled to 9 °C and quenched by addition of a solution of sodium
thiosulfate (400 g) and 50% sodium hydroxide (9.1 kg) in water (130 L). The mixture was warmed to 20 °C and the layers were separated and the organics were washed with 12% brine (40 L). The aqueous layers were sequentially re-extracted with DCM (4 x 50 kg). The organics were combined and 40 L distilled to azeotrope water, then the solution was filtered through a pad of silica (15 kg, bottom) and magensol (15 kg, top), washing with DCM (180 kg). The filtrate was concentrated to a thick slurry (-32 L volume) then hexane (15 kg) was added. Additional hexane (15 kg) was added while continuing the distillation at constant volume (final distillate temperature 52 °C). The slurry was cooled to 16 °C, filtered and washed with hexane (25 kg) to afford Compound 5a (25.6 kg, 69.3 mol, 97%). 1H NMR (CDC13, 300 MHz): δ 8.34-8.33 (m, 1H); 8.07 (d, J= 8.2Hz, 2H); 7.85 (s, 1H); 7.52-7.49 (m, 1H); 7.32-7.28 (m, 2H); 2.40 (s, 3H) ppm.
Preparation of Compound 6a: BEFTA1 Reaction

6a
To an inert 400-L reactor was charged Compound 5a (25.6 kg, 69.3 mol), bis(pinacolato)diboron (19 kg, 74.8 mol), potassium acetate (19 kg, 194 mol), palladium acetate (156 g, 0.69 mol) and triphenylphosphine (564 g, 2.15 mol), followed by dioxane (172 kg), that had been separately de-oxygenated using vacuum-nitrogen cycles (x 3). The mixture was stirred and de-oxygenated using vacuum-nitrogen cycles (x 2), then heated to 100 °C for 15 hours. The mixture was cooled to 35 °C then filtered, washing with 30 °C THF (75 kg). The filtrate was evaporated and the residue dissolved in DCM (-90 L). The solution was stirred with 1 kg carbon and 2 kg magnesol for 45 minutes then filtered through a pad of silica (22 kg, bottom) and magensol (10 kg, top), washing with DCM (160 kg). The filtrate was concentrated to a thick slurry (-40 L volume) then triturated at 35 °C and hexane (26 kg) was added. The slurry was cooled to 20 °C, filtered and washed with a mix of DCM (5.3 kg) and hexane (15 kg), then hexane (15 kg) and dried under nitrogen on the filter to afford Compound 6a (23.31 kg, 56.0 mol, 81%) as a white solid. 1H-NMR consistent with desired product, HPLC 99.5%, palladium assay 2 ppm. 1H NMR (CDC13, 300 MHz): δ 8.25 (s, 1H); 8.18 (s, 1H); 8.09-8.02 (m, 2H); 7.91-7.83 (m, 1H); 7.30-7.23 (m, 2H); 2.39 (s, 3H); 1.38 (s, 12H) ppm.
Preparation of Compounds 8a and 9a

