
Home » 2016 (Page 29)
Yearly Archives: 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
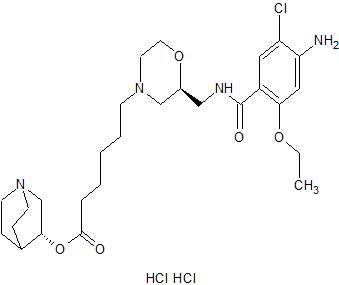
Quisapride Hydrochloride
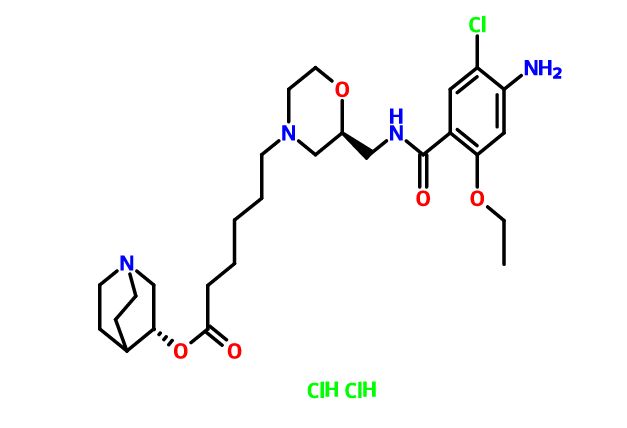
Quisapride Hydrochloride
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate
IND Filed china
A 5-HT4 agonist potentially for the treatment of gastrointestinal motility disorders.
![]()
SHR-116 958, SHR 116958
CAS 1132682-83-7 (Free)
| Shanghai Hengrui Pharmaceutical Co., Ltd. |

CAS 1274633-87-2 (dihcl)
- (3R)-1-Azabicyclo[2.2.2]oct-3-yl (2S)-2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-4-morpholinehexanoate hydrochloride (1:2)
- SHR 116958
-
C27 H41 Cl N4 O5 . 2 Cl H,4-Morpholinehexanoic acid, 2-[[(4-amino-5-chloro-2-ethoxybenzoyl)amino]methyl]-, (3R)-1-azabicyclo[2.2.2]oct-3-yl ester, hydrochloride (1:2), (2S)-
5-HT is a neurotransmitter Chong, widely distributed in the central nervous system and peripheral tissues, 5-HT receptor subtypes at least seven, and a wide variety of physiological functions of 5-HT receptor with different interactions related. Thus, the 5-HT receptor subtypes research is very necessary.
The study found that the HT-5 4 receptor agonists useful for treating a variety of diseases, such as gastroesophageal reflux disease, gastrointestinal disease, gastric motility disorder, non-ulcer dyspepsia, functional dyspepsia, irritable bowel syndrome, constipation, dyspepsia, esophagitis, gastroesophageal disease, nausea, postoperative intestinal infarction, central nervous system disorders, Alzheimer’s disease, cognitive disorder, emesis, migraine, neurological disease, pain, cardiovascular disease, heart failure , arrhythmias, intestinal pseudo-obstruction, gastroparesis, diabetes and apnea syndrome.
The HT-5 4 receptor agonists into benzamides, benzimidazole class and indole alkylamines three kinds, which benzamides derivatives act on the neurotransmitter serotonin in the central nervous system by modulation, It showed significant pharmacological effect. The role of serotonin and benzamides derivatives and pharmacologically related to many diseases. Therefore, more and more people will focus on the human body produce serotonin, a storage position and the position of serotonin receptors, and to explore the relationship between these positions with a variety of diseases.
Commonly used in clinical cisapride (cisapride) and Mosapride (Tony network satisfied) is one of the novel benzamides drugs.

These drugs mainly through the intestinal muscle between the excited 5-HT neurofilament preganglionic and postganglionic neurons 4 receptor to promote the release of acetylcholine and enhancing cholinergic role in strengthening the peristalsis and contraction of gastrointestinal smooth muscle. In large doses, it can antagonize the HT-53 receptors play a central antiemetic effect, when typical doses, through the promotion of gastrointestinal motility and antiemetic effect. These drugs can increase the lower esophageal smooth muscle tension and promote esophageal peristalsis, improving the antrum and duodenum coordinated motion, and promote gastric emptying, but also promote the intestinal movement and enhanced features, increase the role of the proximal colon emptying, It is seen as the whole digestive tract smooth muscle prokinetic effect of the whole gastrointestinal drugs.
Mainly used for reflux esophagitis, functional dyspepsia, gastroparesis, postoperative gastrointestinal paralysis, functional constipation and intestinal pseudo-obstruction patients. Since there is slight antagonism cisapride the HT-5 3 and anti-D2 receptor, can cause cardiac adverse reactions, prolonged QT occurs, and therefore, patients with severe heart disease, ECG QT prolonged, low potassium, and low blood magnesium prohibited drug. Liver and kidney dysfunction, lactating women and children is not recommended. Due to increase between drug diazepam, ethanol, acenocoumarol, cimetidine and ranitidine the absorption of anticholinergic drugs may also antagonize the effect of this product to promote peristalsis of the stomach, should be aware of when using these, such as when diarrhea should reduce, anticoagulant therapy should pay attention to monitoring the clotting time. Mosapride selective gastrointestinal tract the HT-5 4 receptor agonists, there is no antagonism of D2 receptors, does not cause QT prolonged, reduce adverse reactions, mainly fatigue, dizziness, loose stools, mild abdominal pain , the efficacy of cisapride equivalent clinical effect broader Puka cisapride (prucalopride, Pru) of benzimidazole drugs, with high selectivity and specificity of the HT-5 4 receptor, increasing cholinergic neurotransmitters quality release, stimulate peristalsis reflex, enhance colon contraction, and accelerate gastric emptying, gastrointestinal motility to promote good effect, can effectively relieve the patient’s symptoms of constipation, constipation and for treatment of various gastrointestinal surgery peristalsis slow and weak, and intestinal pseudo-obstruction.
WO2005068461 discloses as the HT-5 4 receptor agonists benzamides compounds, particularly discloses compounds represented by the formula:

ATI-7505
ATI-7505 is stereoisomeric esterified. Cisapride analogs, safe and effective treatment of various gastrointestinal disorders, including gastroparesis, gastroesophageal reflux disease and related disorders. The drug can also be used to treat a variety of central nervous system disorders. ATI-7505 for the treatment or prevention of gastroesophageal reflux disease, also taking cisapride significantly reduced side effects. These side effects include diarrhea, abdominal cramps and blood pressure and heart rate rise.
Further, the compounds and compositions of this patent disclosure also useful in treating emesis and other diseases. Such as indigestion, gastroesophageal reflux, constipation, postoperative ileus, and intestinal pseudo-obstruction. In the course of treatment, but also taking cisapride reduce the side effects.
ΑΉ-7505 as the HT-5 4 receptor ligands may be mediated by receptors to treat the disease. These receptors are located in several parts of the central nervous system, modulate the receptor can be used to affect the CNS desired modulation.
ATI-7505 contained in the ester moiety does not detract from the ability of the compounds to provide treatment, but to make the compound easier to serum and / or cytosolic esterases degraded, so you can avoid the drug cytochrome P450 detoxification system, and this system with cisapride cause side effects related, thus reducing side effects.
The HT-Good 5 4 receptor agonists and should the HT-5 4 receptor binding powerful, while the other hardly shows affinity for the receptor, and show functional activity as agonists. They should be well absorbed from the gastrointestinal tract, metabolically stable and possess desirable pharmacokinetic properties. When targeting the receptor in the central nervous system, they should cross the blood-free, selectively targeting peripheral nervous system receptors, they should not pass through the blood-brain barrier. They should be non-toxic, and there is little proof of side effects. Furthermore, the ideal drug candidate will be a stable, non-hygroscopic and easily formulated in the form of physical presence.
Based on the HT-5 4 receptor agonists current developments, the present invention relates to a series of efficacy better, safer, less side effects of the benzamide derivatives.

