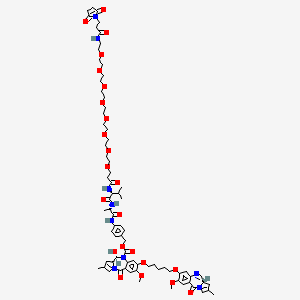
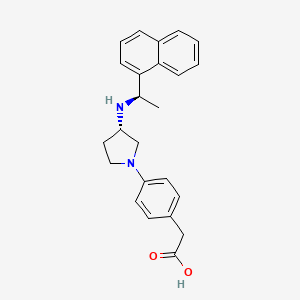
Patisiran
| Sense strand: |
 |
GUAACCAAGAGUAUUCCAUdTdT |
|
Anti-sense strand: |
|
AUGGAAUACUCUUGGUUACdTdT |
RNA, (A-U-G-G-A-A-Um-A-C-U-C-U-U-G-G-U-Um-A-C-dT-dT), complex with RNA (G-Um-A-A-Cm-Cm-A-A-G-A-G-Um-A-Um-Um-Cm-Cm-A-Um-dT-dT) (1:1),
ALN-18328, 6024128 , ALN-TTR02 , GENZ-438027 , SAR-438037 , 50FKX8CB2Y (UNII code)
Patisiran (trade name Onpattro®) is a medication for the treatment of polyneuropathy in people with hereditary transthyretin-mediated amyloidosis. It is the first small interfering RNA-based drug approved by the FDA. Through this mechanism, it is a gene silencing drug that interferes with the production of an abnormal form of transthyretin.
Chemical structure of Patisiran.
During its development, patisiran was granted orphan drug status, fast track designation, priority review and breakthrough therapy designation due to its novel mechanism and the rarity of the condition it is designed to treat.[1][2] It was approved by the FDA in August 2018 and is expected to cost around $345,000 to $450,000 per year.[3]
Patisiran was granted orphan drug designation in the U.S. and Japan for the treatment of familial amyloid polyneuropathy. Fast track designation was also granted in the U.S. for this indication. In the E.U., orphan drug designation was assigned to the compound for the treatment of transthyretin-mediated amyloidosis (initially for the treatment of familial amyloid polyneuropathy)
Hereditary transthyretin-mediated amyloidosis is a fatal rare disease that is estimated to affect 50,000 people worldwide. Patisiran is the first drug approved by the FDA to treat this condition.[4]
Patisiran is a second-generation siRNA therapy targeting mutant transthyretin (TTR) developed by Alnylam for the treatment of familial amyloid polyneuropathy. The product is delivered by means of Arbutus Biopharma’s (formerly Tekmira Pharmaceuticals) lipid nanoparticle technology
“A lot of people think it’s winter out there for RNAi. But I think it’s springtime.” — Alnylam CEO John Maraganore, NYT, February 7, 2011.
Patisiran — designed to silence messenger RNA and block the production of TTR protein before it is made — is number 6 on Clarivate’s list of blockbusters set to launch this year, with a 2022 sales forecast of $1.22 billion. Some of the peak sales estimates range significantly higher as analysts crunch the numbers on a disease that afflicts only about 30,000 people worldwide.
PATENT
WO 2016033326
https://patents.google.com/patent/WO2016033326A2
Transthyretin (TTR) is a tetrameric protein produced primarily in the liver.
Mutations in the TTR gene destabilize the protein tetramer, leading to misfolding of monomers and aggregation into TTR amyloid fibrils (ATTR). Tissue deposition results in systemic ATTR amyloidosis (Coutinho et al, Forty years of experience with type I amyloid neuropathy. Review of 483 cases. In: Glenner et al, Amyloid and Amyloidosis, Amsterdam: Excerpta Media, 1980 pg. 88-93; Hou et al., Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of
neurodegeneration. FEBS J 2007, 274: 1637-1650; Westermark et al, Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA 1990, 87: 2843-2845). Over 100 reported TTR mutations exhibit a spectrum of disease symptoms.
