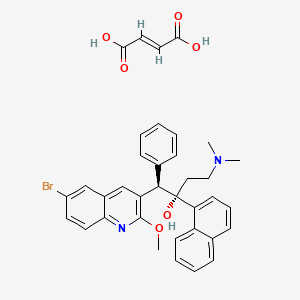
Bedaquiline fumarate; Bedaquiline (fumarate); Cas 845533-86-0; UNII-P04QX2C1A5; P04QX2C1A5;
Bedaquiline fumarate
(1R,2S)-1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-2-naphthalen-1-yl-1-phenylbutan-2-ol;(E)-but-2-enedioic acid
| Molecular Formula: |
C36H35BrN2O6 |
| Molecular Weight: |
671.588 g/mol |
| Product |
|
| Formula |
C32H31BrN2O2. C4H4O4
|
| CAS
|
845533-86-0 FUMARATE
FREE FORM 843663-66-1
|
| Mol weight |
671.5769
|
| 2018/1/19 |
PMDA
JAPAN
APPROVED |
|
|
Bedaquiline fumarate |
Sirturo |
Janssen Pharmaceutical |
|
ベダキリンフマル酸塩
Bedaquiline Fumarate

C32H31BrN2O2▪C4H4O4 : 671.58
[845533-86-0]
FREE FORM
- Saga, Yutaka; Journal of the American Chemical Society 2010, Vol132(23), Pg 7905-7907 , -168.0 ° Conc: 0.8 g/100mL; Solv:DMF; Wavlenght: 589.3 nm; Temp: 25 °C
- Chandrasekhar, Srivari; European Journal of Organic Chemistry 2011, (11), PG 2057-2061, S2057/1-S2057/18 -165.2 ° Conc: 0.8 g/100mL; Solv: DMF ; Wavlenght: 589.3 nm; Temp: 25 °, MP 104 °C
EMA
http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002614/WC500163215.pdf
The applicant Janssen-Cilag International N.V. submitted on 28 August 2012 an application for Marketing Authorisation to the European Medicines Agency (EMA) for SIRTURO, through the centralised procedure falling within the Article 3(1) and point 4 of Annex of Regulation (EC) No 726/2004. The eligibility to the centralised procedure was agreed upon by the EMA/CHMP on 21 July 2011. SIRTURO was designated as an orphan medicinal product EU/3/05/314 on 26 August 2005. SIRTURO was designated as an orphan medicinal product in the following indication: treatment of tuberculosis. The applicant applied for the following indication: SIRTURO is indicated in adults (≥ 18 years) as part of combination therapy of pulmonary tuberculosis due to multi-drug resistant Mycobacterium tuberculosis.
Disease to be treated About a third of the global population, more than 2 billion people, is infected with M. tuberculosis, of which the majority is latent. The life time risk to fall ill in overt TB is around 10% in general, but many times higher (around 10% annual risk) in untreated HIV-positive individuals. Tuberculosis is the leading cause of death in the latter population. It was estimated that a total of 8.8 million new TB cases occurred in 2010, including 1.1 million people co infected with HIV, and that about 1.45 million people died due to TB. During more recent years the burden of TB resistant to first line therapy has increased rapidly. Such multidrug resistant tuberculosis (defined later in this assessment report) has been reported in all regions of the world. Presently around 500.000 of new MDR cases are estimated to emerge every year, which is close to 5% of all new TB cases. China and India carried nearly 50% of the total burden of incident MDR-TB cases in 2008, followed by the Russian Federation (9%). The incidence of MDR-TB in US and EU was reported to be 1.1% and 2.4%, respectively. Within the EU, the incidence is much higher in certain Eastern European countries, with the largest burden in Romania, Latvia and Lithuania. MDR TB is an orphan disease in the EU, US and in Japan.
Current TB therapy and definitions Treatment of pulmonary drug susceptible TB typically takes 6 months resulting in cure rates in well over 90% of cases with good treatment adherence. The two most important drugs in this treatment are isoniazid (INH) and rifampicin (RIF). TB with resistance to at least both INH and RIF is called multidrug resistant (MDR) TB. The two most important “classes” of second-line TB drugs to be used in such cases are injectable drugs (the aminoglycosides amikacin and kanamycin, and the related agent capreomycin) and fluoroquinolones. Apart from these agents a number of miscellaneous drugs are used in addition, as part of combination therapy. The effectiveness of these latter miscellaneous drugs is generally lower, the tolerability is problematic and established breakpoints for resistance determination are lacking.
The term pre-XDR (pre-extensively drug resistant) TB is used when resistance is present also to one of the two main second-line class agents (injectables or any of the fluoroquinolones), and XDR-TB when resistance is present to INH+RIF + injectables + fluoroquinolones. The WHO standard treatment for MDR-TB is commonly divided into 2 phases: • a 4 to 6-month intensive treatment phase in which an injectable drug plus 3-4 other drugs, including a fluoroquinolone, • a continuation phase without the injectable drug and often without pyrazinamide (PZA) for a total duration of 18-24 months. Using this approach, cure rates in MDR-TB are much lower than those seen in DS-TB (ranging from less than 50% to around 75%), despite the higher number of agents and longer treatment duration. Hence, MDR TB is associated with a high mortality and is considered an important major threat to public health. More recent approaches to evaluate various MDR TB regimens have yielded somewhat more optimistic outcomes, despite shorter treatment durations. In these non-randomised studies (with low number of patients) cure rates in the range of 90% were achieved by including a fourth generation fluoroquinolone and by increasing the number of agents even further, to include up to 7 agents in the intensive phase, and still 4-5 agents in a second phase.
