
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Evobrutinib
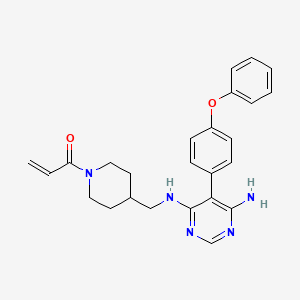
Evobrutinib
429.5 g/mol,C25H27N5O2
- Evobrutinib
- 1415823-73-2
- Evobrutinib [INN]
- 1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one
- MSC2364447C
- MSC2364447C
- M-2951
- MSC-2364447C
- ZA45457L1K
- 1-[4-[[[6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl]amino]methyl]piperidin-1-yl]prop-2-en-1-one
- M2951
Evobrutinib is under investigation in clinical trial NCT03934502 (Effect of Meal Composition and Timing on Evobrutinib Bioavailability).
Evobrutinib is an inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, evobrutinib inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
Evobrutinib is in clinical development to investigate its potential as a treatment for multiple sclerosis (MS). It is an oral, highly selective inhibitor of Bruton’s tyrosine kinase (BTK) which is important in the development and functioning of various immune cells including B lymphocytes and macrophages.
Evobrutinib is designed to inhibit primary B cell responses such as proliferation and antibody and cytokine release, without directly affecting T cells. BTK inhibition is thought to suppress autoantibody-producing cells, which preclinical research suggests may be therapeutically useful in certain autoimmune diseases.
U.S. Patent No. 9073947 discloses a pyrimidine derivative of Evobrutinib which chemically named as l-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)
piperidin-l-yl)prop-2-en-l-one and pharmaceutically acceptable salts, solvates and pharmaceutical compositions thereof.
U.S. Patent No. 9073947 and ‘Journal of Medicinal Chemistry 2019, 62(17), 7643-7655’ discloses process for the preparation of Evobrutinib which involves column purifications and lyophilisation methods to provide Evobrutinib with low yield, which is not viable at large scale production.
https://www.frontiersin.org/articles/10.3389/fnume.2021.820235/full
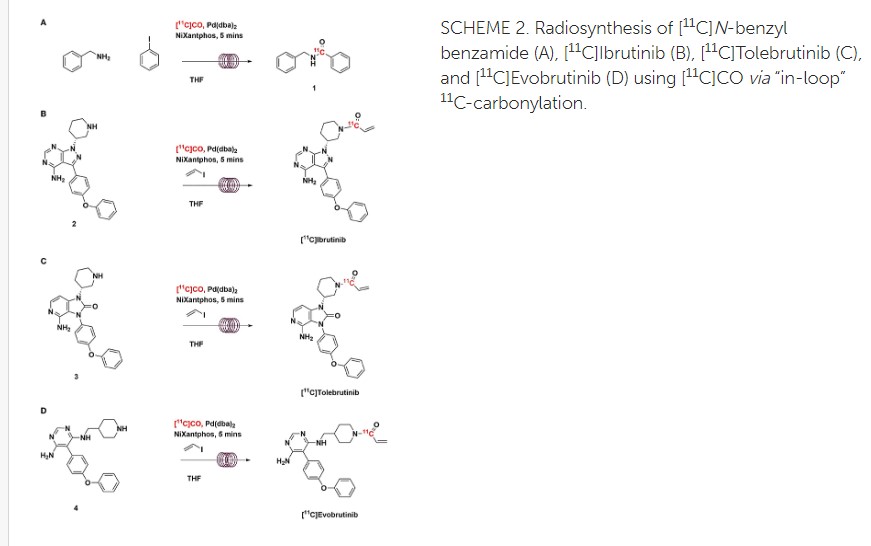

Radiosynthesis of [11C]Evobrutinib. [11C]Evobrutinib was synthesized similarly to the Tolebrutinib example above with the following exceptions. First, the precursor 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (4) (1 mg, 2.7 μmol) was used and the crude reaction mixture after the carbonylation reaction was purified by semi-preparative HPLC (column: Luna C18(2), 5 μ (250 x 9.6 mm); mobile phase: 44% MeCN in 200 mM ammonium formate; flow rate: 5 ml/min; UV: 254 nm). The [11C]1-(4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl)piperidin-1-yl)prop-2-en-1-one ([11C]evobrutinib) was isolated between the 15.5 and 18 min mark of the chromatogram and this sample was collected into a dilution flask that contained 50 ml of a 2 mg/ml sodium ascorbate aqueous solution. This solution was transferred to an HLB light (30 mg) SPE cartridge. After transfer, the cartridge was eluted with 1 ml of ethanol into the sterile product vial that contained 4 ml of sterile saline. Using this method, 2.2 ± 0.6 GBq (81.4 ± 22.2 mCi) [11C]evobrutinib was isolated (n = 3), and the product was analyzed via reverse phase HPLC using the following methods. Method A described above and Method B (Isocratic and molar activity): column: Luna C18(2) 3-μm (250×4.6 mm); mobile phase Isocratic: 36% acetonitrile in aqueous 0.1% TFA; flow rate: 1.3 ml/min; UV: 254 nm. Method A was used to confirm chemical identity using a co-injection of non-radioactive standard. Radiochemical purity and molar activity were determined by Method B. [11C]Evobrutinib was confirmed by co-injection with a verified non-radioactive reference standard. Am was determined using a 4-point standard curve (analytical HPLC peak area) (Y) vs. standard concentration (X: in nmol) by comparison with an evobrutinib reference standard of known concentration (2.3 mg in 1 ml). The isolated [11C] evobrutinib was co-eluted with a non-radioactive reference standard. The sample was >99% radiochemically pure, >95% chemically pure (HPLC, UV: 254 nm), with a molar activity of 496.5 ± 74 GBq/μmol (13.4 Ci/μmol) The overall synthesis time from the end of cyclotron bombardment was 37–46 min.
Patent
U.S. Patent No. 9073947
PAPER
Journal of Medicinal Chemistry 2019, 62(17), 7643-7655
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b00794
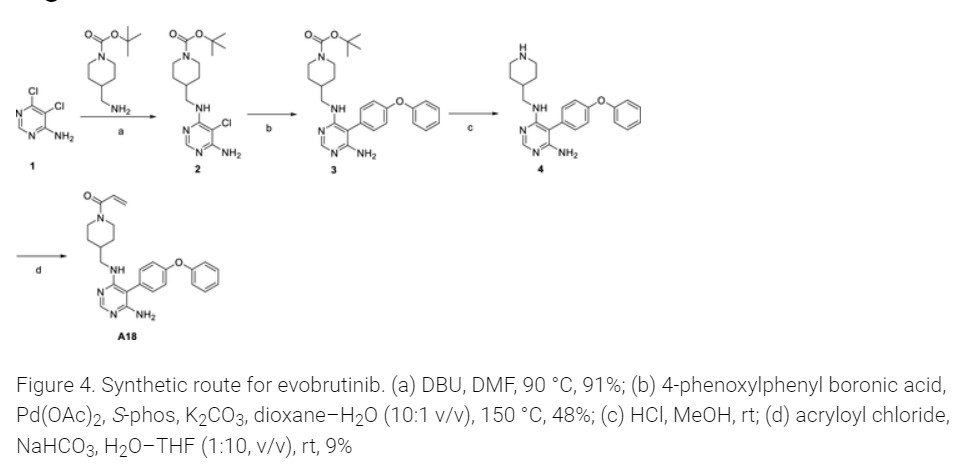