[0004] TTR amyloidosis manifests in various forms. When the peripheral nervous system is affected more prominently, the disease is termed familial amyloidotic
polyneuropathy (FAP). When the heart is primarily involved but the nervous system is not, the disease is called familial amyloidotic cardiomyopathy (FAC). A third major type of TTR amyloidosis is called leptomeningeal/CNS (Central Nervous System) amyloidosis.
[0005] The most common mutations associated with familial amyloid polyneuropathy
(FAP) and ATTR-associated cardiomyopathy, respectively, are Val30Met (Coelho et al, Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012, 79: 785-792) and Vall22Ile (Connors et al, Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin VI 221 amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J 2009, 158: 607-614). [0006] Current treatment options for FAP focus on stabilizing or decreasing the amount of circulating amyloidogenic protein. Orthotopic liver transplantation reduces mutant TTR levels (Holmgren et al, Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet 1991, 40: 242-246), with improved survival reported in patients with early-stage FAP, although deposition of wild-type TTR may continue (Yazaki et al, Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs into myocardium in FAP patients. Am J Transplant 2007, 7:235-242; Adams et al, Rapid progression of familial amyloid polyneuropathy: a multinational natural history study Neurology 2015 Aug 25; 85(8) 675-82; Yamashita et al, Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology 2012, 78: 637-643; Okamoto et al., Liver
transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl 2009, 15: 1229-1235; Stangou et al, Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation 1998, 66:229-233; Fosby et al, Liver transplantation in the Nordic countries – An intention to treat and post-transplant analysis from The Nordic Liver Transplant Registry 1982-2013. Scand J Gastroenterol. 2015 Jun; 50(6):797-808.
Transplantation, in press).
[0007] Tafamidis and diflunisal stabilize circulating TTR tetramers, which can slow the rate of disease progression (Berk et al, Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013, 310: 2658-2667; Coelho et al., 2012; Coelho et al, Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol 2013, 260: 2802-2814; Lozeron et al, Effect on disability and safety of Tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol 2013, 20: 1539-1545). However, symptoms continue to worsen on treatment in a large proportion of patients, highlighting the need for new, disease-modifying treatment options for FAP.
[0008] Description of dsRNA targeting TTR can be found in, for example,
International patent application no. PCT/US2009/061381 (WO2010/048228) and
International patent application no. PCT/US2010/05531 1 (WO201 1/056883). Summary
[0009] Described herein are methods for reducing or arresting an increase in a
Neuropathy Impairment Score (NIS) or a modified NIS (mNIS+7) in a human subject by administering an effective amount of a transthyretin (TTR)-inhibiting composition, wherein the effective amount reduces a concentration of TTR protein in serum of the human subject to below 50 μg/ml or by at least 80%. Also described herein are methods for adjusting a dosage of a TTR- inhibiting composition for treatment of increasing NIS or Familial Amyloidotic Polyneuropathy (FAP) by administering the TTR- inhibiting composition to a subject having the increasing NIS or FAP, and determining a level of TTR protein in the subject having the increasing NIS or FAP. In some embodiments, the amount of the TTR- inhibiting composition subsequently administered to the subject is increased if the level of TTR protein is greater than 50 μg/ml, and the amount of the TTR- inhibiting composition subsequently administered to the subject is decreased if the level of TTR protein is below 50 μg/ml. Also described herein are formulated versions of a TTR inhibiting siRNA.

PATENT
WO 2016203402
PAPERS
Annals of Medicine (Abingdon, United Kingdom) (2015), 47(8), 625-638.