Synthesis
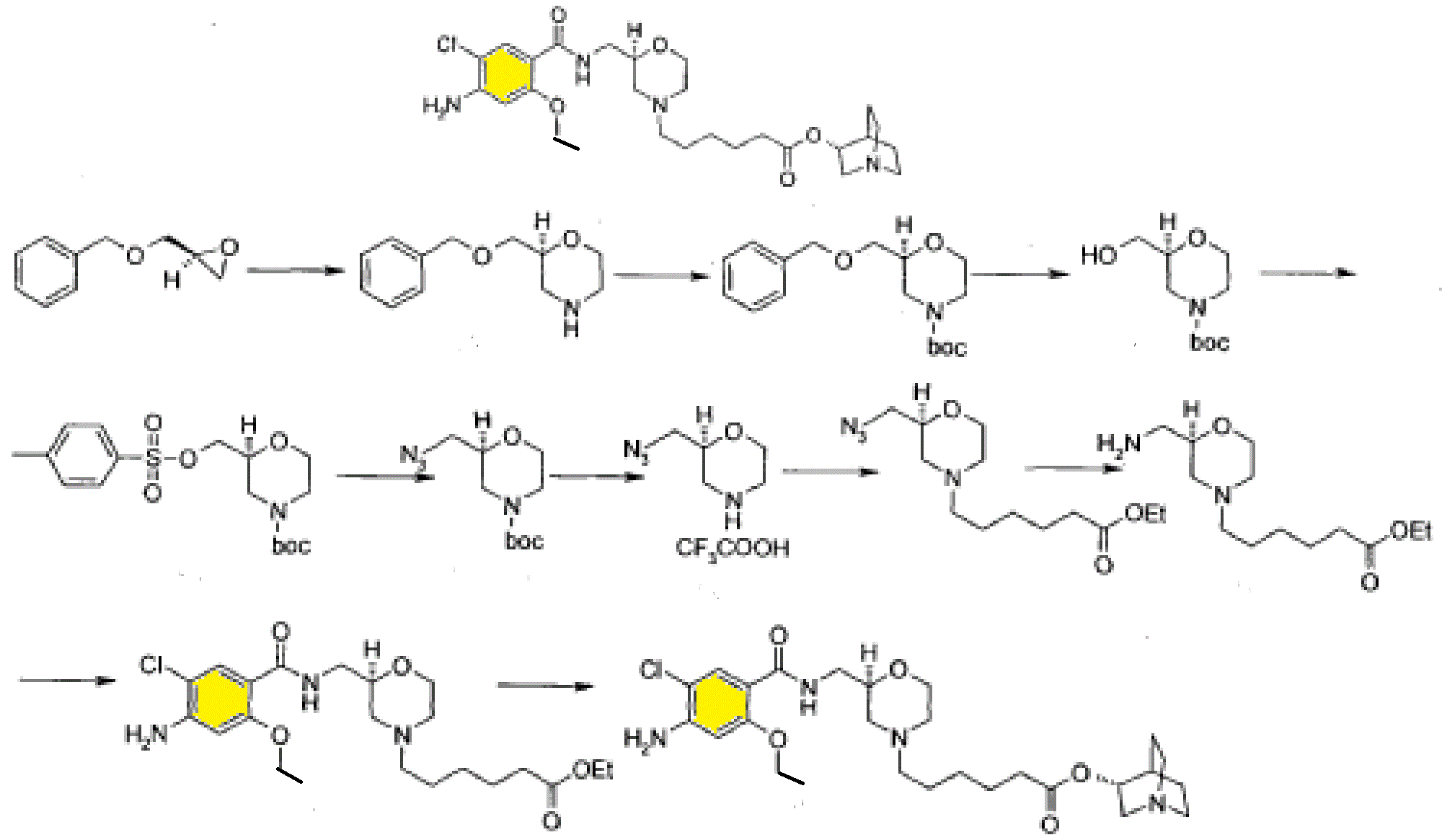
PATENT
Example 3
(R) – quinuclidine-3-5 – ((S) -2 – (( 4 – amino-5-chloro-2-ethoxy benzoylamino) methyl) morpholino) hexanoate



REFERENCES
China Pharmaceuticals: Asia Insight: China Has R&D
Nov 6, 2012 – levofolinate, sevoflurane inhalation, ambroxol hydrochloride, ioversol, etc ….. dextromethorphan hydrochloride 复方沙芬那敏. 3.2 …… quisapride.
//////SHR-116 958, SHR 116958, Quisapride Hydrochloride, preclinical
Cl.Cl.Clc1cc(c(OCC)cc1N)C(=O)NC[C@H]4CN(CCCCCC(=O)O[C@H]3CN2CCC3CC2)CCO4
PDE4 inhibitor , Sumitomo Dainippon Pharma Company

2-[2-Methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-1,3-benzoxazole Hemifumarate
Sumitomo Dainippon Pharma Company,
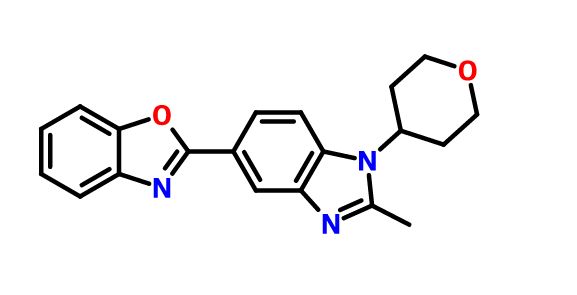
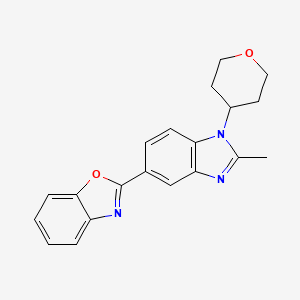
CAS FREE FORM 1256966-65-0
Benzoxazole, 2-[2-methyl-1-(tetrahydro-2H-pyran-4-yl)-1H-benzimidazol-5-yl]-
1H NMR (400 MHz, DMSO-d6)
13C NMR (100 MHz, DMSO-d6)


5- (benzoxazol-2-yl) -2-methyl -1-(tetrahydropyran-4-yl) benzimidazole eggplant flask (100 mL), 2- methyl-1- (tetrahydropyran – 4-yl) reference benzimidazole-5-carboxylic acid (example 4-3) (0.64 g, 2.46 mmol ), 2- amino-phenol (0.32 g, 2.95 mmol), and polyphosphoric acid (about 18 g) put, heated to 160 ℃, and the mixture was stirred for 17 hours. After cooling, ice was added, and the mixture was about pH 9 the liquid with concentrated aqueous ammonia (28%). Extraction with chloroform (about 50 mL X 3 times), dried over anhydrous magnesium sulfate, the crude product obtained by distilling off the solvent (0.08 g) PTLC (CHCl 3 by weight deploy purified), the title compound ( 0.002 g, 0.2% yield) was obtained as a yellow-brown semi-solid. 1H-NMR (CDCl 3 ) Deruta (Ppm): 1.88-1.92 (M, 2 H), 2.58-2.68 (M, 2 H), 2.70 (S, 3 H), 3.57-3.64 (M , 2 H), 4.21-4.25 (m , 2 H), 4.43-4.49 (m, 1 H), 7.29 (d, 1H, J = 9.2 Hz), 7.33-7.35 (m, 2 H ), 7.59-7.62 (m, 1 H ), 7.76-7.78 (m, 1 H), 8.18 (dd, 1 H, J = 8.6, 1.6 Hz), 8.57 (d, 1 H, J = 1.4 Hz).

PAPER

A short and practical synthetic route of a PDE4 inhibitor (1) was established by using Pd–Cu-catalyzed C–H/C–Br coupling of benzoxazole with a heteroaryl bromide. The combination of Pd(OAc)2-Cu(OTf)2-PPh3 was found to be effective for this key step. Furthermore, telescoping methods were adopted to improve the yield and manufacturing time, and a two-step synthesis of1 was accomplished in 71% overall yield.
Direct Synthesis of a PDE4 Inhibitor by Using Pd–Cu-Catalyzed C–H/C–Br Coupling of Benzoxazole with a Heteroaryl Bromide
///////////PDE4 inhibitor , Sumitomo Dainippon Pharma Company
Cc1nc3cc(ccc3n1C2CCOCC2)c4nc5ccccc5o4
ICH M7
DRUG REGULATORY AFFAIRS INTERNATIONAL

ICH M7


View original post 1,776 more words
EMA publishes Q A on data required for sterilized primary packaging materials used in aseptic manufacturing processes
DRUG REGULATORY AFFAIRS INTERNATIONAL