Step 4
To a 20 mL vial was added 5-(4-phenoxyphenyl)-N-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (210.00 mg, 0.56 mmol, 1.00 equiv), sodium bicarbonate (70.48 mg, 0.84 mmol, 1.50 equiv), THF (8.00 mL, 98.74 mmol, 176.55 equiv), and water (0.80 mL, 44.41 mmol, 79.40 equiv). The mixture was cooled to 0 °C on an ice bath. Acryloyl chloride (0.15 mL, 1.83 mmol) was then added dropwise. The ice bath was removed, and the reaction was stirred at room temperature for 12 h before it was purified by silica gel chromatography (25 g KPNH silica, 0–100% methanol/ethyl acetate) to afford the title compound (A18) (21 mg, 8.7% yield) was synthesized with a similar protocol to prepared as described in the main body of the article. 1H NMR (DMSO-d6) δ 7.93 (s, 1 H), 7.40–7.08 (m, 9H), 6.76 (dd, J = 4 Hz, 1 H), 6.04 (d, J = 4 Hz, 1 H), 5.61 (d, J = 4 Hz, 1 H), 5.43 (s, 2H), 4.34 (d, J = 12 Hz, 1 H), 3.98 (d, J = 8 Hz, 1 H), 3.12 (m, 2H), 2.95 (m, 1 H), 2.56 (m, 1 H), 1.81 (m, 1 H), 1.59 (m, 2H), 0.92 (m, 2H). [ES-MS] (ESI+): m/z calcd for C25H28N5O2 [M + H]+ 430, found 430.
PATENT
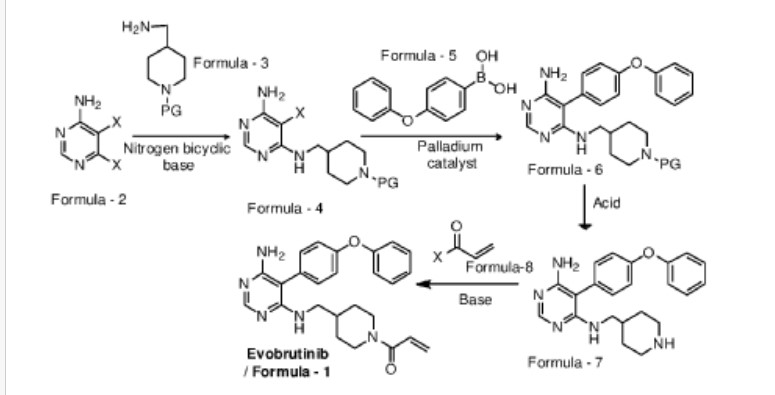

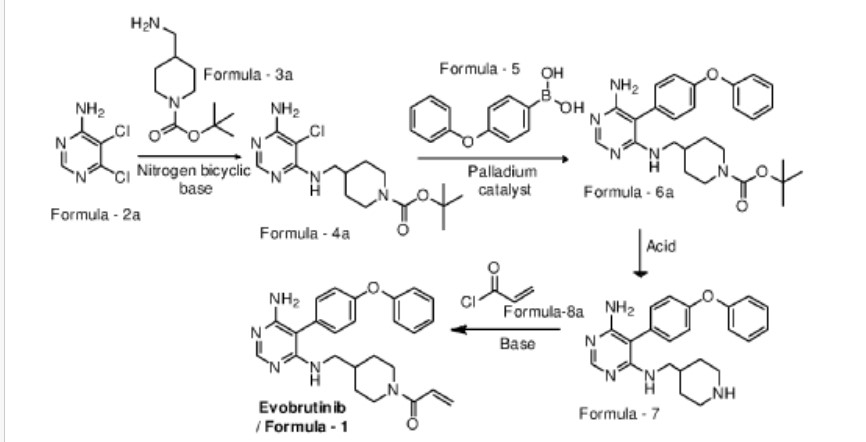

Examples:
Example-1: Preparation of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino) met hy 1 jpiperid ine- 1 -carboxylate
Tert-butyl-4-(aminomethyl)piperidine-l -carboxylate (81 ml) and 1,8-diazabicyclo [5.4.0]undec-7-ene (60.34 g) were added to a mixture of 5,6-dichloropyrimidin-4-amine (50 g) in N,N-dimethylformamide (500 ml) at 25-35°C. Heated the mixture to 90-95°C and stirred for 22 hrs. Cooled the mixture to 25-30°C. Water was added to the mixture at 25-35°C and stirred for 5 hrs. Filtered the precipitated solid, washed with water and n-heptane and dried to get the title compound. Yield: 73.0 gms; Purity by HPLC: 98.7%
Example-2: Preparation of tert-butyl 4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl) amino)methyl)piperidine-l-carboxylate
(4-Phenoxyphenyl)boronic acid (75.12 g) was added to a mixture of tert-butyl 4-(((6-amino-5-chloropyrimidin-4-yl)amino)methyl)piperidine-l-carboxylate(100 g), 2-di cyclo hexylphosphino-2′,6′-dimethoxybiphenyl (12 g) and potassium carbonate (121.28 g) in 1,4-di oxane (1000 ml) at 25-30°C and stirred for 30 minutes under nitrogen atmosphere. Palladium acetate (1.96 g) was added to the mixture at 25-30°C. Heated the mixture to 100-105°C and stirred for 3 hrs. Cooled the mixture to 25-30°C. Water and ethyl acetate were added to the mixture at 25-35°C and stirred for 30 minutes. Filtered the mixture by using hyflow bed. Organic layer was separated from the filtrate. Organic layer was treated with carbon powder and distilled-off the solvent under reduced pressure, n-heptane (800 ml) was added to the obtained compound. Heated the mixture to 60-65°C and stirred for 90 minutes. Cooled the mixture to 25-30°C and stirred for 2 hrs. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 120 gms, Purity by HPEC: 97.6% Example-3: Preparation of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine
Tert-butyl-4-(((6-amino-5-(4-phenoxyphenyl)pyrimidin-4-yl)amino)methyl) piperidine- 1 -carboxylate (200 g) in methanol (600 ml) was cooled to 0-5°C. Hydrochloric acid in ethyl acetate (500 ml) was slowly added to the mixture at 0-5°C. Mixture allowed to warm to 25-30°C and stirred for 20 hours. Water was added to the mixture and treated the mixture with aqueous ammonia solution. Dichloromethane was added to the mixture at 25-30°C and stirred for 10 minutes. Layers were separated and distilled-off the organic layer under reduce pressure. Obtained compound was treated with isopropyl ether and dried to get the title compound. Yield: 150 gms, Purity by HPLC: 76.4%
Example-4: Preparation of Evobrutinib
Sodium bicarbonate (23.86 g) and water (301 ml) were added to the mixture of 5-(4-phenoxyphenyl)-N4-(piperidin-4-ylmethyl)pyrimidine-4,6-diamine (70 g) in tetrahydrofuran (2800 ml). Cooled the mixture to 0-5°C. Acryloyl chloride (23.62 g) was slowly added to the mixture. Mixture allowed to warm to 25-30°C and stirred for 20 hrs. Distilled-off the solvent from the mixture under reduced pressure. Ethyl acetate and water were added to the mixture and stirred for 10 minutes. Both the layers were separated. Organic layer was treated with aqueous hydrochloric acid solution and carbon powder. Distilled-off the organic layer under reduced pressure. Isopropyl ether was added to the mixture at 25-30°C and stirred for 14 hrs. Filtered the mixture and washed with isopropyl ether. Dried to get the title compound.
Yield: 41.8 gms, Purity by HPLC: 97.6%


AS ON AUG2023 4,071,221 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
//////////PHASE 3, MSC2364447C, M-2951, MSC-2364447C, ZA45457L1K, M2951, M 2951, Evobrutinib

