
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

BMS-520
CAS: 1236188-38-7
MF: C23H17F3N4O4
MW: 470.1202
Synonym: BMS-520; BMS 520; BMS520.
INNOVATOR Bristol-Myers Squibb Company
INVENTORS
Scott Hunter Watterson, Alaric J. Dyckman,William J. Pitts, Steven H. Spergel
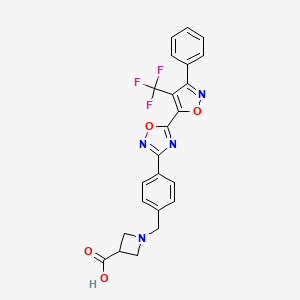
1-[4-[5-[3-Phenyl-4-(trifluoromethyl)isoxazol-5-yl]-1,2,4-oxadiazol-3-yl]benzyl]azetidine-3-carboxylic acid
1-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylic acid
1H NMR (500 MHz, DMSO-d6) δ: 3.20–3.46 (m, 5H), 3.66 (s, 2H), 7.53 (d, J = 8.25 Hz, 2H), 7.60–7. 70 (m, 5H), and 8.06 (d, J = 7. 70 Hz, 2H);
MS m/e 471(M+H+);
HPLC (XBridge 5 μ C18 4.6 × 50 mm, 4 mL/min, solvent A: 10% MeOH/water with 0.2% H3PO4, solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0–100% B over 4 min): 3.14 min;
Anal. Calcd for C23H17N4O4F3•0.01 EtOH: C, 58.72; H, 3.65; N, 11.90. Found: C, 58.63; H, 3.41; N, 11.84.
BMS-520 is a potent and selective S1P1 agonist. BMS-520 demonstrated impressive efficacy when administered orally in a rat model of arthritis and in a mouse experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis. Agonism of S1P1, in particular, has been shown to play a significant role in lymphocyte trafficking from the thymus and secondary lymphoid organs, resulting in immunosuppression.
Sphingosine-1 -phosphate (SlP) has been demonstrated to induce many cellular effects, including those that result in platelet aggregation, cell proliferation, cell morphology, tumor cell invasion, endothelial cell and leukocyte chemotaxis, endothelial cell in vitro angiogenesis, and lymphocyte trafficking. SlP receptors are therefore good targets for a wide variety of therapeutic applications such as tumor
15 growth inhibition, vascular disease, and autoimmune diseases. SlP signals cells in part via a set of G protein-coupled receptors named SlPi or SlPl, SIP2 or S1P2, SIP3 or S1P3, SlP4 Or S1P4, and SlP5 or S1P5 (formerly called EDG-I, EDG-5, EDG-3, EDG-6, and EDG-8, respectively). [0003] SlP is important in the entire human body as it is also a major regulator of
20 the vascular and immune systems. In the vascular system, SlP regulates angiogenesis, vascular stability, and permeability. In the immune system, SlP is recognized as a major regulator of trafficking of T- and B-cells. SlP interaction with its receptor SlPi is needed for the egress of immune cells from the lymphoid organs (such as thymus and lymph nodes) into the lymphatic vessels. Therefore, modulation
25 of SlP receptors was shown to be critical for immunomodulation, and SlP receptor modulators are novel immunosuppressive agents.
The SlPi receptor is expressed in a number of tissues. It is the predominant family member expressed on lymphocytes and plays an important role in lymphocyte trafficking. Downregulation of the SlPi receptor disrupts lymphocyte
30 migration and homing to various tissues. This results in sequestration of the lymphocytes in lymph organs thereby decreasing the number of circulating lymphocytes that are capable of migration to the affected tissues. Thus, development of an SlPi receptor agent that suppresses lymphocyte migration to the target sites associated with autoimmune and aberrant inflammatory processes could be efficacious in a number of autoimmune and inflammatory disease states. [0005] Among the five SlP receptors, SlPi has a widespread distribution and is highly abundant on endothelial cells where it works in concert with S IP3 to regulate cell migration, differentiation, and barrier function. Inhibition of lymphocyte