
Home » 2016 (Page 36)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
PF-05387552
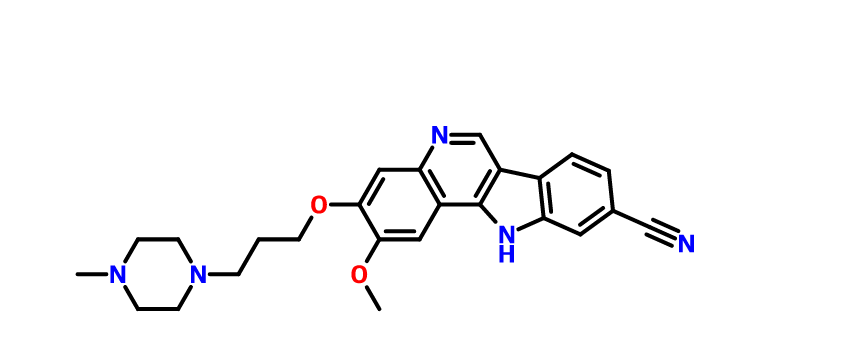
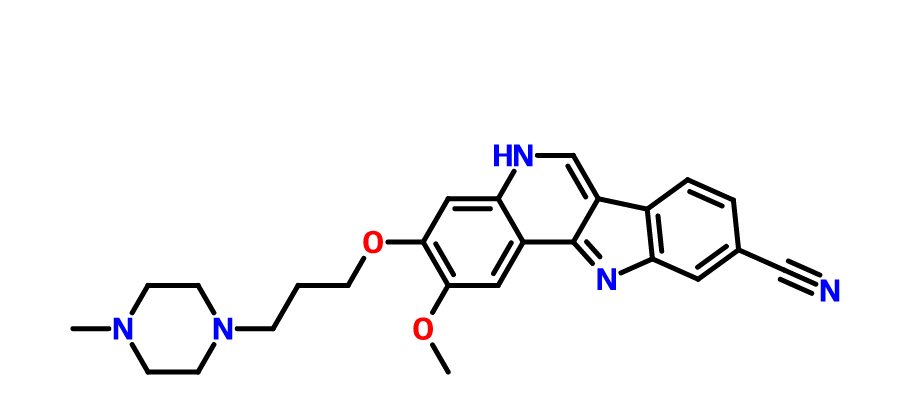
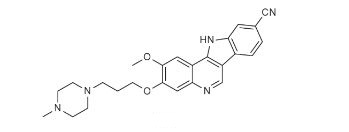

PF-05387552
IRAK4
- Molecular Weight429.51
| Molecular Formula: | C25H27N5O2 |
|---|---|
| Molecular Weight: | 429.51418 g/mol |
![]()
Synthesis
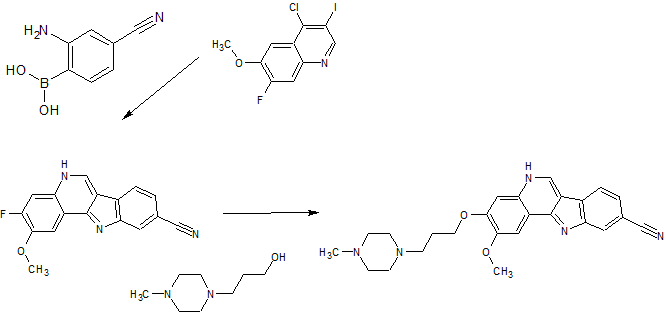
PAPER
Bioorganic & Medicinal Chemistry Letters (2014), 24(9), 2066-2072
Volume 24, Issue 9, 1 May 2014, Pages 2066–2072

Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4
- L. Nathan Tumeya, , ,
- Diane H. Boschellia,
- Niala Bhagiratha,
- Jaechul Shima,
- Elizabeth A. Murphyb,
- Deborah Goodwinb,
- Eric M. Bennettc,
- Mengmeng Wangd,
- Lih-Ling Linb,
- Barry Pressa,
- Marina Shenb,
- Richard K. Frisbiea,
- Paul Morganb,
- Shashi Mohanb,
- Julia Shinb,
- Vikram R. Raob
- b Pfizer Global R&D, 200 Cambridge Park Dr., Cambridge, MA 02140, USA
- c Pfizer Global R&D, 87 Cambridgepark Dr., Cambridge, MA 02140, USA
- d Pfizer Global R&D, 1 Burtt Rd., Andover, MA 01810, USA
http://www.sciencedirect.com/science/article/pii/S0960894X14002832?np=y

IRAK4 is responsible for initiating signaling from Toll-like receptors (TLRs) and members of the IL-1/18 receptor family. Kinase-inactive knock-ins and targeted deletions of IRAK4 in mice cause reductions in TLR induced pro-inflammatory cytokines and these mice are resistant to various models of arthritis.
Herein we report the identification and optimization of a series of potent IRAK4 inhibitors. Representative examples from this series showed excellent selectivity over a panel of kinases, including the kinases known to play a role in TLR-mediated signaling. The compounds exhibited low nM potency in LPS- and R848-induced cytokine assays indicating that they are blocking the TLR signaling pathway.
A key compound (26) from this series was profiled in more detail and found to have an excellent pharmaceutical profile as measured by predictive assays such as microsomal stability, TPSA, solubility, and c log P. However, this compound was found to afford poor exposure in mouse upon IP or IV administration. We found that removal of the ionizable solubilizing group (32) led to increased exposure, presumably due to increased permeability. Compounds 26 and 32, when dosed to plasma levels corresponding to ex vivo whole blood potency, were shown to inhibit LPS-induced TNFα in an in vivo murine model.
To our knowledge, this is the first published in vivo demonstration that inhibition of the IRAK4 pathway by a small molecule can recapitulate the phenotype of IRAK4 knockout mice.

 L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
L. Nathan Tumey, Ph.D., Principal Research Scientist, Pfizer Global R&D
REFERENCES

///////////TLR signaling, Indoloquinoline, IRAK4, Kinase inhibitor, Inflammation, PF-05387552, PF 05387552, 1604034-71-0
N#Cc3ccc4c5cnc2cc(OCCCN1CCN(C)CC1)c(OC)cc2c5nc4c3
ETAMICASTAT

Etamicastat HCl salt
CAS: 677773-32-9 (HCl salt)
CAS 760173-05-5 (free base).
Chemical Formula: C14H16ClF2N3OS
Molecular Weight: 347.8088
Synonym: BIA 5-453; BIA5-453; BIA-5-453; Etamicastat
IUPAC/Chemical Name: (R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydro-2H-imidazole-2-thione hydrochloride
5-(2-Aminoethyl)-1-((3R)-6,8-difluoro-3,4-dihydro-2H-chromen-3-yl)-1,3-dihydro-2h-imidazole-2-thione
R)-5-(2-aminoethyl)-1-(6,8-difluorochroman-3-yl)-1,3-dihydroimidazole-2-thione hydrochloride,
PHASE 2, Treatment of Heart Failure Therapy, Hypertension
Bial-Portela and Ca, S.A

is a novel peripherally selective dopamine β-hydroxylase (DBH) inhibitor being developed by Bial-Portela and Ca, S.A. for treatment of hypertension and congestive heart failure.(1) The compound was shown to be well tolerated in healthy volunteers.
Etamicastat, also known as BIA 5-453, is a potent, reversible, peripherally selective dopamine β-hydroxylase inhibitor (DBH inhibitor). Chronic dopamine ß-hydroxylase inhibition with etamicastat effectively decreases blood pressure, although does not prevent the development of hypertension in the spontaneously hypertensive rat.

aReagents and conditions: a) Boc2O, EtOH, rt, 2 h; b) TBDMS-Cl, Et3N, DMAP, DCM, rt, 18 h; c) Dess–Martin periodinane, DCM, rt, 1 h; d) 2, KSCN, AcOH, EtOAc, reflux, 7 h; e) 2 N HCl, EtOAc, rt, 2 h.
Paper
Development of the Asymmetric Hydrogenation Step for Multikilogram Production of Etamicastat

The asymmetric hydrogenation of methyl (6,8-difluoro-2H-chromen-3-yl)carbamate is a key step in the manufacturing route to etamicastat. A development of this step including the ruthenium or rhodium catalyst screening and the influence of the catalyst preparation (isolated, preformed in solution or in situ), solvent, temperature, pressure, additive, and concentration on the performance of the given ligand was discussed. Scale-up experiments for the best catalysts under optimized conditions were described.
PAPER
J Med Chem 2006, 49(3): 1191
in the processes .
(J?) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3-yl) -1, 3-dihydroimidazole-2 -thione hydrochloride (the compound of formula 1, below) is a potent, non-toxic and peripherally selective inhibitor of ϋβΗ, which can be used for treatment of certain cardiovascular disorders. Compound 1 is disclosed in WO2004/033447 , along with processes for its preparation.

1
The process disclosed in WO2004/033447 involves the reaction of ( R) – 6 , 8 -difluorochroman-3 -ylamine hydrochloride (the structure of ( R) -6, 8-difluorochroman-3 -ylamine is shown below as compound QA) , [4 – ( tert-butyldimethylsilanyloxy) -3 -oxobutyl] carbamic acid tert-butyl ester and potassium thiocyanate .

QA
(R) -6 , 8-difluorochroman- 3 -ylamine (compound QA) is a key intermediate in the synthesis of compound 1. The stereochemistry at the carbon atom to which the amine is attached gives rise to the stereochemistry of compound 1, so it is advantageous that compound QA is present in as pure enantiomeric form as possible. In other words, the (R) -enantiomer of compound QA should be in predominance, with little or no (S) enantiomer present. Thus, the process for preparing compound QA will advantageously produce compound QA with as high enantiomeric excess (ee) as possible.
Advantageous processes for preparing, for example, the compound of formula QA have now been found. In one aspect, the processes involve a biotransformation step. In another aspect, the processes involve chemical transformation. The processes may also be employed in the preparation of similar precursors useful in the production of other peripherally-selective inhibitors of dopamine -β -hydroxylase .
WO2008/136695 discloses a compound of formula YA, its (R) or (S) enantiomer, a mixture of its (R) and (S) enantiomers, or pharmaceutically acceptable salts thereof.

YA
The (R) -enantiomer of the compound of formula YA has been found to be a potent dopamines-hydroxylase inhibitor having high potency and significantly reduced brain access.
As disclosed in WO2008/136695 , the compound of formula YA may be prepared by reacting the compound of formula 1 with benzaldehyde under reductive alkylation conditions. In particular, (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) – 1 , 3 -dihydroimidazole-2 -thione and benzaldehyde may be reacted in the presence of a solvent or mixture of solvents, and a reducing agent such as sodium cyanoborohydride or sodium triacetoxyborohydride .
The compound of formula W may be prepared using a process as disclosed herein from the nitro chromene compound M.
The compound of formula WA may also be prepared using a process comprising bromination of 2 , 4 -difluorophenol to give bromophenol, alkylation of bromophenol with 4 -chloro-3 -oxo butanoate to give ketone followed by cyclization and decarboxylation to produce compound WA.

WA
According to an aspect of the present invention, there is provided the following 2 -part synthetic route from the starting material 2 , 4 -difluorophenol to (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) -1 , 3 -dihydroimidazole-2 – thione
hydrochloride :
Part (1)


Preferred reagents and conditions:
a) HMTA, CF3COOH, 115°C, 18 hours
b) CH2CHCN, DABCO, DMF, water, 70°C, 16 hours
c) H2S04, AcOH, 100°C, 1 hour
d) NaClO, NaOH, MeOH, 25°C, 24 hours
e) (R) -C3 -TunePhosRu (acac) 2 S/C 3000, 30 bar H2, MeOH, 80°C, 20 hours
f) Water, 2-propanol, reflux to 20°C
g) 40% KOH, MeOH, reflux, 24 hours
h) L-tartaric acid, ethanol, water, RT, 1 hour
Part (2)

![]()

Preferred reagents and conditions
a’) methyl vinyl ketone, t-BuONa, EtOAc, EtOH, 40-50°C, 2-3 hours
Br2, MeOH, 20-25°C, 5 hours
water, reflux, 1 hour
KOH, AcOH, reflux, 1 hour
HCl, water, 2-propanol, 75 °C, 4 hours
KSCN, AcOH, 100°C, 2-4 hours
NaHC03, water, EtOH
NaBH4, 2-propanol, THF, water, 20-25°C, 16 hours
HCl, 2-propanol, water, reflux, 1-2 hours
The ( R ) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3 -yl) -1,3-dihydroimidazole-2 – thione hydrochloride
EXAMPLES
Example 1
Nitro chromene synthesis

To 3 , 5-difluoro-2-hydroxybenzaldehyde (lOg, 63mmol, leq) , di-n-butylamine (4.1g, 32mmol, 0.5eq) , phtalic anhydride (18.7g, 126mmol, 2eq) in toluene (500mL) was added nitroethanol (5.75g, 63mmol, leq) . The round bottomed flask fitted with a dean stark apparatus was refluxed for 18h. The mixture was cooled and nitroethanol (5.75g, 63mmol, leq) was added. The resulting reaction mixture was then reflux for 12h. After cooling, the solution was evaporated down to approximately 150mL and purified over silica gel (eluent ethyl acetate : hexane 1:1) this gave several fractions that contained only the product by TLC, these was evaporated under reduced pressure to yield 1.8g which was 100% pure by HPLC aera. Several more fractions were collected containing a mixture of product and starting material. These were combined and washed with 2% NaOH solution (2x50mL) to remove starting material. The organic layer was washed with water (50mL) , dried over sodium sulfate and evaporated under reduced pressure to give 2.49g of brown solid ( 100% pure by HPLC aera) . More fractions were collected. These were combined, washed with 2% NaOH solution (3xl00mL) , water (lOOmL) and dried over sodium sulfate. This was then filtered and evaporated down in vacuum to yield 6.14g of a brown solid which was 91.3% pure by HPLC aera. 6 , 8 -difluoro-3 -nitro-2H-chromene (9.90g, 73.4%) was obtained as a brown solid.
Example 2
Nitro chromene synthesis with column purification
To a solution of isobenzofuran-1 , 3 -dione (4,68 g, 31,6 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (2,5 g, 15,81 mmol) in Toluene (25 ml) was added 2 -nitroethanol (2,88 g, 31,6 mmol). The resulting mixture was heated to reflux overnight (Dean stark) .
The reaction conversion was checked by TLC (eluent PE/EtOAc 9:1) . A yellow spot was observed and corresponds to the expected product .
Reaction was cooled to room temperature and a plug of silica gel was performed. A pale brown solid (3.9g) was obtained. “””H-NMR showed presence of product and starting material. The solid was dissolved in diethylether and the organic layer was washed with aqueous sodium carbonate, dried over Na2S04, filtered and concentrated under reduced pressure. A pale brown solid (1.7g,) was obtained. The 1H-NMR was indicated no starting material but still polymer from nitroethanol and residue of phtalic anhydride. A second silica plug (eluent: PE/EtOAc 95:5) was done. A pale yellow solid (1.5g) was obtained. 1H-NMR of solid showed only product and polymer. The solid was recrystallized from methanol/water . A pale yellow solid (1.05g, 31.2%) was obtained.
Example 3
Nitro chromene synthesis without column purification
To a solution of isobenzofuran- 1 , 3 -dione (18,74 g, 127 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (10 g, 63,3 mmol) in Toluene (100 ml) was added 2 -nitroethanol (6,86 ml, 95 mmol) . The resulting mixture was heated to reflux for 24h (Dean stark) .
The reaction conversion was checked by HPLC and by 1H-NMR. Only 50% conversion was obtained.
The reaction mixture was cooled to room temperature and diluted with DCM (lOOmL) and 1M NaOH solution (200mL) .
The biphasic system was stirred for 30 minutes and then separated (very difficult to see phase separation) . The aqueous layer was washed with DCM (50mL) and the combined organic layers were washed twice with water (2x50ml) , dried over sodium sulfate. The filtered organic layer was concentrated under reduced pressure. To the residue was added methanol (50mL) . The methanol was then removed by distillation under reduced pressure. A brown solution precipitated when most of the methanol was removed. More methanol was added and more solid crushed out then few drops of water was added to increase the product precipitation. The brown slurry was stirred for 30 minutes and filtered. The brown solid was washed with methanol/water (1:9, 5mL) and dried in a vacuum oven at 40°C for 12h.6, 8-difluoro-3 -nitro-2H-chroraene (4,9 g, 22,99 mmol,) was obtained as brown solid in 36.3% yield.
HPLC showed a purity of 98% and 1H-NMR confirmed the structure and purity around 95%
Example 4
Reduction of nitro chromene to nitro-alkane (racemic mixture)