Pharmaceutical Research (2017), 34(7), 1339-1363
Annual Review of Pharmacology and Toxicology (2017), 57, 81-105
CLIP

Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO™ (patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults
− First and Only FDA-approved Treatment Available in the United States for this Indication –
− ONPATTRO Shown to Improve Polyneuropathy Relative to Placebo, with Reversal of Neuropathy Impairment Compared to Baseline in Majority of Patients –
− Improvement in Specified Measures of Quality of Life and Disease Burden Demonstrated Across Diverse, Global Patient Population –
− Alnylam to Host Conference Call Today at 3:00 p.m. ET. −
CAMBRIDGE, Mass.–(BUSINESS WIRE)–Aug. 10, 2018– Alnylam Pharmaceuticals, Inc. (Nasdaq: ALNY), the leading RNAi therapeutics company, announced today that the United States Food and Drug Administration (FDA) approved ONPATTRO™ (patisiran) lipid complex injection, a first-of-its-kind RNA interference (RNAi) therapeutic, for the treatment of the polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. ONPATTRO is the first and onlyFDA-approved treatment for this indication. hATTR amyloidosis is a rare, inherited, rapidly progressive and life-threatening disease with a constellation of manifestations. In addition to polyneuropathy, hATTR amyloidosis can lead to other significant disabilities including decreased ambulation with the loss of the ability to walk unaided, a reduced quality of life, and a decline in cardiac functioning. In the largest controlled study of hATTR amyloidosis, ONPATTRO was shown to improve polyneuropathy – with reversal of neuropathy impairment in a majority of patients – and to improve a composite quality of life measure, reduce autonomic symptoms, and improve activities of daily living.

This press release features multimedia. View the full release here:https://www.businesswire.com/news/home/20180810005398/en/
 ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)
ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)
“Alnylam was founded on the vision of harnessing the potential of RNAi therapeutics to treat human disease, and this approval heralds the arrival of an entirely new class of medicines. We believe today draws us ever-closer to achieving our Alnylam 2020 goals of becoming a fully integrated, multi-product biopharmaceutical company with a sustainable pipeline,” said John Maraganore, Ph.D., Chief Executive Officer of Alnylam. “With the potential for the sequential launches of several new medicines in the coming years, we believe we have the opportunity to meaningfully impact the lives of people around the world in need of new approaches to address serious diseases with significant unmet medical needs.”
“Today’s historic approval marks the arrival of a first-of-its kind treatment option for a rare and devastating condition with limited treatment options,” said Akshay Vaishnaw, M.D., Ph.D., President of R&D at Alnylam. “We extend our deepest gratitude to the patients who participated in the ONPATTRO clinical trials and their families and caregivers who supported them. We are also grateful for the tireless efforts of the investigators and study staff, without whom this important milestone would not have been possible. We also look forward to working with the FDA to potentially expand the ONPATTRO indication in the future.”
The FDA approval of ONPATTRO was based on positive results from the randomized, double-blind, placebo-controlled, global Phase 3 APOLLO study, the largest-ever study in hATTR amyloidosis patients with polyneuropathy. Results from the APOLLO study were published in the July 5, 2018, issue of The New England Journal of Medicine.
In APOLLO, the safety and efficacy of ONPATTRO were evaluated in a diverse, global population of hATTR amyloidosis patients in 19 countries, with a total of 39 TTR mutations. Patients were randomized in a 2:1 ratio to receive intravenous ONPATTRO (0.3 mg per kg of body weight) or placebo once every 3 weeks for 18 months. The study showed that ONPATTRO improved measures of polyneuropathy, quality of life, activities of daily living, ambulation, nutritional status and autonomic symptoms relative to placebo in adult patients with hATTR amyloidosis with polyneuropathy. The primary endpoint of the APOLLO study was the modified Neuropathy Impairment Score +7 (mNIS+7), which assesses motor strength, reflexes, sensation, nerve conduction and postural blood pressure.
- Patients treated with ONPATTRO had a mean 6.0-point decrease (improvement) in mNIS+7 score from baseline compared to a mean 28.0-point increase (worsening) for patients in the placebo group, resulting in a mean 34.0-point difference relative to placebo, after 18 months of treatment.
- While nearly all ONPATTRO-treated patients experienced a treatment benefit relative to placebo, 56 percent of ONPATTRO-treated patients at 18 months of treatment experienced reversal of neuropathy impairment (as assessed by mNIS+7 score) relative to their own baseline, compared to four percent of patients who received placebo.
- Patients treated with ONPATTRO had a mean 6.7-point decrease (improvement) in Norfolk Quality of Life Diabetic Neuropathy (QoL-DN) score from baseline compared to a mean 14.4-point increase (worsening) for patients in the placebo group, resulting in a mean 21.1-point difference relative to placebo, after 18 months of treatment.