recirculation by non-selective SlP receptor modulation produces clinical immunosuppression preventing transplant rejection, but such modulation also results in transient bradycardia. Studies have shown that SlPi activity is significantly correlated with depletion of circulating lymphocytes. In contrast, SIP3 receptor agonism is not required for efficacy. Instead, SIP3 activity plays a significant role in the observed acute toxicity of nonselective SlP receptor agonists, resulting in the undesirable cardiovascular effects, such as bradycardia and hypertension. (See, e.g., Hale et al, Bioorg. Med. Chem. Lett., 14:3501 (2004); Sanna et al, J. Biol. Chem., 279: 13839 (2004); Anliker et al., J. Biol. Chem., 279:20555 (2004); Mandala et al., J. Pharmacol. Exp. Ther., 309:758 (2004).)
An example of an SlPi agonist is FTY720. This immunosuppressive compound FTY720 (JPI 1080026-A) has been shown to reduce circulating lymphocytes in animals and humans, and to have disease modulating activity in animal models of organ rejection and immune disorders. The use of FTY720 in humans has been effective in reducing the rate of organ rejection in human renal transplantation and increasing the remission rates in relapsing remitting multiple sclerosis (see Brinkman et al., J. Biol. Chem., 277:21453 (2002); Mandala et al., Science, 296:346 (2002); Fujino et al., J. Pharmacol, and Exp. Ther., 305:45658 (2003); Brinkman et al., Am. J. Transplant, 4: 1019 (2004); Webb et al., J.
Neuroimmunol, 153: 108 (2004); Morris et al., Eur. J. Immunol, 35:3570 (2005); Chiba, Pharmacology & Therapeutics, 108:308 (2005); Kahan et al., Transplantation, 76: 1079 (2003); and Kappos et al., N. Engl. J. Med, 335: 1124 (2006)). Subsequent to its discovery, it has been established that FTY720 is a prodrug, which is phosphorylated in vivo by sphingosine kinases to a more biologically active agent that has agonist activity at the SlPi, SIP3, SlP4, and SIP5 receptors. It is this activity on the SlP family of receptors that is largely responsible for the pharmacological effects of FTY720 in animals and humans.
Clinical studies have demonstrated that treatment with FTY720 results in bradycardia in the first 24 hours of treatment (Kappos et al., N. Engl. J. Med., 335: 1124 (2006)). The observed bradycardia is commonly thought to be due to agonism at the SIP3 receptor. This conclusion is based on a number of cell based and animal experiments. These include the use of SIP3 knockout animals which, unlike wild type mice, do not demonstrate bradycardia following FTY720 administration and the use of SlPi selective compounds. (Hale et al., Bioorg. Med. Chem. Lett., 14:3501 (2004); Sanna et al., J. Biol. Chem., 279: 13839 (2004); and Koyrakh et al., Am. J. Transplant., 5:529 (2005)).
The following applications have described compounds as SlPi agonists: WO 03/061567 (U.S. Publication No. 2005/0070506), WO 03/062248 (U.S. Patent No. 7,351,725), WO 03/062252 (U.S. Publication No. 2005/0033055), WO 03/073986 (U.S. Patent No. 7,309,721), WO 03/105771, WO 05/058848, WO
06/047195, WO 06/100633, WO 06/115188, WO 06/131336, WO 2007/024922, WO 07/116866, WO 08/023783 (U.S. Publication No. 2008/0200535), and WO 08/074820. Also see Hale et al., J. Med. Chem., 47:6662 (2004). [0009] There still remains a need for compounds useful as SlPi agonists and yet having selectivity over Sl P3.
BMS 520
Paper
Journal of Medicinal Chemistry (2016), 59(6), 2820-2840
Potent and Selective Agonists of Sphingosine 1-Phosphate 1 (S1P1): Discovery and SAR of a Novel Isoxazole Based Series
Abstract