To a suspension of 6 , 8 -difluoro-3 -nitro-2H-chromene (213mg, 0,999 mmol) and silica (0,8 g, 0,999 mmol) in a mixture of CHC13 (10 ml) and IPA (3,4 ml) at 0°C was added portion wise sodium borohydride (95 mg, 2,498 mmol). The resulting mixture was stirred at 0°C for 45 minutes. Reaction conversion was checked by HPLC. 1 mL of acetic acid was added at 0°C and the resulting mixture was stirred for 30 minutes at room temperature. The slurry was filtered and the silica was washed with DCM. The filtrate was diluted with ethyl acetate and water and the biphasic system was separated. The aqueous layer was back extracted with ethyl acetate. The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated under reduced pressure.
6 , 8-difluoro-3 -nitrochroman (196mg, 0,911 mmol, 91 % yield) was obtained as a pale yellow oil.
Example 5
Preparation of 6 , 8 -difluorochroman-3 -one from nitro chromene

A solution of 6, 8-difluoro-3 -nitro-2H-chromene (lOOmg, 0,469 mmol) in acetic acid (0.5 ml) is added slowly to a stirred slurry of iron (262 mg, 4,69 mmol) in acetic acid (1 ml) at 60.deg. C. The reaction mixture is stirred at 60. °C for 2 hour then allowed to cool to room temperature and stirred overnight. The reaction mixture is poured onto ice-water (30 ml) and filtered through Celite. The solid was wash with dichloromethane (DCM) (50 ml) . The organic portion is separated and washed with water (2 x 30 ml) and brine (30 ml) , dried over MgS04, filtered and concentrated in vacuo to give a brown oil. 6,8-difluorochroman-3 -one (75 mg, 0,407 mmol, 87 % yield) was obtained as a brown oil.
Example 6
Preparation of 6 , 8-difluorochroman-3 -one from methyl 6,8-difluoro-2H-chromen-3 -yl-carbamate

Methanol (1000m ml) was added to a slurry of methyl fluoro-2H-chromen-3 -yl -carbamate (250 g, 1.037 mol) hydrogen chloride 6N (2000 ml, 12 mol) at room temperature. The resulting mixture was reflux and stirred for 2 hours. Reaction monitored by HPLC.
Reaction was not complete but was stopped in order to avoid degradation of the product. The yellow solution was cooled to room temperature. A slurry (two type of solid) was observed and diluted with diethyl ether (300mL) . The resulting slurry was stirred at 5°C for 1 hour then filtered. The yellow solid was washed with water. The resulting wet yellow solid was suspended in diethylether (400mL) and petroleum ether (PE) (400mL) was added. Slight yellow solid was stirred at room temperature overnight, filtered and washed with PE (300mL) , dried in a vacuum oven at 30 °C for 4h. The wet sample was checked by NMR. No starting material was detected. A pale yellow solid (72.5g, solid 1) was obtained. The mother liquors were concentrated to dryness. A yellow solid was obtained, suspended in diethyl ether and PE. The slurry was then stirred for 4 hours, filtered, washed with PE . A dark yellow solid (4.5g, solid 2) was obtained. Solid 1 (2g) was diluted in DCM and washed with water (pH =6). The organic layer was then dried over Na2S04, filtered, concentrated to dryness. A crystalline pale yellow solid (1.9g, solid 3) was obtained. NMR showed the same purity for solid 3 as for solid 1. The remaining part of solid 1 was then diluted in DCM. The resulting organic layer was washed with water, dried over Na2S04, filtered and then concentrated to dryness. Slight yellow crystalline solid (68.5g, solid 4) was obtained. NMR confirmed high quality material.
Loss on Drying (LOD) : 1.03% .
Example 7
Biotransformation: Transaminases

Codexis transaminases ATA-025, ATA-251 and ATA-P2-A07 recognized 6 , 8 -difluorochroman-3 -one as the substrate and produced the corresponding 6 , 8 -difluorochroman-3 -amine .
References
1: Igreja B, Wright LC, Soares-da-Silva P. Sustained high blood pressure reduction with etamicastat, a peripheral selective dopamine β-hydroxylase inhibitor. J Am Soc Hypertens. 2015 Dec 19. pii: S1933-1711(15)00838-4. doi: 10.1016/j.jash.2015.12.011. [Epub ahead of print] PubMed PMID: 26803288.
2: Loureiro AI, Bonifácio MJ, Fernandes-Lopes C, Pires N, Igreja B, Wright LC, Soares-da-Silva P. Role of P-glycoprotein and permeability upon the brain distribution and pharmacodynamics of etamicastat: a comparison with nepicastat. Xenobiotica. 2015;45(9):828-39. doi: 10.3109/00498254.2015.1018985. Epub 2015 Jun 10. PubMed PMID: 25915108.
3: Loureiro AI, Soares-da-Silva P. Distribution and pharmacokinetics of etamicastat and its N-acetylated metabolite (BIA 5-961) in dog and monkey. Xenobiotica. 2015;45(10):903-11. doi: 10.3109/00498254.2015.1024780. Epub 2015 Apr 14. PubMed PMID: 25869244.
4: Pires NM, Igreja B, Moura E, Wright LC, Serrão MP, Soares-da-Silva P. Blood pressure decrease in spontaneously hypertensive rats folowing renal denervation or dopamine β-hydroxylase inhibition with etamicastat. Hypertens Res. 2015 Sep;38(9):605-12. doi: 10.1038/hr.2015.50. Epub 2015 Apr 9. PubMed PMID: 25854989.
5: Bonifácio MJ, Sousa F, Neves M, Palma N, Igreja B, Pires NM, Wright LC, Soares-da-Silva P. Characterization of the interaction of the novel antihypertensive etamicastat with human dopamine-β-hydroxylase: comparison with nepicastat. Eur J Pharmacol. 2015 Mar 15;751:50-8. doi: 10.1016/j.ejphar.2015.01.034. Epub 2015 Jan 29. PubMed PMID: 25641750.
6: Pires NM, Loureiro AI, Igreja B, Lacroix P, Soares-da-Silva P. Cardiovascular safety pharmacology profile of etamicastat, a novel peripheral selective dopamine-β-hydroxylase inhibitor. Eur J Pharmacol. 2015 Mar 5;750:98-107. doi: 10.1016/j.ejphar.2015.01.035. Epub 2015 Jan 30. PubMed PMID: 25641747.
7: Igreja B, Pires NM, Bonifácio MJ, Loureiro AI, Fernandes-Lopes C, Wright LC, Soares-da-Silva P. Blood pressure-decreasing effect of etamicastat alone and in combination with antihypertensive drugs in the spontaneously hypertensive rat. Hypertens Res. 2015 Jan;38(1):30-8. doi: 10.1038/hr.2014.143. Epub 2014 Oct 9. PubMed PMID: 25298210.
8: Loureiro AI, Bonifácio MJ, Fernandes-Lopes C, Igreja B, Wright LC, Soares-da-Silva P. Etamicastat, a new dopamine-ß-hydroxylase inhibitor, pharmacodynamics and metabolism in rat. Eur J Pharmacol. 2014 Oct 5;740:285-94. doi: 10.1016/j.ejphar.2014.07.027. Epub 2014 Jul 21. PubMed PMID: 25058908.
9: Almeida L, Nunes T, Costa R, Rocha JF, Vaz-da-Silva M, Soares-da-Silva P. Etamicastat, a novel dopamine β-hydroxylase inhibitor: tolerability, pharmacokinetics, and pharmacodynamics in patients with hypertension. Clin Ther. 2013 Dec;35(12):1983-96. doi: 10.1016/j.clinthera.2013.10.012. Epub 2013 Dec 2. PubMed PMID: 24296323.
10: Loureiro AI, Rocha JF, Fernandes-Lopes C, Nunes T, Wright LC, Almeida L, Soares-da-Silva P. Human disposition, metabolism and excretion of etamicastat, a reversible, peripherally selective dopamine β-hydroxylase inhibitor. Br J Clin Pharmacol. 2014 Jun;77(6):1017-26. doi: 10.1111/bcp.12274. PubMed PMID: 24168152; PubMed Central PMCID: PMC4093927.
11: Loureiro AI, Fernandes-Lopes C, Bonifácio MJ, Wright LC, Soares-da-Silva P. N-acetylation of etamicastat, a reversible dopamine-β-hydroxylase inhibitor. Drug Metab Dispos. 2013 Dec;41(12):2081-6. doi: 10.1124/dmd.113.053736. Epub 2013 Sep 6. PubMed PMID: 24013186.
12: Nunes T, Rocha JF, Vaz-da-Silva M, Falcão A, Almeida L, Soares-da-Silva P. Pharmacokinetics and tolerability of etamicastat following single and repeated administration in elderly versus young healthy male subjects: an open-label, single-center, parallel-group study. Clin Ther. 2011 Jun;33(6):776-91. doi: 10.1016/j.clinthera.2011.05.048. PubMed PMID: 21704242.
13: Vaz-da-Silva M, Nunes T, Rocha JF, Falcão A, Almeida L, Soares-da-Silva P. Effect of food on the pharmacokinetic profile of etamicastat (BIA 5-453). Drugs R D. 2011;11(2):127-36. doi: 10.2165/11587080-000000000-00000. PubMed PMID: 21548660; PubMed Central PMCID: PMC3585837.
14: Rocha JF, Vaz-Da-Silva M, Nunes T, Igreja B, Loureiro AI, Bonifácio MJ, Wright LC, Falcão A, Almeida L, Soares-Da-Silva P. Single-dose tolerability, pharmacokinetics, and pharmacodynamics of etamicastat (BIA 5-453), a new dopamine β-hydroxylase inhibitor, in healthy subjects. J Clin Pharmacol. 2012 Feb;52(2):156-70. doi: 10.1177/0091270010390805. PubMed PMID: 21343348.
15: Nunes T, Rocha JF, Vaz-da-Silva M, Igreja B, Wright LC, Falcão A, Almeida L, Soares-da-Silva P. Safety, tolerability, and pharmacokinetics of etamicastat, a novel dopamine-β-hydroxylase inhibitor, in a rising multiple-dose study in young healthy subjects. Drugs R D. 2010;10(4):225-42. doi: 10.2165/11586310-000000000-00000. PubMed PMID: 21171669; PubMed Central PMCID: PMC3585840.
16: Beliaev A, Learmonth DA, Soares-da-Silva P. Synthesis and biological evaluation of novel, peripherally selective chromanyl imidazolethione-based inhibitors of dopamine beta-hydroxylase. J Med Chem. 2006 Feb 9;49(3):1191-7. PubMed PMID: 16451083.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1995007284A1 * | Aug 29, 1994 | Mar 16, 1995 | Smithkline Beecham Plc | Phosphinic acid derivatives with anti-hyper glycemic and/or anti-obesity activity |
| WO2006044293A2 * | Oct 11, 2005 | Apr 27, 2006 | Pharmacopeia Drug Discovery, Inc. | Bicyclic compounds as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| WO2013002660A2 * | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| GR1005093B * | Title not available |
| Reference | ||
|---|---|---|
| 1 | * | AL NEIRABEYEH M. ET AL.: “Methoxy and hydroxy derivatives of 3,4-dihydro-3-(di-n-propylamino)-2H-1-benzopyrans: new synthesis and dopaminergic activity“, EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 26, no. 5, 1991, EDITIONS SCIENTIFIQUE ELSEVIER, PARIS; FR, pages 497 – 504, XP023870436, ISSN: 0223-5234, DOI: 10.1016/0223-5234(91)90145-D |
| 2 | * | BELIAEV, A. ET AL.: “Process Research for Multikilogram Production of Etamicastat: A Novel Dopamine ß-Hydroxylase Inhibitor“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, no. 16, 2012, American Chemical Society, Washington; US, pages 704 – 709, XP002731798, DOI: 10.1021/op300012d |
| 3 | * | BOYE, S. ET AL.: “N,N-Disubstituted aminomethyl benzofuran derivatives: synthesis and preliminary binding evaluation“, BIOORGANIC & MEDICINAL CHEMISTRY, no. 7, 1999, ELSEVIER SCIENCE LTD; GB, pages 335 – 341, XP002731795, ISSN: 0968-0896, DOI: 10.1016/S0968-0896(98)00239-9 |
| 4 | * | COMOY, C. ET AL.: “3-Amino-3,4-dihydro-2H-1-benzopyran Derivatives as 5-HT1A Receptor Ligandsand Potential Anxiolytic Agents. 2. Synthesis and QuantitativeStructure-Activity Relationship Studies of Spiro[pyrrolidine- andpiperidine-2,3′(2’H)-benzopyrans]“, JOURNAL OF MEDICINAL CHEMISTRY., vol. 39, no. 21, 1996, AMERICAN CHEMICAL SOCIETY. WASHINGTON; US, pages 4285 – 4298, XP002731797, ISSN: 0022-2623, DOI: 10.1021/JM950861W |
| 5 | * | SHIN, C. ET AL.: “Total Synthesis of Bistratamide G, a Metabolite of the PhilippinesAscidian Lissoclinum bistratum, from Dehydrotripeptides“, CHEMISTRY LETTERS, vol. 33, no. 6, 2004, Chemical Society of Japan, Tokyo; JP, pages 664 – 665, XP002731799, ISSN: 0366-7022, DOI: 10.1246/cl.2004.664 |
| 6 | * | VASSE, J. L. ET AL.: “New efficient conditions for the reduction with NADH models“, SYNLETT, October 1998 (1998-10-01), THIEME INTERNATIONAL, STUTTGART; DE, pages 1144 – 1146, XP002731796, ISSN: 0936-5214, DOI: 10.1055/s-1998-1876 |
| 7 | * | XIAO, G.-Q. ET AL.: “3-Nitro-2H-chromenes as a New Class of Inhibitors against Thioredoxin Reductase and Proliferation of Cancer Cells“, ARCHIV DER PHARMAZIE, no. 345, 2012, VCH VERLAGSGESELLSCHAFT MBH, WEINHEIM; DE, pages 767 – 770, XP002731794, ISSN: 0365-6233, DOI: 10.1002/ardp.201200121 |
////////Etamicastat, BIA-5-453 , PHASE 2, Treatment, Heart Failure Therapy, Hypertension, Bial-Portela and Ca, S.A
SMILES Code: FC1=CC(F)=C(OC[C@H](N2C(CCN)=CNC2=S)C3)C3=C1.[H]Cl
c1c(cc(c2c1C[C@H](CO2)n3c(c[nH]c3=S)CCN)F)F
Zamicastat

- BIAL – PORTELA & CA., S.A. [PT/PT]; À Avenida da Siderurgia Nacional P-4745-457 S. Mamede do Coronado (PT)

- Zamicastat is a dopamine beta-monooxygenase inhibitor in phase I clinical studies at BIAL for the treatment of hypertension and heart failure.
- Zamicastat is a potent and selective dopamine β-mono-oxygenase inhibitor. Zamicastat Prevents the Deterioration of Cardiometabolic and Inflammatory Biomarkers in a Genetic Model of Salt-sensitive Hypertension. Chronic high salt intake deteriorates several cardiometabolic and inflammatory biomarkers in Dahl/SS rats, which can be prevented by dopamine β-hydroxylase inhibition with zamicastat.
- crystalline forms of l-[(3R)-6,8-difluoro- 3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino]ethyl]-2H- imidazole-2-thione, i.e. the Renantiomer of

and processes for preparing the same. Background and prior art:Interest in the development of inhibitors of dopamines-hydroxylase (ϋβΗ) has centred on the hypothesis that inhibition of this enzyme may provide significant clinical improvements in patients suffering from cardiovascular disorders such as hypertension or chronic heart failure. The rationale for the use of ϋβΗ inhibitors is based on their capacity to inhibit the biosynthesis of noradrenaline, which is achieved via enzymatic hydroxylation of dopamine. Activation of neurohumoral systems, chiefly the sympathetic nervous system, is the principal clinical manifestation of congestive heart failure (Parmley, W.W., Clinical Cardiology, 18: 440-445, 1995). Congestive heart failure patients have elevated concentrations of plasma noradrenaline (Levine, T.B. et al., Am. J. Cardiol., 49: 1659-1666, 1982), increased central sympathetic outflow (Leimbach, W.N. et al., Circulation, 73: 913- 919, 1986) and augmented cardiorenal noradrenaline spillover (Hasking, G.J. et al., Circulation, 73:615-621, 1966). Prolonged and excessive exposure of the myocardium to noradrenaline may lead to down-regulation of cardiac β] -adrenoceptors, remodelling of the left ventricle, arrhythmias and necrosis, all of which can diminish the functional integrity of the heart. Congestive heart failure patients who have high plasma concentrations of noradrenaline also have the most unfavourable long-term prognosis (Cohn, J.N. et al., N. Engl. J. Med., 311 :819-823, 1984). Of greater significance is the observation that plasma noradrenaline concentrations are already elevated in asymptomatic patients with no overt heart failure and can predict ensuing mortality and morbidity (Benedict, C.R. et al., Circulation, 94:690-697, 1996). An activated sympathetic drive is not therefore merely a clinical marker of congestive heart failure, but may contribute to progressive worsening of the disease.
Potent dopamines-hydroxylase inhibitors having high potency and significantly reduced brain access are disclosed in WO 2008/136695. WO 2008/136695 describes compounds of formula I:

I where Rls R2 and R3 are the same or different and signify hydrogens, halogens, alkyl, nitro, amino, alkylcarbonylamino, alkylamino or dialkylamino group; R4 signifies -alkylaryl or – alkylheteroaryl; X signifies CH2, oxygen atom or sulphur atom; n is 2 or 3; including the individual (R)- and (S)-enantiomers or mixtures of enantiomers thereof; and including pharmaceutically acceptable salts and esters thereof, wherein the term alkyl means hydrocarbon chains, straight or branched, containing from one to six carbon atoms, optionally substituted by aryl, alkoxy, halogen, alkoxycarbonyl or hydroxycarbonyl groups; the term aryl means a phenyl or naphthyl group, optionally substituted by alkyl, alkyloxy, halogen or nitro group; the term halogen means fluorine, chlorine, bromine or iodine; the term heteroaryl means heteroaromatic group. In particular, WO 2008/136695 describes l-[(3R)-6,8-difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2- [(phenylmethyl)amino]ethyl]-2H-Imidazole-2-thione.
Processes for the preparation of compounds of formula I, and in particular l-[(3R)-6,8- difluoro-3,4-dihydro-2H-l-benzopyran-3-yl]-l,3-dihydro-5-[2-[(phenylmethyl)amino] ethyl] -2H-Imidazole-2-thione, are described in WO 2008/136695 and are incorporated by reference herein. It is known that polymorphic forms of the same drug may have substantially different pharmaceutically important properties such as dissolution characteristics and bioavailability as well as stability of the drug. Furthermore, different forms may have different particle size, hardness and glass transition temperature. Thus, one form may provide significant advantages over other forms of the same drug in solid dosage form manufacture processes, such as accurate measurement of the active ingredients, easier filtration, or improved stability during granulation or storage. Furthermore, a particular process suitable for one form may also provide drug manufacturers several advantages such as economically or environmentally suitable solvents or processes, or higher purity or yield of the desired product.