About the product SIRTURO (bedaquiline, formerly known as TMC 207) is a new agent of a unique class, specific for mycobacteria, and seemingly without cross-resistance to available TB agents. A large number of pre-clinical studies showed promising results for bedaquiline. For example, in animal models bedaquiline + pyrazinamide cured TB at a higher rate than the traditional first line combination, even when therapy was shortened for the former combination. The clinical program for bedaquiline has been aimed at treating MDR-TB, and data is now available from phase 2b studies of moderate size, both placebo-controlled and non-controlled studies. The treatments given in these studies were similar to those recommended by the WHO, although the number of agents used was slightly higher (five agents in the preferred background regimens). Bedaquiline (versus placebo in the controlled study) was added during the first (intensive) treatment phase, while the background regimens were generally unchanged throughout the complete course of therapy (18-24 months). On the basis of these studies, the applicant submitted an application for a conditional approval for bedaquiline, with the proposed indication: treatment of adult patients infected with pulmonary tuberculosis due to MDR M. tuberculosis, as part of combination therapy. In line with the approach in the phase 2 studies, Sirturo is only to be used during the first 6 months of therapy. However the planned pivotal study (as a specific obligation) will test for 40 weeks of bedaquiline treatment.
In 2009, the drug candidate was licensed to Global Alliance TB Drug Development by Tibotec worldwide for the treatment of tuberculosis.
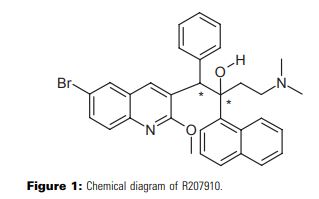
Bedaquiline (INN) is chemically designated as (1R,2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4- (dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol with fumaric acid (1:1), and has the following structure:

Bedaquiline fumarate is a white to almost white powder. It contains two asymmetric carbon atoms, C-1 (R), C-2 (S) and exhibits ability to rotate the orientation of linearly polarized light (optical rotation). The substance is non-hygroscopic. It is practically insoluble in aqueous media over a wide pH range and very slightly soluble in 0.01 N HCl. The substance is soluble in a variety of organic solvents. Due to the low solubility Log KD (log P) could not be determined experimentally. In Biopharmaceutics Classification System (BCS) bedaquiline is classified as a Class 2 compound (expressing low solubility and high permeability). Bedaquiline exists in only one non-solvated crystalline form: Form A. In addition 2 pseudopoly-morphs were found: Form B and Form C. The substance can also be made amorphous. Sufficient evidence was provided to demonstrate that Form A is obtained by the employed manufacturing process of the active substance. Particle size was considered a critical quality attribute of the active substance as bedaquiline is not dissolved in the dosage form. Therefore an appropriate test on particle size determination was included in the active substance specification. The acceptance criteria are based upon the capabilities of the milling process, batch and stability data, and the known impact of the particle size on manufacturability, in-vitro release, and in-vivo performance
Bedaquiline is a bactericidal antimycobacterial drug. Chemically it is a diarylquinoline. FDA approved on December 28, 2012.

Bedaquiline is indicated as part of combination therapy in adults (≥ 18 years) with pulmonary multi-drug resistant tuberculosis (MDR-TB).
Bedaquiline, sold under the brand name Sirturo, is a medication used to treat active tuberculosis.[1] It is specifically used to treat multi-drug-resistant tuberculosis(MDR-TB) when other treatment cannot be used.[1][5] It should be used along with at least three other medications for tuberculosis.[1][5] It is used by mouth.[5]
Common side effects include nausea, joint pains, headaches, and chest pain.[1] Serious side effects include QT prolongation, liver dysfunction, and an increased risk of death.[1] While harm during pregnancy has not been found, it has not been well studied in this population.[6] It is in the diarylquinoline antimycobacterialclass of medications.[1] It works by blocking the ability of M. tuberculosis to make adenosine 5′-triphosphate (ATP).[1]
Bedaquiline was approved for medical use in the United States in 2012.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[7] The cost for six months is approximately $900 USD in low income countries, $3,000 USD in middle income countries, and $30,000 USD in high income countries.[5]
SIRTURO (bedaquiline) for oral administration is available as 100 mg strength tablets. Each tablet contains 120.89 mg of bedaquiline fumarate drug substance, which is equivalent to 100 mg of bedaquiline. Bedaquiline is a diarylquinoline antimycobacterial drug.