- As measured by Norfolk QoL-DN, 51 percent of patients treated with ONPATTRO experienced improvement in quality of life at 18 months relative to their own baseline, compared to 10 percent of the placebo-treated patients.
- Over 18 months of treatment, patients treated with ONPATTRO experienced significant benefit vs. placebo for all other secondary efficacy endpoints, including measures of activities of daily living, walking ability, nutritional status, and autonomic symptoms.
- The most common adverse events that occurred more frequently with ONPATTRO than with placebo were upper respiratory tract infections and infusion-related reactions. To reduce the risk of infusion-related reactions, patients received premedications prior to infusion.
“FDA approval of ONPATTRO represents an entirely new approach to treating patients with polyneuropathy in hATTR amyloidosis and shows promise as a new era in patient care,” said John Berk, M.D., Associate Professor of Medicine at Boston University School of Medicine and assistant director of the Amyloidosis Center at Boston University School of Medicine. “Given the strength of the APOLLO data, including data showing the possibility of halting or improving disease progression in many patients, ONPATTRO holds tremendous promise for people living with this disease.”
“For years I have witnessed the tragic impact of hATTR amyloidosis on generations of families. Today, we celebrate the FDA approval of ONPATTRO,” said Muriel Finkel, President of Amyloidosis Support Groups. “It’s extremely gratifying to see promising science translate into a treatment option that will allow patients to potentially experience an improvement in their disease and an improvement in their overall quality of life.”
“Today’s approval is significant in so many respects. It means the hATTR amyloidosis community of patients, families, caregivers and healthcare professionals in the United States now has a treatment option that offers renewed hope,” said Isabelle Lousada, Founder and Chief Executive Officer of the Amyloidosis Research Consortium. “With an FDA-approved treatment now available, I am more optimistic than ever that we can increase awareness of this rare disease and encourage more people to get tested and receive the proper diagnosis.”
ONPATTRO is expected to be available for shipment to healthcare providers in the U.S. within 48 hours.
Alnylam is committed to helping people access the medicines they are prescribed and will be offering comprehensive support services for people prescribed ONPATTRO through Alnylam Assist™. Visit AlnylamAssist.com for more information or call 1-833-256-2748.
ONPATTRO was reviewed by the FDA under Priority Review and had previously been granted Breakthrough Therapy and Orphan Drug Designations. On July 27, patisiran received a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) for the treatment of hereditary transthyretin-mediated amyloidosis in adults with stage 1 or stage 2 polyneuropathy under accelerated assessment by the European Medicines Agency. The recommended Summary of Product Characteristics (SmPC) for the European Union (EU) includes data on secondary and exploratory endpoints. Expected in September, the European Commission will review the CHMP recommendation to make a final decision on marketing authorization, applicable to all 28 EU member states, plus Iceland, Liechtenstein and Norway. Regulatory filings in other markets, including Japan, are planned beginning in mid-2018.
Visit ONPATTRO.com for more information,
About ONPATTRO™ (patisiran) lipid complex injection
ONPATTRO was approved by the U.S. Food and Drug Administration (FDA) for the treatment of the polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults. ONPATTRO is the first and only RNA interference (RNAi) therapeutic approved by the FDA for this indication. ONPATTRO utilizes a novel approach to target and reduce production of the TTR protein in the liver via the RNAi pathway. Reducing the TTR protein leads to a reduction in the amyloid deposits that accumulate in tissues. ONPATTRO is administered through intravenous (IV) infusion once every 3 weeks following required premedication and the dose is based on actual body weight. Home infusion may be an option for some patients after an evaluation and recommendation by the treating physician, and may not be covered by all insurance plans. Regardless of the setting, ONPATTRO infusions should be performed by a healthcare professional. For more information about ONPATTRO, visit ONPATTRO.com.
About hATTR Amyloidosis
Hereditary transthyretin (TTR)-mediated amyloidosis (hATTR) is an inherited, progressively debilitating, and often fatal disease caused by mutations in the TTR gene. TTR protein is primarily produced in the liver and is normally a carrier of vitamin A. Mutations in the TTR gene cause abnormal amyloid proteins to accumulate and damage body organs and tissue, such as the peripheral nerves and heart, resulting in intractable peripheral sensory neuropathy, autonomic neuropathy, and/or cardiomyopathy, as well as other disease manifestations. hATTR amyloidosis represents a major unmet medical need with significant morbidity and mortality. The median survival is 4.7 years following diagnosis. Until now, people living with hATTR amyloidosis in the U.S. had no FDA-approved treatment options.