PATENT
http://www.google.com/patents/WO2012087174A2?cl=en
Preparation of compound 2
[0090] Six lots of compound 2 (designated as lots 1, 2, 3, 4, 5 and 6) were prepared. The starting materials were prepared according to the following experimental protocols.
Lot 1 (Form A)
To a suspension of (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (6.23 g, 20 mmol) in a mixture of Dichloromethane (DCM – 40 ml) and Methanol (40.0 ml) was added BENZALDEHYDE (2.230 ml, 22.00 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (1.9 g, 28.7 mmol) was added in portions at 20-25°C to avoid intensive foaming and the solution was stirred at 20- 25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (35 ml), neutralised with 3N NaOH (35 ml), the mixture was extracted with DCM (200 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The oily residue crystallised from 2-propanol (40 ml) at 20-25°C over a week-end. The crystals were collected, washed with 2-propanol, dried to give 5.2 g of the crude product. Re- crystallisation from 2-propanol-DCM hasn’t removed all impurities. Everything collected, evaporated with silica, applied on a column, eluted with Ethyl Acetate (EA)->EA-MeOH 9:1->4: 1, fractions 8-25 collected to give 3.8 g. Re-crystallised from 2-propanol (45 ml) and DCM (120 ml, removed on a rotavap) to give 2.77 g => initial lot (a) (HPLC 98.3% area) and 0.3 g of undissolved filtered off, by TLC right product. Initial lot (a) re- crystallised from 2-propanol (35 ml) and DCM (95 ml, removed on a rotavap) to give 2.51 g => initial lot (b) (HPLC 98.3% area). Combined with the above undissolved, re- crystallised from acetonitrile (200 ml, reflux to ice bath) to give 2.57 g => initial lot (c) (HPLC 98.8% area). Re-crystallised from acetonitrile (180 ml, reflux to 15°C) to give 2.25 g => Lot 1 (HPLC 99.2% area), mp 190-92°C. Lot 2 (Form A)
[0092] (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole- 2(3H)-thione (12 g, 29.9 mmol) was dissolved with heating to reflux in Tetrahydrofuran (300 ml), the solution was cooled to 5-10°C, Water (510 ml) was added slowly (approx 10 min) with stirring. The mixture was stirred for 1 h, solid was collected, washed with water, dried to give 11.73 g of product, by HPLC 1% of (R)-5-(2-Aminoethyl)-l-(6,8- difluorochroman-3-yl)-l,3-dihydroimidazole-2-thione hydrochloride and 1% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred in ice for 1.5 h. Solid was collected, washed with 2- propanol, dried to give 11.2 g of product, by HPLC 0.8% of (R)-5-(2-aminoethyl)-l-(6,8- difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride and 0.5% of less polar impurity. The product was dissolved in Tetrahydrofuran (300 ml) with heating to reflux, 2- Propanol (150 ml) was added, the solution was concentrated to approx 100 ml (crystallisation occured), stirred at 20-25°C for 1 h. Solid was collected, washed with 2- propanol, dried to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (10.22 g, 25.5 mmol, 85 % yield).,
Lot 3 (form B)
To (R)-5-(2-aminoethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (2.36 g, 7.58 mmol) in a mixture of Methanol (15.00 ml) and Dichloromethane (15 ml) was added BENZALDEHYDE (0.845 ml, 8.34 mmol). To the resulting clear solution SODIUM CYANOBOROHYDRIDE (0.702 g, 10.61 mmol) was added in portions at 20- 25°C to avoid intensive foaming and the solution was stirred at 20-25°C for 40 h. The solution was quenched at 20-25°C with IN HC1 (12 ml), neutralised with 3N NaOH (12 ml), the mixture was extracted with DCM (100 ml). The organic phase was washed with brine, dried (MgS04), evaporated to dryness. The residue was purified on a column with EA-MeOH 9: 1 as eluent, fractions collected, concentrated to approx 20 ml, cooled in ice. The precipitate collected, washed with Ethyl Acetate-Petroleum Ether 1 : 1, dried on air to give (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)- thione (1.55 g, 3.86 mmol, 50.9 % yield). Lot 4 (Form A)
To a 500 mL flask set up for atmospheric distillation was added (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (20 g, 49,8 mmol) and Tetrahydrofuran (400 ml) to afford a suspension. The suspension was heated until full dissolution was achieved (61°C) whereupon it was filtered. The resulting solution was then heated to 66°C in order to commence the distillation. A mixture of Water (125 ml) & 2-Propanol (125 ml) was added at the same rate as the distillate was collected. The distillation was continued until 400 mL of distillate was collected. Crystallisation commenced after ~320 mL of distillate was collected. The suspension was cooled to 20°C and aged for 45 min. before filtering and washing with additional 2- propanol (80 mL) and then dried under vacuum at 50°C overnight to give (R)-5-(2- (benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione (18.79 g, 94%). Lot 5 (Form A)
To a mixture of Methanol (66 L) and Water (10 L) at 20°C was added purified (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH-imidazole-2(3H)-thione hydrochloride (4.37 kg, 9.98 mol) to afford a suspension. The reaction mixture was then heated to 67°C to affect complete dissolution, whereupon IN Sodium hydroxide (10.48 Ls 10.48 mol, 1.05 eq) was added in a single portion. The reaction mixture was adjusted back to 67°C and held at 67°C for 30 min. The reaction mixture was then cooled to 20°C and aged at 20°C for at least 30 min. The reaction was then filtered and the filter cake washed with aqueous Methanol (1 : 1 v/v, 20 L), sucked down for 15 min. and then dried at 45°C under vacuum, to afford (R)-5-(2-(benzylamino)ethyl)-l-(6,8-difluorochroman-3-yl)-lH- imidazole-2(3H)-thione (3.855 kg, 96%) as a pale tan crystalline solid.
PATENT
WO 2015038022
http://www.google.com/patents/WO2015038022A1?cl=en
processes .
(J?) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3-yl) -1, 3-dihydroimidazole-2 -thione hydrochloride (the compound of formula 1, below) is a potent, non-toxic and peripherally selective inhibitor of ϋβΗ, which can be used for treatment of certain cardiovascular disorders. Compound 1 is disclosed in WO2004/033447 , along with processes for its preparation.
1
The process disclosed in WO2004/033447 involves the reaction of ( R) – 6 , 8 -difluorochroman-3 -ylamine hydrochloride (the structure of ( R) -6, 8-difluorochroman-3 -ylamine is shown below as compound QA) , [4 – ( tert-butyldimethylsilanyloxy) -3 -oxobutyl] carbamic acid tert-butyl ester and potassium thiocyanate .
QA
(R) -6 , 8-difluorochroman- 3 -ylamine (compound QA) is a key intermediate in the synthesis of compound 1. The stereochemistry at the carbon atom to which the amine is attached gives rise to the stereochemistry of compound 1, so it is advantageous that compound QA is present in as pure enantiomeric form as possible. In other words, the (R) -enantiomer of compound QA should be in predominance, with little or no (S) enantiomer present. Thus, the process for preparing compound QA will advantageously produce compound QA with as high enantiomeric excess (ee) as possible.
Advantageous processes for preparing, for example, the compound of formula QA have now been found. In one aspect, the processes involve a biotransformation step. In another aspect, the processes involve chemical transformation. The processes may also be employed in the preparation of similar precursors useful in the production of other peripherally-selective inhibitors of dopamine -β -hydroxylase .
WO2008/136695 discloses a compound of formula YA, its (R) or (S) enantiomer, a mixture of its (R) and (S) enantiomers, or pharmaceutically acceptable salts thereof.
YA
The (R) -enantiomer of the compound of formula YA has been found to be a potent dopamines-hydroxylase inhibitor having high potency and significantly reduced brain access.
As disclosed in WO2008/136695 , the compound of formula YA may be prepared by reacting the compound of formula 1 with benzaldehyde under reductive alkylation conditions. In particular, (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) – 1 , 3 -dihydroimidazole-2 -thione and benzaldehyde may be reacted in the presence of a solvent or mixture of solvents, and a reducing agent such as sodium cyanoborohydride or sodium triacetoxyborohydride .
process comprises the following steps:

The route from 2 , 4-difluorophenol may be as described 9/064210.
Preferably, the reagents and conditions are:
(i) H2S04, acetic acid
(ii) NaOCl, MeOH/water
(iii) Ru-based catalyst, H2, 30 bars, MeOH
(iv) aqueous KOH, MeOH, L-tartaric acid
(v) KSCN, AcOH/lPA
(vi) NaBH4, BF3.THF complex, THF then IPA
n one aspect, the process comprises the following steps


i. KOH, Thioglycolic acid or cysteine
ii. MEK
According to an aspect of the present invention, there is provided the following 2 -part synthetic route from the starting material 2 , 4 -difluorophenol to (R) -5- (2 -aminoethyl ) -1- (6 , 8-difluorochroman-3 -yl) -1 , 3 -dihydroimidazole-2 – thione
hydrochloride :
Part (1)
Preferred reagents and conditions:
a) HMTA, CF3COOH, 115°C, 18 hours
b) CH2CHCN, DABCO, DMF, water, 70°C, 16 hours
c) H2S04, AcOH, 100°C, 1 hour
d) NaClO, NaOH, MeOH, 25°C, 24 hours
e) (R) -C3 -TunePhosRu (acac) 2 S/C 3000, 30 bar H2, MeOH, 80°C, 20 hours
f) Water, 2-propanol, reflux to 20°C
g) 40% KOH, MeOH, reflux, 24 hours
h) L-tartaric acid, ethanol, water, RT, 1 hour
Part (2)
![]()
Preferred reagents and conditions
a’) methyl vinyl ketone, t-BuONa, EtOAc, EtOH, 40-50°C, 2-3 hours
Br2, MeOH, 20-25°C, 5 hours
water, reflux, 1 hour
KOH, AcOH, reflux, 1 hour
HCl, water, 2-propanol, 75 °C, 4 hours
KSCN, AcOH, 100°C, 2-4 hours
NaHC03, water, EtOH
NaBH4, 2-propanol, THF, water, 20-25°C, 16 hours
HCl, 2-propanol, water, reflux, 1-2 hours
The ( R ) -5- (2-Aminoethyl) -1- (6, 8-difluorochroman-3 -yl) -1,3-dihydroimidazole-2 – thione hydrochloride may then be used to
prepare (R) -5- (2- (benzylamino) ethyl) -1- (6, 8-difluorochroman-3 -yl) -lH-imidazole-2 (3H) -thione as follows.