9a
[0247] Compound 8a: Anhydride 7a (24.6 kgs, Apex) and quinine (49.2 kgs, Buchler) were added to a reactor followed by the addition of anhydrous PhMe (795.1 kgs). The reactor was then cooled to -16 °C and EtOH (anhydrous, 41.4 kgs) was added at such a rate to maintain the internal reactor temperature < -12 °C. The maximum reaction temp recorded for this experiment was -16 °C. The reaction mixture was then stirred for 16 h at -16 °C. A sample was removed and filtered. The solid was dried and evaluated by 1H-NMR which showed that no anhydride remained. The contents of the reactor were filtered. The reactor and subsequent wet cake were washed with PhMe (anhydrous, 20 kgs). The resulting solid was placed in a tray dryer at < 45 °C with a N2 sweep for at least 48 h. In this experiment, the actual temperature was 44 °C and the vacuum was -30 inHG. Material was sampled after 2.5 d drying and showed 3% PhMe by NMR. After an additional 8 hrs, the amt of PhMe analyzed showed the same 3% PhMe present and the drying was stopped. The weight of the white solid was 57.7 kgs, 76% yield. 1 H-NMR showed consistent with structure and Chiral SFC analysis showed material >99% ee.
Compound 9a: The reactor was charged with quinine salt 8a (57.7 kgs) and PhMe (250.5 kgs, Aldrich ACS grade, >99.5%) and the agitator was started. The contents were cooled to <15 °C and was treated with 6N HCI (18 kgs H20 were treated with 21.4 kgs of cone. HCI) while keeping the temperature <25 °C. The mixture was stirred for 40 min and visually inspected to verify that no solids were present. Stirring was stopped and the phases were allowed to settle and phases were separated. The aqueous phases were extracted again with PhMe (160 kgs; the amount typically used was much less, calc. 43 kgs. However, for efficient stirring due to minimal volume, additional PhMe was added. The organic phases were combined. Sample the organic phase and run HPLC analysis to insure product is present; for information only test.
To the organic phases were cooled to <5 °C (0-5 °C) and was added sodium sulfate (anhydrous, 53.1 kgs) with agitation for 8 hrs (in this instance 12 hrs). The contents of the reactor containing the organic phase were passed through a filter containing sodium sulfate (31 kgs, anhydrous) and into a cleaned and dried reactor. The reactor was rinsed with PhMe (57.4 kgs), passed through the filter into reactor 201. The agitator was started and an additional amount of PhMe (44 kgs) was added and the reaction mixture cooled to -20 °C. At that temperature PhMe solution of potassium tert-pentoxide was added over 2 h while keeping the temperature between -15 and -22 °C. The reaction mixture was held at -20 °C for an additional 30 min before being sampled. Sampling occurred by removing an aliquat with immediate quenching into 6N HC1. The target ratio here is 96:4 (trans is).
Having achieved the target ratio, the reactor was charged with acetic acid (2.8 kgs) over 6 min. The temperature stayed at – 20 °C. The temperature was then adjusted to -5 °C and aqueous 2N HC1 (65.7 kgs water treated with 15.4 kgs of cone HC1) was added. The contents were warmed to 5 °C +/- 5 °C, agitated for 45 min before warming to 20 °C +/- 5 °C with stirring for 15 min. The agitator was stopped and the phases allowed to settle. The aqueous layer was removed (temporary hold). The organic phase was washed with water (48 kgs, potable), agitated for 15 min and phases allowed to settle (at least 15 min) and the aqueous layer was removed and added to the aqueous layer. 1/3 of a buffer solution (50 L) that was prepared (7.9 kgs NaH2P04, 1.3 kgs of Na2HP04 and 143.6 kgs water) was added to the organic phase and stirred for at least 15 min. Agitation was stopped and phases were allowed to separate for at least 15 min. The lower layer was discarded. Another portion of the buffered solution (50 L) was used to wash the organic layer as previously described. The wash was done a third time as described above.
Vacuum distillation of the PhMe phase (150 L) was started at 42 °C/-13.9 psig and distilled to an oil of 20 L volume. After substantial reduction in volume the mixture was transferred to a smaller vessel to complete the distillation. Heptanes (13.7 kgs) was added and the mixture warmed to 40 +/- 5 °C for 30 min then the contents were cooled to 0-5 °C over 1.5 h. The solids were filtered and the reactor washed with approximately 14 kgs of cooled (0-5 °C) heptanes. The solids were allowed to dry under vacuum before placing in the oven at <40 °C under house vac (-28 psig) until LOD is <1%. 15.3 kgs, 64%, 96% HPLC purity. 1H NMR (400 MHz, CDC13) δ 11.45 (br. s, 1H), 6.41 (t, J= 7.2 Hz, 1H), 6.25 (t, J=
7.2 Hz, 1H), 4.18 (m, 2H), 3.27 (m, 1H), 3.03 (m, 1H), 2.95 (m, 1H), 2.77 (m, 1H), 1.68 (m,
1H), 1.49 (m, 1H), 1.25 (t, J= 7.2Hz), 1.12 (m, 1H).
Preparation of Compound 10a

9a 10a
A three neck flask equipped with a mechanical stirrer, temperature probe, reflux condenser, addition funnel and nitrogen inlet was charged with Compound 9a (145.0 g, 1 equiv) and anhydrous toluene (Aldrich, cat# 244511) (1408 g, 1655 ml) under an atmosphere of nitrogen. Then triethylamine (Aldrich, cat# 471283) (140 g, 193 ml,
2.14 equiv) was added in portions over 5 minutes to the stirred solution during which an exotherm to a maximum temperature of 27 °C was observed. Data acquisition by ReactIR was started. The reaction mixture was then heated to 95 °C over 70 minutes. Then diphenyl phosphoryl azide (Aldrich, cat# 178756) (176.2 g; 138.0 ml, 0.99 equiv) was added by addition funnel in portions over a total time of 2.25 hours.
Following completion of the addition of diphenyl phosphoryl azide (addition funnel rinsed with a small amount of toluene), the resulting mixture was heated at 96 °C for an additional 50 minutes. A sample of the reaction mixture diluted in toluene was analyzed by GC/MS which indicated consumption of diphenyl phosphoryl azide. Then benzyl alcohol (Aldrich, cat# 108006) (69.9 g, 67.0 ml, 1.0 equiv) was added by addition funnel over 5-10 minutes. The resulting mixture was then heated at 97 °C overnight (for approximately 19 hours). A sample of the reaction mixture diluted in toluene by GC/MS indicated formation of product (m/e =330). The reaction mixture was then cooled to 21 °C after which water (870 g, 870 ml) was added in portions (observed slight exotherm to maximum temperature of 22 °C). The reaction mixture was first quenched by addition of 500 g of water and mechanically stirred for 10 minutes. The mixture was then transferred to the separatory funnel containing the remaining 370 g of water and then manually agitated. After agitation and phase separation, the organic and aqueous layers were separated (aqueous cut at pH of -10). The organic layer was then washed with an additional portion of water (870 g; 1 x 870 ml). The organic and aqueous layers were separated (aqueous cut at pH of ~10). The collected organic phase was then concentrated to dryness under reduced pressure (water bath at 45-50 °C) affording 215 g of crude Compound 10a (approximate volume of 190 ml). The 1H NMR and GC/MS conformed to compound 10a (with residual toluene and benzyl alcohol).
Preparation o Compound 11a