Alnylam Assist™
As part of Alnylam’s commitment to making therapies available to those who may benefit from them, Alnylam Assist will offer a wide range of services to guide patients through treatment with ONPATTRO, including financial assistance options for eligible patients, benefit verification and claims support, and ordering assistance and facilitation of delivery via specialty distributor or specialty pharmacy. Patients will have access to dedicated Case Managers who can provide personalized support throughout the treatment process and Patient Education Liaisons to help patients gain a better understanding of the disease. Visit AlnylamAssist.com for more information.
About RNAi
RNAi (RNA interference) is a natural cellular process of gene silencing that represents one of the most promising and rapidly advancing frontiers in biology and drug development today. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and was recognized with the award of the 2006 Nobel Prize for Physiology or Medicine. RNAi therapeutics are a new class of medicines that harness the natural biological process of RNAi. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, function upstream of today’s medicines by potently silencing messenger RNA (mRNA) – the genetic precursors – that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach in developing medicines to improve the care of patients with genetic and other diseases.
About Alnylam
Alnylam (Nasdaq: ALNY) is leading the translation of RNA interference (RNAi) into a whole new class of innovative medicines with the potential to improve the lives of people afflicted with rare genetic, cardio-metabolic, and hepatic infectious diseases. Based on Nobel Prize-winning science, RNAi therapeutics represent a powerful, clinically validated approach for the treatment of a wide range of severe and debilitating diseases. Founded in 2002, Alnylam is delivering on a bold vision to turn scientific possibility into reality, with a robust discovery platform. ONPATTRO, available in the U.S. for the treatment of the polyneuropathy of hereditary transthyretin-mediated (hATTR) amyloidosis in adults, is Alnylam’s first U.S. FDA-approved RNAi therapeutic. Alnylam has a deep pipeline of investigational medicines, including three product candidates that are in late-stage development. Looking forward, Alnylam will continue to execute on its “Alnylam 2020” strategy of building a multi-product, commercial-stage biopharmaceutical company with a sustainable pipeline of RNAi-based medicines to address the needs of patients who have limited or inadequate treatment options. Alnylam employs over 800 people worldwide and is headquartered in Cambridge, MA. For more information about our people, science and pipeline, please visit www.alnylam.com and engage with us on Twitter at @Alnylam or on LinkedIn.

FDA approves first-of-its kind targeted RNA-based therapy to treat a rare disease
First treatment for the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adult patients
The U.S. Food and Drug Administration today approved Onpattro (patisiran) infusion for the treatment of peripheral nerve disease (polyneuropathy) caused by hereditary transthyretin-mediated amyloidosis (hATTR) in adult patients. This is the first FDA-approved treatment for patients with polyneuropathy caused by hATTR, a rare, debilitating and often fatal genetic disease characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart and other organs. It is also the first FDA approval of a new class of drugs called small interfering ribonucleic acid (siRNA) treatment
Continue reading…
https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/UCM616518.htm?utm_campaign=08102018_PR_FDA%20approves%20new%20drug%20for%20rare%20disease%2C%20hATTR&utm_medium=email&utm_source=Eloqua
The U.S. Food and Drug Administration today approved Onpattro (patisiran) infusion for the treatment of peripheral nerve disease (polyneuropathy) caused by hereditary transthyretin-mediated amyloidosis (hATTR) in adult patients. This is the first FDA-approved treatment for patients with polyneuropathy caused by hATTR, a rare, debilitating and often fatal genetic disease characterized by the buildup of abnormal amyloid protein in peripheral nerves, the heart and other organs. It is also the first FDA approval of a new class of drugs called small interfering ribonucleic acid (siRNA) treatment.