Preferred reaction conditions/reagents:
q) NaBH(OAc)3, PhCHO, IPA;
t) NaOH, MeOH , H20
Either r) and s) :
r) HCI aq;
s) MeOH/Toluene;
Or n) , o) and p) :
n) HCI aq;
o) MeOH, toluene;
p) IPA.
EXAMPLES
Example 1
Nitro chromene synthesis
To 3 , 5-difluoro-2-hydroxybenzaldehyde (lOg, 63mmol, leq) , di-n-butylamine (4.1g, 32mmol, 0.5eq) , phtalic anhydride (18.7g, 126mmol, 2eq) in toluene (500mL) was added nitroethanol (5.75g, 63mmol, leq) . The round bottomed flask fitted with a dean stark apparatus was refluxed for 18h. The mixture was cooled and nitroethanol (5.75g, 63mmol, leq) was added. The resulting reaction mixture was then reflux for 12h. After cooling, the solution was evaporated down to approximately 150mL and purified over silica gel (eluent ethyl acetate : hexane 1:1) this gave several fractions that contained only the product by TLC, these was evaporated under reduced pressure to yield 1.8g which was 100% pure by HPLC aera. Several more fractions were collected containing a mixture of product and starting material. These were combined and washed with 2% NaOH solution (2x50mL) to remove starting material. The organic layer was washed with water (50mL) , dried over sodium sulfate and evaporated under reduced pressure to give 2.49g of brown solid ( 100% pure by HPLC aera) . More fractions were collected. These were combined, washed with 2% NaOH solution (3xl00mL) , water (lOOmL) and dried over sodium sulfate. This was then filtered and evaporated down in vacuum to yield 6.14g of a brown solid which was 91.3% pure by HPLC aera. 6 , 8 -difluoro-3 -nitro-2H-chromene (9.90g, 73.4%) was obtained as a brown solid.
Example 2
Nitro chromene synthesis with column purification
To a solution of isobenzofuran-1 , 3 -dione (4,68 g, 31,6 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (2,5 g, 15,81 mmol) in Toluene (25 ml) was added 2 -nitroethanol (2,88 g, 31,6 mmol). The resulting mixture was heated to reflux overnight (Dean stark) .
The reaction conversion was checked by TLC (eluent PE/EtOAc 9:1) . A yellow spot was observed and corresponds to the expected product .
Reaction was cooled to room temperature and a plug of silica gel was performed. A pale brown solid (3.9g) was obtained. “””H-NMR showed presence of product and starting material. The solid was dissolved in diethylether and the organic layer was washed with aqueous sodium carbonate, dried over Na2S04, filtered and concentrated under reduced pressure. A pale brown solid (1.7g,) was obtained. The 1H-NMR was indicated no starting material but still polymer from nitroethanol and residue of phtalic anhydride. A second silica plug (eluent: PE/EtOAc 95:5) was done. A pale yellow solid (1.5g) was obtained. 1H-NMR of solid showed only product and polymer. The solid was recrystallized from methanol/water . A pale yellow solid (1.05g, 31.2%) was obtained.
Example 3
Nitro chromene synthesis without column purification
To a solution of isobenzofuran- 1 , 3 -dione (18,74 g, 127 mmol) , 3 , 5-difluoro-2 -hydroxybenzaldehyde (10 g, 63,3 mmol) in Toluene (100 ml) was added 2 -nitroethanol (6,86 ml, 95 mmol) . The resulting mixture was heated to reflux for 24h (Dean stark) .
The reaction conversion was checked by HPLC and by 1H-NMR. Only 50% conversion was obtained.
The reaction mixture was cooled to room temperature and diluted with DCM (lOOmL) and 1M NaOH solution (200mL) .
The biphasic system was stirred for 30 minutes and then separated (very difficult to see phase separation) . The aqueous layer was washed with DCM (50mL) and the combined organic layers were washed twice with water (2x50ml) , dried over sodium sulfate. The filtered organic layer was concentrated under reduced pressure. To the residue was added methanol (50mL) . The methanol was then removed by distillation under reduced pressure. A brown solution precipitated when most of the methanol was removed. More methanol was added and more solid crushed out then few drops of water was added to increase the product precipitation. The brown slurry was stirred for 30 minutes and filtered. The brown solid was washed with methanol/water (1:9, 5mL) and dried in a vacuum oven at 40°C for 12h.6, 8-difluoro-3 -nitro-2H-chroraene (4,9 g, 22,99 mmol,) was obtained as brown solid in 36.3% yield.
HPLC showed a purity of 98% and 1H-NMR confirmed the structure and purity around 95%
Example 4
Reduction of nitro chromene to nitro-alkane (racemic mixture)
To a suspension of 6 , 8 -difluoro-3 -nitro-2H-chromene (213mg, 0,999 mmol) and silica (0,8 g, 0,999 mmol) in a mixture of CHC13 (10 ml) and IPA (3,4 ml) at 0°C was added portion wise sodium borohydride (95 mg, 2,498 mmol). The resulting mixture was stirred at 0°C for 45 minutes. Reaction conversion was checked by HPLC. 1 mL of acetic acid was added at 0°C and the resulting mixture was stirred for 30 minutes at room temperature. The slurry was filtered and the silica was washed with DCM. The filtrate was diluted with ethyl acetate and water and the biphasic system was separated. The aqueous layer was back extracted with ethyl acetate. The combined organic layers were washed with brine, dried over MgS04, filtered and concentrated under reduced pressure.
6 , 8-difluoro-3 -nitrochroman (196mg, 0,911 mmol, 91 % yield) was obtained as a pale yellow oil.
Example 5
Preparation of 6 , 8 -difluorochroman-3 -one from nitro chromene
A solution of 6, 8-difluoro-3 -nitro-2H-chromene (lOOmg, 0,469 mmol) in acetic acid (0.5 ml) is added slowly to a stirred slurry of iron (262 mg, 4,69 mmol) in acetic acid (1 ml) at 60.deg. C. The reaction mixture is stirred at 60. °C for 2 hour then allowed to cool to room temperature and stirred overnight. The reaction mixture is poured onto ice-water (30 ml) and filtered through Celite. The solid was wash with dichloromethane (DCM) (50 ml) . The organic portion is separated and washed with water (2 x 30 ml) and brine (30 ml) , dried over MgS04, filtered and concentrated in vacuo to give a brown oil. 6,8-difluorochroman-3 -one (75 mg, 0,407 mmol, 87 % yield) was obtained as a brown oil.
Example 6
Preparation of 6 , 8-difluorochroman-3 -one from methyl 6,8-difluoro-2H-chromen-3 -yl-carbamate
Methanol (1000m ml) was added to a slurry of methyl fluoro-2H-chromen-3 -yl -carbamate (250 g, 1.037 mol) hydrogen chloride 6N (2000 ml, 12 mol) at room temperature. The resulting mixture was reflux and stirred for 2 hours. Reaction monitored by HPLC.
Reaction was not complete but was stopped in order to avoid degradation of the product. The yellow solution was cooled to room temperature. A slurry (two type of solid) was observed and diluted with diethyl ether (300mL) . The resulting slurry was stirred at 5°C for 1 hour then filtered. The yellow solid was washed with water. The resulting wet yellow solid was suspended in diethylether (400mL) and petroleum ether (PE) (400mL) was added. Slight yellow solid was stirred at room temperature overnight, filtered and washed with PE (300mL) , dried in a vacuum oven at 30 °C for 4h. The wet sample was checked by NMR. No starting material was detected. A pale yellow solid (72.5g, solid 1) was obtained. The mother liquors were concentrated to dryness. A yellow solid was obtained, suspended in diethyl ether and PE. The slurry was then stirred for 4 hours, filtered, washed with PE . A dark yellow solid (4.5g, solid 2) was obtained. Solid 1 (2g) was diluted in DCM and washed with water (pH =6). The organic layer was then dried over Na2S04, filtered, concentrated to dryness. A crystalline pale yellow solid (1.9g, solid 3) was obtained. NMR showed the same purity for solid 3 as for solid 1. The remaining part of solid 1 was then diluted in DCM. The resulting organic layer was washed with water, dried over Na2S04, filtered and then concentrated to dryness. Slight yellow crystalline solid (68.5g, solid 4) was obtained. NMR confirmed high quality material.
Loss on Drying (LOD) : 1.03% .
Example 7
Biotransformation: Transaminases
Codexis transaminases ATA-025, ATA-251 and ATA-P2-A07 recognized 6 , 8 -difluorochroman-3 -one as the substrate and produced the corresponding 6 , 8 -difluorochroman-3 -amine .
PATENT
WO 2014077715
WO 2013002660
WO 2008136695
REFERNCES
International Journal of Pharmaceutics (Amsterdam, Netherlands) (2016), 501(1-2), 102-111.
| WO2012087174A2 | Dec 21, 2011 | Jun 28, 2012 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2012087174A3 * | Dec 21, 2011 | May 10, 2013 | BIAL – PORTELA & Cª., S.A. | Crystalline forms and processes for their preparation |
| WO2013002660A2 | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| WO2014077715A1 * | Nov 14, 2013 | May 22, 2014 | BIAL – PORTELA & Cª, S.A. | 1,3-dihydroimidazole-2-thione derivatives for use in the treatment of pulmonary arterial hypertension and lung injury |
| US8481582 | May 6, 2008 | Jul 9, 2013 | Bial-Portela & Ca, S.A. | 1,3-dihydroimidazole-2-thione derivatives as inhibitors of dopamine-beta-hydroxylase |
| US8865913 | Jun 19, 2013 | Oct 21, 2014 | Bial-Portela & Ca, S.A. | Crystalline forms and processes for their preparation |
| WO1995007284A1 * | Aug 29, 1994 | Mar 16, 1995 | Smithkline Beecham Plc | Phosphinic acid derivatives with anti-hyper glycemic and/or anti-obesity activity |
| WO2006044293A2 * | Oct 11, 2005 | Apr 27, 2006 | Pharmacopeia Drug Discovery, Inc. | Bicyclic compounds as selective melanin concentrating hormone receptor antagonists for the treatment of obesity and related disorders |
| WO2012007548A1 * | Jul 14, 2011 | Jan 19, 2012 | Dsm Ip Assets B.V. | (r)-selective amination |
| WO2013002660A2 * | Jun 29, 2012 | Jan 3, 2013 | BIAL – PORTELA & Cª, S.A. | Process |
| GR1005093B * | Title not available |
///////Zamicastat, BIA-5-1058, dopamine beta-monooxygenase inhibitor, phase I, clinical studies, BIAL, treatment of hypertension , heart failure.
S=C4NC=C(CCNCc1ccccc1)N4[C@@H]2Cc3cc(F)cc(F)c3OC2
FDA approves new drug Venclexta (venetoclax) for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality

April 11, 2016
Release
The U.S. Food and Drug Administration today approved Venclexta (venetoclax) for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a chromosomal abnormality called 17p deletion and who have been treated with at least one prior therapy. Venclexta is the first FDA-approved treatment that targets the B-cell lymphoma 2 (BCL-2) protein, which supports cancer cell growth and is overexpressed in many patients with CLL.
According to the National Cancer Institute, CLL is one of the most common types of leukemia in adults, with approximately 15,000 new cases diagnosed each year. CLL is characterized by the progressive accumulation of abnormal lymphocytes, a type of white blood cell. Patients with CLL who have a 17p deletion lack a portion of the chromosome that acts to suppress cancer growth. This chromosomal abnormality occurs in approximately 10 percent of patients with untreated CLL and in approximately 20 percent of patients with relapsed CLL.
“These patients now have a new, targeted therapy that inhibits a protein involved in keeping tumor cells alive,” said Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option for their specific condition.”
The efficacy of Venclexta was tested in a single-arm clinical trial of 106 patients with CLL who have a 17p deletion and who had received at least one prior therapy. Trial participants took Venclexta orally every day, beginning with 20 mg and increasing over a five-week period to 400 mg. Results showed that 80 percent of trial participants experienced a complete or partial remission of their cancer.
Venclexta is indicated for daily use after detection of 17p deletion is confirmed through the use of the FDA-approved companion diagnostic Vysis CLL FISH probe kit.
The most common side effects of Venclexta include low white blood cell count (neutropenia), diarrhea, nausea, anemia, upper respiratory tract infection, low platelet count (thrombocytopenia) and fatigue. Serious complications can include pneumonia, neutropenia with fever, fever, autoimmune hemolytic anemia, anemia and metabolic abnormalities known as tumor lysis syndrome. Live attenuated vaccines should not be given to patients taking Venclexta.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Venclexta also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Venclexta is manufactured by AbbVie Inc. of North Chicago, Illinois, and marketed by AbbVie and Genentech USA Inc. of South San Francisco, California. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular of Des Plaines, Illinois.
Curis and Aurigene’s CA 4948, AU 4948
![]()


Example 13 WO2015104688
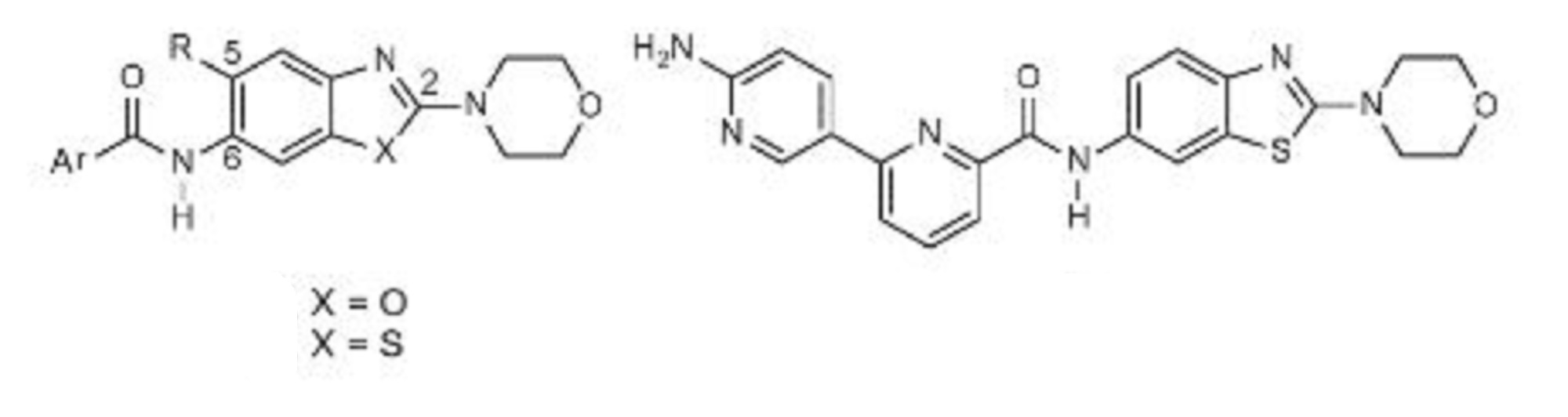
6-(6-aminopyridin-3-yl)-N-(2-morpholin-4-yl-1,3-benzothiazol-6-yl)pyridine-2-carboxamide
| Molecular Formula: | C22H20N6O2S |
|---|---|
| Molecular Weight: | 432.4982 g/mol |
PROBABLE STRUCTURE

Example 1 ……..6′-amino-N-(2-morpholinooxazolo[4,5-b]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamideWO2015104688

Compound-6: 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.WO2013042137
PROBABLE CA 4948, AU 4948,AU-4948, CA-4948
STRUCTURE AND SYNTHESIS COMING……..
| Latest Stage of Development | Preclinical |
| Standard Indication | B cell lymphoma |
| Indication Details | Treat diffuse large B cell lymphoma (DLBCL) |
| Regulatory Designation | |
| Partner | Curis Inc. |
Interleukin-1 Receptor Associated Kinase-4 (IRAK-4) is a serine/threonine protein kinase belonging to tyrosine like kinase (TLK) family. IRAK-4 is one of the important signalling components downstream of IL-1/Toll family of receptors (IL-1R, IL-18R, IL-33R, Toll-like receptors). Recent studies have reported occurrence of oncogenic mutations in MYD88 in 30% of ABC diffuse large B cell lymphomas (ABC DLBCL) and 90% of Waldenstrom’s macroglobulinemia (WM). Most of ABC DLBCLs have a single amino acid substitution of proline for the leucine at position 265 (L265P) in the TIR domain of MYD88 protein resulting in constitutive activation of IRAK-4. Thus, IRAK4 is an attractive therapeutic target for the treatment of B-cell lymphomas with activating MYD88 L265P mutation. We have designed, synthesized and tested small molecule IRAK-4 inhibitors based on hits originating from Aurigene’ s compound library. These novel compounds were profiled for IRAK4 kinase inhibition, anti-proliferative activity, kinase selectivity, and drug-like properties. Furthermore, selected compounds were tested in a proliferation assay and pIRAK1 mechanistic assay using ABC-DLBCL cell lines with activating MYD88 L265P mutation, OCI-lLy10 and OCI-lLy3. We have identified a series of novel bicyclic heterocycles as potent inhibitors of IRAK-4. Aurigene Lead compound exhibited potent inhibitory activity for IRAK-4 with an IC50 of 3nM in biochemical assay. Aurigene Lead compound inhibited pIRAK1 levels, and proliferation of OCI-Ly3 and OCI-Ly10 cells with an IC501of 132nM and 52nM respectively. To the best of our knowledge, Aurigene Lead compound represents the most potent IRAK4 inhibitor reported for target modulation and anti-proliferative activity in DLBCL cell lines with activating MYD88 L265P mutation. Aurigene Lead compound has good oral pharmacokinetic profile in mice and has demonstrated excellent pharmacodynamic effect in an in vivo LPS induced TNF-α model with an ED50 of 3.8 mg/Kg in mice. Preliminary in vitro tox studies indicated clean safety profile. Demonstration of efficacy in OCI-lLy10 mouse tumor model is ongoing. In summary, a series of potent IRAK-4 inhibitors belonging to 3 different chemical series have been discovered and are being evaluated for treatment of B-cell lymphomas.
Curis with the option to exclusively license Aurigene’s orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers.
![]()
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About IRAK4:
Interleukin-1 receptor-associated kinase 4, or IRAK4 is a signaling kinase that becomes inappropriately activated in certain cancers including activated B cell-diffuse large B cell lymphoma (ABC-DLBCL), an aggressive form of lymphoma with poor prognosis. There appears to be a mechanistic link with IRAK4 in ABC-DLBCL where these tumors from approximately 35% of patients harbor oncogenic mutations in the MYD88 gene, which encodes an adaptor protein that interacts directly with IRAK4. MYD88 mutations appear to constitutively activate the IRAK4 kinase complex, driving pro-survival pathways in ABC-DLBCL disease. Oncogenic MYD88 mutations have also been identified in other cancers, including in over 90% of patients with Waldenström’s Macroglobulinemia as well as in a subset of patients with chronic lymphocytic leukemia (CLL).
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
Small Molecule IRAK4 Kinase Inhibitor)
Innate immune responses mediated through Toll-like receptors or certain interleukin receptors are important mediators of the body’s initial defense against foreign antigens, while their dysregulation is associated with certain inflammatory conditions. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. More recently, components of this pathway are recognized to be genetically altered and have important roles in specific human cancers. Toll-like receptor and interleukin receptor signaling through the adaptor protein MYD88, results in the assembly and activation of IRAK4, initiating a signaling cascade that induces cytokine and survival factor expression mediated by the transcription factor NFκB. MYD88 gene mutations are shown to occur in approximately 30% of Activated B-Cell (ABC) subtype of diffuse large B-cell lymphomas (DLBCL)1,2 and in over 90% of the B-cell malignancy Waldenstrom’s macroglobulinemia.3 Due to IRAK4’s central role in these signaling pathways, it is considered an attractive target for generation of therapeutics to treat these B-cell malignancies as well as certain inflammatory diseases.
As part of the collaboration with Aurigene, in October 2015 we exercised our option to exclusively license a program of orally-available, small molecule inhibitors of IRAK4 kinase, including the development candidate, CA-4948. Curis expects to file an IND and initiate clinical testing of CA-4948 in patients with advanced hematologic cancers during the second half of 2016.
1Nature. 2011; 470(7332):115–1192Immunology and Cell Biology. 2011; 89(6):659–6603N Engl J Med. 30, 2012; 367(9):826–833
CLIP
In November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
Aurigene Collaboration (IRAK4 Inhibitor):
In October 2015, Curis exercised its option to exclusively license a program of orally available small molecule inhibitors of IRAK4 kinase, a serine/threonine kinase involved in innate immune responses as well as in certain hematologic cancers. The Company has since designated the development candidate as CA-4948 and expects to file an IND application for this molecule during 2016.
In November 2015, Curis’ collaborator Aurigene presented preclinical data from the IRAK4 program at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA. This presentation included data from chemically distinct series of small molecule compounds with potent IRAK4 inhibitory activity in biochemical assays as well as in in vivo preclinical models, including MYD88 mutant DLBCL xenograft tumor models as well as a model of inflammatory disease.
CLIP
In April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA, showed the compounds in vivo to have activity down to 10 mg/kg .
CLIP

 Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Research Director, Medicinal Chemistry, Aurigene Discovery Technologies LimitedApril 24-25 2014
Drug Discovery Chemistry – CHI’s Ninth Annual Conference: Fifth Annual Kinase inhibitor Chemistry, San Diego, CA, USA
Novel IRAK4 inhibitors
Susanta Samajdar from Aurigene Discovery Technologies presented the discovery of new IRAK4 (IL-1 receptor-associated kinase 4) inhibitors. Research began with a HTS campaign using two types of libraries: rationally designed novel scaffolds by hopping and morphing of known IRAK4 inhibitors and novel scaffolds identified by virtual screening of drug-like commercial library. A benzoxazol series was identified and crystallography was used to help their design. Lead optimization culminated in the identification of very potent compounds (AU-2807 and AU-2202) in cell assay (inflammation pathway and oncology pathway, respectively). The compounds were also active against Flt3 and KDR. Some PD in vivo data using LPS and TNFalpha release were presented in which the compound showed activity down to 10 mg/kg: no other in vivo model data were disclosed, but it was mentioned that studies in the CIA (collagen induced arthritis) model was ongoing. Dr Samajdar answered to three questions, one related to IRAK1 selectivity (the answer was that the compound is fully selective against IRAK1 and IRAK2). It was also mentioned that the compounds have a PBB higher than 98%. And the last question was related to the synergetic effect with BTK inhibitor in activated B-cell like diffuse large B-cell lymphoma, and this effect was observed with these compounds.