The European Medicines Agency, EMA, recently published questions and answers on what data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process. Read more about “What data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process?“.
The European Medicines Agency, EMA, recently published questions and answers on quality of packaging materials (H+V April 2016):
“3. What data is required for sterilisation processes of primary packaging materials subsequently used in an aseptic manufacturing process?
Terminal sterilisation of the primary packaging, used subsequently during aseptic processing of the finished product, is a critical process and the sterility of the primary container is a critical quality attribute to ensure the sterility of the finished product. Both need to be assured for compliance with relevant Pharmacopoeial requirements for the finished product and product approval.
The site where sterilisation…
View original post 556 more words
FDA´s new policy regarding grouping of supplements for CMC changes
DRUG REGULATORY AFFAIRS INTERNATIONAL
![]()
The US Food and Drug Administration’s (FDA) Office of Pharmaceutical Quality (OPQ) released a new document outlining how supplements can be grouped together and submitted concurrently for the same chemistry, manufacturing and controls (CMC) changes. Find out more about Policy and Procedures regarding the Review of Grouped Product Quality Supplements.
On April 19, 2016 the US Food and Drug Administration’s (FDA) Office of Pharmaceutical Quality (OPQ) released a new document outlining how supplements can be grouped together and submitted concurrently for the same chemistry, manufacturing and controls (CMC) changes to multiple approved new drug applications (NDAs), abbreviated new drug applications (ANDAs) and biological license applications (BLAs) submitted by the same applicant.
The agency says the goal of its new policy is to make the process more efficient and consistent when reviewing grouped supplements.The term “grouped supplements” is used to describe two or more supplements reviewed and processed using…
View original post 343 more words
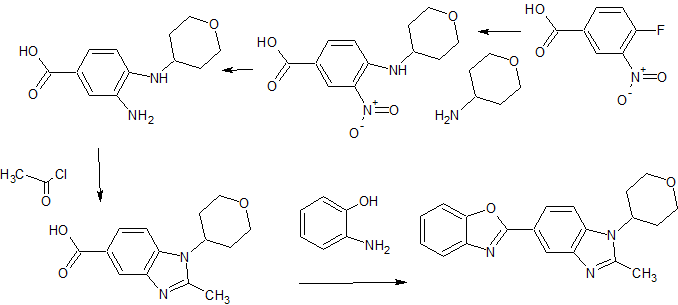
MK-7145
MK-7145,
cas 1255204-84-2
1(3H)-Isobenzofuranone, 5,5′-(1,4-piperazinediylbis((1R)-1-hydroxy-2,1-ethanediyl))bis(4-methyl-
The Renal Outer Medullary Potassium (ROMK) channel (KM .1 ) (see e.g., Ho,K., et al., Cloning and expression of an inwardly rectifying ATP -regulated potassium channel, Nature, 1993, 362(6415): p. 31-8.1, 2; and Shuck, M.E., et al., Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel, J Biol Chem, 1994, 269(39): p. 24261-70) is a member of the inward rectifier family of potassium channels expressed in two regions of the kidney: thick ascending loop of Henle (TALH) and cortical collecting duct (CCD) (see Hebert, S. C, et al., Molecular diversity and regulation of renal potassium channels, Physiol Rev, 2005, 85(1): p. 319-713). At the TALH, ROMK participates in potassium recycling across the luminal membrane which is critical for the function of the Na+/K+/2CF co-transporter, the rate-determining step for salt reuptake in this part of the nephron. At the CCD, ROMK provides a pathway for potassium secretion that is tightly coupled to sodium uptake through the amiloride-sensitive sodium channel (see Reinalter, S. C, et al., Pharmacotyping of hypokalemic salt-losing tubular disorders, Acta. Physiol Scand, 2004, 181(4): p. 513-21 ; and Wang, W., Renal potassium channels: recent developments, Curr Opin Nephrol Hypertens, 2004, 13(5): p. 549-55). Selective inhibitors of the ROMK channel (also referred to herein as inhibitors of ROMK or ROMK inhibitors) are predicted to represent novel diuretics for the treatment of hypertension and other conditions where treatment with a diuretic would be beneficial with potentially reduced liabilities (i.e., hypo- or hyperkalemia, new onset of diabetes, dyslipidemia) over the currently used clinical agents (see Lifton, R.P., A.G. Gharavi, and D.S. Geller, Molecular mechanisms of human hypertension, Cell, 2001, 104(4): p. 545-56). Human genetics (Ji, W., et al., Rare independent mutations in renal salt handling genes contribute to blood pressure variation, Nat Genet, 2008, 40(5): p. 592-9; and Tobin, M.D., et al., Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population, Hypertension, 2008, 51(6): p. 1658-64) and genetic ablation of ROMK in rodents (see Lorenz, J.N., et al., Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome, J Biol Chem, 2002, 277(40): p. 37871-80 and Lu, M., et al.s Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Banter’s) knockout mice, J Biol Chem, 2002, 277(40): p. 37881-7) support these expectations. To our knowledge, the first small molecule selective inhibitors of ROMK were reported from work done at Vanderbilt University as described in Lewis, L.M., et al., High-Throughput Screening Reveals a Small-Molecule Inhibitor of the Renal Outer Medullary Potassium Channel and KirJ.l, MoI Pharmacol, 2009, 76(5): p. 1094-1103.
PATENT
http://www.google.com/patents/WO2010129379A1?cl=ko
SCHEME 1
![]()

![]()
SCHEME 2


SCHEME 3

SCHEME 5


SCHEME 6
![]()
SCHEME 7
![]()



SCHEME 8
![]()
14 15
The preparation of compounds 16 can be achieved following the sequence detailed in Scheme 9. Treating epoxide 2-1 with commercially available 1-Boc piperazine at elevated temperatures gives rise to alcohol 2-2 (Nomura, Y. et al. Chemical & Pharmaceutical Bulletin, 1995, 43(2), 241-6). The hydroxyl group of 2-2 can be converted to the fluoride by treatment of such fluorinating reagent as DAST (Hudlicky, M. Organic Reactions, 1988, 35). Removal of the Boc group of 3-1 under acidic conditions such as TFA gives rise to piperazine 3-2. Piperazine 3-2 can be washed with an aqueous base solution followed by extraction with organic solvents to generate the free base form. The free base of 3-2 can be coupled to epoxide 5-1 at elevated temperatures to afford compound 16. The Ar-CHF- and Ar’-CHOH- groups in 16 represent examples of either Z1 or Z2.
SCHEME 9


16 General Procedures.
INTERMEDIATE (Ry-H (free base)

5-\(lR)-l -hγdroxγ-2-piperazio- 1 -ylethyl] -4-methyl-2-benzofuran- 1 f 3/f)-one To a 20 mL microwave tube charged with 4-methyl-5-[(2jS)-oxiran-2-yl]-2-benzofuran-l(3H)-one (1020 mg, 5.40 mmol) and a stir bar was added 1-Boc Piperazine (800mg, 4.3 mmol) and EtOH (15 mL). The tube was sealed and heated in a microwave apparatus to 150 0C for 1 hour. The crude product was adsorbed onto silica gel, and purified by flash chromatography (Hexanes-EtOAc with 10% EtOH: 0 – 100% gradient), and solvent removed to afford terl-butyl~4-[(2R-2-hydroxy-2-(4-methyl-l -oxo-1 ,3-dihydro-2-bers2θfuran-5-yl) ethyl}piperazine-l-carboxylate. LCMS M+l (calc. 377.20, found 377.13). This product was treated with neat TFA for 15 minutes to remove the Boc group. After removal of TFA under reduced pressure, the residue was taken into aq NaHCO3, and back-extracted with CHCl3-IPA (3:1). The organic layers were combined, dried over sodium sulfate, and concentrated to afford 5 – [( 1 R)- 1 -hydroxy-2-piperazin- 1 -ylethyl] -4-methyl-2-benzofuran- 1 (3H)-one. 1H NMR (OMSO-d6, 500 MHz) δ 7.68 (d, J= 8.0 Hz, IH), 7.65 (d, J= 8.0 Hz, IH)5 5.38, 5.35 (AB system, J- 15.4, J= 16.7, 2H), 5.06 (dd5 J- 3.9 Hz, J= 3.7 Hz, IH), 3.76 (m, IH)5 2.72 (m, 4H), 2.42 (m, 4H), 2.34 (d, J= 3.8 Hz5 IH), 2.32 (d, J= 3.8 Hz, IH), 2.24 (s, 3H); LC/MS: (IE, m/z) [M +I]+ = 277.03.
EXAMPLE 2A