Bedaquiline fumarate is a white to almost white powder and is practically insoluble in aqueous media. The chemical name of bedaquiline fumarate is (1R, 2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4- (dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol compound with fumaric acid (1:1). It has a molecular formula of C32H31BrN2O2 · C4H4O4 and a molecular weight of 671.58 (555.50 + 116.07). The molecular structure of bedaquiline fumarate is the following:
SIRTURO (bedaquiline) contains the following inactive ingredients: colloidal silicon dioxide, corn starch, croscarmellose sodium, hypromellose 2910 15 mPa.s, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 20, purified water (removed during processing).
Medical uses
Its use was formally approved (Dec 2012) by the U.S. Food and Drug Administration (FDA) for use in tuberculosis (TB) treatment, as part of a Fast-Trackaccelerated approval, for use only in cases of multidrug-resistant tuberculosis, and the more resistant extensively drug resistant tuberculosis.[8]
As of 2013 Both the World Health Organization (WHO) and US Centers for Disease Control (CDC) have recommended (provisionally) that bedaquiline be reserved for patients with multidrug-resistant tuberculosis when an otherwise recommended regimen cannot be designed.[9][10]
Clinical trials
Bedaquiline has been studied in phase IIb studies for the treatment of multidrug-resistant tuberculosis while phase III studies are currently underway.[11] It has been shown to improve cure rates of smear-positive multidrug-resistant tuberculosis, though with some concern for increased rates of death (further detailed in the Adverse effects section).[12]
Small studies have also examined its use as salvage therapy for non-tuberculous mycobacterial infections.[11]
It is a component of the experimental BPaMZ combination treatment (bedaquiline + pretomanid + moxifloxacin + pyrazinamide).[13][14]
Side effects
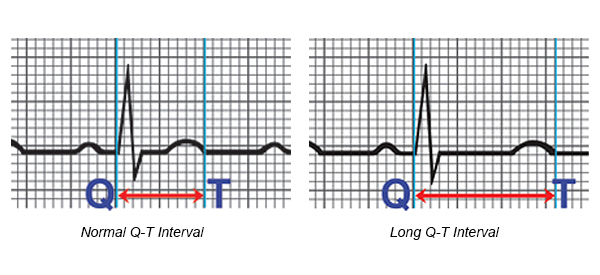
The most common side effects of bedaquiline in studies were nausea, joint and chest pain, and headache. The drug also has a black-box warning for increased risk of death and arrhythmias, as it may prolong the QT interval by blocking the hERG channel.[15] All patients on bedaquiline should have monitoring with a baseline and repeated ECGs.[16] If a patient has a QTcF of > 500ms or a significant ventricular arrythmia, bedaquiline and other QT prolonging drugs should be stopped.
There is considerable controversy over the approval for the drug, as one of the largest studies to date had more deaths in the group receiving bedaquiline that those receiving placebo.[17] 10 deaths occurred in the bedaquiline group out of 79, while 2 occurred in the placebo group, out of 81.[12] Of the 10 deaths on bedaquiline, 1 was due to a motor vehicle accident, 5 were judged as due to progression of the underlying tuberculosis and 3 were well after the patient had stopped receiving bedaquiline.[17] However, there is still significant concern for the higher mortality in patients treated with bedaquiline, leading to the recommendation to limit its use to situations where a 4 drug regimen cannot otherwise be constructed, limit use with other medications that prolong the QT interval and the placement of a prominent black box warning.[17][11]
Drug interactions
Bedaquiline should not be co-administered with other drugs that are strong inducers or inhibitors of CYP3A4, the hepatic enzyme responsible for oxidative metabolism of the drug.[16] Co-administration with rifampin, a strong CYP3A4 inducer, results in a 52% decrease in the AUC of the drug. This reduces the exposure of the body to the drug and decreases the antibacterial effect. Co-administration with ketoconazole, a strong CYP3A4 inhibitor, results in a 22% increase in the AUC, and potentially an increase in the rate of adverse effects experienced[16]
Since bedaquiline can also prolong the QT interval, use of other QT prolonging drugs should be avoided.[9] Other medications for tuberculosis that can prolong the QT interval include fluoroquinolones and clofazimine.
Mode of action
Bedaquiline blocks the proton pump for ATP synthase of mycobacteria. ATP production is required for cellular energy production and its loss leads to cell death, even in dormant or nonreplicating mycobacteria.[18] It is the first member of a new class of drugs called the diarylquinolines.[18] Bedaquiline is bactericidal.[18]
Resistance
The specific part of ATP synthase affected by bedaquiline is subunit c which is encoded by the gene atpE. Mutations in atpE can lead to resistance. Mutations in drug efflux pumps have also been linked to resistance.[19]
History
Bedaquiline was described for the first time in 2004 at the Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) meeting, after the drug had been in development for over seven years.[20] It was discovered by a team led by Koen Andries at Janssen Pharmaceutica.[21]
Bedaquiline was approved for medical use in the United States in 2012.[1]
It is manufactured by Johnson & Johnson (J&J), who sought accelerated approval of the drug, a type of temporary approval for diseases lacking other viable treatment options.[22] By gaining approval for a drug that treats a neglected disease, J&J is now able to request expedited FDA review of a future drug.[23]
When it was approved by the FDA on the 28th December 2012, it was the first new medicine for TB in more than forty years.[24][25]
Bedaquiline, formally called (1R, 2S)-1-(6-Bromo-2-methoxy-3-quinolinyl)-4-(dimethylamino)-2-(1-naphthyl)-1-phenyl-2-butanol in chemistry and known as Sirturo in commercial, is a new anti-mycobacterial medicine of diarylquinolines. It impinges on the
ATP synthesis of Mycobacterium tuberculosis by inhibiting the activity of proton pump on the cell’s ATP synthetase, and thereby eliminates M. tuberculosis (TB). It’s used for adult multi-drug resistant tuberculosis (MDR-PTB).