Research Director at Aurigene Discovery Technologies
PATENT
http://www.google.com/patents/WO2013042137A1?cl=en
Compound-6: Synthesis of 6′-amino-N-(5-(cyclopropyIamino)-2-morpholinobenzo [d]oxazoI-6-yl)-[2,3′-bipyridine]-6-carboxamide.
Step_l^N-cyclopropyl-2-morpholino-6-nitrobenzo[d]oxazol-5-amine.

N-cyclopropyl-2-moφholino-6-nitrobenzo[d]oxazol-5-amine(0.7g,70%) was prepared from 5-fluoro-2-mo holino-6-nitrobenzo[d]oxazole(lg,Intermediate-2) by treating with cyciopropanamine in sealed tube at 100°C for 8-14h. The progress of the reaction was monitored by TLC. After the reaction was completed, it was extracted with water (15ml) and dichioromethane (2x 15ml). The organic layer was collected, washed with brine, dried over sodium sulfate and concentrated under reduced pressure to get the crude. MS (ES) m/e 305(M+1, 50%).
Steg2:6-bromo-N-(5-(cyclopropylamino)-2-morpholinobenzo[d]oxazol-6-yl)
picolinamide.

Step Π and ii):The process of these steps are adopted from step 2 and step 3 of compound- 1.
Step3:6′-amino-N-(5-(cvclopropvlamino)-2-morpholinobenzord]oxazol-6-yl)-r2,3′- bipyridine]-6-carboxamide.

(i) N-(4-methoxybenzyl)-5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin
Na2C03, Pd(dppf)Cl2, ACN, H20, 80-100°C, 8-14h; TFA, 60-70°C, 8-14h.
6′-amino-N-(5-(cyclopropylamino)-2-mo holinobenzo[d]oxazol-6-yl)-[2,3′-bipyridine]-6- carboxamide (0.03g,61%) was prepared from 6-bromo-N-(5-(cyclopropyIamino)-2- moφholinobenzo[d]o azoI-6-yl)picolinamide(0.07g, step-3) by following the same process used in step-1 and 2 of compound-3.
Ή NMR (400 MHz, DMSO-< ):6 1 1.63 (s, IH), 8.90 (s, IH), 8.61 (s, IH), 8.55 (s, IH), 8.37- 8.03 (m, 2H), 7.39 (s, IH), 6.80-6.62 (s, IH), 3.80-3.59 (m, 15H), 2.88-2.64 (m, 2H). MS (ESI): 472 (M+l , 60%).
PATENT
Example 13
6′-amino-N-(2-morphol ne]-6-carboxamide

Step-1: Synthesis of 6-chloro thiazolo[4,5-c]pyridine-2(3H)-thione
Using the same reaction conditions as described in step 1 of example 1, 4,6-dichloropyridin-3-amine (1.3 g, 7 mmol) was cyclised using potassium ethyl xanthate (2.55 g, 15 mmol) in DMF (25mL) at 150°C for 8h to afford the title compound (1.3 g, 86.6 %) as a light brown solid.
1HNMR (400 MHz, DMSO-d6): δ 14.2-14.0 (b, 1H), 8.274 (s, 1H), 7.931 (s, 1H); LCMS: 100%, m/z = 201.3 (M+l)+.
Step-2: Synthesis of 4-(6-chloro thiazolo[4,5-c]pyridin-2-yl) morpholine
To a suspension of 6-chlorothiazolo[4,5-c]pyridine-2(3H)-thione (0.3 g, 1.16 mmol) in
DCM (4 mL), oxalyl chloride (0.2 mL, 2.38 mmol) and DMF (1.5 mL) were added at 0°C. The resulting mixture was slowly allowed to warm to room temperature and stirred there for 1 h. The reaction mixture was again cooled to 0°C and triethyl amine (0.66 mL, 4.76 mmol) and morpholine (0.13 mL, 1.75 mmol) were added. The reaction mixture was stirred at RT for 1 h and quenched with water and extracted with ethyl acetate. The combined organic layers were washed with water, brine, dried over sodium sulphate and concentrated under reduced pressure. The crude material was purified by column chromatography (EtOAc/n-hexanes 3:7) to afford the title compound (0.14 g, 39.6 %) as a light brown solid.
1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.04 (s, 1H), 3.74-3.72 (m, 4H), 3.61-3.59 (m, 4H); LCMS: m/z = 256.1 (M+l)+.
Step-3: Synthesis of 6′-amino-/V-(2-morpholino thiazolo [4,5-c]pyridin-6-yl)-[2,3′-bipyridine]-6-carboxamide
Using the same reaction conditions as described in step 4 of example 12, 4-(6-chlorothiazolo[4,5-c] pyridin-2-yl) morpholine (0.081 g, 0.32 mmol), was coupled with tert-butyl (6-carbamoyl-[2,3′-bipyridin]-6′-yl)carbamate (intermediate 2) (0.1 g, 0.32 mmol) using cesium carbonate (0.21 g, 0.64 mmol), XantPhos (0.028g, 0.047mmol) and Pd2(dba)3 (0.015 mg, 0.015 mmol) in toluene : dioxane (2:2mL) to get the crude product. The resultant crude was purified by 60-120 silica gel column chromatography using 2% methanol in DCM as eluent. Further the resultant crude was purified by prep HPLC to afford title compound (0.01 g, 6 %) as an off-white solid.
1H NMR (400 MHz, DMSO-d6): δ 10.65 (s, 1H), 8.88 (d, 1H), 8.85 (dd, 1H), 8.71 (s, 1H), 8.55 (s, 1H), 8.22-8.13 (m, 4 H), 7.09 (d, 1H), 3.73 (t, 4H), 3.58 (t, 4H). LCMS: 100%, m/z = 434.2 (M+l)+.
Example 11
(S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide

Step l:Preparation of (S)-tert-butyl 3-((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
A solution of 5-chloro-2-morpholino-6-nitrooxazolo[4,5-b]pyridine (300mg, 1.0563 mmol) (S)-tert-butyl 3 -aminopyrrolidine- 1 -carboxylate (237mg, 1.267 mmol) and potassium carbonate (292mg, 2.112 mmol) in DMF (2mL) was heated at 100°C for 2h. Reaction was quenched with ice water and filtered the solid. The resultant crude was purified by 60-120 silica gel column chromatography using 1 % methanol in DCM as eluent to obtain the title compound (350mg, 76.25%). LCMS: m/z: 435.4 (M+l)+.
Step 2:Preparation of (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine- 1 -carboxylate
Using the same reaction conditions as described in step 5 of example 1, (S)-tert-butyl 3- ((2-morpholino-6-nitrooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (350mg, 0.806 mmol) was reduced with zinc dust (422mg, 6.451 mmol) and ammonium chloride (691mg, 12.903 mmol) in THF/methanol/H20 (10mL/2mL/lmL) to get the title compound (240mg, 71.8%). LCMS: m/z: 405.2 (M+l)+.
Step 3:Preparation of (S)-tert-butyl 3-((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l-carboxylate
Using the same reaction conditions as described in step 6 of example 1, (S)-tert-butyl 3-((6-amino-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (115mg, 0.284 mmol), was coupled with 2-(2-methylpyridin-4-yl)oxazole-4-carboxylic acid (70mg, 0.341 mmol) using EDCI.HCl (82mg, 0.426 mmol), HOBt (58mg, 0.426 mmol), DIPEA (0.199mL, 1.138 mmol) in DMF (2mL) to afford the title compound (lOOmg, 59.52%). LCMS: m/z: 591.4 (M+l)+.
Step 4: Preparation of (S)-2-(2-methylpyridin-4-yl)-N-(2-morpholino-5-(pyrrolidin-3-ylamino)oxazolo[4,5-b]pyridin-6-yl)oxazole-4-carboxamide
Using the same reaction conditions as described in step 8 of example 1, (S)-tert-butyl 3- ((6-(2-(2-methylpyridin-4-yl)oxazole-4-carboxamido)-2-morpholinooxazolo[4,5-b]pyridin-5-yl)amino)pyrrolidine-l -carboxylate (lOOmg, 0.169 mmol) was deprotected using methanolic HC1 (5mL) to get the crude product. This was then purified by prep HPLC to get the title compound (9mg, 10.84%).
1HNMR (CDCI3, 400MHz): δ 9.91 (s, 1H), 8.78 (s, 1H), 8.74-8.73 (d, 1H), 8.45 (s, 1H), 7.82 (s, 1H), 7.76-7.74 (d, 1H), 4.50 (s, 1H), 4.04-4.03 (d, 4H), 3.30-3.00 (m, 7H), 2.70 (s, 3H), 2.40-1.80 (m, 4H), 1.00-0.08 (m, 1H). LCMS: 100%, m/z = 491.3 (M+l)+.
REFERENCES
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR-NCI-EORTC_2015.pdf
http://www.curis.com/images/stories/pdfs/posters/Aurigene_IRAK4_AACR_20150421.pdf
1Nature. 2011; 470(7332):115–119
2Immunology and Cell Biology. 2011; 89(6):659–660
3N Engl J Med. 30, 2012; 367(9):826–833
April 2014, preclinical data presented at the CHI’s Ninth Drug Discovery Chemistry Conference in San Diego, CA
November 2015, preclinical data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
http://pubs.acs.org/doi/abs/10.1021/jm5016044
http://cancerres.aacrjournals.org/content/75/15_Supplement/3646
2015 Apr 18-22; Philadelphia, PA. Philadelphia (PA): AACR; Cancer Res 2015;75(15 Suppl):Abstract nr 3646. doi:10.1158/1538-7445.AM2015-3646
////////IRAK4 Kinase Inhibitor, Curis, Aurigene, CA 4948, AU 4948, CA-4948, AU-4948, 1428335-77-6
c21ccc(cc1sc(n2)N3CCOCC3)NC(c4nc(ccc4)c5ccc(nc5)N)=O
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on Facebook 