5, 5 ‘-{ piperazine- 1 ,4-diylbis[( 1 R)- 1 -hydroxy ethane-2 , 1 -diyl] } bis(4-methyl-2-benzofuran- 1 (3H)-one)
Method 1: To a 20 mL microwave tube charged with 4-methyl-5-[(2i?)-oxiran-2-yl]-2-benzofuran-l(3H)-one (972 mg, 5.11 mmol) and piperazine (200 mg, 2.3 mmol) was added a stir bar and EtOH (16 mL). The tube was sealed and heated in a microwave apparatus to 150 0C for 90 minutes. The crude product was adsorbed onto silica gel, and purified by flash chromatography (MeOΗ-DCM 0 ~ 7% gradient). After removal of solvents, 5»5′-{piperazine-1 ,4-diyIbi s [( 1 R)- 1 -hydroxyethane-2, 1 -diyl] } bis(4-methyl-2-benzofuran- 1 (3 H)-one) was collected. 1H-NMR (500 MHz9 CDCl3) δ ppm 7.80 (s, 4H), 5.25 (s, 4H), 5.11 (d, J= 10.5 Hz5 2H), 4.00 (broad, 2H), 2.90 (broad, 4H)3 2.69-2.50 (m, 6H), 2.44 (t, J= 11 Hz, 2H), 2.29 (s, 6H); LCMS M+l (calc. 467, found 467).
Method 2: Piperazine (4.51 g, 52.4 mmol) and 4-methyl-5-[(2Λ)-oxiran-2-yl]-2-benzofuran-1 (3//)-one (20.0 g, 105 mmol) were charged to a 3-neck 500-mL roundbottom flask, equipped with a reflux condensor, under nitrogen. Toluene (80.0 mL, 751 mmol) and N,N-dimethylacetamide (80 mL, 854 mmol) were added to provide a suspension. The reaction mixture was warmed to 110 0C, becoming homogeneous at 25 0C. After stirring for 4.5 h at 110 0C, the temperature was increased to 115 °C to drive the reaction forward. After stirring for 48 h, the reaction mixture was cooled to RT. On cooling, crystallization occurred. Water was added via addition funnel (45 mL), generating a thick slurry. The suspension was filtered and the solids were washed with 4:1 water :DMA (60 mL), followed by water (2 x 35 mL). The solid was dried on the funnel under vacuum with a nitrogen sweep to constant mass. 5,5′-{Piperazine-l,4-diylbis[(li?)-l-hydroxyethane-2,l-diyl]}bis(4-methyl-2-beiizofurari-l(3H)-one) was isolated. 1H-NMR (500 MHz, CDCl3) δ ppm 7.80 (s, 4H), 5.25 (s, 4H), 5.11 (d, J- 11 Hz, 2H), 4.30-3.51 (broad, 2H), 2.90 (broad, 4H), 2.69-2.50 (m, 6H), 2.44 (t, J- 11 Hz, 2H), 2.30 (s, 6H).
Compounds of the present invention are amines and can therefore be converted to a variety of salts by treatment with any of a number of acids. For example, the compound of Example 2A can be converted to several different salt forms as shown in the following representative examples. These are selected examples and are not meant to be an exhaustive list; numerous additional salts can be prepared in a similar fashion using a variety of acids. EXAMPLE 2A-1 (di-HCl salt): 5,5t-{piperazme-l,4-diylbis[(17?)-l-hydroxyethane-2,l- diyl] } bis(4-methyl-2-benzofuran- 1 (3H)-one) dihydrochloride To a 250 mL pear shape flask charged with the free base (1.2 g, 2.6 mmol) and a stir bar was added DCM. The solution was stirred until all solids were gone. To this solution was added 4N HCl in dioxane (2.6 mL, 4.0 eq), and the mixture was allowed to stir for another 15 minutes. The solvent was removed on a rotary evaporator, and the product was left dry on a high vacuum pump until there was no weight change. The product was determined to be 5, 5 ‘-{piperazine- 1,4-diylbis [( 1 R)- 1 ~hydroxyethane-2, 1 -diyl] } bis(4-methyl-2-benzofuran- 1 (3i?)-one) dihydrochloride. EXAMPLE 2A-2 (HCl salt): 5,5’-{piperazine-l,4-diylbis[(l^)-l-hydroxyethane-2,l- diyl] } bis(4-methyl-2-benzofuran- 1 QHVone) hvdrochl oride
To a 20 dram vial charged with the free base (160 mg, 0.34 mmol) and a stir bar was added 0.1 M HCl in IPA. The solution was allowed to stir at RT for 30 minutes, and then heated to 400C for 1 hour. The solvent was removed under vacuum, and the resulting product was left on a high vacuum pump for 16 hours. The product corresponded to 5,5′-{piperazine-l,4-diylbis[(li?)-l-hydroxyethane~2, 1 -diyl] } bis(4-methyl-2-benzofuran- 1 (3 H)-one) hydrochloride.
EXAMPLE 2A-3 (mono-hydrate of the di-HCl salt): 5, 5′- {piperazine- l,4-diylbis[( Ii?)- 1-hydroxyethane-2,l-diyl] Ibis^-niethyl-g-benzofuran-lfS/^-one) dihydrochloride hydrate To a flask charged with the free base (1.0 g, 2.1 rnmol) and a stir bar was added 1 N HCl (50 mL). The mixture was allowed to stir until all solids dissolved. The solvent was removed on a rotary evaporator, and the resulting product was left on a high vacuum pump for 16 hours. The product was determined to be 5,5′-{piperazine-l ,4-diylbis[(li?)-l-hydroxyethane-2,l-diyl]}bis(4-methyl-2-benzofuran-l(3H)-one) dihydrochloride hydrate.
EXAMPLE 2A-4 (H2SO4 salt): 5.5′-{piperaziiie-l>4-diylbis[(lJΪ)-l-hydioxyethane-2,l- diyl] }bis(4-methyl-2-benzofuran-l(3/f)-one) sulfate (salt) To a 100 mL flask charged with a solution of the free base (154 mg, 0.330 mmol) in DMF : MeOH (3 : 1) (20 mL) and a stir bar was added 0.1 M H2SO4 (3.3 mL). The solution was allowed to stir at RT for 30 minutes, and then heated to 40 0C for 2 hours. A lot of solids formed during that time. The solvent was removed under vacuum, and the white solids were left on high vacuum for 16 hours to afford 5)5l-{piperazine-l,4-diylbis[(lJ?)~l-hydroxyethane-2,l-diyl] }bis(4-methyl-2-benzofuran-l(3H)-one) sulfate (salt).
Paper

ROMK, the renal outer medullary potassium channel, is involved in potassium recycling at the thick ascending loop of Henle and potassium secretion at the cortical collecting duct in the kidney nephron. Because of this dual site of action, selective inhibitors of ROMK are expected to represent a new class of diuretics/natriuretics with superior efficacy and reduced urinary loss of potassium compared to standard-of-care loop and thiazide diuretics. Following our earlier work, this communication will detail subsequent medicinal chemistry endeavors to further improve lead selectivity against the hERG channel and preclinical pharmacokinetic properties. Pharmacological assessment of highlighted inhibitors will be described, including pharmacodynamic studies in both an acute rat diuresis/natriuresis model and a subchronic blood pressure model in spontaneous hypertensive rats. These proof-of-biology studies established for the first time that the human and rodent genetics accurately predict the in vivo pharmacology of ROMK inhibitors and supported identification of the first small molecule ROMK inhibitor clinical candidate, MK-7145.
Discovery of MK-7145, an Oral Small Molecule ROMK Inhibitor for the Treatment of Hypertension and Heart Failure
////////
Cc1c(ccc2c1COC2=O)[C@H](CN3CCN(CC3)C[C@@H](c4ccc5c(c4C)COC5=O)O)O
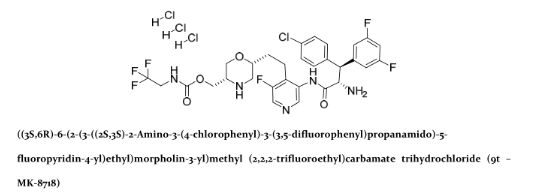
MK 8718