NEW DRUG APPROVALS
ONE TIME
$10.00
Zuranolone
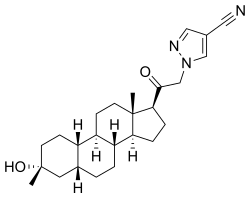
Zuranolone
CAS 1632051-40-1
FDA APPROVED 8/4/2023, To treat postpartum depression
Press Release
WeightAverage: 409.574
Monoisotopic: 409.272927379Chemical FormulaC25H35N3O2
- SAGE 217
- SAGE-217
- SAGE217
Zuranolone, sold under the brand name Zurzuvae, is a medication used for the treatment of postpartum depression.[1][2] It is taken by mouth.[1]
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis, and urinary tract infection.[1][2] An orally active inhibitory pregnane neurosteroid, zuranolone acts as a positive allosteric modulator of the GABAA receptor.[6][7][8]
Zuranolone was approved for medical use in the United States for the treatment of postpartum depression in August 2023.[2] It was developed by Sage Therapeutics and Biogen.[9]
Medical uses
Zuranolone is indicated for the treatment of postpartum depression.[1][2]
Adverse effects
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis (cold-like symptoms), and urinary tract infection.[2]
The US FDA label contains a boxed warning noting that zuranolone can impact a person’s ability to drive and perform other potentially hazardous activities.[2] Use of zuranolone may cause suicidal thoughts and behavior.[2] Zuranolone may cause fetal harm.[2]
History
Zuranolone was developed as an improvement on the intravenously administered neurosteroid brexanolone, with high oral bioavailability and a biological half-life suitable for once-daily administration.[7][10] Its half-life is around 16 to 23 hours, compared to approximately 9 hours for brexanolone.[4][5]
The efficacy of zuranolone for the treatment of postpartum depression in adults was demonstrated in two randomized, double-blind, placebo-controlled, multicenter studies.[2] The trial participants were women with postpartum depression who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode and whose symptoms began in the third trimester or within four weeks of delivery.[2] In study 1, participants received 50 mg of zuranolone or placebo once daily in the evening for 14 days.[2] In study 2, participants received another zuranolone product that was approximately equal to 40 mg of zuranolone or placebo, also for 14 days.[2] Participants in both studies were monitored for at least four weeks after the 14-day treatment.[2] The primary endpoint of both studies was the change in depressive symptoms using the total score from the 17-item Hamilton depression rating scale (HAMD-17), measured at day 15.[2] Participants in the zuranolone groups showed significantly more improvement in their symptoms compared to those in the placebo groups.[2] The treatment effect was maintained at day 42—four weeks after the last dose of zuranolone.[2]
Society and culture
Zuranolone is the international nonproprietary name.[11]
Legal status
Zuranolone was approved by the US Food and Drug Administration (FDA) for the treatment of postpartum depression in August 2023.[2][12] The FDA granted the application for zuranolone priority review and fast track designations.[2] Approval of Zurzuvae was granted to Sage Therapeutics, Inc.[2]
Zuranolone has also been under development for the treatment of major depressive disorder, but the application for this use was given a Complete Response Letter (CRL) by the FDA due to insufficient evidence of effectiveness.[13]
Research
In a randomized, placebo-controlled phase III trial to assess its efficacy and safety for the treatment of major depressive disorder, subjects in the zuranolone group (50 mg oral zuranolone once daily for 14 days) experienced statistically significant and sustained improvements in depressive symptoms (as measured by HAM-D score) throughout the treatment and follow-up periods of the study.[14]
Other investigational applications include insomnia, bipolar depression, essential tremor, and Parkinson’s disease.[15][6][16]
syn
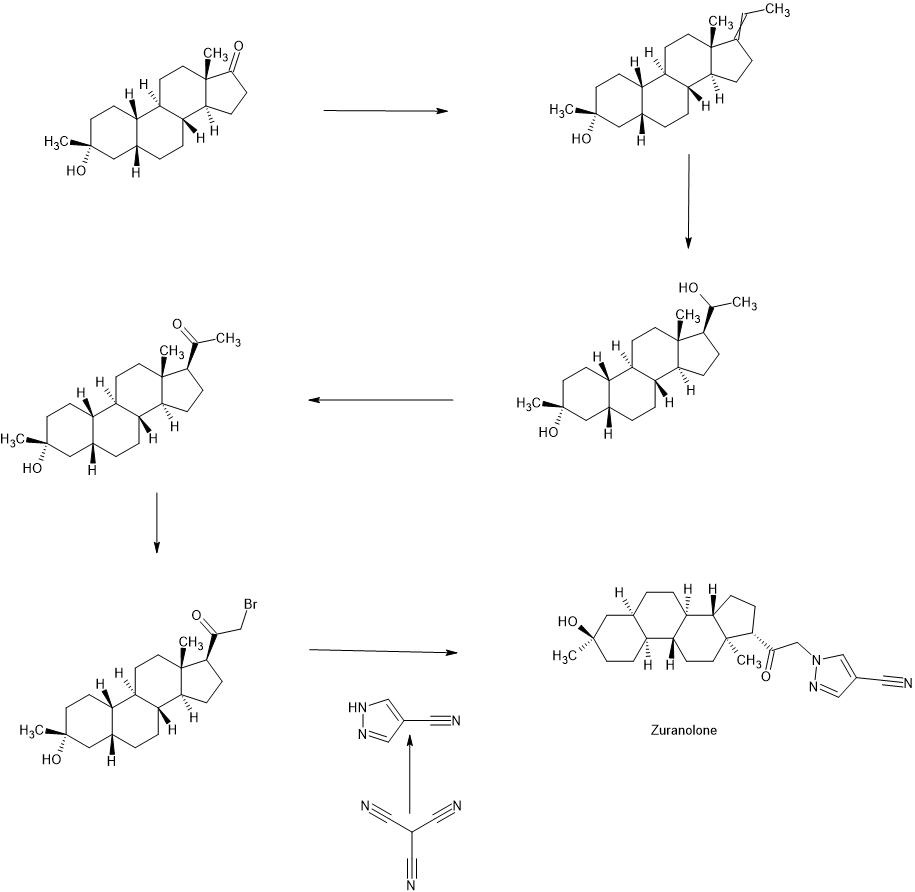

PATENT
WO2022020363
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022020363&_cid=P11-LLRZ9A-38538-1
Example 1. Synthesis of 1-(2-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl)-1H-pyrazole-4-carbonitrile (Compound 1).
[00488] To a suspension of K2CO3 (50 mg, 0.36 mmol) in THF (5 mL) was added 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol) and 2-bromo-1-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[ ^]phenanthren-17-yl)ethan-1-one (50 mg, 0.12 mmol). The mixture was stirred at room temperature for 15 hours. The reaction mixture was poured into 5 mL H2O and extracted with ethyl acetate (2×10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified by reverse-phase preparative HPLC to afford Compound 1 as a white solid (9 mg, 17.4% yield).1H NMR (500 MHZ, CDCl3) δ (ppm) 7.87 (1H, s), 7.82 (1H, s), 5.02 (1H, AB), 4.2 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dxt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt=2.24 min, m/z=410.1 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2017), 60(18), 7810-7819
https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b00846
Certain classes of neuroactive steroids (NASs) are positive allosteric modulators (PAM) of synaptic and extrasynaptic GABAA receptors. Herein, we report new SAR insights in a series of 5β-nor-19-pregnan-20-one analogues bearing substituted pyrazoles and triazoles at C-21, culminating in the discovery of 3α-hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217, 3), a potent GABAA receptor modulator at both synaptic and extrasynaptic receptor subtypes, with excellent oral DMPK properties. Compound 3 has completed a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial and is currently being studied in parallel phase 2 clinical trials for the treatment of postpartum depression (PPD), major depressive disorder (MDD), and essential tremor (ET).