10a 11a
HCI in ethanol preparation: A three neck flask equipped with a temperature probe, nitrogen inlet and magnetic stirrer was charged with ethanol (1000 ml, 773 g) under a
nitrogen atmosphere. The solution was stirred and cooled in a dry ice/acetone bath until an internal temperature of- 12 °C was reached. Then anhydrous HC1 (~ 80 g, 2.19 moles) was slowly bubbled in the cooled solution (observed temperature of -24 to -6 °C during addition) over 2 hours. Following the addition, the solution was transferred to a glass bottle and allowed to warm to ambient temperature. A sample of the solution was submitted for titration giving a concentration of 2.6 M. The solution was then stored in the cold room (approximately 5 °C) overnight.
Hydrogenation/HCl salt formation: A glass insert to a 2 gallon Parr autoclave was charged with palladium on carbon (Pd/C (Aldrich, cat# 330108), 10 % dry basis; (50 % wet), 13.11 g, 0.01 equiv on the basis of Compound 10a) under a nitrogen atmosphere and then moistened with ethanol (93 g; 120 ml). Then a solution of crude Compound 10a (212 g, 1 eq) in ethanol (1246 g; 1600 ml) was added to the glass insert (small rinse with ethanol to aid with transfer). The glass insert was placed in the autoclave after which HC1 in ethanol (prepared as described above; 2.6 M; 1.04 equiv based on Compound 10a; 223 g; 259 ml) was added. The autoclave was sealed and then purged with hydrogen (3 x at 20 psi). The hydrogenation was then started under an applied pressure of hydrogen gas (15 psi) for 3 hours at which time the pressure of hydrogen appeared constant. Analysis of an aliquot of the reaction mixture by 1H NMR and GC/MS indicated consumption of starting
material/formation of product. The resulting mixture was then filtered over a bed of Celite (192 g) after which the Celite bed was washed with additional ethanol (3 x; a total of 1176 g of ethanol was used during the washes). The filtrate (green in color) was then concentrated under reduced pressure (water bath at 45 °C) to ~ 382 g ((-435 ml; 2.9 volumes based on theoretical yield of Compound 11a. Then isopropyl acetate (1539 g; 1813 ml (12 volumes based on theoretical yield of Compound 11a was added to the remainder. The resulting solution was distilled under vacuum with gradual increase in temperature.
The distillation was stopped after which the remaining solution (370 g, -365 ml total volume; brownish in color) was allowed to stand at ambient temperature over the weekend. The mixture was filtered (isopropyl acetate used to aid with filtration) and the collected solids were washed with additional isopropyl acetate (2 x 116 ml; each wash was approximately 100 g). The solid was then dried under vacuum at 40 °C (maximum observed temperature of 42 °C) overnight to afford 1 18 g (78.1 % over two steps) of Compound 11a. The 1H NMR of the material conformed to the structure of Compound 11a, and GC/MS indicated 99% purity.
Preparation of Compound 13a