PATENT
US 20050148581
WO 2005117875
WO 2006125769
CA 2529265
WO 2006131519
JP 2011168519
CN 105017147
CN 105085395
CN 105198808
CN 105085396
WO 2016116076
WO 2016058564
WO 2016116075
WO 2016116073
WO 2016198031
CN 106866525
CN 106279017
CN 107602464
PATENT
WO 2017015793
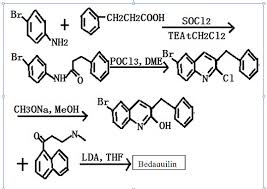
The chemical name of beidaquinoline is (1R,2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4-dimethylamino-2-(1-naphthyl)-1 -Phenyl-2-butanol, the first drug developed by Johnson & Johnson in the United States to inhibit mycobacterium adenosine triphosphate (ATP) synthetase, was first introduced in the United States in December 2012 for the treatment of adult multidrug-resistant tuberculosis. The trade name is Sirturo. Beidaquinoline shows strong selectivity for Mycobacterium tuberculosis ATP synthase. Its novel mechanism of action makes it not cross-resistance with other anti-tuberculosis drugs, which will greatly reduce the drug resistance of Mycobacterium tuberculosis. It shows good activity against MDR-TB in macrophages, suggesting that it has the effect of shortening treatment time.
The synthesis of beidaquinoline has been reported in the literature. The specific synthesis route is as follows:

The patent WO2004011436 mentions the use of column chromatography to separate and purify the crude product, but this method is not conducive to industrialization; in addition, a method for isolating and purifying beraquinoline diastereomer A is disclosed in Step C of the Example of WO2006125769. . However, although the purity of the diastereomer A obtained by the separation and purification method disclosed in this patent is 82%, it is actually only possible to achieve the reaction conversion rate of more than 80%. The actual study found that due to the difficult control of the reaction conditions for the preparation of bedaquino, the control conditions for water, temperature, and drip rate are harsh and the reaction is unstable, and it cannot be ensured that the conversion rate reaches more than 80% per batch, and the conversion is usually When the rate is between 60-80%, the ratio of diastereomer B to diastereomer A obtained by this method is between 1:1 and 1:3, and the next step is chiral separation. It has an impact; even the conversion rate is sometimes as low as about 50%. When the conversion rate is as low as 50%, since the amount of the product in the reaction liquid is small, as in the method using patent WO 2006125769, the isolated product can hardly be purified even if the product is separated and purified by the purification method disclosed in this patent. The resulting diastereomer A is also of low purity.
Reaction material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA (20g) reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 56%. After quenching the reaction, n-heptane (40 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 0° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 50% acetic acid aqueous solution to remove 3-dimethylamino-1-(naphthalene-5-yl)acetone as a raw material, and 15% hydrochloric acid aqueous solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by layering the filtrate, adjusted to alkaline with aqueous ammonia, extracted with toluene and free, and then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure to obtain a product that is not correct. Enantiomer A (4.9 g), purity 89%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetylene (40 ml), DMSO (4.9 ml), and R-binaphthol phosphate (2.62 g) were added to diastereomer A (4.9 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitates are separated out; at room temperature, the filter cake is washed with acetone and dried under vacuum at 50-60° C. to give a resolution salt (2.07 g);
Split salt (2.07g), toluene (37ml), potassium carbonate (1.51g) and water (13ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, organic layer was treated with 10% potassium carbonate aqueous solution ( (5ml) was washed once, at this time organic layer TLC monitoring; washed with purified water to neutral pH (20ml × 3 times); organic layer was concentrated under reduced pressure to give a colorless oil (1.5g); add toluene (1ml) to heat the whole Dissolve, add ethanol (12ml) and stir at room temperature for 0.5h. Precipitate the solid, and stir in ice water bath for 1h. Filter and wash the filter cake with ethanol. Dry it in vacuo at 50-60°C to give bedaquinoline (1.07g). The HPLC purity is >99%. .
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 65%. After quenching the reaction, diisopropyl ether (160 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 5° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 10% aqueous formic acid to remove 3-dimethylamino-1-(naphthalene-5-yl)acetone as a raw material, and 5% aqueous sulfuric acid solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. Filtration, filtration of the filtrate, the product was transferred to the aqueous layer, the raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer, and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. The product was diastereoisomer A (5.7 g), purity 92%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (45 ml), DMSO (5.7 ml), and R-binaphthol phosphate (3.04 g) were added to diastereomer A (5.7 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. Cooling, precipitated salt precipitation; filtered at room temperature, washed with acetone cake, 50-60 ° C vacuum drying salt (2.6g);
The resolved salt (2.41 g), toluene (39 ml), potassium carbonate (1.58 g) and water (14 ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, the organic layer was treated with 10% aqueous potassium carbonate solution ( (5ml) was washed once, washed with purified water until the pH was neutral (20ml × 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.6g); toluene was added (1ml) to heat the solution and ethanol was added (12ml) The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedalquinoline (1.19 g) with an HPLC purity of >99%.