“This approval is part of a broader wave of advances that allow us to treat disease by actually targeting the root cause, enabling us to arrest or reverse a condition, rather than only being able to slow its progression or treat its symptoms. In this case, the effects of the disease cause a degeneration of the nerves, which can manifest in pain, weakness and loss of mobility,” said FDA Commissioner Scott Gottlieb, M.D. “New technologies like RNA inhibitors, that alter the genetic drivers of a disease, have the potential to transform medicine, so we can better confront and even cure debilitating illnesses. We’re committed to advancing scientific principles that enable the efficient development and review of safe, effective and groundbreaking treatments that have the potential to change patients’ lives.”
RNA acts as a messenger within the body’s cells, carrying instructions from DNA for controlling the synthesis of proteins. RNA interference is a process that occurs naturally within our cells to block how certain genes are expressed. Since its discovery in 1998, scientists have used RNA interference as a tool to investigate gene function and its involvement in health and disease. Researchers at the National Institutes of Health, for example, have used robotic technologies to introduce siRNAs into human cells to individually turn off nearly 22,000 genes.
This new class of drugs, called siRNAs, work by silencing a portion of RNA involved in causing the disease. More specifically, Onpattro encases the siRNA into a lipid nanoparticle to deliver the drug directly into the liver, in an infusion treatment, to alter or halt the production of disease-causing proteins.
Affecting about 50,000 people worldwide, hATTR is a rare condition. It is characterized by the buildup of abnormal deposits of protein fibers called amyloid in the body’s organs and tissues, interfering with their normal functioning. These protein deposits most frequently occur in the peripheral nervous system, which can result in a loss of sensation, pain, or immobility in the arms, legs, hands and feet. Amyloid deposits can also affect the functioning of the heart, kidneys, eyes and gastrointestinal tract. Treatment options have generally focused on symptom management.
Onpattro is designed to interfere with RNA production of an abnormal form of the protein transthyretin (TTR). By preventing the production of TTR, the drug can help reduce the accumulation of amyloid deposits in peripheral nerves, improving symptoms and helping patients better manage the condition.
“There has been a long-standing need for a treatment for hereditary transthyretin-mediated amyloidosis polyneuropathy. This unique targeted therapy offers these patients an innovative treatment for their symptoms that directly affects the underlying basis of this disease,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Onpattro was shown in a clinical trial involving 225 patients, 148 of whom were randomly assigned to receive an Onpattro infusion once every three weeks for 18 months, and 77 of whom were randomly assigned to receive a placebo infusion at the same frequency. The patients who received Onpattro had better outcomes on measures of polyneuropathy including muscle strength, sensation (pain, temperature, numbness), reflexes and autonomic symptoms (blood pressure, heart rate, digestion) compared to those receiving the placebo infusions. Onpattro-treated patients also scored better on assessments of walking, nutritional status and the ability to perform activities of daily living.
The most common adverse reactions reported by patients treated with Onpattro are infusion-related reactions including flushing, back pain, nausea, abdominal pain, dyspnea (difficulty breathing) and headache. All patients who participated in the clinical trials received premedication with a corticosteroid, acetaminophen, and antihistamines (H1 and H2 blockers) to reduce the occurrence of infusion-related reactions. Patients may also experience vision problems including dry eyes, blurred vision and eye floaters (vitreous floaters). Onpattro leads to a decrease in serum vitamin A levels, so patients should take a daily Vitamin A supplement at the recommended daily allowance.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Onpattro also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
Approval of Onpattro was granted to Alnylam Pharmaceuticals, Inc.