3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19- nor-5β-pregnan-20-one (3). Yield: 28 g (49%) as an off-white solid. LC-MS: tR = 1.00 min, m/z = 410 (M + 1). 1 H NMR (400 MHz, CDCl3): δ 7.86 (s, 1H), 7.80 (s, 1H), 5.08−4.84 (m, 2H), 2.70−2.55 (m, 1H), 2.25−2.15 (m, 1H), 2.10−2.00 (m, 1H), 1.88−1.59 (m, 7H), 1.53−1.30 (m, 15H), 1.25−1.00 (m, 3H), 0.67 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 13.92 (CH3), 23.20, 24.44, 25.54, 25.78, 26.15 (5 × CH2), 26.69 (CH3), 31.43, 34.61 (2 × CH2), 34.77, 37.71 (2 × CH), 39.26 (CH2), 40.35 (CH), 41.21 (CH2), 41.75 (CH), 45.56 (C), 56.04, 61.24 (2 × CH), 61.78 (CH2), 72.14 (C), 93.25 (C), 113.35 (CN), 136.16, 142.49 (2 × CH), 202.23 (CO). HRMS m/z 410.2803 calcd for C25H36N3O2 + 410.2802
PATENT
WO2014169833
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014169833&_cid=P11-LLRZJ9-40598-1
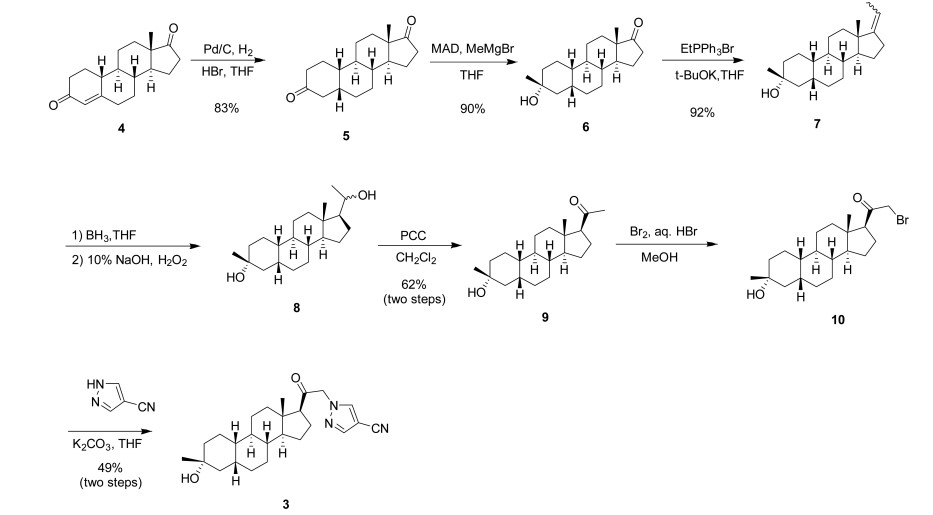
Synthetic Procedures
The compounds of the invention can be prepared in accordance with methods described in the art (Upasmi et al., J. Med. Chem. 1997, 40:73-84; and Hogenkamp et al., J. Med. Chem. 1997, 40:61- 72) and using the appropriate reagents, starting materials, and purification methods known to those skilled in the art. In some embodiments, compounds described herein can be prepared using methods shown in general Schemes 1-4, comprising a nucleophilic substitution of 19-nor pregnane bromide with a neucleophile. In certain embodiments, the nucleophile reacts with the 19-nor pregnane bromide in the presence of K2CO3 in THF.
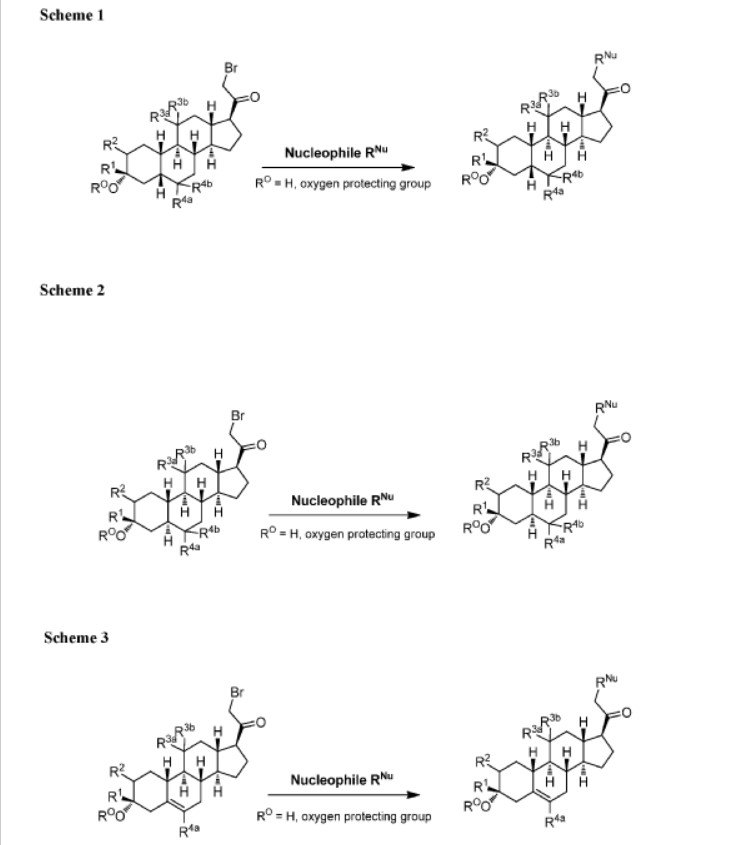

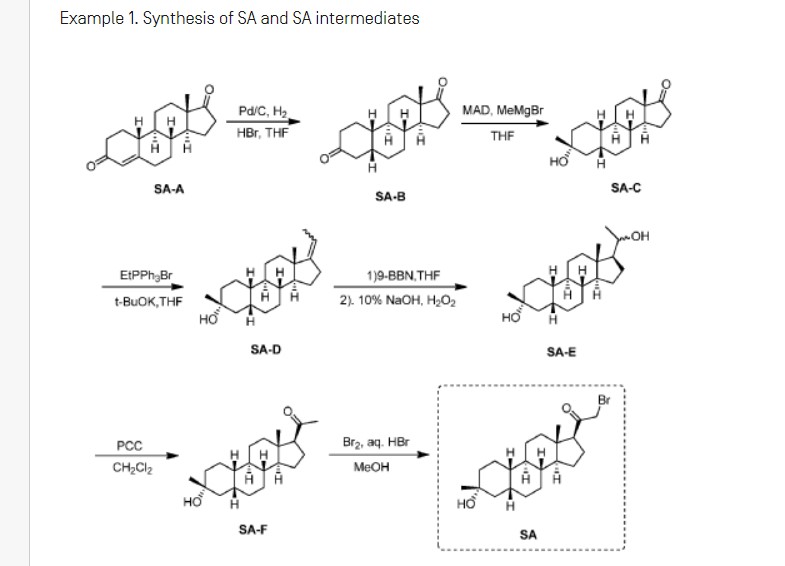

Synthesis of compound SA-B. Compound SA (50 g, 184 mmol) and palladium black (2.5 g) in tetrahydrofuran (300 mL) and concentrated hydrobromic acid (1.0 mL) was hydrogenated with 10 atm hydrogen. After stirring at room temperature for 24h, the mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to afford the crude compound. Recrystallization from acetone gave compound SA-B (42.0 g, yield: 83.4%) as white powder.
1H NMR: (400 MHz, CDCl3) δ 2.45-2.41 (m, 1H), 2.11-3.44 (m, 2H), 3.24 (s, 3H), 2.18-2.15 (m, 1H), 2.01-1.95 (m, 1H), 1.81-1.57 (m, 7H), 1.53-1.37 (m, 7H), 1.29-1.13 (m, 3H), 1.13-0.90 (m, 2H), 0.89 (s, 3H).
Synthesis of compound SA-C. A solution of SA-B (42.0 g, 153.06 mmol) in 600 mL anhydrous toluene was added dropwise to the methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD) (459.19 mmol, 3.0 eq, freshly prepared) solution under N2 at -78°C. After the addition was completed, the reaction mixture was stirred for 1 hr at -78°C. Then 3.0 M MeMgBr (153.06 mL, 459.19 mmol) was slowly added dropwise to the above mixture under N2 at -78°C. Then the reaction mixture was stirred for 3 hr at this temperature. TLC (Petroleum ether/ethyl acetate = 3:1) showed the reaction was completed. Then saturated aqueous NH4Cl was slowly added dropwise
to the above mixture at -78°C. After the addition was completed, the mixture was filtered, the filter cake was washed with EtOAc, the organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated, purified by flash Chromatography on silica gel (Petroleum ether/ ethyl acetate20:1 to 3:1) to afford compound SA-C (40.2 g, yield: 90.4%) as white powder. 1H NMR: (400 MHz, CDCl3) δ 2.47-2.41 (m, 1H), 2.13-2.03 (m, 1H), 1.96-1.74 (m, 6H), 1.70-1.62 (m, 1H), 1.54-1.47 (m, 3H), 1.45-1.37 (m, 4H), 1.35-1.23 (m, 8H), 1.22-1.10 (m, 2H), 1.10-1.01 (m, 1H), 0.87 (s, 3H).
Synthesis of compound SA-D. To a solution of PPh3EtBr (204.52 g, 550.89 mmol) in THF (500 mL) was added a solution of t-BuOK (61.82 g, 550.89 mmol) in THF (300 mL) at 0°C. After the addition was completed, the reaction mixture was stirred for 1 h 60 °C, then SA-C (40.0 g, 137.72 mmol) dissolved in THF (300 mL) was added dropwise at 60°C. The reaction mixture was heated to 60 °C for 18 h. The reaction mixture was cooled to room temperature and quenched with Sat. NH4Cl, extracted with EtOAc (3*500 mL). The combined organic layers were washed with brine, dried and concentrated to give the crude product, which was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 10:1) to afford compound SA-D (38.4 g, yield:92%) as a white powder. 1H NMR: (400 MHz, CDCl3) δ 5.17-5.06 (m, 1H), 2.42-2.30 (m, 1H), 2.27-2.13 (m, 2H), 1.89-1.80 (m, 3H), 1.76-1.61 (m, 6H), 1.55-1.43 (m, 4H), 1.42-1.34 (m, 3H), 1.33-1.26 (m, 6H), 1.22-1.05 (m, 5H), 0.87 (s, 3H).
Synthesis of compound SA-E. To a solution of SA-D (38.0 g, 125.62 mmol) in dry THF (800 mL) was added dropwise a solution of BH3.Me2S (126 mL, 1.26 mol) under ice-bath. After the addition was completed, the reaction mixture was stirred for 3 h at room temperature (14-20 °C). TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed. The mixture was cooled to 0 °C and 3.0 M aqueous NaOH solution (400 mL) followed by 30% aqueous H2O2 (30%, 300 mL) was added. The mixture was stirred for 2 h at room temperature (14-20 °C), and then filtered, extracted with EtOAc (3*500 mL). The combined organic layers were washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product (43 g , crude) as colorless oil. The crude product was used in the next step without further purification.
Synthesis of compound SA-F. To a solution of SA-E (43.0 g, 134.16 mmol) in dichloromethane (800 mL) at 0 °C and PCC (53.8 g, 268.32 mmol) was added portion wise. Then the reaction mixture was stirred at room temperature (16-22 °C) for 3 h. TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed, then the reaction mixture was filtered, washed with DCM. The organic phase was washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product. The crude product was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 8:1) to afford compound SA-F (25.0 g, yield:62.5%, over two steps) as a white powder. 1H NMR (SA-F): (400 MHz, CDCl3) δ 2.57-2.50 (m, 1H), 2.19-2.11 (m, 4H), 2.03-1.97 (m, 1H), 1.89-1.80 (m, 3H), 1.76-1.58 (m, 5H), 1.47-1.42 (m, 3H), 1.35-1.19 (m, 10H), 1.13-1.04 (m, 3H), 0.88-0.84 (m, 1H), 0.61 (s, 3H).
Synthesis of compound SA. To a solution of SA-F (10 g, 31.4 mmol) and aq. HBr (5 drops, 48% in water) in 200 mL of MeOH was added dropwise bromine (5.52 g, 34.54 mmol). The reaction mixture was stirred at 17 °C for 1.5 h. The resulting solution was quenched with saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (150 mLx2). The combined organic layers were dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (PE: EA=15:1 to 6:1) to afford compound SA (9.5 g, yield: 76.14%) as a white solid. LC/MS: rt 5.4 mm ; m/z 379.0, 381.1, 396.1.