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 75%. After quenching the reaction, diisopropyl ether (400 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 2° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 60% aqueous solution of propionic acid to remove 3-dimethylamino-1-(naphthalen-5-yl)acetone as a raw material, and 40% methanesulfonic acid aqueous solution was added to the organic layer for stirring to make the product salified. Precipitated in the water layer. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (6.0 g), purity 94%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (48 ml), DMSO (6.0 ml), and R-binaphthol phosphate (3.09 g) were added to diastereomeric A (6.0 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give the resolved salt (2.59 g).
The resolved salt (2.59g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved; while hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5 ml) was washed once, washed with purified water until the pH was neutral (20 ml × 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.7 g); toluene (1 ml) was added and heated to complete dissolution, and ethanol (12 ml) was added. The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedaquinoline (1.20 g) with an HPLC purity of >99%.
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one step reaction to obtain the racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 70%. After quenching the reaction, petroleum ether (16 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 3° C. and filtered to remove diastereoisomer B. The obtained filtrate was washed with 30% acetic acid aqueous solution to remove 3-methylamino-1-(naphthalen-5-yl)acetone as a raw material, and 25% phosphoric acid aqueous solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be slightly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, and then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (5.72 g), purity 88%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetylene (45 ml), DMSO (5.7 ml), and R-binaphthol phosphate (3.04 g) were added to diastereomer A (5.72 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated out; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give a resolution salt (2.43 g);
Split salt (2.43g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5 ml) was washed once, washed with purified water until the pH was neutral (20 ml x 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.5 g); toluene (1 ml) was added for heating and ethanol was added (12 ml) The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, and the filter cake was washed with ethanol. Drying in vacuo at 50-60° C. gave bedaquinoline (1.16 g) with an HPLC purity of >99%.
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one step reaction to obtain the racemic bedaquiline reaction solution. The conversion of this reaction was 80% by HPLC analysis. After quenching the reaction, n-hexane (80 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 1° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 40% aqueous acetic acid to remove 3-dimethylamino-1-(naphthalen-5-yl)acetone as starting material, and 20% aqueous hydrochloric acid solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (6.1 g), purity 96%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (48 ml), DMSO (6.1 ml), and R-binaphthol phosphate (3.09 g) were added to diastereomer A (6.1 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated out; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give the resolution salt (2.69 g).
The resolved salt (2.69g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved; while hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5ml) was washed once, washed with purified water until the pH was neutral (20ml×3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.8g); toluene (1ml) was added to heat to dissolve and ethanol (12ml) was added. The precipitated solid was stirred for 0.5 h at room temperature, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedalquinoline (1.28 g) with an HPLC purity of >99%.
PAPER
ACS Medicinal Chemistry Letters (2017), 8(10), 1019-1024
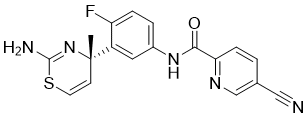
6-Cyano Analogues of Bedaquiline as Less Lipophilic and Potentially Safer Diarylquinolines for Tuberculosis
Amy S. T. Tong†, Peter J. Choi†, Adrian Blaser†, Hamish S. Sutherland†, Sophia K. Y. Tsang†, Jerome Guillemont‡, Magali Motte‡, Christopher B. Cooper§, Koen Andries∥, Walter Van den Broeck∥, Scott G. Franzblau⊥, Anna M. Upton§, William A. Denny*†  , Brian D. Palmer†, and Daniel Conole†
, Brian D. Palmer†, and Daniel Conole†
† Auckland Cancer Society Research Centre, School of Medical Sciences, The University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
‡ Medicinal Chemistry Department (Infectious Diseases), Janssen Pharmaceuticals, Campus de Maigremont, BP315, 27106 Val de Reuil Cedex, France
§ Global Alliance for TB Drug Development, 40 Wall Street, New York, New York 10005, United States
∥ Infectious Diseases BVBA, Janssen Pharmaceuticals, Beerse, Belgium
⊥ Institute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, 833 South Wood Street, Chicago, Illinois 60612, United States
ACS Med. Chem. Lett., 2017, 8 (10), pp 1019–1024
DOI: 10.1021/acsmedchemlett.7b00196
Publication Date (Web): September 22, 2017
Copyright © 2017 American Chemical Society
Abstract

Bedaquiline (1) is a new drug for tuberculosis and the first of the diarylquinoline class. It demonstrates excellent efficacy against TB but induces phospholipidosis at high doses, has a long terminal elimination half-life (due to its high lipophilicity), and exhibits potent hERG channel inhibition, resulting in clinical QTc interval prolongation. A number of structural ring A analogues of bedaquiline have been prepared and evaluated for their anti-M.tb activity (MIC90), with a view to their possible application as less lipophilic second generation compounds. It was previously observed that a range of 6-substituted analogues of 1 demonstrated a positive correlation between potency (MIC90) toward M.tb and drug lipophilicity. Contrary to this trend, we discovered, by virtue of a clogP/M.tb score, that a 6-cyano (CN) substituent provides a substantial reduction in lipophilicity with only modest effects on MIC values, suggesting this substituent as a useful tool in the search for effective and safer analogues of 1.