MK 8718
Cas 1582729-24-5 (free base); 1582732-29-3 (HCl).
MF: C30H30ClF6N5O4
MW: 673.1891
| INNOVATOR | Merck Sharp & Dohme Corp., Merck Canada Inc. |
((3S,6R)-6-(2-(3-((2S,3S)-2-amino-3-(4-chlorophenyl)-3-(3,5-difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)morpholin-3-yl)methyl (2,2,2-trifluoroethyl)carbamate
MK-8718 is a potent, selective and orally bioavailable HIV protease inhibitor with a favorable pharmacokinetic profile with potential for further development.
A retrovirus designated human immunodeficiency virus (HIV), particularly the strains known as HIV type-1 (HIV-1) virus and type-2 (HIV-2) virus, is the etiological agent of acquired immunodeficiency syndrome (AIDS), a disease characterized by the destruction of the immune system, particularly of CD4 T-cells, with attendant susceptibility to opportunistic infections, and its precursor AIDS-related complex (“ARC”), a syndrome characterized by symptoms such as persistent generalized lymphadenopathy, fever and weight loss. This virus was previously known as LAV, HTLV-III, or ARV. A common feature of retrovirus replication is the extensive post-translational processing of precursor polyproteins by a virally encoded protease to generate mature viral proteins required for virus assembly and function. Inhibition of this processing prevents the production of normally infectious virus. For example, Kohl et al., Proc. Nat’l Acad. Sci. 1988, 85: 4686, demonstrated that genetic inactivation of the HIV encoded protease resulted in the production of immature, non-infectious virus particles. These results indicated that inhibition of the HIV protease represents a viable method for the treatment of AIDS and the prevention or treatment of infection by HIV.
Nucleotide sequencing of HIV shows the presence of a pol gene in one open reading frame [Ratner et al, Nature 1985, 313: 277]. Amino acid sequence homology provides evidence that the pol sequence encodes reverse transcriptase, an endonuclease, HIV protease and gag, which encodes the core proteins of the virion (Toh et al, EMBO J. 1985, 4: 1267; Power et al, Science 1986, 231 : 1567; Pearl et al, Nature 1987, 329: 351].
Several HIV protease inhibitors are presently approved for clinical use in the treatment of AIDS and HIV infection, including indinavir (see US 5413999), amprenavir (US5585397), saquinavir (US 5196438), ritonavir (US 5484801) and nelfmavir (US 5484926). Each of these protease inhibitors is a peptide-derived peptidomimetic, competitive inhibitor of the viral protease which prevents cleavage of the HIV gag-pol polyprotein precursor. Tipranavir (US 5852195) is a non-peptide peptidomimetic protease inhibitors also approved for use in treating HIV infection. The protease inhibitors are administered in combination with at least one and typically at least two other HIV antiviral agents, particularly nucleoside reverse transcriptase inhibitors such as zidovudine (AZT) and lamivudine (3TC) and/or non-nucleoside reverse transcriptase inhibitors such as efavirenz and nevirapine. Indinavir, for example, has been found to be highly effective in reducing HIV viral loads and increasing CD4 cell counts in HIV-infected patients, when used in combination with nucleoside reverse transcriptase inhibitors. See, for example, Hammer et al, New England J. Med. 1997, 337: 725-733 and Gulick et al, New England J. Med. 1997, 337: 734-739.
The established therapies employing a protease inhibitor are not suitable for use in all HIV-infected subjects. Some subjects, for example, cannot tolerate these therapies due to adverse effects. Many HIV-infected subjects often develop resistance to particular protease inhibitors. Furthermore, the currently available protease inhibitors are rapidly metabolized and cleared from the bloodstream, requiring frequent dosing and use of a boosting agent.
Accordingly, there is a continuing need for new compounds which are capable of inhibiting HIV protease and suitable for use in the treatment or prophylaxis of infection by HIV and/or for the treatment or prophylaxis or delay in the onset or progression of AIDS.

PATENT
https://www.google.co.in/patents/WO2014043019A1?cl=en
INTERMEDIATE 1
Synthesis of morpholine intermediate (tert-butyl ( ^S^-S-d tert- butyl(dimethyl)silylloxy|methyl)-2-(hydroxymethyl)morpholine-4-carboxylate)



Scheme 1

EXAMPLE 97
( S)- -(4-Chlorophenyl)-3,5-difiuoro-N-(5-fiuoro-4-{2-[(2R,5S)-5-({[(2,2,2- trifluoroethyl)carbamoyl]oxy}methyl)morpholin-2-yl]ethyl}pyridin-3-yl)-L-phenylalaninamide

Step 1. (2S,3S)-2-Azido-3-(4-chlorophenyl)-3-(3,5-difluorophenyl)propanoic acid

The title compound was prepared from 4-chlorocinnamic acid and 3,5- difluorophenylmagnesium bromide using the procedures given in steps 1-4 of Example 92.
Step 2. (2R,5S)-tert-butyl 2-(2-(3-((2S,3S)-2-azido-3-(4-chlorophenyl)-3-(3,5- difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)-5-((((2,2,2- trifluoroethyl)carbamoyl)oxy)methyl)morpholine-4-carboxylate

The product from step 1 (105 mg, 0.31 mmol) and the product from step 4 of Example 89 (150 mg, 0.31 mmol) were dissolved in pyridine (1 mL) and the stirred solution was cooled to -10 °C in an ice/acetone bath. To the cold solution was added POCI3 dropwise (0.035 mL, 0.38 mmol). The mixture was stirred at -10 °C for 30 min. The reaction was quenched by the addition of saturated aqueous NaHC03 solution (1 mL) and the mixture was allowed to warm to ambient temperature. The mixture was diluted with water (10 mL) and extracted with dichloromethane (3 x 10 mL). The combined dichloromethane phases were dried (Na2S04), filtered, and the filtrate solvents were removed in vacuo. The residue was purified on a 12 g silica gel column using a gradient elution of 0-70% EtOAc:hexanes. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 800.6.
Step 3. (2R,5S)-tert-butyl 2-(2-(3-((2S,3S)-2-amino-3-(4-chlorophenyl)-3-(3,5- difluorophenyl)propanamido)-5-fluoropyridin-4-yl)ethyl)-5-((((2,2,2- trifluoroethyl)carbamoyl)oxy)methyl)morpholine-4-carboxylate
The product from step 2 (150 mg, 0.19 mmol) and triphenylphosphine (74 mg, 0.28 mmol) were dissolved in THF (4 mL) and to the solution was added water (1 mL). The mixture was heated to reflux under a nitrogen atmosphere for 12 h. The mixture was cooled to ambient temperature and the solvents were removed in vacuo. The residue was purified on a 12 g silica gel column eluting with a gradient of 0-10% methanol: chloroform. Fractions containing product were combined and the solvents were removed in vacuo to give the title compound as a gum. (M+H)+ = 774.7. Step 4. ( S)- -(4-Chlorophenyl)-3,5-difluoro-N-(5-fluoro-4-{2-[(2R,5S)-5-({[(2,2,2- trifluoroethyl)carbamoyl]oxy}methyl)morpholin-2-yl]ethyl}pyridin-3-yl)-L-phenylala
The product from step 3 (60 mg, 0.078 mmol) was dissolved in a solution of 4M HCl in dioxane (1 mL, 4 mmol) and the solution was stirred at ambient temperature for 1 h. The solvent was removed under reduced pressure and the residue was dried in vacuo for 12 h to give an HCl salt of the title compound as a solid. LCMS: RT = 0.95 min (2 min gradient), MS (ES) m/z = 674.6 (M+H)+.
PAPER