To a suspension of K2CO3 (50 mg, 0.36mmol) in THF (5 mL) was added ethyl 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol ) and SA (50 mg,0.12 mmol). The mixture was stirred at rt for 15h. The reaction mixture was poured in to 5 mL H2O and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified with by reverse-phase prep-HPLC to afford the title compound as a white solid (9mg, 17.4%). 1H NMR (500 MHz, CDCl3), δ (ppm) 7.87 (1H, s),
7.82 (1H, s), 5.02 (1H, AB), 4.92 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dXt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt = 2.24 mm, m/z = 410.1 [M+H]+.
PATENT
WO2020150210
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
References
- ^ Jump up to:a b c d e “Zurzuvae (zuranolone) capsules, for oral use, [controlled substance schedule pending]” (PDF). Archived (PDF) from the original on 5 August 2023. Retrieved 5 August 2023.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t “FDA Approves First Oral Treatment for Postpartum Depression”. U.S. Food and Drug Administration (FDA) (Press release). 4 August 2023. Retrieved 4 August 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “Zuranolone”. DrugBank Online.
- ^ Jump up to:a b Cerne R, Lippa A, Poe MM, Smith JL, Jin X, Ping X, et al. (2022). “GABAkines – Advances in the discovery, development, and commercialization of positive allosteric modulators of GABAA receptors”. Pharmacology & Therapeutics. 234: 108035. doi:10.1016/j.pharmthera.2021.108035. PMC 9787737. PMID 34793859. S2CID 244280839.
- ^ Jump up to:a b Faden J, Citrome L (2020). “Intravenous brexanolone for postpartum depression: what it is, how well does it work, and will it be used?”. Therapeutic Advances in Psychopharmacology. 10: 2045125320968658. doi:10.1177/2045125320968658. PMC 7656877. PMID 33224470.
- ^ Jump up to:a b “SAGE 217”. AdisInsight. Archived from the original on 29 March 2019. Retrieved 10 February 2018.
- ^ Jump up to:a b Blanco MJ, La D, Coughlin Q, Newman CA, Griffin AM, Harrison BL, et al. (2018). “Breakthroughs in neuroactive steroid drug discovery”. Bioorganic & Medicinal Chemistry Letters. 28 (2): 61–70. doi:10.1016/j.bmcl.2017.11.043. PMID 29223589.
- ^ Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. (2017). “Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor”. Journal of Medicinal Chemistry. 60 (18): 7810–7819. doi:10.1021/acs.jmedchem.7b00846. PMID 28753313.
- ^ Saltzman J (4 August 2023). “FDA approves postpartum depression pill from two Cambridge drug firms”. The Boston Globe. Archived from the original on 6 August 2023. Retrieved 5 August 2023.
- ^ Althaus AL, Ackley MA, Belfort GM, Gee SM, Dai J, Nguyen DP, et al. (2020). “Preclinical characterization of zuranolone (SAGE-217), a selective neuroactive steroid GABAA receptor positive allosteric modulator”. Neuropharmacology. 181: 108333. doi:10.1016/j.neuropharm.2020.108333. PMC 8265595. PMID 32976892.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
- ^ “FDA Approves Zurzuvae (zuranolone), the First and Only Oral Treatment Approved for Women with Postpartum Depression, and Issues a Complete Response Letter for Major Depressive Disorder” (Press release). Biogen Inc. 4 August 2023. Retrieved 4 August 2023 – via GlobeNewswire.
- ^ McKenzie H. “Sage Hints at Difficult Decisions After Zuranolone’s Rejection in MDD”.
- ^ Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. (May 2023). “Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial”. The American Journal of Psychiatry: appiajp20220459. doi:10.1176/appi.ajp.20220459. PMID 37132201. S2CID 258461851.
- ^ Deligiannidis KM, Meltzer-Brody S, Gunduz-Bruce H, Doherty J, Jonas J, Li S, et al. (2021). “Effect of Zuranolone vs Placebo in Postpartum Depression: A Randomized Clinical Trial”. JAMA Psychiatry. 78 (9): 951–959. doi:10.1001/jamapsychiatry.2021.1559. PMC 8246337. PMID 34190962.
- ^ Bullock A, Kaul I, Li S, Silber C, Doherty J, Kanes SJ (2021). “Zuranolone as an oral adjunct to treatment of Parkinsonian tremor: A phase 2, open-label study”. Journal of the Neurological Sciences. 421: 117277. doi:10.1016/j.jns.2020.117277. PMID 33387701. S2CID 229333842.
External links
- Clinical trial number NCT04442503 for “A Study to Evaluate the Efficacy and Safety of SAGE-217 in Participants With Severe Postpartum Depression (PPD)” at ClinicalTrials.gov
- Clinical trial number NCT02978326 for “A Study to Evaluate SAGE-217 in Participants With Severe Postpartum Depression” at ClinicalTrials.gov
/////////Zuranolone, FDA 2023, APPROVALS 2023, Zurzuvae, postpartum depression , SAGE 217, SAGE-217, SAGE217
[H][C@@]1(CC[C@@]2([H])[C@]3([H])CC[C@]4([H])C[C@](C)(O)CC[C@]4([H])[C@@]3([H])CC[C@]12C)C(=O)CN1C=C(C=N1)C#N
EIDD-2173, ATI-2173, Fosclevudine alafenamide
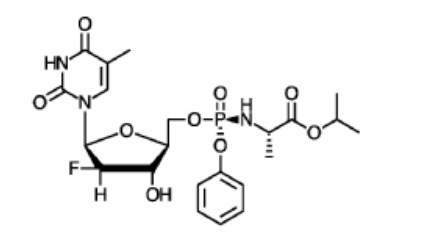