2a
Procedure A: A mixture of 5-fluoro-2,4-dichloropyrimidine (12a, 39.3 g, 235 mmol, 1.1 equiv), and HCI amine salt (11a, 50 g, 214 mmol) was treated with CH2C12(169 mL) and the mixture was warmed to 30 °C. The mixture was then treated slowly with DIEA (60.8 g, 82 mL, 471 mmol, 2.2 equiv) via syringe pump over 3 h. Peak temp was up to 32 °C. The reaction was stirred for 20 h, the reaction mixture was judged complete by HPLC and cooled to rt. The resulting reaction mixture was washed sequentially with water (21 1 mL, pH = 8-9), 5% NaHS04 (21 1 mL, pH = 1-2) then 5% aq. NaCl (211 mL, pH = 5-6).
The organic phase was then distilled under reduced pressure to 190 mL. PhMe was charged (422 mL) and temperature set at 70 -80 °C and internal temp at 60-65 °C until vol back down to 190 mL. The mixture was allowed to cool to approximately 37 °C with stirring – after approximately 10 min, crystallization began to occur and the temperature was observed to increase to approximately 41 °C. After equilibrating at 37 “C, the suspension was charged with n-heptane (421 mL) over 3.5 h followed by cooling to 22 °C over 1 h. The mixture was allowed to stir overnight at that temperature before filtering. The resulting solid on the filter was washed with a 10% PhMe in n-heptane solution (2 x 210 mL). The solid was then dried in the oven under vacuum with an N2 purge at 50 °C overnight. The resulting solid weighed 62 g (88% yield).
Procedure B: A three neck flask equipped with a mechanical stirrer, temperature probe, reflux condenser, nitrogen inlet and addition funnel was charged with Compound 11a (51.2 g) and Compound 12a (40.2 g) under an atmosphere of nitrogen. Dichloromethane (173 ml, 230 g) was added and the resulting mixture was stirred while warming to an internal temperature of 30 °C. Then N,N-diisopropylethylamine (85 ml, 63.09 g) was slowly added by addition funnel over 2.5-3 hours during which time an exotherm to a maximum observed temperature of 33.5 °C was observed. After complete addition, the resulting solution was stirred at 30-31 °C overnight under a nitrogen atmosphere (for approximately 19 hours).
A 100 μΐ sample of the reaction mixture was diluted with dichloromethane up to a total volume of 10 ml and the solution mixed well. A sample of the diluted aliquot was analyzed by GC/MS which indicated the reaction to be complete by GC/MS; observed
formation of product (m/e = 328)). The reaction mixture was cooled to 26 °C and transferred to a separatory funnel (aided with dichloromethane). The mixture was then sequentially washed with water (211 ml, 211 g; pH of aqueous cut was -8; small rag layer was transferred with aqueous cut), 5 % aqueous NaHS04 ((prepared using 50 g of sodium bisulfate monohydrate (Aldrich cat. # 233714) and 950 g water) 211 ml, 216 g; pH of aqueous cut was ~2) and then 5 % aqueous NaCl ((prepared using 50 g of sodium chloride (Aldrich cat. # S9888) and 950 g water) 211 ml, 215 g; pH of aqueous cut was -4-5). The collected organic phase was then concentrated under reduced pressure (water bath at 35 °C) to -190 ml (2.7 volumes based on theoretical yield of Compound 13a after which toluene (Aldrich cat. # 179418, 422 ml, 361 g) was added. The resulting mixture was concentrated under reduced pressure (water bath at 55-65 °C) to -190 ml (2.7 volumes based on theoretical yield of Compound 13a. Analysis of a sample of the solution at this stage by 1H NMR indicated the absence of dichloromethane. The remaining mixture was allowed to cool to 37 °C (using water bath at 37 °C on rotovap with agitation). During this time pronounced crystallization was observed. The mixture was then mechanically stirred and heated to approximately 37 °C (external heat source set to 38 °C) after which n-heptane (430 ml, 288 g; Aldrich cat# H2198) was slowly added by addition funnel over 3 hours. Following the addition, heating was stopped and the resulting slurry mechanically stirred while cooling to ambient temperature overnight. The resulting mixture was then filtered and the collected solids were washed with 10 % toluene in n-heptane (2 x 210 ml; each wash was prepared by mixing 21 ml (16 g) of toluene and 189 ml (132 g) of n-heptane). Vacuum was applied until very little filtrate was observed. The solids were then further dried under vacuum at 50 °C under a nitrogen bleed to constant weight (3.5 hours) giving 64.7 g (90 %) of Compound 13a. Analysis of a sample of the solid by Ή NMR showed the material to conform to structure and LC analysis indicated 99.8 % purity using the supplied LC method.
Preparation of Compound 14a

The ethyl ester 13a (85 g, 259 mmol) was dissolved in THF (340 mL) and treated with a solution of LiOH (2M, 389 mL, 778 mmol) over 10 min (temp from 21 to 24 °C). The mixture was warmed to 45 °C with stirring for 17 h at which time the reaction was judged complete by HPLC (no SM observed). The reaction mixture was cooled to rt and CH2C12 was added (425 mL). A solution of citric acid (2 M, 400 mL) was then added slowly over 45 min (temp up to 26 °C). It was noted that during the charge some white solids were formed but quickly dissolved with stirring. The reaction mixture was stirred for an additional 15 min before phases were allowed to separate. After the phases were split, the aqueous phase pH was measured pH = 4.0. The organic phase was washed (15 min stir) with water (255 mL) -phases were allowed to separate. The lower layer (organic) containing the desired product was then stored in the fridge overnight.
The organic phase was concentrated under reduced pressure (pot set to 65 °C) to 150 mL (est. 1.76 vol wrt SM). IPA (510 mL) was charged and distilled under reduced pressure (85 °C chiller temp setting) to 255 mL (3 vol). The level of solvent was brought to approximately 553 mL (6.5 vol) by the addition of IPA (298 mL). Water (16 mL) was then added and the reaction mixture warmed to reflux (77 °C) with good agitation which dissolved solids precipitated on the walls of the vessel. Reaction mixture was then cooled slowly to 65 °C (over 60 min) and held there – all material still in solution (sample pulled for residual solvent analysis). The reaction was further cooled to 60 °C and the reaction mixture appeared slightly opaque. After stirring for 15 min further cooled to 55 °C. While more product precipitates, the mixture is still thin and easily stirred. Water (808 mL) was added very slowly (2.5-3 hrs) while maintaining the temperature around 55 C. The mixture was then cooled to 22 °C over 2 h and allowed to stir overnight. Material was then filtered and washed with a mixture of water: IPA (75:25, 2 x 255 mL). The acid was dried in a vac oven at 55 °C overnight. Obtained 69 g of acid 14a, 88% yield of a white solid. The material analyzed >99% purity by HPLC.
Preparation o f Compound 15a: Suzuki Coupling