PAPER
Chinese Chemical Letters (2015), 26(6), 790-792
PAPER
Organic & Biomolecular Chemistry (2016), 14(40), 9622-9628.
http://pubs.rsc.org/en/content/articlelanding/2016/ob/c6ob01893a/unauth#!divAbstract
New synthetic approaches towards analogues of bedaquiline
Abstract
Multi-drug resistant tuberculosis (MDR-TB) is of growing global concern and threatens to undermine increasing efforts to control the worldwide spread of tuberculosis (TB). Bedaquiline has recently emerged as a new drug developed to specifically treat MDR-TB. Despite being highly effective as a result of its unique mode of action, bedaquiline has been associated with significant toxicities and as such, safety concerns are limiting its clinical use. In order to access pharmaceutical agents that exhibit an improved safety profile for the treatment of MDR-TB, new synthetic pathways to facilitate the preparation of bedaquiline and analogues thereof have been discovered.
PAPER
Topics in Organometallic Chemistry (2011), 37(Bifunctional Molecular Catalysis), 1-30
PAPER
Saga, Yutaka; Journal of the American Chemical Society 2010, Vol132(23), Pg 7905-7907
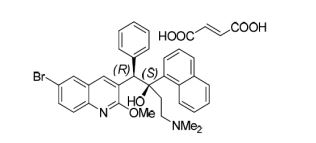
Catalytic Asymmetric Synthesis of R207910
Graduate School of Pharmaceutical Sciences, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
J. Am. Chem. Soc., 2010, 132 (23), pp 7905–7907
DOI: 10.1021/ja103183r
Publication Date (Web): May 20, 2010
Copyright © 2010 American Chemical Society
Abstract

The first asymmetric synthesis of a very promising antituberculosis drug candidate, R207910, was achieved by developing two novel catalytic transformations; a catalytic enantioselective proton migration and a catalytic diastereoselective allylation of an intermediate α-chiral ketone. Using 2.5 mol % of a Y-catalyst derived from Y(HMDS)3 and the new chiral ligand 9, 1.25 mol % of p-methoxypyridine N-oxide (MEPO), and 0.5 mol % of Bu4NCl, α-chiral ketone 3 was produced from enone 4 with 88% ee. This reaction proceeded through a catalytic chiral Y-dienolate generation via deprotonation at the γ-position of 4, followed by regio- and enantioselective protonation at the α-position of the resulting dienolate. Preliminary mechanistic studies suggested that a Y: 9: MEPO = 2: 3: 1 ternary complex was the active catalyst. Bu4NCl markedly accelerated the reaction without affecting enantioselectivity. Enantiomerically pure 3 was obtained through a single recrystallization. The second key catalytic allylation of ketone 3 was promoted by CuF•3PPh3•2EtOH (10 mol %) in the presence of KOtBu (15 mol %), ZnCl2 (1 equiv), and Bu4PBF4 (1 equiv), giving the desired diastereomer 2 in quantitative yield with a 14: 1 ratio without any epimerization at the α-stereocenter. It is noteworthy that conventional organometallic addition reactions did not produce the desired products due to the high steric demand and a fairly acidic α-proton in substrate ketone 3. This first catalytic asymmetric synthesis of R207910 includes 12 longest linear steps from commercially available compounds with an overall yield of 5%.


Click to access ja103183r_si_001.pdf
1 in 62 % yield (6.5 mg, 0.012 mmol ). 1H NMR (500 MHz, CDCl3) : 1.91-1.95 (m, 1H), 1.98 (s, 6H), 1.99-2.10 (m, 2H), 2.52 (d, J = 14.1 Hz, 1H), 4.21 (s, 3H), 5.89 (s, 1H), 6.87-6.89 (m, 3H), 7.10-7.15 (m, 2H), 7.31 (t, J = 7.6 Hz, 2H), 7.48 (t, J = 8.5 Hz, 1H), 7.61 (t, J = 8.5 Hz, 1H), 7.63-7.67 (m, 2H), 7.72 (d, J = 8.9 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 2.2 Hz, 1H), 8.60 (d, J = 8.5 Hz, 1H), 8.89 (s, 1H); 13C NMR (126 MHz, CDCl3) : 29.7, 33.5, 44.7, 49.5, 54.2, 56.4, 82.6, 117.0, 124.5, 125.0, 125.1, 125.3, 125.8, 126.9, 127.1, 127.4, 127.9, 128.0, 128.1, 128.5, 129.8, 129.9, 131.9, 132.7, 133.3, 134.7, 138.8, 140.6, 141.8, 143.8, 161.4; IR (KBr, cm-1 ): 3443; MS (ESI) m/z 555 (M+H) + ; HRMS (FAB) calcd for C32H32N2O2Br (M+H) + 555.1647. Found 555.1644; [] 26 D – (c = 0.3, DMF).