 LIONEL MY SON
LIONEL MY SON
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
Read all about Organic Spectroscopy on ORGANIC SPECTROSCOPY INTERNATIONAL
Curis and Aurigene’s AUPM 170, CA 170
1,2,4-oxadiazole and 1 ,2,4-thiadiazole compounds of formula (I):
ONE EXAMPLE
EXAMPLES
PREDICTED AUPM 170, CA 170, AUPM-170, CA-170
Synthesis coming………….
WATCH THIS SPACE
Aurigene Discovery Technologies Limited INNOVATOR
Curis with the option to exclusively license Aurigene’s orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field
Addressing immune checkpoint pathways is a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients.
Through its collaboration with Aurigene, Curis is now engaged in the discovery and development of the first ever orally bioavailable, small molecule antagonists that target immune checkpoint receptor-ligand interactions, including PD-1/PD-L1 interactions. In the first half of 2016, Curis expects to file an IND application with the U.S. FDA to initiate clinical testing of CA-170, the first small molecule immune checkpoint antagonist targeting PD-L1 and VISTA. The multi-year collaboration with Aurigene is focused on generation of small molecule antagonists targeting additional checkpoint receptor-ligand interactions and Curis expects to advance additional drug candidates for clinical testing in the coming years. The next immuno-oncology program in the collaboration is currently targeting the immune checkpoints PD-L1 and TIM3.
In November 2015, preclinical data were reported. Data demonstrated tha the drug rescued and sustained activation of T cells functions in culture. CA-170 resulted in anti-tumor activity in multiple syngeneic tumor models including melanoma and colon cancer. Similar data were presented at the 2015 AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Conference in Boston, MA
By August 2015, preclinical data had been reported. Preliminary data demonstrated that in in vitro studies, small molecule PD-L1 antagonists induced effective T cell proliferation and IFN-gamma production by T cells that were specifically suppressed by PD-L1 in culture. The compounds were found to have effects similar to anti-PD1 antibodies in in vivo tumor models
(Oral Small Molecule PD-L1/VISTAAntagonist)
Certain human cancers express a ligand on their cell surface referred to as Programmed-death Ligand 1, or PD-L1, which binds to its cognate receptor, Programmed-death 1, or PD-1, present on the surface of the immune system’s T cells. Cell surface interactions between tumor cells and T cells through PD-L1/PD-1 molecules result in T cell inactivation and hence the inability of the body to mount an effective immune response against the tumor. It has been previously shown that modulation of the PD-1 mediated inhibition of T cells by either anti-PD1 antibodies or anti-PD-L1 antibodies can lead to activation of T cells that result in the observed anti-tumor effects in the tumor tissues. Therapeutic monoclonal antibodies targeting the PD-1/PD-L1 interactions have now been approved by the U.S. FDA for the treatment of certain cancers, and multiple therapeutic monoclonal antibodies targeting PD-1 or PD-L1 are currently in development.
In addition to PD-1/PD-L1 immune regulators, there are several other checkpoint molecules that are involved in the modulation of immune responses to tumor cells1. One such regulator is V-domain Ig suppressor of T-cell activation or VISTA that shares structural homology with PD-L1 and is also a potent suppressor of T cell functions. However, the expression of VISTA is different from that of PD-L1, and appears to be limited to the hematopoietic compartment in tissues such as spleen, lymph nodes and blood as well as in myeloid hematopoietic cells within the tumor microenvironment. Recent animal studies have demonstrated that combined targeting/ blockade of PD-1/PD-L1 interactions and VISTA result in improved anti-tumor responses in certain tumor models, highlighting their distinct and non-redundant functions in regulating the immune response to tumors2.
As part of the collaboration with Aurigene, in October 2015 Curis licensed a first-in-class oral, small molecule antagonist designated as CA-170 that selectively targets PD-L1 and VISTA, both of which function as negative checkpoint regulators of immune activation. CA-170 was selected from the broad PD-1 pathway antagonist program that the companies have been engaged in since the collaboration was established in January 2015. Preclinical data demonstrate that CA-170 can induce effective proliferation and IFN-γ (Interferon-gamma) production (a cytokine that is produced by activated T cells and is a marker of T cell activation) by T cells that are specifically suppressed by PD-L1 or VISTA in culture. In addition, CA-170 also appears to have anti-tumor effects similar to anti-PD-1 or anti-VISTA antibodies in multiple in vivo tumor models and appears to have a good in vivo safety profile. Curis expects to file an IND and initiate clinical testing of CA-170 in patients with advanced tumors during the first half of 2016.
![]()
Curis and Aurigene Announce Collaboration, License and Option Agreement to Discover, Develop and Commercialize Small Molecule Antagonists for Immuno-Oncology and Precision Oncology Targets
— Agreement Provides Curis with Option to Exclusively License Aurigene’s Antagonists for Immuno-Oncology, Including an Antagonist of PD-L1 and Selected Precision Oncology Targets, Including an IRAK4 Kinase Inhibitor —
— Investigational New Drug (IND) Application Filings for Both Initial Collaboration Programs Expected this Year —
— Curis to issue 17.1M shares of its Common Stock as Up-front Consideration —
— Management to Host Conference Call Today at 8:00 a.m. EST —
LEXINGTON, Mass. and BANGALORE, India, Jan. 21, 2015 (GLOBE NEWSWIRE) — Curis, Inc. (Nasdaq:CRIS), a biotechnology company focused on the development and commercialization of innovative drug candidates for the treatment of human cancers, and Aurigene Discovery Technologies Limited, a specialized, discovery stage biotechnology company developing novel therapies to treat cancer and inflammatory diseases, today announced that they have entered into an exclusive collaboration agreement focused on immuno-oncology and selected precision oncology targets. The collaboration provides for inclusion of multiple programs, with Curis having the option to exclusively license compounds once a development candidate is nominated within each respective program. The partnership draws from each company’s respective areas of expertise, with Aurigene having the responsibility for conducting all discovery and preclinical activities, including IND-enabling studies and providing Phase 1 clinical trial supply, and Curis having responsibility for all clinical development, regulatory and commercialization efforts worldwide, excluding India and Russia, for each program for which it exercises an option to obtain a license.
The first two programs under the collaboration are an orally-available small molecule antagonist of programmed death ligand-1 (PD-L1) in the immuno-oncology field and an orally-available small molecule inhibitor of Interleukin-1 receptor-associated kinase 4 (IRAK4) in the precision oncology field. Curis expects to exercise its option to obtain exclusive licenses to both programs and file IND applications for a development candidate from each in 2015.
“We are thrilled to partner with Aurigene in seeking to discover, develop and commercialize small molecule drug candidates generated from Aurigene’s novel technology and we believe that this collaboration represents a true transformation for Curis that positions the company for continued growth in the development and eventual commercialization of cancer drugs,” said Ali Fattaey, Ph.D., President and Chief Executive Officer of Curis. “The multi-year nature of our collaboration means that the parties have the potential to generate a steady pipeline of novel drug candidates in the coming years. Addressing immune checkpoint pathways is now a well validated strategy to treat human cancers and the ability to target PD-1/PD-L1 and other immune checkpoints with orally available small molecule drugs has the potential to be a distinct and major advancement for patients. Recent studies have also shown that alterations of the MYD88 gene lead to dysregulation of its downstream target IRAK4 in a number of hematologic malignancies, including Waldenström’s Macroglobulinemia and a subset of diffuse large B-cell lymphomas, making IRAK4 an attractive target for the treatment of these cancers. We look forward to advancing these programs into clinical development later this year.”
Dr. Fattaey continued, “Aurigene has a long and well-established track record of generating targeted small molecule drug candidates with bio-pharmaceutical collaborators and we have significantly expanded our drug development capabilities as we advance our proprietary drug candidates in currently ongoing clinical studies. We believe that we are well-positioned to advance compounds from this collaboration into clinical development.”
CSN Murthy, Chief Executive Officer of Aurigene, said, “We are excited to enter into this exclusive collaboration with Curis under which we intend to discover and develop a number of drug candidates from our chemistry innovations in the most exciting fields of cancer therapy. This unique collaboration is an opportunity for Aurigene to participate in advancing our discoveries into clinical development and beyond, and mutually align interests as provided for in our agreement. Our scientists at Aurigene have established a novel strategy to address immune checkpoint targets using small molecule chemical approaches, and have discovered a number of candidates that modulate these checkpoint pathways, including PD-1/PD-L1. We have established a large panel of preclinical tumor models in immunocompetent mice and can show significant in vivo anti-tumor activity using our small molecule PD-L1 antagonists. We are also in the late stages of selecting a candidate that is a potent and selective inhibitor of the IRAK4 kinase, demonstrating excellent in vivo activity in preclinical tumor models.”
In connection with the transaction, Curis has issued to Aurigene approximately 17.1 million shares of its common stock, or 19.9% of its outstanding common stock immediately prior to the transaction, in partial consideration for the rights granted to Curis under the collaboration agreement. The shares issued to Aurigene are subject to a lock-up agreement until January 18, 2017, with a portion of the shares being released from the lock-up in four equal bi-annual installments between now and that date.
The agreement provides that the parties will collaborate exclusively in immuno-oncology for an initial period of approximately two years, with the option for Curis to extend the broad immuno-oncology exclusivity.
In addition Curis has agreed to make payments to Aurigene as follows:
- for the first two programs: up to $52.5 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any;
- for the third and fourth programs: up to $50 million per program, including $42.5 million per program for approval and commercial milestones, plus specified approval milestone payments for additional indications, if any; and
- for any program thereafter: up to $140.5 million per program, including $87.5 million per program in approval and commercial milestones, plus specified approval milestone payments for additional indications, if any.
Curis has agreed to pay Aurigene royalties on any net sales ranging from high single digits to 10% in territories where it successfully commercializes products and will also share in amounts that it receives from sublicensees depending upon the stage of development of the respective molecule.
About Immune Checkpoint Modulation and Programmed Death 1 Pathway
Modulation of immune checkpoint pathways has emerged as a highly promising therapeutic approach in a wide range of human cancers. Immune checkpoints are critical for the maintenance of self-tolerance as well as for the protection of tissues from excessive immune response generated during infections. However, cancer cells have the ability to modulate certain immune checkpoint pathways as a mechanism to evade the immune system. Certain immune checkpoint receptors or ligands are expressed by various cancer cells, targeting of which may be an effective strategy for generating anti-tumor activity. Some immune-checkpoint modulators, such as programmed death 1 (PD-1) protein, specifically regulate immune cell effector functions within tissues. One of the mechanisms by which tumor cells block anti-tumor immune responses in the tumor microenvironment is by upregulating ligands for PD-1, such as PD-L1. Hence, targeting of PD-1 and/or PD-L1 has been shown to lead to the generation of effective anti-tumor responses.
About Curis, Inc.
Curis is a biotechnology company focused on the development and commercialization of novel drug candidates for the treatment of human cancers. Curis’ pipeline of drug candidates includes CUDC-907, a dual HDAC and PI3K inhibitor, CUDC-427, a small molecule antagonist of IAP proteins, and Debio 0932, an oral HSP90 inhibitor. Curis is also engaged in a collaboration with Genentech, a member of the Roche Group, under which Genentech and Roche are developing and commercializing Erivedge®, the first and only FDA-approved medicine for the treatment of advanced basal cell carcinoma. For more information, visit Curis’ website at www.curis.com.
About Aurigene
Aurigene is a specialized, discovery stage biotechnology company, developing novel and best-in-class therapies to treat cancer and inflammatory diseases. Aurigene’s Programmed Death pathway program is the first of several immune checkpoint programs that are at different stages of discovery and preclinical development. Aurigene has partnered with several large- and mid-pharma companies in the United States and Europe and has delivered multiple clinical compounds through these partnerships. With over 500 scientists, Aurigene has collaborated with 6 of the top 10 pharma companies. Aurigene is an independent, wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (NYSE:RDY). For more information, please visit Aurigene’s website at http://aurigene.com/.
POSTER


WO2011161699, WO2012/168944, WO2013144704 and WO2013132317 report peptides or peptidomimetic compounds which are capable of suppressing and/or inhibiting the programmed cell death 1 (PD1) signaling pathway.
PATENT
Example 5: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 4 (compound 4) using D-amino acids are linked up in reverse order. Boc-D-Thr(‘Bu)-OH was used in place of Boc-Ser(‘Bu)-OH, Fmoc-D-Asn(trt)-OH in place of Fmoc-Asn(trt)-OH and H-D-Ser(‘Bu)-0’Bu was used in place of H-Thr^Bu^O’Bu to yield 0.3 g crude material of the title compound. The cmde solid material was purified using preparative HPLC described under experimental conditions. LCMS: 361.3 (M+H)+. HPLC: tR = 13.58 min.
Example 8: Synthesis of

The compound was synthesised using similar procedure as depicted in Example 2 (compound 2) using Fmoc-Glu(0’Bu)-OH instead of Fmoc-Asn(Trt)-OH to get 0.4 g crude material of the title compound. The crude solid material was purified using preparative HPLC described under experimental conditions. LCMS: 362.1 (M+H)+. HPLC: tR = 13.27 min.
PATENT
Example 3: Synthesis of compound 3
Step 3a:

3a
Lawesson’s reagent (2.85 g, 7.03 mmol) was added to a solution of compound 2e (4 g, 4.68 mmol) in THF (40 mL) and stirred at 75°C for 4 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced
pressure and the obtained residue was partitioned between ice water and ethyl acetate. The organic layer was washed with NaHCC>3 solution followed brine solution. The organic layer was dried over Na2S04, filtered and evaporated under reduced pressure to get residue which was further purified by silica gel column chromatography (eluent: 0-5% ethyl acetate in hexane) to afford 2.7 g of compound 3a (Yield: 67.66%). LCMS: 852.3 (M+H)+,
Step 3

3a 3b
Fmoc group on compound 3a was deprotected by adding diethylamine (3.8 mL) to the solution of compound 3a (1 g, 1.17 mmol) in CH2CI2 (3.8 mL). The reaction mixture was stirred at room temperature for 30 min. The resulting solution was concentrated in vacuum to get a thick gummy residue. The crude compound was purified by neutral alumina column chromatography (eluent: 0-50% ethyl acetate in hexane then 0-5% methanol in chloroform) to attain 0.62 g of compound 3b. LCMS: 630.5 (M+H)+.
Step 3c

To a solution of compound 3b (0.6 g) in CH2CI2 (7.5 mL), trifluoroacetic acid (2.5 mL) and catalytic amount of triisopropylsilane were added and stirred at room temperature for 3 h. The resulting solution was concentrated in vacuum to get 0.13 g of compound 3 which was purified by preparative HPLC method described under experimental conditions. LCMS: 232.3 (M+H)+.
Example 1: Synthesis of compound 1
Step la:

Potassium carbonate (7.9 g, 57.39 mmol) and Methyl iodide (1.3 mL, 21.04 mmol) were added to a solution of compound la (5.0 g, 19.13 mmol) in DMF (35 mL) and stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get 5.0 g of compound lb (Yield: 96.1%). LCMS: 176.1 (M-Boc)+.
Step lb:

Hydrazine hydrate (7.2 mL) was added to a solution of compound lb (5.0 g, 18.16 mmol) in methanol (30 mL) and stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction mixture was evaporated under reduced pressure, the residue obtained was partitioned between water and ethyl acetate. Organic layer was washed with water, brine, dried over Na2S04 and evaporated under reduced pressure to get 4.0 g of compound lc (Yield: 80.0%). LCMS: 276.3 (M+H)+. Step lc:

NMM (0.67 ml, 6.52 mmol) was slowly added to a stirred solution of lc (1.2 g, 4.35 mmol), Id (1.43 g, 4.35 mmol), HOBt (0.7 g, 5.22 mmol) and EDC.HC1 (0.99 g, 5.22 mmol) in DMF (15 mL) at 0°C. The reaction mixture was stirred at room temperature for 12 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice and the solid precipitated was filtered and dried under vacuum to obtain 2.0 g of pure product le (Yield: 83.3%). LCMS: 591.5 (M+Na)+.
St

1 e
1f
To a stirred solution of le (1.5 g, 2.63 mmol) in dry THF (15.0 mL) and DMF (5.0 mL) triphenylphosphine (1.38 g, 5.27 mmol) and iodine (1.33 g, 5.27 mmol) were added at 0°C. After the iodine was completely dissolved, Et3N (1.52 mL, 10.54 mmol) was added to this reaction mixture at ice cold temperature. Reaction mixture was allowed to attain room temperature and stirred for 4 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice water and extracted with ethyl acetate. Organic layer was washed with saturated sodium thiosulphate and brine solution.
The separated Organic layer was dried over Na2SC>4 and evaporated under reduced pressure to get residue, which was further purified by silica gel column chromatography (eluent: 30% ethyl acetate in hexane) to afford 0.8 g of compound If (Yield: 55%). LCMS: 551.3 (M+H)+.
Step le:

1f i g
Fmoc group was deprotected by the addition of diethylamine (20.0 mL) to a solution of compound If (0.8 g, 1.45 mmol) in CH2CI2 (20.0 mL) at 0°C. The reaction was stirred at room temperature for 2 h. The resulting solution was concentrated in vacuum to get a thick gummy residue. The crude compound was purified by neutral alumina column chromatography (eluent: 2% methanol in chloroform) to afford 0.38 g of compound lg (Yield: 80.0%): LCMS: 329.4 (M+H)+.
Step If:

ig 1 i
Compound lg (0.38 g, 1.16 mmol), TEA (0.33 mL, 2.32 mmol) dissolved in DMF (10 mL) were added drop wise to a solution of lh (0.55 g, 1.39 mmol) at 0°C for urea bond formation and the mixture was stirred at room temperature for 2 h. The completeness of the reaction was confirmed by TLC analysis. The reaction was quenched with ice water, the solid precipitated was filtered and dried under vacuum to get crude compound, which was further purified by silica gel column chromatography (eluent: 0-35% ethyl acetate in hexane) to get 0.4 g of product li (Yield: 59.7%). LCMS: 586.4 (M+H)+.
Step lg:
BocHN’ IJ, H LT Y~™
![]()
1
To a solution of compound li (0.4 g, 0.68 mmol) in CH2CI2 (5 m L), trifluoro acetic acid (5 mL) and catalytic amount of triisopropylsilane were added and stirred at room temperature for 3 h to remove the acid sensitive protecting groups. The resulting solution was concentrated under nitrogen and the solid material was purified by preparative HPLC method as described under experimental conditions (Yield: 0.05 g). LCMS: 318.0 (M+H)+; HPLC: tR= 10.96 min.
Synthesis of compound lh (N02-C6H4-OCO-Thr(tBu)- 0¾u):

To a solution of 4-nitrophenylchloroformate (4.79 g, 23.77 mmol) in DCM (25.0 mL) was added a solution of H-Thr(tBu)-OtBu (5.0 g, 21.61 mmol) TEA (6.2 mL, 43.22 mmol) in CH2CI2 (25 mL) slowly at 0°C and allowed to stir for 30 min. The completion of the reaction was confirmed by TLC analysis. After completion of reaction it was diluted with DCM and washed with 1.0 M of citric acid followed by 1.0 M sodium carbonate solution. The organic layer was dried over Na2S04 and evaporated under reduced pressure to afford crude compound 1 h, which was further purified by silica gel column chromatography (eluent: 0-5% ethyl acetate in hexane) to get 3.0 g of product lh. jH NMR (CDCI3, 400 MHz): £1.17 (s, 9H), 1 .28 (d, 3H), .50 (s, 9H), 4.11 (m, 1 H), 4.28 (m, 1H , 5.89 (d, 1H), 7.37 (d, 2H), 8.26 (d, 2H).


Brahma Reddy V, Thomas Antony, Murali Ramachandra, Venkateshwar Rao G, Wesley Roy Balasubramanian, Kishore Narayanan, Samiulla DS, Aravind AB, and Shekar Chelur.
REFERENCES
US20150073024
| WO2011161699A2 | 27 Jun 2011 | 29 Dec 2011 | Aurigene Discovery Technologies Limited | Immunosuppression modulating compounds |
| WO2012168944A1 | 21 Dec 2011 | 13 Dec 2012 | Aurigene Discovery Technologies Limited | Therapeutic compounds for immunomodulation |
| WO2013132317A1 | 4 Mar 2013 | 12 Sep 2013 | Aurigene Discovery Technologies Limited | Peptidomimetic compounds as immunomodulators |
| WO2013144704A1 | 28 Mar 2013 | 3 Oct 2013 | Aurigene Discovery Technologies Limited | Immunomodulating cyclic compounds from the bc loop of human pd1 |
http://www.curis.com/images/stories/pdfs/posters/Aurigene_PD-L1_VISTA_AACR-NCI-EORTC_2015.pdf
////////Curis and Aurigene, AUPM 170, CA 170, AUPM-170, CA-170, PD-L1, VISTA antagonist
ND 2158

(2S)-2-hydroxy-3-[(3R)-12-{[(1r,4r)-4-(morpholin-4-yl)cyclohexyl]oxy}-7-thia-9,11-diazatricyclo[6.4.0.0²,⁶]dodeca-1(12),2(6),8,10-tetraen-3-yl]propanamide
S)-2-hydroxy-3-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7-dihydro-5H-cyclopenta [4,5] thieno [2,3-d] pyrimidin-5-yl)propanamide
- Molecular Weight446.56
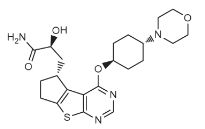
ND 2158
IRAK4, 446.2

ND-2158 is a potent and selective experimental inhibitor of IRAK4 described in patent WO2013106535 [2] and in a poster presented at the American College of Rheumatology meeting in 2012 (Abstract #1062 in Supplement: Abstracts of the American College of Rheumatology & Association of Rheumatology Health Professionals, Annual Scientific Meeting, November 9-4, 2012 Washington DC, Volume 64, Issue S10, Page S1-S1216).