To 14a (91.4 g, 305 mmol), 6a (158.6 g, 381 mmol, 1.25 equiv.), Pd(OAc)2 (0.34 g, 1.5 mmol, 0.5 mol%), X-Phos (1.45 g, 3.0 mmol, 1.0 mol%), and K2C03 (168.6 g,
1220 mmol, 4 equiv.) was added THF (731 mL, 8 volumes) and water (29 mL, 0.32 vol). The reaction mixture was sparged with N2 for 30 min, then warmed to 65-70 °C and stirred for 5 h. HPLC analysis of the reaction mixture showed 99.3% conversion. The reaction mixture was cooled to 22-25 °C and water was added. The mixture was stirred, the phases
were allowed to separate, and the aqueous phase was decanted. A solution of 18 wt% NaCl in water (half-saturated aqueous NaCl) was added to the organic phase and the pH of the mixture was adjusted to 6.0-6.5 using 2N HC1. The phases were allowed to separate and the aqueous phase was decanted. The organic phase was concentrated to a minimum volume and acetonitrile was added. The process was repeated one more time and acetonitrile was added to bring the final volume to 910 mL (10 vol). The slurry was warmed to 80-85 °C for 6 h, then cooled to 20-25 °C. The slurry was stirred for 2 h, then filtered. The solids were rinsed with acetonitrile to give 15a (161 g, 89% yield).
Preparation of Compound (1): Detosylation Step

To 15a (25 g, 45.2 mmol) was added THF (125 ml, 5 vol), then MP-TMT resin (6.25 g, 25 wt%). The mixture was stirred at 20-25 °C for 16 h and filtered, rinsing with 1 vol THF. The resin treatment process and filtration were repeated. The THF solution was concentrated to 5 vol. To the mixture at 22-25 °C was added an aqueous solution of 2M LiOH (90.3 mL, 4 equiv). The reaction mixture was warmed to 40-45 °C and stirred for 5 h. HPLC analysis showed 99.7% conversion. The reaction mixture was cooled to 22-25 °C and MTBE (50 mL, 2 vol) was added. Phase separation occurred. The lower aqueous phase was collected. The aqueous phase was extracted with MTBE. The lower aqueous phase was collected. To the aqueous phase was added 2-MeTHF and the mixture was stirred. The pH of the mixture was adjusted to 6.0-6.5, and the lower aq. phase was decanted. The organic phase was washed with pH 6.5 buffer. The organic phase was concentrated to 85 mL, diluted with 2-MeTHF (150 mL), and concentrated to a final volume of 180 mL. The resultant slurry was warmed to 70-75 °C and stirred until complete dissolution, then cooled to 45-50 °C to give slurry. The slurry was stirred for 1 h, then heptane (180 mL) was added. The slurry was cooled to 20-25 °C over 1 h and stirred for 16 h. The batch was filtered, rinsing the solids with heptane. The solids were dried to give crude Compound (l)-2-MeTHF solvate, 79% yield.
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015073476
Preparation of Compound (1): Detosylation Step