Sphingosine 1-phosphate (S1P) is the endogenous ligand for the sphingosine 1-phosphate receptors (S1P1–5) and evokes a variety of cellular responses through their stimulation. The interaction of S1P with the S1P receptors plays a fundamental physiological role in a number of processes including vascular development and stabilization, lymphocyte migration, and proliferation. Agonism of S1P1, in particular, has been shown to play a significant role in lymphocyte trafficking from the thymus and secondary lymphoid organs, resulting in immunosuppression. This article will detail the discovery and SAR of a potent and selective series of isoxazole based full agonists of S1P1. Isoxazole 6d demonstrated impressive efficacy when administered orally in a rat model of arthritis and in a mouse experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis.
SEE…..http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b00089
PAPER

This article reports an efficient scale-up synthesis of 1-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylic acid (BMS-520), a potent and selective isoxazole-containing S1P1 receptor agonist. This process features a highly regioselective cycloaddition leading to a key intermediate, ethyl 3-phenyl-4-(trifluoromethyl)isoxazole-5-carboxylate, a chemo-selective hydrolysis of its regioisomers, as well as an improved method for 1,2,4-oxadiazole formation, relative to the original synthesis. The improved process was applied to the preparation of multiple batches of BMS-520 for preclinical toxicological studies.
An Efficient Scale-Up Synthesis of BMS-520, a Potent and Selective Isoxazole-Containing S1P1 Receptor Agonist
PATENT
Scheme 1

Scheme 2

Scheme 3

Scheme 4


Scheme 5

Scheme 6

Example 1
l-(4-(5-(3-Phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3- yl)benzyl)azetidine-3-carboxylic acid

1-A. 4,4,4-Trifluorobut-2-yn-l-ol
To a solution of diisopropylamine (24.7 mL, 176 mmol) in ether (100 mL) at -78 0C was added a 1OM solution of butyllithium in ether (17.6 mL, 176 mmol) over 5 min. After 10 min. at -78°C, 2-bromo-3,3,3-trifluoroprop-l-ene (14.0 g, 80 mmol) was added to the pale yellow solution. After an additional 10 min., paraformaldehyde (2.40 g, 80 mmol) was added, the dry-ice bath was removed, and the reaction mixture was stirred at room temperature overnight. As the reaction mixture approached room temperature, it became dark in color. The reaction was quenched with a IN aqueous solution of hydrochloric acid (100 mL), diluted with ether (500 mL), washed with a IN aqueous solution of hydrochloric acid (2 x 100 mL), washed with brine 100 mL, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded a dark liquid which was distilled under low-vacuum (-50 Torr, ~50 0C) to give 4,4,4-trifluorobut-2-yn-l-ol (7.1 g, 57.2 mmol, 72 % yield) as a pale yellow liquid. 1H NMR (500 MHz, CDCl3) δ ppm 2.31 (br. s., IH) and 4.38 – 4.42 (m, 2H).An Alternative Preparation of 1 -A: 4,4,4-Trifluorobut-2-yn- 1 -ol
HO
-CF, (1-A) [00117] To an ether (pre-dried over magnesium sulfate) solution of phenanthroline (2.16 mg, 0.012 mmol) (indicator) at -78°C under nitrogen was added a 2M solution of n-butyl lithium in pentane. An orange color immediately appeared. Trifluoromethylacetylene gas was bubbled through the solution at -78°C. After ~4 min. of gas introduction, the orange color almost completely disappeared, the reaction solution became cloudy (due to some precipitation), and a pale light orange color persisted. Paraformaldehyde was added, and the dry ice/isopropanol bath was removed after 5 min. and replaced with a 00C ice-bath. Stirring was continued for 45 min., the ice bath was removed, and stirring was continued for an additional 1.25 h. The reaction flask was immersed in a 00C ice bath, and a saturated aqueous solution of ammonium chloride (20.0 mL) was added. The layers were separated, and the organic layer was washed with water (2x), washed with brine, and dried over anhydrous sodium sulfate. Concentration under low-vacuum (~50 Torr) without heat afforded a dark brown liquid which was purified by vacuum distillation (~50 Torr, -50 0C) to give 4,4,4-trifluorobut-2-yn-l-ol (7.1 g, 57.2 mmol, 72 % yield) as a colorless liquid.
1-B. N-Hydroxybenzimidoyl chloride