A novel HIV protease inhibitor was designed using a morpholine core as the aspartate binding group. Analysis of the crystal structure of the initial lead bound to HIV protease enabled optimization of enzyme potency and antiviral activity. This afforded a series of potent orally bioavailable inhibitors of which MK-8718 was identified as a compound with a favorable overall profile.
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
References
Discovery of MK-8718, an HIV Protease Inhibitor Containing a Novel Morpholine Aspartate Binding Group
Christopher J. Bungard*†, Peter D. Williams†, Jeanine E. Ballard†, David J. Bennett†, Christian Beaulieu‡, Carolyn Bahnck-Teets†, Steve S. Carroll†, Ronald K. Chang†, David C. Dubost†, John F. Fay†, Tracy L. Diamond†, Thomas J. Greshock†, Li Hao§, M. Katharine Holloway†, Peter J. Felock, Jennifer J. Gesell†, Hua-Poo Su†, Jesse J. Manikowski†, Daniel J. McKay‡, Mike Miller†, Xu Min†, Carmela Molinaro†, Oscar M. Moradei‡, Philippe G. Nantermet†, Christian Nadeau‡, Rosa I. Sanchez†, Tummanapalli Satyanarayana§, William D. Shipe†, Sanjay K. Singh§, Vouy Linh Truong‡, Sivalenka Vijayasaradhi§, Catherine M. Wiscount†, Joseph P. Vacca‡, Sheldon N. Crane‡, and John A. McCauley†
† Merck Research Laboratories, 770 Sumneytown Pike, PO Box 4, West Point, Pennsylvania 19486, United States
‡ Merck Frosst Centre for Therapeutic Research, 16711 TransCanada Highway, Kirkland, Quebec H9H 3L1, Canada
§ Albany Molecular Research Singapore Research Center, 61 Science Park Road #05-01, The Galen Singapore Science Park II, Singapore 117525
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00135
Publication Date (Web): May 09, 2016
////MK-8718, HIV, protease, inhibitor
O=C(OC[C@H]1NC[C@@H](CCC(C(F)=CN=C2)=C2NC([C@@H](N)[C@@H](C3=CC=C(Cl)C=C3)C4=CC(F)=CC(F)=C4)=O)OC1)NCC(F)(F)F
PDE4 Inhibitors, Boehringer Ingelheim Pharmaceuticals
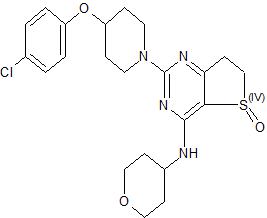 R CONF SHOWN
R CONF SHOWN
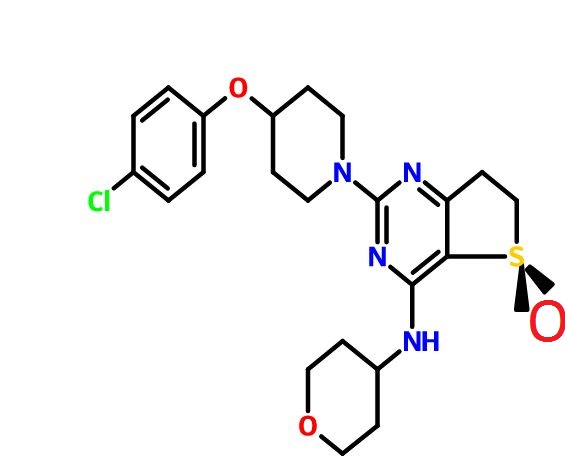
BI ?
(R)-2-(4-(4-Chlorophenoxy)piperidin-1-yl)-4-((tetrahydro-2H-pyran-4-yl)amino)-6,7-dihydrothieno[3,2-d]pyrimidine 5-Oxide
1H NMR (400 MHz, CDCl3) δ 1.49 (dq, J = 4.2, 11.8 Hz, 1H), 1.62 (dq, J = 4.2, 11.8 Hz, 1H), 1.74–1.89 (m, 3H), 1.90–2.02 (m, 3H), 2.96–3.07 (m, 2H), 3.29 (dt, J = 13.6, 8.4 Hz, 1H), 3.44 (ddd, J = 19.2, 11.2, 2.0 Hz, 2H), 3.62 (dt, J = 17.2, 7.8 Hz, 1H), 3.76 (m, 2H), 3.96 (dd, J = 15.6, 12.8 Hz, J = 2H), 4.09–3.99 (m, 3H), 4.51 (m, 1H), 6.21 (br d, J = 6.0 Hz, 1H), 6.86 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.8 Hz, 2H);
13C NMR (100 MHz, CDCl3) δ 30.4, 32.5, 32.7, 41.0, 47.2, 49.6, 66.9, 66.9, 72.9, 107.8, 117.5, 125.9, 129.5, 155.8, 158.9, 163.0, 174.6.
The use of phosphodiesterase type 4 (PDE4) inhibitors for the treatment of COPD (chronic obstructive pulmonary disease) by reducing inflammation and improving lung function is well documented. Given the potential therapeutic benefit offered by these compounds, a number of PDE4-selective inhibitors containing a dihydrothieno[3,2-d]pyrimidine core were identified as preclinical candidates in Boehringer Ingelheim Pharmaceuticals discovery laboratories

While the pathogenesis of chronic obstructive pulmonary disease (COPD) is incompletely understood, chronic inflammation is a major factor. In fact, the inflammatory response is abnormal, with CD8+ T-cells, CD68+ macrophages, and neutrophils predominating in the conducting airways, lung parenchyma, and pulmonary vasculature. Elevated levels of the second messenger cAMP can inhibit some inflammatory processes. Theophylline has long been used in treating asthma; it causes bronchodilation by inhibiting cyclic nucleotide phosphodiesterase (PDE), which inactivates cAMP. By inhibiting PDE, theophylline increases cAMP, inhibiting inflammation and relaxing airway smooth muscle. Rather than one PDE, there are now known to be more than 50, with differing activities, substrate preferences, and tissue distributions. Thus, the possibility exists of selectively inhibiting only the enzyme(s) in the tissue(s) of interest. PDE 4 is the primary cAMP-hydrolyzing enzyme in inflammatory and immune cells (macrophages, eosinophils, neutrophils). Inhibiting PDE 4 in these cells leads to increased cAMP levels, down-regulating the inflammatory response. Because PDE 4 is also expressed in airway smooth muscle and, in vitro, PDE 4 inhibitors relax lung smooth muscle, selective PDE 4 inhibitors are being developed for treating COPD. Clinical studies have been conducted with PDE 4 inhibitors;