[0214] To 15a (25 g, 45.2 mmol) was added THF (125 ml, 5 vol), then MP-TMT resin (6.25 g, 25 wt%). The mixture was stirred at 20 °C – 25 °C for 16 h and filtered, rinsing with 1 vol. THF. The resin treatment process and filtration were repeated. The THF solution was concentrated to 5 vol. To the mixture at 22 °C – 25 °C was added an aqueous solution of 2M LiOH (90.3 mL, 4 equiv.). The reaction mixture was warmed to 40 °C – 45 °C and stirred for 5 h. HPLC analysis showed 99.7% conversion. The reaction mixture was cooled to 22 °C -25 °C and MTBE (50 mL, 2 vol) was added. Phase separation occurred. The lower aqueous phase was collected. The aqueous phase was extracted with MTBE. The lower aqueous phase was collected. To the aqueous phase was added 2-MeTHF and the mixture was stirred. The pH of the mixture was adjusted to 6.0 – 6.5, and the lower aq. phase was decanted. The organic phase was washed with pH 6.5 buffer. The organic phase was concentrated to 85 mL, diluted with 2-MeTHF (150 mL), and concentrated to a final volume of 180 mL. The resultant slurry was warmed to 70 °C – 75 °C and stirred until complete dissolution, then cooled to 45 °C – 50 °C to give slurry. The slurry was stirred for 1 h, then heptane (180 mL) was added. The slurry was cooled to 20 °C – 25 °C over 1 h and stirred for 16 h. The batch was filtered, rinsing the solids with heptane. The solids were dried to give crude Compound (l 2-MeTHF solvate, 79% yield.
PAPER
Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2
J. Med. Chem., 2014, 57 (15), pp 6668–6678
DOI: 10.1021/jm5007275
http://pubs.acs.org/doi/abs/10.1021/jm5007275
Vertex Pharmaceuticals Inc
1H NMR (300 MHz, DMSO-d6) δ 12.71 (br s, 1H), 8.58 (s, 1H), 8.47 (dd, J = 9.6, 2.8 Hz, 1H), 8.41 (d, J = 4.8 Hz, 1H), 8.39–8.34 (m, 1H), 4.89–4.76 (m, 1H), 2.94 (d, J = 6.9 Hz, 1H), 2.05 (br s, 1H), 1.96 (br s, 1H), 1.68 (complex m, 7H);
13C NMR (300 MHz, DMSO-d6) δ 174.96, 157.00, 155.07, 153.34, 152.97, 145.61, 142.67, 140.65, 134.24, 133.00, 118.02, 114.71, 51.62, 46.73, 28.44, 28.00, 24.90, 23.78, 20.88, 18.98;
LCMS gradient 10–90%, 0.1% formic acid, 5 min, C18/ACN, tR = 2.24 min, (M + H) 400.14;
HRMS (ESI) of C20H20F2N5O2 [M + H] calcd, 400.157 95; found, 400.157 56.
Article
Vertex Licenses VX-787 to Janssen Pharmaceuticals for the Treatment of Influenza
Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that it has entered into a licensing agreement with Janssen Pharmaceuticals, Inc. for the worldwide development and commercialization of VX-787, a novel medicine discovered by Vertex for the treatment of influenza. As part of the agreement, Vertex will receive an up-front payment of $30 million from Janssen and has the potential to receive additional development and commercial milestone payments as well as royalties on future product sales. Vertex completed a Phase 2a study of VX-787 in 2013 that showed statistically significant improvements in viral and clinical measurements of influenza infection. VX-787 is designed to directly inhibit replication of the influenza virus.
“With a deep history in developing new medicines for viral infections and diseases, Janssen is well-positioned to advance the global development of VX-787 for the treatment of influenza,” said Jeffrey Leiden, M.D., Ph.D., Chairman, President and Chief Executive Officer of Vertex. “This collaboration provides important support for the continued development of VX-787 in influenza and contributes to our financial strength to enable continued investment in our key development programs for cystic fibrosis and in research aimed at discovering new medicines.”
About the Collaboration
Under the terms of the collaboration, Janssen will have full global development and commercialization rights to VX-787. Vertex will receive a $30 million up-front payment from Janssen and could receive additional development and commercial milestone payments as well as royalties on future product sales. The collaboration, and the related $30 million up-front payment, is subject to the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act.
About VX-787
VX-787 is an investigational medicine that is designed to directly inhibit replication of influenza A, including recent H1 (pandemic) and H5 (avian) influenza strains, based on in-vitro data. VX-787’s mechanism represents a new class of potential medicines for the treatment of influenza, distinct from neuraminidase inhibitors, the current standard of care for the treatment of influenza. VX-787 is intended to provide a rapid onset of action and an expanded treatment window.
In a Phase 2a influenza challenge study, statistically significant improvements in viral and clinical measurements of influenza infection were observed after treatment with VX-787. The study met its primary endpoint and showed a statistically significant decrease in the amount of virus in nasal secretions (viral shedding) over the seven-day study period. In addition, at the highest dosing regimen evaluated in the study, there was a statistically significant reduction in the severity and duration of influenza-like symptoms. In this study, VX-787 was generally well-tolerated, with no adverse events leading to discontinuation. Those who took part in the study volunteered to be experimentally exposed to an attenuated form of live H3N2 influenza A virus. H3N2 is a common type of influenza virus and was the most common type observed in the 2012/2013 influenza season in the United States.