This compound was prepared according to the method of Liu, K.C. et al, J. Org. Chem., 45:3916-1918 (1980).To a colorless, homogeneous solution of (E)-benzaldehyde oxime (24.4 g, 201 mmol) in N,N-dimethylformamide (60 mL) at room temperature was added N- chlorosuccinimide (26.9 g, 201 mmol) portion-wise over 30 min. During each addition, the reaction mixture would turn yellow and then gradually return to near colorlessness. Additionally, an exotherm was noted with each portion added to ensure that the reaction initiated after the addition of N-chlorosuccinimide. An ice bath was available, if required, to cool the exotherm. After the addition was complete, the homogeneous reaction mixture was stirred overnight at room temperature. The reaction mixture was diluted with 250 mL of water and extracted with ether (3 x 100 mL). The organic layers were combined, washed with water (2 x 100 mL), washed with a 10% aqueous solution of lithium chloride (2 x 100 mL), and washed with brine (100 mL). The aqueous layers were back extracted with ether (100 mL), and the combined organic layers (400 mL) were dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded (Z)-N-hydroxybenzimidoyl chloride (30.84 g, 198 mmol, 98 % yield) as a fluffy, pale yellow solid. The product had an HPLC ret. time = 1.57 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 155.8. 1H NMR (500 MHz, DMSO-d6) δ ppm 7.30 – 7.64 (m, 3H), 7.73 – 7.87 (m, 2H), and 12.42 (s, IH).
l-C. 3-Phenyl-4-(trifluoromethyl)isoxazol-5-yl)methanol

To a pale yellow, homogeneous mixture of N-hydroxybenzimidoyl chloride (5.50 g, 35.4 mmol) and 4,4,4-trifluorobut-2-yn-l-ol (5.46 g, 39.6 mmol) in dichloroethane (85 mL) in a 250 mL round bottom flask at 700C was added triethylamine (9.85 mL, 70.7 mmol) in 22 mL of dichloroethane over 2.5 h via an addition funnel (the first -50% over 2 h and the remaining 50% over 0.5 h). After the addition was complete, the reaction mixture was complete by HPLC (total time at 700C was 3 h). The reaction mixture was stirred at room temperature overnight. [00121] The reaction mixture was diluted with dichloromethane (100 mL), washed with water (100 mL), and the organic layer was collected. The aqueous layer was extracted with dichloromethane (2 x 50 mL), and the combined organic layers were dried over anhydrous sodium sulfate. The solvent was removed under reduced pressure. Analysis indicated that the product mixture was composed of a 86: 14 mixture of the desired regioisomer (1-C), (3-phenyl-4-(trifluoromethyl)isoxazol-5- yl)methanol, and the undesired regioisomer, (3-phenyl-5-(trifluoromethyl)isoxazol-4- yl)methanol. The mixture was purified by silica gel chromatography using a mixture of ethyl acetate and hexane (1% to pack and load – 5% – 9% – 12%) to afford (3- phenyl-4-(trifluoromethyl)isoxazol-5-yl)methanol (5.34 g, 21.96 mmol, 62.1 % yield) as a pale yellow oil. The compound had an HPLC ret. time = 1.91 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 =244.2. 1H NMR (500 MHz, CDCl3) δ ppm 2.21 (br. s., IH), 4.97 (s, 2H), 7.47 – 7.56 (m, 3H), and 7.65 (d, J=6.60 Hz, 2H).
1-D. 3-Phenyl-4-(trifluoromethyl)isoxazole-5-carboxylic acid