Chronic obstructive pulmonary disease (COPD) is a serious and increasing global public health problem; physiologically, it is characterized by progressive, irreversible airflow obstruction and pathologically, by an abnormal airway inflammatory response to noxious particles or gases (MacNee 2005a). The COPD patient suffers a reduction in forced expiratory volume in 1 second (FEV1), a reduction in the ratio of FEV1 to forced vital capacity (FVC), compared with reference values, absolute reductions in expiratory airflow, and little improvement after treatment with an inhaled bronchodilator. Airflow limitation in COPD patients results from mucosal inflammation and edema, bronchoconstriction, increased secretions in the airways, and loss of elastic recoil. Patients with COPD can experience ‘exacerbations,’ involving rapid and prolonged worsening of symptoms (Seneff et al 1995; Connors et al 1996; Dewan et al 2000; Rodriguez-Roisin 2006; Mohan et al 2006). Many are idiopathic, though they often involve bacteria; airway inflammation in exacerbations can be caused or triggered by bacterial antigens (Murphy et al 2000; Blanchard 2002; Murphy 2006;Veeramachaneni and Sethi 2006). Increased IL-6, IL-1β, TNF-α, GRO-α, MCP-1, and IL-8 levels are found in COPD patient sputum; their levels increase further during exacerbations. COPD has many causes and significant differences in prognosis exist, depending on the cause (Barnes 1998; Madison and Irwin 1998).
COPD is already the fourth leading cause of death worldwide, according to the World Health Organization (WHO); the WHO estimates that by the year 2020, COPD will be the third-leading cause of death and the fifth-leading cause of disability worldwide (Murray and Lopez 1997). COPD is the fastest-growing cause of death in developed nations and is responsible for over 2.7 million deaths per year worldwide. In the US, there are currently estimated to be 16 million people with COPD. There are estimated to be up to 20 million sufferers in Japan, which has the world’s highest per capita cigarette consumption and a further 8–12 million in Europe. In 2000, COPD accounted for over 20 million outpatient visits, 3.4 million emergency room visits, 6 million hospitalizations, and 116,500 deaths in the US (National Center for Health Statistics 2002). Factors associated with COPD, including immobility, often lead to secondary health consequences (Polkey and Moxham 2006).
Risk factors for the development of COPD include cigarette smoking, and occupational exposure to dust and chemicals (Senior and Anthonisen 1998; Anthonisen et al 2002; Fabbri and Hurd 2003; Zaher et al 2004). Smoking is the most common cause of COPD and the underlying inflammation typically persists in ex-smokers. Oxidative stress from cigarette smoke is also an issue in COPD (Domej et al 2006). Despite this, relatively few smokers ever develop COPD (Siafakas and Tzortzaki 2002).
While many details of the pathogenesis of COPD remain unclear, chronic inflammation is now recognized as a major factor, predominantly in small airways and lung parenchyma, characterized by increased numbers of macrophages, neutrophils, and T-cells (Barnes 2000; Stockley 2002). As recently as 1995, the American Thoracic Society issued a statement defining COPD without mentioning the underlying inflammation (American Thoracic Society 1995). Since then, the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines have made it clear that chronic inflammation throughout the airways, parenchyma, and pulmonary vasculature plays a central role (Pauwels et al 2001; GOLD 2003). The comparatively recent realization of the role of airway inflammation in COPD has altered thinking with regard to potential therapies (Rogers and Giembycz 1998; Vignola 2004).
Most pharmacological therapies available for COPD, including bronchodilator and anti-inflammatory agents, were first developed for treating asthma. The mainstays of COPD treatment are inhaled corticosteroids (McEvoy and Niewoehner 1998; Borron and deBoisblanc 1998; Pauwels 2002; Gartlehner et al 2006;D’Souza 2006), supplemental oxygen (Petty 1998; Austin and Wood-Baker 2006), inhaled bronchodilators (Costello 1998; Doherty and Briggs 2004), and antibiotics (Taylor 1998), especially in severely affected patients (Anthonisen et al 1987; Saint et al 1995; Adams et al 2001; Miravitlles et al 2002; Donnelly and Rogers 2003; Sin et al 2003; Rabe 2006), though the use of antibiotics remains controversial (Ram et al 2006). Long-acting β2-agonists (LABAs) improve the mucociliary component of COPD. Combination therapy with LABAs and anticholinergic bronchodilators resulted in modest benefits and improved health-related quality of life (Buhl and Farmer 2005; Appleton et al 2006). Treatment with mucolytics reduced exacerbations and the number of days of disability (Poole and Black 2006). The combined use of inhaled corticosteroids and LABAs has been demonstrated to produce sustained improvements in FEV1 and positive effects on quality of life, number of hospitalizations, distance walked, and exacerbations (Mahler et al 2002;Szafranski et al 2003; Sin et al 2004; Miller-Larsson and Selroos 2006; van Schayck and Reid 2006). However, all of these treatments are essentially palliative and do not impact COPD progression (Hay 2000;Gamble et al 2003; Antoniu 2006a).
A further complication in drug development and therapy is that it can be difficult to determine the efficacy of therapy, because COPD has a long preclinical stage, is progressive, and patients generally do not present for treatment until their lung function is already seriously impaired. Moreover, because COPD involves irreversible loss of elasticity, destruction of the alveolar wall, and peribronchial fibrosis, there is often little room for clinical improvement.
Smoking cessation remains the most effective intervention for COPD. Indeed, to date, it is the only intervention shown to stop the decline in lung function, but it does not resolve the underlying inflammation, which persists even in ex-smokers. Smoking cessation is typically best achieved by a multifactor approach, including the use of bupropion, a nicotine replacement product, and behavior modification (Richmond and Zwar 2003).
In COPD, there is an abnormal inflammatory response, characterized by a predominance of CD8+ T-cells, CD68+ macrophages, and neutrophils in the conducting airways, lung parenchyma, and pulmonary vasculature (Soto and Hanania 2005; O’Donnell et al 2006; Wright and Churg 2006). Inflammatory mediators involved in COPD include lipids, inflammatory peptides, reactive oxygen and nitrogen species, chemokines, cytokines, and growth factors. COPD pathology also includes airway remodeling and mucociliary dysfunction (mucus hypersecretion and decreased mucus transport). Corticosteroids reduce the number of mast cells, but CD8+ and CD68+ cells, and neutrophils, are little affected (Jeffery 2005). Inflammation in COPD is not suppressed by corticosteroids, consistent with it being neutrophil-, not eosinophil-mediated. Corticosteroids also do not inhibit the increased concentrations of IL-8 and TNF-α (both neutrophil chemoattractants) found in induced sputum from COPD patients. Neutrophil-derived proteases, including neutrophil elastase and matrix metalloproteinases (MMPs), are involved in the inflammatory process and are responsible for the destruction of elastin fibers in the lung parenchyma (Mercer et al 2005; Gueders et al 2006). MMPs play important roles in the proteolytic degradation of extracellular matrix (ECM), in physiological and pathological processes (Corbel, Belleguic et al 2002). PDE 4 inhibitors can reduce MMP activity and the production of MMPs in human lung fibroblasts stimulated with pro-inflammatory cytokines (Lagente et al 2005). In COPD, abnormal remodeling results in increased deposition of ECM and collagen in lungs, because of an imbalance of MMPs and TIMPs (Jeffery 2001). Fibroblast/myofibroblast proliferation and activation also occur, increasing production of ECM-degrading enzymes (Crouch 1990; Segura-Valdez et al 2000). Additionally, over-expression of cytokines and growth factors stimulates lung fibroblasts to synthesize increased amounts of collagen and MMPs, including MMP-1 (collagenase-1) and MMP-2 and MMP-9 (gelatinases A and B) (Sasaki et al 2000; Zhu et al 2001).
It is now generally accepted that bronchial asthma is also a chronic inflammatory disease (Barnes et al 1988;Barnes 1995). The central role of inflammation of the airways in asthma’s pathogenesis is consistent with the efficacy of corticosteroids in controlling clinical symptoms. Eosinophils are important in initiating and continuing the inflammatory state (Holgate et al 1987; Bruijnzeel 1989; Underwood et al 1994; Teixeira et al 1997), while other inflammatory cells, including lymphocytes, also infiltrate the airways (Holgate et al 1987;Teixeira et al 1997). The familiar acute symptoms of asthma are the result of airway smooth muscle contraction. While recognition of the key role of inflammation has led to an emphasis on anti-inflammatory therapy in asthma, a significant minority of patients remains poorly controlled and some exhibit accelerated declines in lung function, consistent with airway remodeling (Martin and Reid 2006). Reversal or prevention of structural changes in remodeling may require additional therapy (Burgess et al 2006).
There is currently no cure for asthma; treatment depends primarily on inhaled glucocorticoids to reduce inflammation (Taylor 1998; Petty 1998), and inhaled bronchodilators to reduce symptoms (Torphy 1994;Costello 1998; Georgitis 1999; DeKorte 2003). Such treatments, however, do not address disease progression.
COPD and asthma are both characterized by airflow obstruction, but they are distinct in terms of risk factors and clinical presentation. While both involve chronic inflammation and cellular infiltration and activation, different cell types are implicated and there are differences in the inflammatory states (Giembycz 2000;Fabbri and Hurd 2003; Barnes 2006). In COPD, neutrophil infiltration into the airways and their activation appear to be key (Stockley 2002); in asthma, the inflammatory response involves airway infiltration by activated eosinophils and lymphocytes, and T-cell activation of the allergic response (Holgate et al 1987;Saetta et al 1998; Barnes 2006). While macrophages are present in both conditions, the major controller cells are CD8+ T-cells in COPD (O’Shaughnessy et al 1997; Saetta et al 1998) and CD4+ T-cells in asthma. IL-1, IL-8, and TNF-α are the key cytokines in COPD, while in asthma, IL-4, IL-5, and IL-13 are more important. There are differences in histopathological features of lung biopsies between COPD patients and asthmatics; COPD patients have many fewer eosinophils in lung tissue than asthmatics.
While the early phases of COPD and asthma are distinguishable, there are common features, including airway hyper-responsiveness and mucus hypersecretion. MUC5AC is a major mucin gene expressed in the airways; its expression is increased in COPD and asthmatic patients. At least in vitro, epidermal growth factor stimulates MUC5AC mRNA and protein expression; this can be reversed by PDE 4 inhibitors, which may contribute to their clinical efficacy in COPD and asthma (Mata et al 2005). Similar structural and fibrotic changes make COPD and asthma much less distinguishable in extreme cases; the chronic phases of both involve inflammatory responses, alveolar detachment, mucus hypersecretion, and subepithelial fibrosis. The two conditions have been linked epidemiologically; adults with asthma are up to 12 times more likely to develop COPD over time than those without (Guerra 2005).