EIDD-2173; also known as ATI-2173
Fosclevudine alafenamide
Phase 2
| Molecular Weight | 529.45 |
|---|---|
| Formula | C22H29FN3O9P |
| CAS No. | 1951476-79-1 |
Hepatitis B virus (HBV) is an infectious disease that targets the liver resulting in either an acute infection, with symptoms arising in 45 to 160 days, or a chronic infection, which 350 million people worldwide are affected by. Estimates indicate that 600,000 deaths occur each year as a result of consequences related to HBV infection. HBV possesses a 3.2- kb relaxed circular DNA (rcDNA) genome that is used to form covalently closed circular DNA (cccDNA) in a host cell. The cccDNA is then transcribed by RNA polymerase II, a host DNA-dependent RNA polymerase, to produce pregenomic RNA (pgRNA). The pgRNA is then used by the virally encoded reverse transcriptase to form rcDNA. The goals of current treatments for chronic HBV infections are to reduce HBV replication and reduce liver damage.
Current treatments for chronic HBV infections include pegylated alpha interferon and nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs). NRTIs are converted to their corresponding 5 ‘-triphosphate, or diphosphate in the case of phosphonate containing NRTIs, and reduce viral replication by inhibiting the HBV encoded polymerase. Clevudine is an NRTI that is no longer being developed for the treatment of chronic HBV because of drug-related skeletal myopathy that was a result of mitochondrial dysfunction in patients.
Interestingly, clevudine triphosphate has been shown to be a competitive nonsubstrate inhibitor of the HBV encoded polymerase, and due to its long intracellular half-life, is able to suppress HBV replication for an extended period of time after drug withdrawal.
The discovery and synthesis of the (S,S) and (S,R) diastereomers of clevudine phosphoramidate has been previously reported. These studies were undertaken to address the myopathy concerns associated with clevudine. The phosphoramidate moiety was utilized to deliver clevudine, as its 5 ‘-monophosphate, to the liver reducing 1) systemic exposure to clevudine and 2) the possibility of skeletal myopathy. Both phosphoramidates showed anti-HBV activity similar to clevudine with the (S,S) diastereomer being slightly more potent.
SYN
See U.S. Patent No. 10,683,319.
PATENT
WO20 17223421
PATENT
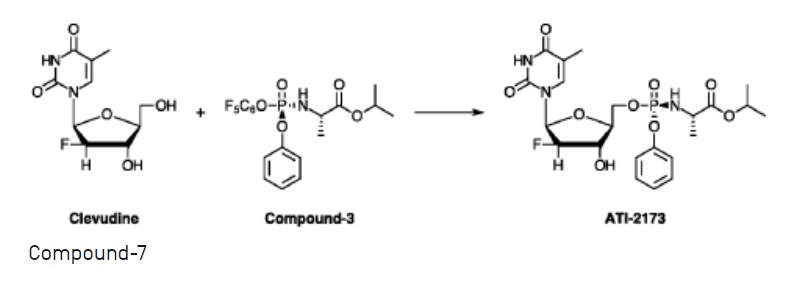

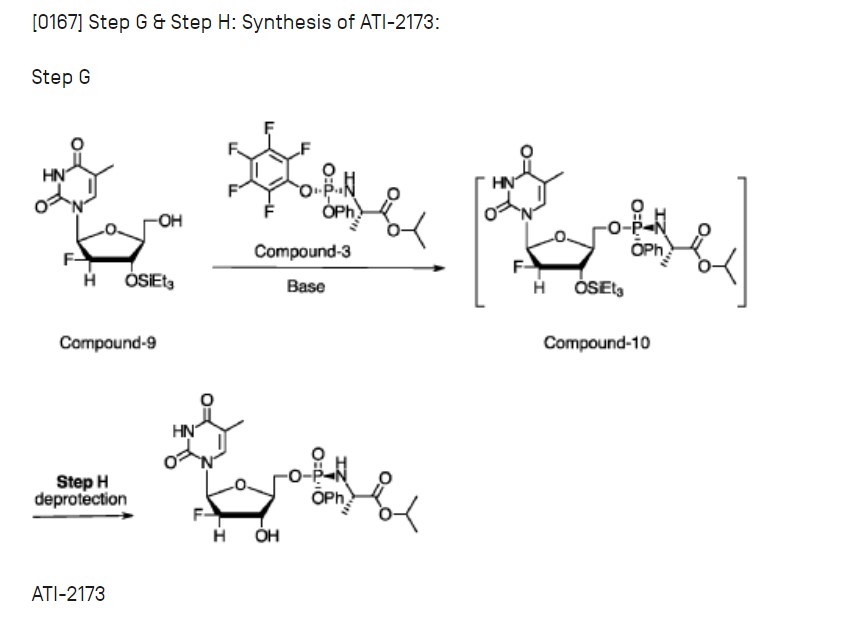

EXAMPLE 11 :
[0374] Preparation of intermediate ATI-2173 from Compound-10.
[0375] Experimental Procedure
[0376] (H-l) Crude Compound-10 in THF (see Example 10) was divided into three aliquots and stirred with 2% HC1 aq. solution at pH 5-6, 4-5, and 3-4; for 16 h at 20-25 °C; the aliquots were combined and stirred at 15-20 °C for 72 h, with no degradation of ATI-2173 observed over the second time period;
[0377] (H-2) the pH of the mixture was adjusted to 7 with 7% NaHCCh;
[0378] (H-3) phase separation was carried out using 2-MeTHF, the organic phase was washed with NA2SO4 aqueous soltion, then concentrated to 1-3 V, and MTBE (5 V) was added; this operation was repeated twice;
[0379] (H-5) ATI-2173 was precipitated gradually upon addition of seed crystal and addition of n-heptane (5 V);
[0380] (H-6) the product was filtered; and
[0381] (H-7) the wetcake was dried, resulting in ATI-2173 in 99.73a% purity and
84.4% yield.
[0382] EXAMPLE 12:
[0383] Crystallization of ATI-2173
[0384] Initial studies examined the use of single or mixed solvent systems to crystalize the amorphous product, ATI-2173. Several solvent conditions were screened, including single solvent and mixed solvent systems, in order to determine the potential for obtaining a crystalline material from the amorphous material. None of the solvents tested worked and all conditions produced an oil product. The results are shown below in Tables 8 and 9.
[0385] Table 8: Single Solvent Systems
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
////////// EIDD-2173, ATI-2173, EIDD 2173, ATI 2173, Hepatitis B virus, ANTI HBV, Fosclevudine alafenamide, PHASE 2

NEW DRUG APPROVALS
ONE TIME
$10.00
Palovarotene

Palovarotene
CAS 410528-02-8
4-[(E)-2-[5,5,8,8-tetramethyl-3-(pyrazol-1-ylmethyl)-6,7-dihydronaphthalen-2-yl]ethenyl]benzoic acid
FDA 8/16/2023
To reduce the volume of new heterotopic ossification in adults and pediatric patients (aged 8 years and older for females and 10 years and older for males) with fibrodysplasia ossificans progressiva
- RG-667
- RO-3300074
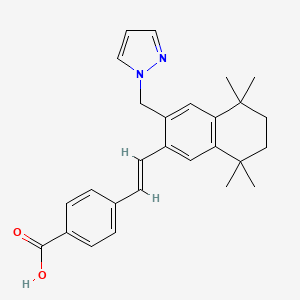
Palovarotene, sold under the brand name Sohonos, is a medication used for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva.[4][5] It is a highly selective retinoic acid receptor gamma (RARγ) agonist.[6]
It was approved for medical use in Canada in June 2022,[4] and in the United States in August 2023.[5]
Medical uses
Palovarotene is indicated for the treatment of heterotopic ossification and fibrodysplasia ossificans progressiva.[4][5]
History
Palovarotene is a retinoic acid receptor gamma (RARγ) agonist licensed to Clementia Pharmaceuticals from Roche Pharmaceuticals. At Roche, palovarotene was evaluated in more than 800 individuals including healthy volunteers and patients with chronic obstructive pulmonary disease (COPD).[7] A one-year trial did not demonstrate a significant benefit on lung density in moderate-to-severe emphysema secondary to severe α(1)-antitrypsin deficiency.[8]
In 2011, animal studies demonstrated that RARγ agonists, including palovarotene, blocked new bone formation in both an injury-induced mouse model of heterotopic ossification (HO) and a genetically modified biological mouse model of fibrodysplasia ossificans progressiva containing a continuously active ACVR1/ALK2 receptor in a dose-dependent manner.[9][10] A 2016 study demonstrated that palovarotene also inhibited spontaneous heterotopic ossification, maintained limb mobility and functioning, and restored skeletal growth in fibrodysplasia ossificans progressiva mouse models.[11]
Society and culture
Legal status
Palovarotene is being developed by Ipsen Biopharmaceuticals and was granted priority review and orphan drug designations by the United States Food and Drug Administration (FDA) for the treatment of fibrodysplasia ossificans progressiva[12][13] and orphan medicinal product designation by the European Medicines Agency (EMA) in 2014.[14][15][16][17] Phase II clinical studies failed to show a significant change in heterotopic bone volume, the main outcome measure, but prompted further investigation in a phase III clinical trial.[18] In December 2022, the FDA declined to approve palovarotene for the fibrodysplasia ossificans progressive without additional clinical trial data.[19] In January 2023, the European Medicines Agency (EMA) recommended the refusal of the marketing authorization for palovarotene for the treatment of fibrodysplasia ossificans progressiva.[20]
Research
Phase II
Clementia submitted a new drug application for palovarotene for the treatment of fibrodysplasia ossificans progressiva after observing positive phase II results.[21]
Phase III
In December 2019, Ipsen issued a partial clinical hold for people under the age of 14, due to reports of early fusion of growth plates.[22] Ipsen acquired Clementia in 2019.[23]
SYN
J. Med. Chem. 2025, 68, 2147−2182
Palovarotene (Sohonos). Palovarotene (7) is a selective retinoic acid receptor γ (RARγ) agonist that was
developed for the treatment of fibrodysplasia ossificans progressiva (FOP), a very rare autosomal dominant disorder, impacting ∼1 in2million individuals worldwide. 54,55 This orally bioavailable agonist reduces the incidence of heterotopic ossification in patients with FOP and was developed by the
French biopharmaceutical company Ipsen. 56 The small Molecule agonist was originally developed by Roche for a different indication, and was later licensed to Clementia Pharmaceuticals, which was ultimately acquired by Ipsen.
AlthoughapprovedbytheUSFDAinAugust2023,palovarotene was first approved by Health Canada in January 2022 for patients with FOP inadults andchildren aged 10 years and older for males and aged 8 years and older for females. With respect to pharmacodynamics, the agonist binds to RARγ and thus inhibits bone morphogenetic protein and Smad 1/5/8 signaling.57 This signaling inhibition permits normal muscle tissue repair and ultimately reduces the incidence of heterotopic ossification. A robust kilogram-scale synthesis of palovarotene has been disclosed in a patent by Roche and is depicted in Scheme 11.58
Starting from 2,5-dimethyl-2,5-hexanediol (7.1), the two tertiary alcohols were chlorinated with concentrated hydro chloric acid in toluene. Without isolation, the resulting