PAPER
Gaurrand, Sandrine; Chemical Biology & Drug Design 2006, VOL 68(2), PG 77-84
http://web.a.ebscohost.com/ehost/pdfviewer/pdfviewer?vid=1&sid=fac0fcc3-2a10-4f5f-8e20-d057adf71ce9%40sessionmgr4006



PAPER
Chandrasekhar, Srivari; European Journal of Organic Chemistry 2011, (11), PG 2057-2061, S2057/1-S2057/18
Srivari Chandrasekhar

|
Dr.Chandrasekhar S
Director
CSIR-Indian Institute of Chemical Technology
(Council of Scientific and Industrial Research)
Ministry of Science & Technology, Government of India
Tarnaka, Hyderabad-500007, Telangana, INDIA
|
|
READ
http://www.iictindia.org/staffprofiles/staffprofile.aspx?qry=1372

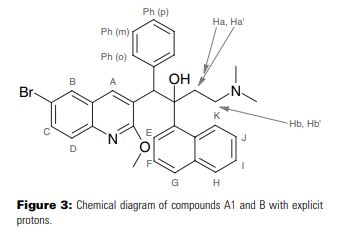
(1R,2S)-1-(6-Bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)- 2-(naphthalen-1-yl)-1-phenylbut-an-2-ol (3a): A solution of 16a and 16b (6.0 g, 10.2 mmol) in Me2NH (200 mL, 8.0 m in THF) was stirred at 45 °C for 24 h. The solution was filtered and the filtrate concentrated under reduced pressure to afford the crude product which on purification by silica gel column chromatography (eluent: ethyl acetate/hexane = 1:6) furnished 3a and 3b as white solids (4.8 g, 90%) (1:1 w/w).
3a: M.p. 104 °C. [α]D 25 = –165.2 (c = 0.8, DMF). (2S)- R207910 (3a)
1 H NMR (300 MHz, CDCl3): δ = 8.89 (s, 1 H, H4), 8.61 (d, J = 8.6 Hz, 1 H, H20), 7.96 (d, J = 2.0 Hz, 1 H, H5), 7.92 (d, J = 7.4 Hz, 1 H, H14), 7.87 (d, J = 8.1 Hz, 1 H, H17), 7.72 (d, J = 8.8 Hz, 1 H, H8), 7.68–7.56 (m, 3 H, H7, H16, H19), 7.48 (t, J = 7.6 Hz, 1 H, H18), 7.30 (t, J = 7.7 Hz, 1 H, H15), 7.17–7.10 (m, 2 H, H24), 6.93–6.83 (m, 3 H, H25, H26), 5.89 (s, 1 H, H11), 4.21 (s, 3 H, H30), 2.60–2.51 (m, 1 H, H27), 2.18–2.02 (m, 2 H, H27, H28), 1.99 (s, 6 H, H29), 1.95–1.85 (m, 1 H, H28) ppm.
13C NMR (75 MHz, CDCl3): δ = 161.3, 143.7, 141.6, 140.5, 138.7, 134.6, 131.9, 129.9, 129.8, 129.7, 128.4, 128.1, 127.8, 127.3, 127.1, 126.8, 125.7, 125.2, 125.1, 125.0, 124.4, 116.9, 82.4, 56.2, 54.1, 49.5, 44.6, 33.4, 29.6 ppm.
IR (KBr): ν˜ = 3441 cm–1.
HRMS (ESI) calcd. for C32H32BrN2O2 [M + H]+ 555.1642; found 555.1671.


(2R)-R207910



References[
- ^ Jump up to:a b c d e f g h i j “Bedaquiline Fumarate”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 8 December 2016.
- Jump up^ Diacon AH, Pym A, Grobusch M, et al. (2009). “The diarylquinoline TMC207 for multidrug-resistant tuberculosis”. N Engl J Med. 360 (23): 2397–405. doi:10.1056/NEJMoa0808427. PMID 19494215.
- ^ Jump up to:a b c “Bedaquiline”. Archived from the original on 20 May 2013. Retrieved 28 April 2014.
- Jump up^ “Sirturo: Clinical Pharmacology”. Archived from the original on 28 February 2015. Retrieved 28 April 2014.
- ^ Jump up to:a b c d The selection and use of essential medicines: Twentieth report of the WHO Expert Committee 2015 (including 19th WHO Model List of Essential Medicines and 5th WHO Model List of Essential Medicines for Children) (PDF). World Health Organization. 2015. p. vii, 29. ISBN 9789241209946. Archived(PDF) from the original on 20 December 2016. Retrieved 10 December 2016.
- Jump up^ “Bedaquiline (Sirturo) Use During Pregnancy”. http://www.drugs.com. Archived from the original on 20 December 2016. Retrieved 10 December 2016.
- Jump up^ “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- Jump up^ “Press Announcements – FDA approves first drug to treat multi-drug resistant tuberculosis”. http://www.fda.gov. Dec 2012. Archivedfrom the original on 2016-12-19.