VX-787 was discovered by Vertex scientists.
About Influenza
Often called “the flu,” seasonal influenza is caused by influenza viruses, which infect the respiratory tract.1 The flu can result in seasonal epidemics2 and can produce severe disease and high mortality in certain populations, such as the elderly.3 Each year, on average 5 to 20 percent of the U.S. population gets the flu4 resulting in more than 200,000 flu-related hospitalizations and 36,000 deaths.5 The overall national economic burden of influenza-attributable illness for adults is $83.3 billion.5 Direct medical costs for influenza in adults totaled $8.7 billion including $4.5 billion for adult hospitalizations resulting from influenza-attributable illness.5 The treatment of the flu consists of antiviral medications that have been shown in clinical studies to shorten the disease and reduce the severity of symptoms if taken within two days of infection.6 There is a significant need for new medicines targeting flu that provide a wider treatment window, greater efficacy and faster onset of action.
About Vertex
Vertex is a global biotechnology company that aims to discover, develop and commercialize innovative medicines so people with serious diseases can lead better lives. In addition to our clinical development programs focused on cystic fibrosis, Vertex has more than a dozen ongoing research programs aimed at other serious and life-threatening diseases.
Founded in 1989 in Cambridge, Mass., Vertex today has research and development sites and commercial offices in the United States, Europe, Canada and Australia. For four years in a row, Science magazine has named Vertex one of its Top Employers in the life sciences. For additional information and the latest updates from the company, please visit www.vrtx.com.
Vertex’s press releases are available at www.vrtx.com.
| WO2002024705A1 |
13 Sep 2001 |
28 Mar 2002 |
Charles Jackson Barnett |
Stereoselective process for preparing cyclohexyl amine derivatives |
| WO2003015798A1 |
13 Aug 2002 |
27 Feb 2003 |
Toyama Chemical Co Ltd |
Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 |
30 Mar 2005 |
13 Oct 2005 |
Vertex Pharma |
Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2006069258A1 * |
20 Dec 2005 |
29 Jun 2006 |
Amgen Inc |
Substituted heterocyclic compounds and methods of use |
| WO2007084557A2 |
17 Jan 2007 |
26 Jul 2007 |
Vertex Pharma |
Azaindoles useful as inhibitors of janus kinases |
| WO2008079346A1 |
21 Dec 2007 |
3 Jul 2008 |
Vertex Pharma |
5-cyan0-4- (pyrrolo [2, 3b] pyridine-3-yl) -pyrimidine derivatives useful as protein kinase inhibitors |
| WO2009073300A1 |
31 Oct 2008 |
11 Jun 2009 |
Vertex Pharma |
[1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 |
22 Jul 2009 |
28 Jan 2010 |
Vertex Pharmaceuticals Incorporated |
Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 |
22 Jul 2009 |
28 Jan 2010 |
Vertex Pharmaceuticals Incorporated |
Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 |
22 Jul 2009 |
28 Jan 2010 |
Vertex Pharmaceuticals Incorporated |
Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * |
17 Jun 2010 |
23 Dec 2010 |
Vertex Pharmaceuticals Incorporated |
Inhibitors of influenza viruses replication |
| WO2011008915A1 * |
15 Jul 2010 |
20 Jan 2011 |
Abbott Laboratories |
Pyrrolopyridine inhibitors of kinases |
| US20100038988 |
12 Aug 2008 |
18 Feb 2010 |
Gannon Ramy |
Stator and Method of Making the Same |
| WO2003015798A1 |
Aug 13, 2002 |
Feb 27, 2003 |
Toyama Chemical Co Ltd |
Novel virus proliferation inhibition/virucidal method and novel pyradine nucleotide/pyradine nucleoside analogue |
| WO2005095400A1 |
Mar 30, 2005 |
Oct 13, 2005 |
Vertex Pharma |
Azaindoles useful as inhibitors of jak and other protein kinases |
| WO2007084557A2 |
Jan 17, 2007 |
Jul 26, 2007 |
Vertex Pharma |
Azaindoles useful as inhibitors of janus kinases |
| WO2009073300A1 |
Oct 31, 2008 |
Jun 11, 2009 |
Vertex Pharma |
[1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2010011756A1 |
Jul 22, 2009 |
Jan 28, 2010 |
Vertex Pharmaceuticals Incorporated |
Pyrazolopyridine kinase inhibitors |
| WO2010011768A1 |
Jul 22, 2009 |
Jan 28, 2010 |
Vertex Pharmaceuticals Incorporated |
Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010011772A2 |
Jul 22, 2009 |
Jan 28, 2010 |
Vertex Pharmaceuticals Incorporated |
Tri-cyclic pyrazolopyridine kinase inhibitors |
| WO2010148197A1 * |
Jun 17, 2010 |
Dec 23, 2010 |
Vertex Pharmaceuticals Incorporated |
Inhibitors of influenza viruses replication |
| US20100038988 |
Aug 12, 2008 |
Feb 18, 2010 |
Gannon Ramy |
Stator and Method of Making the Same |
……
 .
.