(54) Wentworth, K. L.; Masharani, U.; Hsiao, E. C. Therapeutic
advances for blocking heterotopic ossification in fibrodysplasia
ossificans progressiva. Br. J. Clin. Pharmacol. 2019, 85, 1180−1187.
(55) Semler, O.; Rehberg, M.; Mehdiani, N.; Jackels, M.; Hoyer
Kuhn, H. Current and emerging therapeutic options for the
management of rare skeletal diseases. Paediatr. Drugs 2019, 21, 95−
106.
(56) Hoy, S. M. Palovarotene: first approval. Drugs 2022, 82, 711−
716.
(57) Pignolo, R. J.; Pacifici, M. Retinoid agonists in the targeting of
heterotopic ossification. Cells 2021, 10, 3245.
(58) Martin, M. Process for preparing retinoid compounds. US
20070232810, 2007.

.
SYN
Desjardins, C., Grogan, D. R., Packman, J. N., & Harnett, M. (2017). Methods for treating heterotopic ossification (WO2017210792A1). World Intellectual Property Organization. https://patents.google.com/patent/WO2017210792A1
Chemical Communications (Cambridge, United Kingdom) (2019), 55(38), 5420-5422
WO2014105446
US20070232810
Patent
https://patents.google.com/patent/WO2002028810A3/en
WO2002028810
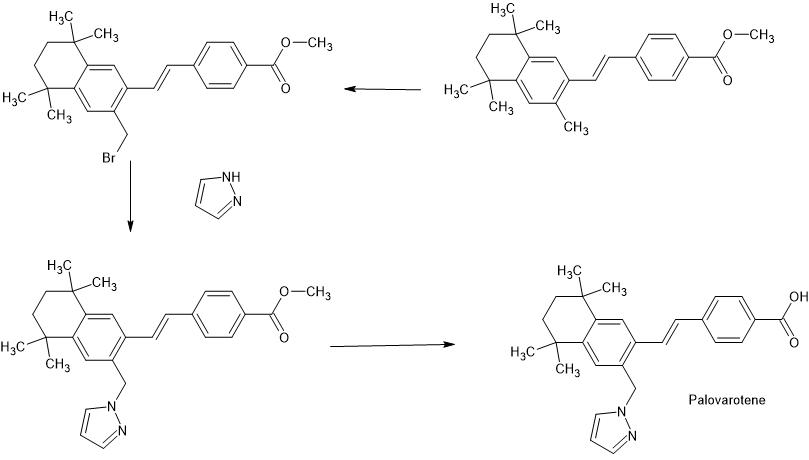