References
////////////// Onpattro, patisiran, fda 2018, Fast Track, Priority Review, Breakthrough Therapy, Orphan Drug designation, Alnylam Pharmaceuticals, ALN-18328, 6024128 , ALN-TTR02 , GENZ-438027 , SAR-438037 , 50FKX8CB2Y
CC1=CN(C2OC(COP(=O)(O)OC3C(O)C(OC3COP(=O)(O)OC4C(O)C(OC4COP(=O)(O)OC5C(O)C(OC5COP(=O)(O)OC6C(O)C(OC6COP(=O)(O)OC7C(O)C(OC7COP(=O)(O)OC8C(O)C(OC8COP(=O)(O)OC9C(O)C(OC9COP(=O)(O)OC%10C(O)C(OC%10COP(=O)(O)OC%11C(O)C(OC%11COP(=O)(O)OC%12C(O)C(OC%12COP(=O)(O)OC%13C(O)C(OC%13COP(=O)(O)OC%14C(O)C(OC%14COP(=O)(O)OC%15C(O)C(OC%15COP(=O)(O)OC%16C(O)C(OC%16COP(=O)(O)OC%17C(O)C(OC%17CO)n%18cnc%19C(=O)NC(=Nc%18%19)N)N%20C=C(C)C(=O)NC%20=O)n%21cnc%22c(N)ncnc%21%22)n%23cnc%24c(N)ncnc%23%24)N%25C=C(C)C(=NC%25=O)N)N%26C=C(C)C(=NC%26=O)N)n%27cnc%28c(N)ncnc%27%28)n%29cnc%30c(N)ncnc%29%30)n%31cnc%32C(=O)NC(=Nc%31%32)N)n%33cnc%34c(N)ncnc%33%34)n%35cnc%36C(=O)NC(=Nc%35%36)N)N%37C=C(C)C(=O)NC%37=O)n%38cnc%39c(N)ncnc%38%39)N%40C=C(C)C(=O)NC%40=O)N%41C=C(C)C(=O)NC%41=O)C(OP(=O)(O)OCC%42OC(C(O)C%42OP(=O)(O)OCC%43OC(C(O)C%43OP(=O)(O)OCC%44OC(C(O)C%44OP(=O)(O)OCC%45OC(CC%45OP(=O)(O)OCC%46OC(CC%46O)N%47C=C(C)C(=O)NC%47=O)N%48C=C(C)C(=O)NC%48=O)N%49C=C(C)C(=O)NC%49=O)n%50cnc%51c(N)ncnc%50%51)N%52C=C(C)C(=NC%52=O)N)C2O)C(=O)N=C1N.CC%53=CN(C%54CC(O)C(COP(=O)(O)OC%55CC(OC%55COP(=O)(O)OC%56C(O)C(OC%56COP(=O)(O)OC%57C(O)C(OC%57COP(=O)(O)OC%58C(O)C(OC%58COP(=O)(O)OC%59C(O)C(OC%59COP(=O)(O)OC%60C(O)C(OC%60COP(=O)(O)OC%61C(O)C(OC%61COP(=O)(O)OC%62C(O)C(OC%62COP(=O)(O)OC%63C(O)C(OC%63COP(=O)(O)OC%64C(O)C(OC%64COP(=O)(O)OC%65C(O)C(OC%65COP(=O)(O)OC%66C(O)C(OC%66COP(=O)(O)OC%67C(O)C(OC%67COP(=O)(O)OC%68C(O)C(OC%68COP(=O)(O)OC%69C(O)C(OC%69COP(=O)(O)OC%70C(O)C(OC%70COP(=O)(O)OC%71C(O)C(OC%71COP(=O)(O)OC%72C(O)C(OC%72COP(=O)(O)OC%73C(O)C(OC%73COP(=O)(O)OC%74C(O)C(OC%74CO)n%75cnc%76c(N)ncnc%75%76)N%77C=CC(=O)NC%77=O)n%78cnc%79C(=O)NC(=Nc%78%79)N)n%80cnc%81C(=O)NC(=Nc%80%81)N)n%82cnc%83c(N)ncnc%82%83)n%84cnc%85c(N)ncnc%84%85)N%86C=C(C)C(=O)NC%86=O)n%87cnc%88c(N)ncnc%87%88)N%89C=CC(=NC%89=O)N)N%90C=CC(=O)NC%90=O)N%91C=CC(=NC%91=O)N)N%92C=CC(=O)NC%92=O)N%93C=CC(=O)NC%93=O)n%94cnc%95C(=O)NC(=Nc%94%95)N)n%96cnc%97C(=O)NC(=Nc%96%97)N)N%98C=CC(=O)NC%98=O)N%99C=C(C)C(=O)NC%99=O)n1cnc2c(N)ncnc12)N3C=CC(=NC3=O)N)N4C=C(C)C(=O)NC4=O)O%54)C(=O)NC%53=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....