Vertex Pharmaceuticals’ Boston Campus, United States of America


Lynette Hopkinson VP Commercial Regulatory Affairs, Global Regulatory Affairs Vertex Pharmaceuticals Incorporated, United States

swati Patel, a lead analyst, shared a toast with Mir Hussain, a systems engineer, at Vertex Pharmaceuticals during the Friday beer hour, which features beer and chips for employees.
On Fridays around 5 o’clock, after a hard week of work, Frank Holland likes to unwind with a beer. And he doesn’t have to leave work to get one.
Holland is a research scientist at Vertex Pharmaceuticals, which every Friday rings in “beer hour,” offering free adult beverages and munchies to its 1,300 Boston employees.
For Holland, the weekly ritual is a chance to escape the bubble of his chemistry lab and bump into colleagues from other departments — as well as Vertex’s top executives, who regularly attend. For those who prefer grapes to hops, there is also wine.
“Some of the other companies I worked at, you really had to go out of your way to meet people,” said Holland, 32. “At Vertex all you have to do is show up in the cafeteria on a Friday afternoon.”
Sure, free beer is common at hip tech offices; some even have their own bars. But Vertex, best known for its treatment for cystic fibrosis, was doing this way before it was cool. The beer-hour tradition goes back to the company’s founding days, in 1989. Back then, it was just two dozen people in a small office in Cambridge. Someone went to a corner store, bought a case of beer and some chips, and beer hour was born.

Virginia Carden Carnahan
Vice President, New Product Planning and Strategy, Vertex Pharmaceuticals

A scientist works in the lab at Boston-based Vertex Pharmaceuticals.

Vertex Pharmaceuticals Headquarters Lobby
REFERENCES
1: Boyd MJ, Bandarage UK, Bennett H, Byrn RR, Davies I, Gu W, Jacobs M, Ledeboer
MW, Ledford B, Leeman JR, Perola E, Wang T, Bennani Y, Clark MP, Charifson PS.
Isosteric replacements of the carboxylic acid of drug candidate VX-787: Effect of
charge on antiviral potency and kinase activity of azaindole-based influenza PB2
inhibitors. Bioorg Med Chem Lett. 2015 May 1;25(9):1990-4. doi:
10.1016/j.bmcl.2015.03.013. Epub 2015 Mar 14. PubMed PMID: 25827523.
2: Byrn RA, Jones SM, Bennett HB, Bral C, Clark MP, Jacobs MD, Kwong AD, Ledeboer
MW, Leeman JR, McNeil CF, Murcko MA, Nezami A, Perola E, Rijnbrand R, Saxena K,
Tsai AW, Zhou Y, Charifson PS. Preclinical activity of VX-787, a first-in-class,
orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit.
Antimicrob Agents Chemother. 2015 Mar;59(3):1569-82. doi: 10.1128/AAC.04623-14.
Epub 2014 Dec 29. PubMed PMID: 25547360; PubMed Central PMCID: PMC4325764.
3: Clark MP, Ledeboer MW, Davies I, Byrn RA, Jones SM, Perola E, Tsai A, Jacobs
M, Nti-Addae K, Bandarage UK, Boyd MJ, Bethiel RS, Court JJ, Deng H, Duffy JP,
Dorsch WA, Farmer LJ, Gao H, Gu W, Jackson K, Jacobs DH, Kennedy JM, Ledford B,
Liang J, Maltais F, Murcko M, Wang T, Wannamaker MW, Bennett HB, Leeman JR,
McNeil C, Taylor WP, Memmott C, Jiang M, Rijnbrand R, Bral C, Germann U, Nezami
A, Zhang Y, Salituro FG, Bennani YL, Charifson PS. Discovery of a novel,
first-in-class, orally bioavailable azaindole inhibitor (VX-787) of influenza
PB2. J Med Chem. 2014 Aug 14;57(15):6668-78. doi: 10.1021/jm5007275. Epub 2014
Jul 24. PubMed PMID: 25019388.
/////////PIMODIVIR , VX-787, JNJ-63623872, JNJ-872, VRT-0928787, 1629869-44-8, VX-787, JNJ-63623872, JNJ-872, VRT-0928787, VX-787, VX 787, VX787, JNJ-872, JNJ 872, JNJ872, VRT-0928787, VRT 0928787, VRT0928787, pimodivir, PHASE 2
O=C([C@H]1C(CC2)CCC2[C@@H]1NC3=NC(C4=CNC5=NC=C(F)C=C54)=NC=C3F)O
O.Cl.Cl.OC(=O)[C@H]1C2CCC(CC2)[C@@H]1Nc3nc(ncc3F)c4c[nH]c5ncc(F)cc45.OC(=O)[C@H]6C7CCC(CC7)[C@@H]6Nc8nc(ncc8F)c9c[nH]c%10ncc(F)cc9%10

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....