PAPER

A practical, safe, and efficient process for the synthesis of PDE4 (phosphodiesterase type 4) inhibitors represented by 1 and 2 was developed and demonstrated on a multi-kilogram scale. Key aspects of the process include the regioselective synthesis of dihydrothieno[3,2-d]pyrimidine-2,4-diol 9 and the asymmetric sulfur oxidation of intermediate 11.
Development of a Practical Process for the Synthesis of PDE4 Inhibitors
PDE 4 in COPD
With regard to COPD, PDE 4 is the primary cAMP-hydrolyzing enzyme in inflammatory and immune cells, especially macrophages, eosinophils, and neutrophils, all of which are found in the lungs of COPD and asthma patients (Torphy et al 1992; Karlsson and Aldous 1997; De Brito et al 1997; Wang et al 1999;Torphy and Page 2000). Inhibition of PDE 4 leads to elevated cAMP levels in these cells, down-regulating the inflammatory response (Dyke and Montana 2002).
PDE 4 has also attracted much attention because it is expressed in airway smooth muscle (Ashton et al 1994;Undem et al 1994; Nicholson et al 1995; Kerstjens and Timens 2003; Mehats et al 2003; Lipworth 2005; Fan Chung 2006). In vitro, PDE 4 inhibitors relax lung smooth muscle (Undem et al 1994; Dent and Giembycz 1995). In COPD and asthma, a selective PDE 4 inhibitor with combined bronchodilatory and anti-inflammatory properties would seem desirable (Nicholson and Shahid 1994; Lombardo 1995; Palfreyman 1995; Cavalia and Frith 1995; Palfreyman and Souness 1996; Karlsson and Aldous 1997; Compton et al 2001; Giembycz 2002; Jacob et al 2002; Soto and Hanania 2005).
PDE 4 inhibitors in COPD
So, because PDE 4 inhibitors suppress inflammatory functions in several cell types involved in COPD and asthma (Huang and Mancini 2006) and because, at least in vitro, PDE 4 inhibitors relax lung smooth muscle, selective PDE 4 inhibitors, originally intended for use in treating depression (Renau 2004), have been developed for the treatment of COPD and asthma (Torphy et al 1999; Spina 2000; Huang et al 2001; Spina 2004; Giembycz 2005a, 2005b; Lagente et al 2005; Boswell-Smith, Spina et al 2006). PDE 4 enzymes are strongly inhibited by the antidepressant drug rolipram (Pinto et al 1993), which decreases the influx of inflammatory cells at sites of inflammation (Lagente et al 1994; Lagente et al 1995; Alves et al 1996). PDE 4 inhibitors down-regulate cytokine production in inflammatory cells, in vivo and in vitro (Undem et al 1994;Dent and Giembycz 1995). TNF-α is an important inflammatory cytokine in COPD; its release is reduced by PDE 4 inhibitors (Souness et al 1996; Chambers et al 1997; Griswold et al 1998; Gonçalves de Moraes et al 1998; Corbel, Belleguic et al 2002). Some PDE 4 inhibitors, including cilomilast and AWD 12-281, can inhibit neutrophil degranulation, a property not shared by theophylline (Ezeamuzie 2001; Jones et al 2005). PDE 4 inhibitors reduce overproduction of other pro-inflammatory mediators, including arachidonic acid and leukotrienes (Torphy 1998). PDE 4 inhibitors also inhibit cellular trafficking and microvascular leakage, production of reactive oxygen species, and cell adhesion molecule expression in vitro and in vivo (Sanz et al 2005). PDE 4 inhibitors, including cilomilast and CI-1044, inhibit LPS-stimulated TNF-α production in whole blood from COPD patients (Burnouf et al 2000; Ouagued et al 2005).
There are now thought to be at least four PDE 4s, A, B, C, and D, derived from four genes (Lobbam et al 1994; Muller et al 1996; Torphy 1998; Conti and Jin 1999; Matsumoto et al 2003). Alternative splicing and alternative promoters add further complexity (Manganiello et al 1995; Horton et al 1995; Torphy 1998). Indeed, the four genes encode more than 16 PDE 4 isoforms, which can be divided into short (∼65–75 kDa) and long forms (∼80–130 kDa); the difference between the short and long forms lies in the N-terminal region (Bolger et al 1997; Huston et al 2006). PDE 4 isoforms are regulated by extracellular signal-related protein kinase (ERK), which can phosphorylate PDE 4 (Houslay and Adams 2003).
The four PDE 4 genes are differentially expressed in various tissues (Silver et al 1988; Lobbam et al 1994;Manganiello et al 1995; Horton et al 1995; Muller et al 1996; Torphy 1998). PDE 4A is expressed in many tissues, but not in neutrophils (Wang et al 1999). PDE 4B is also widely expressed and is the predominant PDE 4 subtype in monocytes and neutrophils (Wang et al 1999), but is not found in cortex or epithelial cells (Jin et al 1998). Upregulation of the PDE 4B enzyme in response to pro-inflammatory agents suggest that it has a role in inflammatory processes (Manning et al 1999). PDE 4C is expressed in lung and testis, but not in circulating inflammatory cells, cortex, or hippocampus (Obernolte et al 1997; Manning et al 1999; Martin-Chouly et al 2004). PDE 4D is highly expressed in lung, cortex, cerebellum, and T-cells (Erdogan and Houslay 1997; Jin et al 1998). PDE 4D also plays an important role in airway smooth muscle contraction (Mehats et al 2003).
A major issue with early PDE 4 inhibitors was their side effect profile; the signature side effects are largely gastrointestinal (nausea, vomiting, increased gastric acid secretion) and limited the therapeutic use of PDE 4 inhibitors (Dyke and Montana 2002). The second generation of more selective inhibitors, such as cilomilast and roflumilast, have improved side effect profiles and have shown clinical efficacy in COPD and asthma (Barnette 1999; Spina 2000; Lagente et al 2005). However, even cilomilast and roflumilast, the most advanced clinical candidates, discussed below, cause some degree of emesis (Spina 2003).
It is now thought that the desirable anti-inflammatory properties and unwanted side effects of nausea and emesis are associated with distinct biochemical activities (Torphy et al 1992; Jacobitz et al 1996; Barnette et al 1996; Souness et al 1997; Souness and Rao 1997). Specifically, the side effects are believed to be associated with the so-called ‘high-affinity rolipram binding site’ (HARBS) (Barnette et al 1995; Muller et al 1996; Jacobitz et al 1996; Kelly et al 1996; Torphy 1998) and/or inhibition of the form of PDE 4 found in the CNS (Barnette et al 1996). The exact nature of HARBS remains unclear, although it has been described as a conformer of PDE 4 (Souness and Rao 1997; Barnette et al 1998). Using mice deficient in PDE 4B or PDE 4D, it appears that emesis is the result of selective inhibition of PDE 4D (Robichaud et al 2002; Lipworth 2005), which is unfortunate, because the most clinically advanced PDE 4 inhibitors are selective for PDE 4D. Also, from animal studies, it appears that the nausea and vomiting are produced via the CNS, though there may also be direct effects on the gastrointestinal system (Barnette 1999).
While beyond the scope of this review, it has been proposed that PDE 4 inhibitors may be useful in treating inflammatory bowel disease (Banner and Trevethick 2004), cystic fibrosis (Liu et al 2005), pulmonary arterial hypertension (Growcott et al 2006), myeloid and lymphoid malignancies (Lerner and Epstein 2006), Alzheimer’s disease (Ghavami et al 2006), rheumatoid arthritis and multiple sclerosis (Dyke and Montana 2002), infection-induced preterm labor (Oger et al 2004), depression (Wong et al 2006), and allergic disease (Crocker and Townley 1999). Varying degrees of in vitro, in vivo, and clinical data exist to support these claims.
So, after that theoretical buildup, we reach the proof of the pudding; clinical studies have been conducted with PDE 4 inhibitors. A potent, but not-very-selective, PDE 4 inhibitor is approved in Japan and is used clinically, including for treating asthma. Another is awaiting approval in the US. One is in advanced clinical development and others are at earlier stages.
REF
Pouzet, P.; Hoenke, C.; Martyres, D.; Nickolaus, P.; Jung, B.; Hamman, H. Dihydrothienopyrimidines for the treatment of inflammatory diseases. PatentWO 2006111549 A1, October 26, 2006.
Ohnacker, G.; Woitun, E. Novel dihydrothieno[3, 2-d]pyrimidines. U.S. Patent US 3,318,881, May 9, 1967.
/////PDE4 Inhibitors, Boehringer Ingelheim Pharmaceuticals, BI ?, PRECLINICAL, 1910076-27-5
Clc1ccc(cc1)OC2CCN(CC2)c4nc(NC3CCOCC3)c5c(n4)CCS5=O
ICH Q3D Implementation Working Group (IWG)—Training Modules
DRUG REGULATORY AFFAIRS INTERNATIONAL

ICH Q3D Implementation Working Group (IWG)—Training Modules
View original post 1,045 more words
ICH M8 “Specification for Submission Formats for eCTD”
DRUG REGULATORY AFFAIRS INTERNATIONAL

This additional specification describes the way files should be constructed for inclusion in the eCTD.
 Key Points:
Key Points:
- It is not necessary to use a product from Adobe or from any specific company to produce PDF documents.
- All ICH regional regulatory authorities are able to read and accept PDF files saved as PDF version 1.4 through 1.7, PDF/A-1, or PDF/A-2 compliant to ISO 32000-1:2008.
- The size of a PDF file should not exceed 500MB.
- Regulatory authorities cannot guarantee the availability of any fonts except Times New Roman, Arial, and Courier and fonts supported in the Acrobat product set itself. Therefore, all additional fonts used in the PDF files should be embedded to ensure that those fonts would always be available to the reviewer.
- Times New Roman, 12-point font, is adequate in size for narrative text and should be used whenever possible. Times New Roman font sizes 9-10 or an equivalent size…
View original post 496 more words