PATENT
http://www.google.com/patents/WO2013106535A1?cl=en


Scheme II

Example 88: (S)-l-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7-dihydro- 5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-5-yl)butan-2-ol (1-64) and Example 89: (R)-l- ((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-

Synthesis of compound 88.1. Note: For the preparation of the starting material compound 29.2, please see Example 29. A solution of
yl)cyclohexyl]oxy]-7-thia-9,l l-diazatricyclo[6.4.0.0[2,6]]dodeca-l(8),2(6),9,l l-tetraen-3- yl]ethan-l-ol (190 mg, 0.47 mmol, 1.00 equiv) in 10 mL of dichloromethane was added Dess- Martin periodinane at 0 °C in a water/ice bath under nitrogen. The resulting mixture was stirred for 2 h at room temperature. After completion of the reaction, the mixture was then diluted with saturated aqueous sodium bicarbonate and extracted with 3 x 30 mL of ethyl acetate. The combined organic layers were dried over sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5 to 1 : 1) to afford 2-[(3Λ)-12-[[4-^ο 1ιο1ϊη-4-γ1)ογο1ο1ιβχγ1]οχγ]-7-ωΕ-9,11- diazatricyclo[6.4.0.0[2,6]]dodeca-l(8),2(6),9,l l-tetraen-3-yl]acetaldehyde (130 mg, 69%) as a colorless oil. MS (ES): m/z 402 [M+H]+.
Synthesis of Compound 1-64 and Compound 1-65. A solution of [(3i?)-12-[[4- (moφholin-4-yl)cyclohexyl]oxy]-7-thia-9,l l-diazatricyclo[6.4.0.0[2,6]]dodeca-l(8),2(6),9,l l- tetraen-3-yl]acetaldehyde (130 mg, 0.32 mmol, 1.00 equiv) in 5 mL of anhydrous THF was added bromo(ethyl)magnesium (1 M in THF, 0.62 mL, 2.0 equiv) dropwise at 0 °C under nitrogen. The resulting solution was stirred for 4 h at room temperature and then quenched by the addition of saturated aqueous NH4CI and extracted with 3 x 50 mL of DCM/i-PrOH (3:1). The combined organic layers was dried over anhydrous sodium sulfate and concentrated under vacuum. The crude product (150 mg) was purified by preparative HPLC under the following conditions (SHIMADZU): column: SunFire Prep C18, 19*150 mm 5um; mobile phase: water with 0.05% NH4CO3 and CH3CN (6.0% CH3CN up to 54.0% in 25 min); UV detection at 254/220 nm to afford (S)-l-((R)-4-(((lr,4R)-4-moφholinocyclohexyl)oxy)-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-d]pyrimidin-5-yl)butan-2-ol (11.8 mg) and (R)-l-((R)-4-(((lr,4R)-4- mo holinocyclohexyl)oxy)-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-5-yl)butan- 2-ol (23.9 mg) as white solids.
Example 88 (1-64): MS: 432 (M+H)+. ¾ NMR (300 MHz, CDC13) S 8.47 (s, 2H), 5.24-5.20 (m, 1H), 3.75-3.58 (m, 5H), 3.06-2.93 (m, 2H), 2.70-2.61 (m, 4H), 2.28-1.98 (m, 3H), 1.59-1.41 (m, 10H), 1.28-1.23 (m, 2H),0.95-0.85 (m, 3H).
Example 89 (1-65): MS: 432 (M+H)+. ¾ NMR (300 MHz, CDC13) S 8.47 (s, 2H), 5.25 (m, 1H), 3.71-3.39 (m, 6H), 3.04-2.90 (m, 2H), 2.67-2.55 (m, 5H), 2.34-2.22 (m, 4H), 2.01- 1.81 (m, 3H), 1.64-1.39 (m, 7H), 0.94-0.92 (m, 3H).
WATCH OUT SYNTHESIS COMING…………
PATENT
WO 2014011906
https://www.google.co.in/patents/WO2014011906A2?cl=en
PATENT
WO-2014194242
https://www.google.com/patents/WO2014194242A2?cl=en
Example 49: Synthesis of Intermediate 49.1.
step 1 step 2
35.1 49.1 49.2
step 3 49 3
] Intermediate 49.3 was prepared from 35.1 in a manner analogous to the synthesis of 36.3. Isolated 150 mg of a white solid in 57% overall yield. MS (ES): m/z 402 [M+H]+.
Example 50: Synthesis of Intermediate 50.4.
49.3 50.1 50.2
50.3 50.4
Intermediate 50.4 was prepared from 49.3 in a manner analogous to the synthesis of 1-25, except that HCl/MeOH rather than TBAF/THF was used in the second step. Isolated 124 mg of a white solid in 48% overall yield. MS (ES): m/z 447 [M+H]+. 1H NMR (400 MHz, CDCls): δ 8.46 (s, 1H), 5.28-5.25 (m, 1H), 4.17-4.06 (m, 51H), 3.74-3.72 (m, 5H), 3.37-2.98 (m, 2H), 2.72-2.28 (m, 10H), 2.11-2.08 (m, 2H), 1.79-1.46 (m, 5H).
Example 51: Synthesis of (S)-2-hydroxy-3-((R)-4-(((lr,4R)-4- morpholinocyclohexyl)oxy)-6,7-dihydro-5H-cyclopenta [4,5] thieno [2,3-d] pyrimidin-5- yl)propanamide (1-34) and Example 52: Synthesis of (R)-2-hydroxy-3-((R)-4-(((lr,4R)-4- morpholinocyclohexyl)oxy)-6,7-dihydro-5H-cyclopenta [4,5] thieno [2,3-d] pyrimidin-5- yl)propanamide (1-44)
The racemic 50.4 (1.6 g, 96.5% purity) was separated by Chiral-HPLC with the following conditions (Gilson G x 281): column: Chiralpak AD-H, 2*25 cm Chiral-P(AD-H); mobile phase: phase A: hex (O. P/oDEA) (HPLC grade), phase B: IPA (HPLC grade), gradient: 30% B in 9 min; flow rate: 20 mL/min; UV detection at 220/254 nm. The former fractions (tR = 4.75 min) were collected and evaporated under reduced pressure and lyophilized overnight to afford 1-44 (520 mg) with 100% ee as a white solid. And the latter fractions (tR = 5.82 min) were handled as the former fractions to give the desired 1-34 (510 mg) with 99.6%> ee as a white solid. The ee values of the two isomers were determined by the chiral-HPLC with the following conditions (SHIMADZU-SPD-20A): column: Chiralpak AD-H, 0.46*25 cm, 5um (DAICEL); mobile phase: hex (0.1% TEA): IPA = 85:15; UV detection at 254 nm. Flow rate: 1.0 mL/min. tR (1-44) = 7.939 min and tR (1-34) = 11.918 min.
[00431] Analytical data for 1-44: MS: (ES, m/z) 447 [M+H]+. 1H NMR (400 MHz, CD3OD+CDCI3): δ 8.47 (s, 1H), 5.32-5.22 (m, 1H), 4.08 (dd, 1H), 4.89-4.62 (m, 5H), 3.20-3.10 (m, 1H), 3.05-2.95 (m, 1H), 2.75-2.55 (m, 5H), 2.44-2.38 (m, 2H), 2.34-2.28 (m, 3H), 2.10 (d, 2H), 1.82-1.62 (m, 3H), 1.58-1.40 (m, 2H).
Analytical data for 1-34: MS: (ES, m/z) 447 [M+H]+. 1H NMR (400 MHz, CDC13): δ 8.46 (s, 1H), 5.32-5.22 (m, 1H), 4.15 (t, 1H), 3.73 (t, 4H), 3.59 (td, 1H), 3.19-3.08 (m, 1H), 3.02- 2.92 (m, 1H), 2.78-2.70 (m, 1H), 2.69-2.60 (m, 4H), 2.58-2.20 (m, 5H), 2.10 (d, 2H), 1.75-1.63 (m, 3H), 1.53-1.40 (m, 2H).
Paper
http://pubs.acs.org/doi/abs/10.1021/jm5016044
Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders
Miniperspective

IRAK4, a serine/threonine kinase, plays a key role in both inflammation and oncology diseases. Herein, we summarize the compelling biology surrounding the IRAK4 signaling node in disease, review key structural features of IRAK4 including selectivity challenges, and describe efforts to discover clinically viable IRAK4 inhibitors. Finally, a view of knowledge gained and remaining challenges is provided.
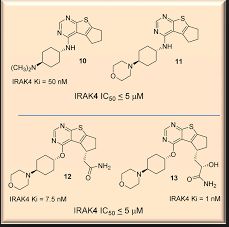
-
78 Romero, D. L.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO201401902, January 16, 2014.
-
Harriman, G. C.; Romero, D. L.; Masse, C. E.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011911A2, January 16, 2014.
-
Harriman, G. C.; Wester, R. T.; Romero, D. L.; Masse, C. E.; Robinson, R.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011906A2, January 16, 2014
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013231328 | 2013-09-05 | IRAK INHIBITORS AND USES THEREOF |
PATENT

WO 2014194242
WO 2013106535
WO 2012097013
| US20070155777 * | Feb 21, 2007 | Jul 5, 2007 | Amgen, Inc. | Antiinflammation agents |
| US20100041676 * | Feb 18, 2010 | Hirst Gavin C | Kinase inhibitors | |
| US20100143341 * | Jun 21, 2006 | Jun 10, 2010 | Develogen Aktiengesellschaft | Thienopyrimidines for pharmaceutical compositions |
| US20120015962 * | Jan 19, 2012 | Nidhi Arora | PYRAZOLO[1,5a]PYRIMIDINE DERIVATIVES AS IRAK4 MODULATORS | |
| US20120283238 * | Nov 8, 2012 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| References |
| 1. Chaudhary D, Robinson S, Romero DL. (2015) Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders. J. Med. Chem., 58 (1): 96-110. [PMID:25479567] |
| 2. Harriman GC, Wester RT, Romero DL, Robinson S, Shelley M, Wessel MD, Greenwood JR, Masse CE, Kapeller-Libermann R. (2013) Irak inhibitors and uses thereof. Patent number: WO2013106535. Assignee: Nimbus Iris, Inc.. Priority date: 18/07/2013. Publication date: 10/01/2012. |
http://nimbustx.com/sites/default/files/uploads/posters/irak4_nimbus_acr_poster_2012_small.pdf
///////ND 2158, IRAK4, ND-2158, NIMBUS, 1388896-07-8
NC(=O)C(CC1CCc2c1c1c(ncnc1s2)OC1CCC(CC1)N1CCOCC1)O
C1CC(CCC1N2CCOCC2)OC3=C4C5=C(CCC5CC(C(=O)N)O)SC4=NC=N3
ND 2110
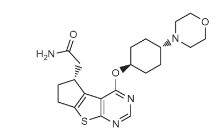
ND -2110
| Molecular Formula: | C21H28N4O3S |
|---|---|
| Molecular Weight: | 416.53702 g/mol |

2-[(3R)-12-{[(1r,4r)-4-(morpholin-4-yl)cyclohexyl]oxy}-7-thia-9,11-diazatricyclo[6.4.0.0²,⁶]dodeca-1(12),2(6),8,10-tetraen-3-yl]acetamide
1388894-17-4
- Molecular Weight416.54
ND-2110 is a potent and selective experimental inhibitor of IRAK4 described in patent WO2013106535 [2] and in a poster presented at the American College of Rheumatology meeting in 2012 (Abstract #1062 in Supplement: Abstracts of the American College of Rheumatology & Association of Rheumatology Health Professionals, Annual Scientific Meeting, November 9-4, 2012 Washington DC, Volume 64, Issue S10, Page S1-S1216).
PATENT
http://www.google.com/patents/WO2013106535A1?cl=en
Example 29: Synthesis of 2-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7-

29.3 1-67
Synthesis of compound 29.1. 4-(Morpholin-4-yl)cyclohexan-l-ol (commercially available; 218 mg, 1.2 mmol, 1.50 equiv) was treated with NaH (60% dispersion in mineral oil, 128 mg, 3.2 mmol, 4 equiv) in freshly distilled tetrahydrofuran (15 mL) for 30 min at 0 °C in a water/ice bath under nitrogen. Then a solution of intermediate 25.1 (289 mg, 0.8 mmol, 1.00 equiv) in 5 mL of THF was added via syringe and the resulting solution was allowed to stir for an additional 3 h at 60 °C in an oil bath. The reaction was then quenched with saturated aqueous NH4CI and extracted with 3 x 50 mL of ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1:5-1:2) and purified to afford compound 29.1 (260 mg, 63%) as a colorless oil.
Synthesis of compound 29.2. To a solution of 29.1 (260 mg, 0.5 mmol, 1.0 equiv) in 10 mL of DCM was added 0.5 mL of concentrated hydrochloric acid in an ice/water bath. The resulting solution was stirred for 2 h and concentrated in vacuo. The residue was neutralized with saturated aqueous Na2C03 and extracted with 3 x 50 mL of ethyl acetate. The organic layers were combined, washed with brine, dried (Na2S04) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with DCM MeOH (15:1) to afford the desired alcohol 29.2 (185 mg, 91%) as a colorless oil. [00416] Synthesis of compound 29.3. Alcohol 29.2 (185 mg, 0.46 mmol, 1.00 equiv) was oxidized with dipyridinium dichromate (752 mg, 2.00 mmol, 4.36 equiv) in 50 mL of DMF for 24 h at room temperature. The resulting solution was diluted with water and extracted with 3 x 50 mL of mixed solutions of CHC¾/iso-PrOH. The organic layers were combined, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (5:1 to 1:1) and purified to afford 105 mg (55%) of acid 29.3 as a yellow oil.
[00417] Synthesis of Compound 1-67. A 50 mL round-bottom flask containing a solution of acid 29.3 (105 mg, 0.25 mmol, 1.00 equiv), NH4C1 (80 mg, 1.50 mmol, 6.00 equiv), EDCI (57 mg, 0.3 mmol, 1.2 equiv), 4-dimethylaminopyridine (37 mg, 0.3 mmol, 1.2 equiv) and HOBt (40 mg, 0.3 mmol, 1.2 equiv) in 5 mL of anhydrous DMF was stirred for 24 h at room temperature. The resulting solution was diluted with water and extracted with 4 x 50 mL of mixed solution of CHCl3:iso-PrOH. The combined organic layers were concentrated under vacuum. The crude product was purified by preparative HPLC (SHIMADZU) under the following conditions: column: SunFire Prep C18, 19*150mm 5um; mobile phase: water (0.05% NH4CO3) and CH3CN (6.0% CH3CN up to 50.0% in 25 min); UV detection at 254/220 nm. The product-containing fractions were collected and concentrated to give Compound 1-67 (22.5 mg) as a white solid. ¾ NMR (300 MHz, CD3OD) δ 8.43 (s, 1H), 5.27-5.20 (m, 1H), 3.80-3.70 (m, 5H), 3.29-3.27 (m, 1H), 3.12-2.90 (m, 2H), 2.73-2.67 (m, 5H), 2.49-2.42 (m, 1H), 2.32-2.19 (m, 4H), 2.10-2.06 (d, 2H), 1.67-1.46 (m, 4H). MS: m/z 417 (M+H)+.
PATENT
http://www.google.com/patents/WO2012097013A1
Example 29: Synthesis of 2-((R)-4-(((lr,4R)-4-morpholinocyclohexyl)oxy)-6,7- dihydro-5H-cyclopenta[4,5]thieno[2,3-d]pyrimidin-5-yl)acetamide.
Page 280 of 407
2009184-0008