- ^ Jump up to:a b Centers for Disease Control and Prevention (2013-10-25). “Provisional CDC guidelines for the use and safety monitoring of bedaquiline fumarate (Sirturo) for the treatment of multidrug-resistant tuberculosis”. MMWR. 62 (RR-09): 1–12. ISSN 1545-8601. PMID 24157696.
- Jump up^ WHO (2013). The use of bedaquiline in the treatment of multidrug-resistant tuberculosis : interim policy guidance. Archived from the original on 2017-09-10.
- ^ Jump up to:a b c Field, Stephen K. (2015-07-01). “Bedaquiline for the treatment of multidrug-resistant tuberculosis: great promise or disappointment?”. Therapeutic Advances in Chronic Disease. 6(4): 170–184. doi:10.1177/2040622315582325. ISSN 2040-6223. PMC 4480545
 . PMID 26137207. Archived from the original on 2015-10-28.
. PMID 26137207. Archived from the original on 2015-10-28.
- ^ Jump up to:a b Diacon, Andreas H.; Pym, Alexander; Grobusch, Martin P.; de los Rios, Jorge M.; Gotuzzo, Eduardo; Vasilyeva, Irina; Leimane, Vaira; Andries, Koen; Bakare, Nyasha (2014-08-21). “Multidrug-Resistant Tuberculosis and Culture Conversion with Bedaquiline”. New England Journal of Medicine. 371 (8): 723–732. doi:10.1056/NEJMoa1313865. ISSN 0028-4793. PMID 25140958.
- Jump up^ BPaMZ @ TB Alliance Archived 2017-02-19 at the Wayback Machine.
- Jump up^ Two new drug therapies might cure every form of tuberculosis. Feb 2017 Archived 2017-02-20 at the Wayback Machine.
- Jump up^ Drugs.com: Sirturo Side Effects Archived 2013-09-23 at the Wayback Machine.
- ^ Jump up to:a b c “Prescribing Information for Bedaquiline” (PDF). Archived (PDF) from the original on 24 August 2013. Retrieved 28 April 2014.
- ^ Jump up to:a b c Cox, Edward; Laessig, Katherine (2014-08-21). “FDA Approval of Bedaquiline — The Benefit–Risk Balance for Drug-Resistant Tuberculosis”. New England Journal of Medicine. 371(8): 689–691. doi:10.1056/NEJMp1314385. ISSN 0028-4793. PMID 25140952.
- ^ Jump up to:a b c Worley, Marylee V.; Estrada, Sandy J. (2014-11-01). “Bedaquiline: A Novel Antitubercular Agent for the Treatment of Multidrug-Resistant Tuberculosis”. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy. 34 (11): 1187–1197. doi:10.1002/phar.1482. ISSN 1875-9114. Archivedfrom the original on 2016-12-20.
- Jump up^ Andries, Koen; Villellas, Cristina; Coeck, Nele; Thys, Kim; Gevers, Tom; Vranckx, Luc; Lounis, Nacer; Jong, Bouke C. de; Koul, Anil (2014-07-10). “Acquired Resistance of Mycobacterium tuberculosis to Bedaquiline”. PLOS ONE. 9 (7): e102135. doi:10.1371/journal.pone.0102135. ISSN 1932-6203. PMC 4092087 . PMID 25010492. Archived from the original on 2017-09-10.
- Jump up^ Protopopova M, Bogatcheva E, Nikonenko B, Hundert S, Einck L, Nacy CA (May 2007). “In search of new cures for tuberculosis”(PDF). Med Chem. 3 (3): 301–16. doi:10.2174/157340607780620626. PMID 17504204.[permanent dead link]
- Jump up^ de Jonge MR, Koymans LH, Guillemont JE, Koul A, Andries K (June 2007). “A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910”. Proteins. 67 (4): 971–80. doi:10.1002/prot.21376. PMID 17387738.
- Jump up^ Walker, Joseph; Tadena, Nathalie (December 31, 2012). “J&J Tuberculosis Drug Gets Fast-Track Clearance”. Wall Street Journal. Archived from the original on September 23, 2015. Retrieved 2013-01-01.
- Jump up^ Edney, Anna (December 31, 2012). “J&J&J Sirturo Wins FDA Approval to Treat Drug-Resistant TB”. Bloomberg. Archivedfrom the original on January 4, 2013. Retrieved 2013-01-01.
- Jump up^ “FDA Approves 1st New Tuberculosis Drug in 40 Years”. ABC News. Archived from the original on 4 January 2013. Retrieved 31 December 2012.
- Jump up^ “F.D.A. Approves New Tuberculosis Drug”. New York Times. Archived from the original on 8 January 2013. Retrieved 31 December 2012.
//////////////Bedaquiline, JAPAN 2018, R 207910, Sirturo, TMC 207, FDA 2012, EMA 2014, ベダキリンフマル酸塩
CN(C)CCC(C1=CC=CC2=CC=CC=C21)(C(C3=CC=CC=C3)C4=C(N=C5C=CC(=CC5=C4)Br)OC)O.C(=CC(=O)O)C(=O)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....