XAMPLE 12: PREPARATION OF 4-r(E)-2-(5,5.8.8-TETRAMETHYL-3-PYRAZOL-l-YLMETHYL -5.6.7.8-TETRAHYDRO-NAPHTHALEN-2-YL VINYLl BENZOIC ACID (6)
A mixture of 2.0 g (4.5 mmol) of (E)- methyl-4-[2-(3-bromomethyl-5,5,8,8-tetramethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 0.65 g (9.5 mmol) of pyrazole in 15 mL of N-methyl pyrrolidine was heated at 100°. After 2 hours, the reaction mixture was cooled to room temperature, poured into brine and extracted with ethyl acetate. The organic extracts were washed with brine, dried over sodium sulfate and concentrated under reduced pressure. The residue was stirred with hexane and the product was filtered off, washed with hexane and dried to give 1.6 g (83%) of methyl-4-[2-(5,5,8,8-Tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate (M+ = 429).
A mixture of 27.6 g (64.4 mmol) of methyl-4-[2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoate and 97 mL (193 mmol) of 2 N sodium hydroxide in 300 mL of ethyl alcohol was heated at reflux. After 1 hour, the reaction mixture was cooled to room temperature and diluted with 900 mL of water. The reaction mixture was acidified with 2 N HCl and the product was isolated by filtration, washed with water and pentane and dried to give 25.9 g (97%) of 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid (m.p. = 246.5-248 °C) 6.
Proceeding as described in the example above but substituting pyrazole with pyrrole, 4-methylpyrazole, 1,2,4-triazole, moφholine, 2-pyrrohdone, 3,5-dimethylpyrzole,
δ – valerolactone, 2-methyhmidazole and 4-methylimidzole gave 4-[(E)-2-(5,5,8,8-tetramethyl-3-pyrrol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 7, 4-{(E)-2-[5,5,8,8-Tetramemyl-3-(4-methylpyrazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 20, 4-[(E)-2-(5,5,8,8-Tetxamethyl-3-[l,2,4]triazol-l-ylmethyl-5,6,7,8Jetrahydro-naphthalen-2-yl]vinyl}benzoic acid 39, 4-[(E)-2-(5,5,8,8-tetramethyl-3-moφhohn-4-ylmethyl- 5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 138, 4-[(E)-2-(5,5,8,8-tetramethyl-3- (2-oxo-pyrrohdin-l-yl-methyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 139, 4-{(E)-2-[5,5,8,8-Tetramet yl-3-(3,5-mmemylpyτazol-l-yhnethyl-5,6,7,8-tetrahydro-napn^ 2-yl)vinyl]benzoic acid 143, 4-[(E)-2-(5,5,8,8-tetramethyl-3-(2-oxo-piperidin-l-yl-methyl-5,6,7,8-tetrahydro-naρhthalen-2-yl)vinyl]benzoic acid 146 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(2-methyhmidazol-l-ylmethyl-5,6,7,8-tetrahydro-naphthalen-2-yl)vinyl]benzoic acid 149and 4-{(E)-2-[5,5,8,8-Tetramethyl-3-(4-methyhmidazol-l-ylmethyl-5,6,7,8-tettahydro-naphthalen-2-yl)vinyl]benzoic acid 150 respectively.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////
| Clinical data | |
|---|---|
| Trade names | Sohonos |
| Other names | R-667, RG-667 |
| License data | US DailyMed: Palovarotene |
| Routes of administration | By mouth |
| Drug class | Retinoic acid receptor gamma agonist |
| ATC code | M09AX11 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1][2][3][4]US: ℞-only[5] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 410528-02-8 |
| PubChem CID | 10295295 |
| DrugBank | DB05467 |
| ChemSpider | 8470763 |
| UNII | 28K6I5M16G |
| KEGG | D09365 |
| ChEBI | CHEBI:188559 |
| Chemical and physical data | |
| Formula | C27H30N2O2 |
| Molar mass | 414.549 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References[
- ^ “Notice: Multiple Additions to the Prescription Drug List (PDL) [2022-01-24]”. Health Canada. 24 January 2022. Archived from the original on 29 May 2022. Retrieved 28 May 2022.
- ^ “Summary Basis of Decision – Sohonos”. Health Canada. 23 October 2014. Archived from the original on 6 August 2022. Retrieved 6 August 2022.
- ^ “Sohonos product information”. Health Canada. 20 June 2022. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ Jump up to:a b c d “Sohonos Product Information”. Health Canada. 22 October 2009. Archived from the original on 18 August 2023. Retrieved 17 August 2023.
- ^ Jump up to:a b c d “Archived copy” (PDF). Archived (PDF) from the original on 18 August 2023. Retrieved 18 August 2023.
- ^ “Health Canada Approves Ipsen’s Sohonos (palovarotene capsules) as the First Approved Treatment for Fibrodysplasia Ossificans Progressiva” (Press release). Ipsen. 24 January 2022. Retrieved 28 May 2022 – via Business Wire.
- ^ Hind M, Stinchcombe S (November 2009). “Palovarotene, a novel retinoic acid receptor gamma agonist for the treatment of emphysema”. Current Opinion in Investigational Drugs. 10 (11): 1243–50. PMID 19876792.
- ^ Stolk J, Stockley RA, Stoel BC, Cooper BG, Piitulainen E, Seersholm N, et al. (August 2012). “Randomised controlled trial for emphysema with a selective agonist of the γ-type retinoic acid receptor”. The European Respiratory Journal. 40 (2): 306–12. doi:10.1183/09031936.00161911. PMID 22282548.
- ^ Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, et al. (April 2011). “Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists”. Nature Medicine. 17 (4): 454–60. doi:10.1038/nm.2334. PMC 3073031. PMID 21460849.
- ^ Kaplan FS, Shore EM (April 2011). “Derailing heterotopic ossification and RARing to go”. Nature Medicine. 17 (4): 420–1. doi:10.1038/nm0411-420. PMC 4913781. PMID 21475232.
- ^ Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al. (September 2016). “Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation”. Journal of Bone and Mineral Research. 31 (9): 1666–75. doi:10.1002/jbmr.2820. PMC 4992469. PMID 26896819.
- ^ “Ipsen announces FDA Priority Review for NDA in patients with FOP”. Ipsen (Press release). 24 August 2022. Retrieved 28 January 2023.
- ^ “Palovarotene Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 January 2013. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ “EU/3/14/1368”. European Medicines Agency (EMA). 17 September 2018. Archived from the original on 27 January 2023. Retrieved 28 January 2023.
- ^ “Public summary of opinion on orphan designation. Palovarotene for the treatment of fibrodysplasia ossificans progressiva” (PDF). European Medicines Agency (EMA). Archived (PDF) from the original on 22 April 2016. Retrieved 11 April 2016.
- ^ “Clementia Pharmaceuticals Receives Fast Track Designation for Palovarotene for Treatment of Fibrodysplasia Ossificans Progressiva (FOP)” (Press release). Clementia Pharmaceuticals. 1 December 2014. Retrieved 11 April 2016 – via PR Newswire.
- ^ “Clementia Pharmaceuticals Receives EMA Orphan Medicinal Product Designation for Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva” (Press release). Clementia Pharmaceuticals. 21 November 2014. Retrieved 11 April 2016 – via PR Newswire.
- ^ Pignolo RJ, Baujat G, Hsiao EC, Keen R, Wilson A, Packman J, et al. (October 2022). “Palovarotene for Fibrodysplasia Ossificans Progressiva (FOP): Results of a Randomized, Placebo-Controlled, Double-Blind Phase 2 Trial”. Journal of Bone and Mineral Research. 37 (10): 1891–1902. doi:10.1002/jbmr.4655. PMC 9804935. PMID 35854638. S2CID 250697248.
- ^ “FDA Tells Ipsen It Won’t Approve Palovarotene for FOP”. Global Genes. 27 December 2022. Archived from the original on 29 January 2023. Retrieved 28 January 2023.
- ^ “Sohonos: Pending EC decision”. European Medicines Agency (EMA). 26 January 2023. Archived from the original on 27 January 2023. Retrieved 28 January 2023.
- ^ “Clementia Announces Plan to Submit a New Drug Application for Palovarotene for the Treatment of FOP Based on Positive Phase 2 Results”. 23 October 2018. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
- ^ “Ipsen Initiates Partial Clinical Hold for Palovarotene IND120181 and IND135403 Studies”. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
- ^ “Ipsen Completes Acquisition of Clementia Pharmaceuticals”. Archived from the original on 15 December 2019. Retrieved 15 December 2019.
External links
Clinical trial number NCT03312634 for “An Efficacy and Safety Study of Palovarotene for the Treatment of Fibrodysplasia Ossificans Progressiva. (MOVE)” at ClinicalTrials.gov
/////////FDA 2023, APPROVALS 2023, Palovarotene, Sohonos, RG-667, RO-3300074
syn
syn
European Journal of Medicinal Chemistry 265 (2024) 116124
Palovarotene (Sohonos)
On February 17, 2022, the FDA granted approval to Palovarotene for the treatment of heterotopic ossification (HO) linked to fibrodysplasia ossificans progressiva (FOP) [64]. FOP, or myositis ossificans pro
gressiva (MOP), is an uncommon hereditary condition marked by atypical bone growth in regions beyond the usual skeletal structure. It is commonly accompanied by recurring episodes of discomfort and abrupt
swelling of soft tissues. This disorder causes restricted mobility and fusion of joints, leading to deformities, limited movement, and premature mortality [65]. Palovarotene is an orally available retinoic acid receptor γ (RARγ) agonist [66]. Palovarotene specifically attaches to RARγ and hinders the phosphorylation process of mothers against decapentaplegic homolog (SMAD)1/5/8. This action results in the suppression of the bone morphogenetic protein (BMP)/ALK2 downstream signaling pathway, leading to a decrease in ALK2/SMAD-dependent chondrogenesis and osteoblast differentiation. Consequently, the over all effect is a reduction in endochondral ossification [67].
The preparation of Palovarotene is shown in Scheme 18 [68].Starting with 2,5-dimethylhexane-2,5-diol (PALO-001), a nucleophilic substitution reaction with HCl, followed by AlClpromoted Friedel-Crafts alkylation with 1-bromo-2-methylbenzene (PALO-003), gave PALO-004. PALO-005 was obtained by substitution with CuCN.The cyano group of PALO-005 was reduced to aldehyde by diisobutylalumium hydride (DIBAL-H) to obtain PALO-006. PALO-006 was subjected to Wittig-Horner reaction with methyl 4-((dimethoxyphosphoryl)methyl)benzoate PALO-007 to obtain olefin PALO-008.
PALO-008 was brominated with N-bromosuccinimide (NBS) to obtain PALO-009. PALO-009 was nucleophilic substituted with 1H-pyrazole (PALO-010) to obtain PALO-011, which was hydrolyzed under alkaline conditions to obtain the final product Palovarotene.
[64] S.M. Hoy, Palovarotene: first approval, Drugs 82 (2022) 711–716.
[65] R.J. Pignolo, E.M. Shore, F.S. Kaplan, Fibrodysplasia ossificans progressiva:
diagnosis, management, and therapeutic horizons, Pediatr. Endocrinol. Rev. 2
(2013) 437–448.
[66] G.J. Pavey, A.T. Qureshi, A.M. Tomasino, C.L. Honnold, D.K. Bishop, S. Agarwal,
S. Loder, B. Levi, M. Pacifici, M. Iwamoto, B.K. Potter, T.A. Davis, J.A. Forsberg,
Targeted stimulation of retinoic acid receptor-γ mitigates the formation of
heterotopic ossification in an established blast-related traumatic injury model,
Bone 90 (2016) 159–167.
[67] H. Kitoh, Clinical aspects and current therapeutic approaches for FOP,
Biomedicines 8 (2020) 325.
[68] J.-M. Lapierre, D.M. Rotstein, E.B. Sjogren, Preparation of New Retinoids for the
Treatment of Emphysema, Cancer and Dermatological Disorders, 2002.
WO2002028810.


NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}