33b
Synthesis of compound 31b. 4-(Morpholin-4-yl)cyclohexan-l-ol (commercially available; 218 mg, 1.2 mmol, 1.50 equiv) was treated with NaH NMR (60% dispersion in mineral oil, 128 mg, 3.2 mmol, 4 equiv) in freshly distilled tetrahydrofuran (15 mL) for 30 min at 0 °C in a water/ice bath under nitrogen. Then a solution of intermediate Hb (289 mg, 0.8 mmol, 1.00 equiv) in 5 mL of THF was added via syringe and the resulting solution was allowed to stir for an additional 3 h at 60 °C in an oil bath. The reaction was then quenched with saturated aqueous NH4CI and extracted with 3 x 50 mL of ethyl acetate. The combined organic layers were washed with brine, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 :5-1 :2) and purified to afford compound 31b (260 mg, 63%) as a colorless oil.
Synthesis of compound 32b. To a solution of 31b (260 mg, 0.5 mmol, 1.0 equiv) in 10 mL of DCM was added 0.5 mL of concentrated hydrochloric acid in an ice/water bath. The resulting solution was stirred for 2 h and concentrated in vacuo. The residue was neutralized with saturated aqueous Na2C( j and extracted with 3 x 50 mL of ethyl acetate. The organic layers were combined, washed with brine, dried (Na2S04) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with DCM/MeOH NMR ( 15: 1 ) to afford the desired alcohol 32b ( 185 mg, 91 %) as a colorless oil.
Synthesis of compound 33b. Alcohol 32b (185 mg, 0.46 mmol, 1.00 equiv) was oxidized with dipyridinium dichromate (752 mg, 2.00 mmol, 4.36 equiv) in 50 mL of DMF for
Page 281 of 407
2009184-0008 24 h at room temperature. The resulting solution was diluted with water and extracted with 3 x 50 mL of mixed solutions of CHCU/iso-PrOH. The organic layers were combined, dried (Na2S04) and concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (5: 1 to 1 : 1 ) and purified to afford 105 mg (55%) of acid 33b as a yellow oil.
Synthesis of Compound. A 50 mL round-bottom flask containing a solution of acid 33b (105 mg, 0.25 mmol, 1.00 equiv), NH4C1 (80 mg, 1.50 mmol, 6.00 equiv), EDCI (57 mg, 0.3 mmol, 1.2 equiv), 4-dimethylaminopyridine (37 mg, 0.3 mmol, 1.2 equiv) and HOBt (40 mg, 0.3 mmol, 1.2 equiv) in 5 mL of anhydrous DMF was stirred for 24 h at room temperature. The resulting solution was diluted with water and extracted with 4 x 50 mL of mixed solution of CHCI3: iso-PrOH. The combined organic layers were concentrated under vacuum. The crude product was purified by preparative HPLC (SHIMADZU) under the following conditions: column: SunFire Prep C I 8, 19* 150mm 5um; mobile phase: water (0.05% Ν¾∞3) and CH3CN (6.0% CH3CN up to 50.0% in 25 min); UV detection at 254/220 nm. The product containing fractions were collected and concentrated to give the product (22.5 mg) as a white solid. Ή MR (300 MHz, CD3OD) δ 8.43 (s, 1H), 5.27-5.20 (m, 1H), 3.80-3.70 (m, 5H), 3.29-3.27 (m, 1 H), 3.12-2.90 (m, 2H), 2.73-2.67 (m, 5H), 2.49-2.42 (m, 1H), 2.32-2.19 (m, 4H), 2.10-2.06 (d, 2H), 1.67- 1.46 (m, 4H). MS: m/z 417 (M+H)+.
Paper
http://pubs.acs.org/doi/abs/10.1021/jm5016044
Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders
Miniperspective
IRAK4, a serine/threonine kinase, plays a key role in both inflammation and oncology diseases. Herein, we summarize the compelling biology surrounding the IRAK4 signaling node in disease, review key structural features of IRAK4 including selectivity challenges, and describe efforts to discover clinically viable IRAK4 inhibitors. Finally, a view of knowledge gained and remaining challenges is provided.
-
78 Romero, D. L.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO201401902, January 16, 2014.
-
Harriman, G. C.; Romero, D. L.; Masse, C. E.; Robinson, S.; Wessel, M. D.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011911A2, January 16, 2014.
-
Harriman, G. C.; Wester, R. T.; Romero, D. L.; Masse, C. E.; Robinson, R.; Greenwood, J. R. IRAK Inhibitors and Uses Thereof. WO2014011906A2, January 16, 2014
WO 2014194245
WO 2014194201
WO 2014194242
WO 2013106535
WO 2012097013
| 1. Chaudhary D, Robinson S, Romero DL. (2015) Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders. J. Med. Chem., 58 (1): 96-110. |
| 2. Harriman GC, Wester RT, Romero DL, Robinson S, Shelley M, Wessel MD, Greenwood JR, Masse CE, Kapeller-Libermann R. (2013) Irak inhibitors and uses thereof. Patent number: WO2013106535C1CC(CCC1N2CCOCC2)OC3=C4C5=C(CCC5CC(=O)N)SC4=NC=N3. Assignee: Nimbus Iris, Inc.. Priority date: 18/07/2013. Publication date: 10/01/2012. |
http://nimbustx.com/sites/default/files/uploads/posters/irak4_nimbus_acr_poster_2012_small.pdf
///////ND-2110, ND 2110. IRAK4, NIMBUS, GTPL8802
NC(=O)CC1CCc2c1c1c(ncnc1s2)OC1CCC(CC1)N1CCOCC1
C1CC(CCC1N2CCOCC2)OC3=C4C5=C(CCC5CC(=O)N)SC4=NC=N3
CN-128 for the treatment of thelassemia and iron overload

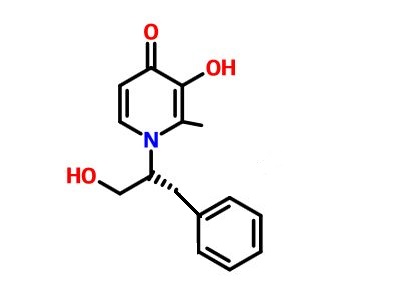
CN-128
(R)-3-Hydroxy-1-(1-hydroxy-3-benzyl propyl-2-)2-methyl pyridine-4(1H)-one
IND Filing
CN-128 is potentially for the treatment of thelassemia and iron overload.
| Zhejiang University, 浙江大学 |
![]()
CAS No. 1335282-04-6
- Molecular Weight, 259.30
Many diseases in humans and animals are caused by excessive accumulated metals, such as iron. Among such diseases, excess iron is accumulated in various tissues, which is called iron overload disorders, formerly known as siderosis Haemorrhagic. Excess iron has the following sources: 1) long-term blood transfusion; 2) the gastrointestinal system absorbing excess iron, because stimulated by diseases such as anemia. It is necessary to repeat transfusion for some patients with severe anemia, for example, β-thalassemia, as well as other anemia requiring transfusion therapy. Excessive iron absorption from the gastrointestinal tract usually occurs in hemochromatosis patients and in anemia patients who do not require blood transfusion, such as thalassemia intermedia. If iron overload disease is not treated, it will result in severe tissue damage, especially the liver, heart and endocrine organs, and ultimately lead to death. Iron chelators can remove and clear excess iron from such organs, relieve symptoms and reduce the corresponding mortality.
Desferrioxamine (DFO) is an effective iron chelator for a long time. However, in the treatment of the diseases mentioned above, the biggest disadvantage regarding DFO and its salts is its poor oral absorption capability. So, administration is achieved with a slow injection method (8∼12h/day), patients need to wear a portable drug delivery device during treatment, such as mounting the syringe on a mechanical pressing device. This method is inconvenient, and also expensive, which largely limits the utilization of DFO, especially for thalassemia-prone areas, such as Mediterranean, Middle East, and India &South East Asia, it plays no role in treatment of malaria in world-wide and sickle cell anemia in some African countries, which is a very serious problem to the populations there.
UK Patent No. 2, 13, 807, US Patent No. 4, 585, 780 and other scientific research have reported the treatment of iron overload symptoms by using 3-hydroxypyridin-4-one derivatives, especially in some pathological symptoms, such as thalassemia, sickle cell anemia, aplastic anemia in children, and idiopathic hemochromatosis, usually, treatment of the first three diseases includes frequent regular blood transfusion. 3-Hydroxypyridin-4-one derivatives, especially CP20 (commercial named Ferriprox) is employed to treat systemic iron overload disorders, and also to treat certain diseases associated with local iron overload distribution, although such patients do not show symptoms of systemic iron overload, i.e. inhibition free radical mediated reactions caused by excess iron ions in certain neurodegenerative diseases and cancer diseases. A serious limitation of CP20 is that the hydroxyl group at 3′ position is vulnerable to glycosylation, which reduces the half-life of this compound (approximately 2∼3 h). So it requires a high dosage, which is associated with obvious side effects.
EP0120669 discloses compounds with a 3-hydroxypyrid-4-one in which the H attached to the N atom is substituted by an aliphatic acyl group, or an aliphatic hydrocarbon group, these groups can be further substituted, but not by aromatic groups and their use against illnesses related to iron overload. Molenda et al. disclose in Journal of Medicinal Chemistry 1994, 37, pages 4363-4370 chiral 3-hydroxy-pyridin-4-one compound 6 as enhancing iron excretion.
US Patent No. 6, 465, 604 described a series of 3,5-diphenyl-1,2,4-triazole compounds, wherein including Exjade (commercial name), which has strong affinity to Fe(III), However, its active groups contain two negatively charged oxygen ions and a carboxyl group; it is a tridentate ligand while chelating Fe(III), which forms a Fe-L2 type complex, possessing three unit negative charges itself, that is bad for their discharge from cells/tissues. Moreover, one of the active groups is a nitrogen atom with a lone pair of electrons, Exjade may have a negative effect on the balance of Zn(II) in vivo, at the same time because it has two phenolic hydroxyl groups in different positions (forming intramolecular hydrogen bonds structure similar to cis/trans isomerization), it can be complexed to several zinc ions to form high molecular weight polymers complexes, which is not conducive to its discharge from the cells either.
Absorption, distribution, metabolism and excretion of chiral medicines are largely related to the 3D structures of their chiral centers. For drug absorption, chiral compounds entering cells via active transport mechanism are usually carried by special transport proteins, their recognition of enantiomers can be different, resulting in different absorption of enantiomers. For drug distribution, the binding effects of plasma protein and tissues are also somewhat stereoselective, leading to different in vivo distribution of enantiomers; for stereoselective of drug metabolism refers to when the substrate is biotransformated, the pathway and speed of enantiomer metabolism by biological systems can be different. One enantiomer may show ascendant metabolism, and therefore it is of great significance to the indicators including drug transformation and in vivo half-life. Glomerular filtration, tubular secretion and reabsorption of chiral drugs to clear the chiral drugs, having stereoselectivity, while the glomerular filtration rate is closely related to drug’s selectivity to binding plasma protein, so discharge style of enantiomers (urine / feces percentage) and the rate is also different.
Therefore, the qualitative difference of the interactions of a pair of enantiomers with various binding sites may exist or not, and the quantitive difference may exist (strong or weak), which results in the different activities between enantiomers. Thus the selection of optical enantiomers for medical use, requires a comprehensive study of metabolic activity, toxicology and pharmacokinetic properties etc. Thus the chiral nature of the 3-hydroxypyridin-4-one derivatives described in this patent has an important role on in vivo iron chelation.
The effectiveness of many oral 3-hydroxy-4-one derivatives drugs are subject to metabolic reaction of the 3-hydroxy moiety, which may be quickly glycosylated (see Reaction I). The hydroxypyridone after glycosylation loses the ability to chelate Fe(III). We can effectively inhibit glycosylation reaction by introducing hydroxyl groups to alkyl substituted residues on the pyridine ring. In addition, the partition coefficient of 3-hydroxy-pyridin-4-one derivatives has a great impact on the in vivo distribution and toxic effects. We have introduced various alkyl groups to the chiral point of the compound, in order to modify their lipophilicity, i.e. a phenyl group connected to the chiral point in compound IV-b while in IV-a it is a methyl group, and thus compound IV-b is relatively more lipophilic, and easier to penetrate through cell membranes of various tissues and critical barriers such as the blood-brain barrier and the placental barrier, thus affecting its in vivo distribution. Thus increase of hydroxyl groups can affect the intestinal absorption capacity, by introducing a large alkyl group, intestinal absorption of 3-hydroxy-pyridin-4-one derivatives can be enhanced.
Reaction I
Example7. (R)-3-Hydroxy-1-(1-hydroxy-3-benzyl propyl-2-)2-methyl pyridine-4(1H)-one, Number: CN128.
60 g 3-phenyloxy-2-methyl-4H-pyran-one(Example 1) was dissolved in 150 mL n-butanol, then 83.7 g D-phenylalaninol was added in. After thoroughly mixing, the solution was refluxed at 118°C for 36 h. After cooling and filtration, products were purified by silica gel column chromatography with Eluent ethanol: acetic ester=1:40. After Elution, light brown solid was obtained after rotary evaporation, which was then dissolved into 150 mL ethanol and 15 mL water, then it was hydrogenated and debenzylated with 5% Pd/C as catalyst, the solvent was removed under rotary evaporation, the remaining solid was recrystallized with methanol and ether, leading to 25.25 g light yellow solid. The yield was 35.1%. The free alkali’s 1HNMR (DMSO-d6): δ 2.00 (s, 3H), 2.98 (dd, J1=14, J2=5.5, 1H), 3.11 (dd, J1=14, J2=5, 1H), 3.73 (m, 2H), 4.54 (m, 1H), 6.21 (d, J=7, 1H), 7.17 (m, 5H), 7.87 (d, J=7.5, 1H).
PATENT
CN 102190644
http://www.google.com/patents/CN102190644B?cl=en
![]()






Debiopharm and Aurigene dual c-src / jak inhibitors

Debio 1142
Jak2 tyrosine kinase inhibitor; Src tyrosine kinase inhibitor
N-[4-methyl-3-[2-[4-(4-methylpiperazin-1-yl)anilino]-5-oxo-7,8-dihydropyrido[4,3-d]pyrimidin-6-yl]phenyl]-3-(trifluoromethyl)benzamide
| Molecular Formula: | C33H32F3N7O2 |
|---|---|
| Molecular Weight: | 615.64809 g/mol |
Debiopharm S.A., Aurigene Discovery Technologies Ltd.
ALLISTER Andrès MC, Maximilien Murone,Saumitra Sengupta, Shankar Jayaram Shetty
https://www.google.co.in/patents/WO2011101806A1?cl=en
Bicyclic compounds and their uses as dual c-src / jak inhibitors

Apr. 14 /PR Newswire/ –Debiopharm and Aurigene Sign Agreement for the Development and Commercialisation of Debio 1142, a Novel Inhibitor of an Undisclosed Oncology Pathway
LAUSANNE, Switzerland and BANGALORE, India, April 14, 2011 /PRNewswire/ — Debiopharm Group(TM) (Debiopharm), a global biopharmaceutical development specialist that focuses on serious medical conditions and particularly oncology, and Aurigene Discovery Technologies Ltd (Aurigene), a Bangalore-based drug discovery company, signed on March 23, 2011 an option and exclusive worldwide license agreement concerning the development and commercialisation of Debio 1142, a novel inhibitor of an undisclosed oncology pathway.
“We are very excited about this new collaboration with Aurigene. Their business model offers a one stop solution for structure guided drug design, lead optimisation and preclinical work. The Debio 1142 project aims at developing inhibitors targeting a key oncology pathway, which plays essential roles in various solid tumours, including resistance to chemotherapy” said Dr Rolland-Yves Mauvernay, president and founder of Debiopharm S.A.
“Coming as it does after a successful collaboration programme we already had with Debiopharm, and as a continuation of our close to 5 year association, the relationship between Debiopharm and Aurigene demonstrates the strategic fit between organisations with complimentary scientific skills. We are happy that we have the opportunity to continue to work with Debiopharm, in a unique business model that has been tailor-made to meet each partners’ needs” added CSN Murthy, CEO of Aurigene.
About Debiopharm Group
Debiopharm Group(TM) (Debiopharm) is a Swiss-based global biopharmaceutical group of companies with a focus on the development of prescription drugs that target unmet medical needs. The group in-licenses, develops and/or co-develops promising biological and small molecule drug candidates having reached clinical development phases I, II or III as well as earlier stage candidates. It develops its products for global registration and maximum commercial potential. The products are out-licensed to pharmaceutical partners for sales and marketing. Debiopharm Group is also active in the field of companion diagnostics with a view to progressing in the area of personalised medicine. Debiopharm independently funds the worldwide development of all of its products while providing expertise in pre-clinical and clinical trials, manufacturing, drug delivery and formulation, and regulatory affairs. For more information on Debiopharm Group(TM), please visit: http://www.debiopharm.com.
About Aurigene
Aurigene Discovery Technologies Limited is a Bangalore-based biotech focused on collaborative drug discovery with pharmaceutical and biotech companies on a risk-sharing basis. Aurigene has fully integrated drug discovery infrastructure, from Target to IND, along with strong in house structural biology and fragment based drug design capabilities. The company is engaged in over 20 discovery collaborations with US and European large and mid-pharma companies in Oncology, Inflammatory disorders and anti-infectives. For more information on Aurigene, please visit: http://www.aurigene.com.
PATENT
WO 2011101806
http://www.google.co.in/patents/WO2011101806A1?cl=en
PAPER
Journal of Chemical and Pharmaceutical Research (2014), 6(4), 1146-1152
http://jocpr.com/vol6-iss4-2014/JCPR-2014-6-4-1146-1152.pdf
REFERENCES
INDIAN PATENTS
7554/CHENP/2012
415/CHE/2010
https://www.debiopharm.com/our-business/pipeline.html
http://www.giiresearch.com/report/labd315710-debiopharm-international-sa-product-pipeline.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US2013143895 | 2013-06-06 | BICYCLIC COMPOUNDS AND THEIR USES AS DUAL C-SRC / JAK INHIBITORS |
| US8440679 | 2013-05-14 | Bicyclic compounds and their uses as dual c-SRC / JAK inhibitors |
///////////Debio 1142, Jak2 tyrosine kinase inhibitor, Src tyrosine kinase inhibitor, Debio-1142, Debiopharm S.A., Aurigene Discovery Technologies Ltd, 1332328-01-4
c21cnc(nc1CCN(C2=O)c3c(ccc(c3)NC(=O)c4cc(ccc4)C(F)(F)F)C)Nc5ccc(cc5)N6CCN(CC6)C