Preparation of Jones’ Reagent
To an orange, homogeneous solution of chromium trioxide (12.4 g, 0.123 mol) in water (88.4 mL) at 00C was added sulfuric acid (10.8 mL) dropwise via addition funnel over 30 min. with stirring. The addition funnel was rinsed with water
(1 mL) to give 1.23 M solution of Jones’ Reagent (0.123 mol of reagent in 100 mL of solvent).
To a solution of (3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)methanol
(5.24 g, 21.6 mmol) in acetone (75 mL) at room temperature (immersed in a water bath) was added Jones’ Reagent (43.8 mL, 53.9 mmol) via addition funnel slowly over 1.5 h. The dark reaction mixture was stirred at room temperature overnight. By HPLC, the reaction was 93% complete. An additional 0.5 equivalents (9 mL) of the Jones’ Reagent was added. After 1 h, the reaction was 95% complete. After an additional 3h, the reaction was 96% complete. An additional 0.5 equivalents (9 mL) of the Jones’ Reagent was added. The reaction mixture was stirred for an additional 2.5 h. By HPLC, the reaction was 97% complete. Isopropyl alcohol (6 mL) was added, and the mixture was stirred for 90 min, resulting in a dark green precipitate. The mixture was diluted with ether (600 mL), washed with a 2% aqueous solution of sodium hydrogen sulfite (5 x 100 mL), and the organic layer was collected. The aqueous layer was back-extracted with ether (2 x 100 mL). By HPLC, there was no additional product in the aqueous layer. The combined organic layers were washed with water (100 mL), washed with a saturated aqueous solution of brine (100 mL), and dried over anhydrous sodium sulfate. The aqueous layer was back-extracted with ether (100 mL), and the organic layer was added to the previous organic layers. The solution was concentration under reduced pressure to give 3-phenyl-4-
(trifluoromethyl)isoxazole-5-carboxylic acid as an off-white solid. The solid was diluted with dichloromethane (200 mL), washed with a 2% aqueous solution of sodium hydrogen sulfite, washed with brine, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 3-phenyl-4- (trifluoromethyl)isoxazole-5-carboxylic acid (3.84 g, 14.93 mmol, 69.3 % yield) as a pale yellow solid. The product was 96% pure by HPLC with a ret. time = 1.60 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 258.2. [00124] The sodium hydrogen sulfite aqueous layer still contained a significant amount of product. The brine layer contained no additional product and was discarded. The aqueous layer was saturated with sodium chloride, the pH was adjusted to -3.5, and the solution was extracted with ether (3 x 100 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to afford additional 3-phenyl-4-(trifluoromethyl)isoxazole-5-carboxylic acid (1.12 g, 4.36 mmol, 20.21 % yield) as a white solid. The product was >99% pure by HPLC with a ret. time = 1.60 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 258.1. 1H NMR (500 MHz, DMSO-(I6) δ ppm 7.55 – 7.63 (m, 5H). The products were combined to give 4.96 g (90% yield) of 3-phenyl-4- (trifluoromethyl)isoxazole-5-carboxylic acid.
An Alternative Preparation of 1-D: 3 -Phenyl -4-(trifluoromethyl)isoxazole-5- carboxylic acid starting with (3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)methanol

A mixture of (3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)methanol (2.1 g, 8.64 mmol), TEMPO (0.094 g, 0.604 mmol), and a sodium phosphate buffer (0.67M) (32.2 mL, 21.59 mmol) was heated to 35°C. A solution of sodium phosphate buffer (40 mL, pH -6.5) consisting of a 1: 1 solution OfNaH2PO4 (20 mL, 0.67M) and Na2HPO4 (20 mL, 0.67M) was prepared in acetonitrile (30 mL) was prepared prior to use. Solutions of sodium chlorite (3.91 g, 34.5 mmol) in water (4.5 mL) and bleach (4.3 mL, 6% wt.) were added simultaneously over 40min. The reaction was monitored by HPLC, and after 2 h, -30% of the starting material remained. After 6 h, 10% remained. Additional bleach (100 μL) was added, and the reaction mixture was left at room temperature overnight. [00127] Additional bleach (100 μL) was added. The resulting mixture was allowed to stir at 35°C for additional 2 h. HPLC indicated complete conversion. The reaction was quenched by the slow addition of a solution of sodium sulfite (2.07 mL, 43.2 mmol) in water (90 mL) at 00C, resulting in the disappearance of the brown reaction color. The solvent was removed under reduced pressure, and the remaining aqueous residue was extracted with ethyl acetate (3 x 40 mL). The organic layers were combined, washed with water (8 mL), washed with brine (8 mL), and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded 3 -phenyl – 4-(trifluoromethyl)isoxazole-5-carboxylic acid (2.2 g, 8.55 mmol, 99 % yield) as a pale yellow solid. An alternative procedure for the for the preparation of 3-phenyl-4-(trifluoromethyl) isoxazole-5-carboxylic acid starting with 4,4,4-trifluorobut-2ynoate (1-D)

Alt.1 -D- 1. Ethyl 3 -phenyl-4-(trifluoromethyl)isoxazole-5-carboxylate

To a pale yellow mixture of (Z)-N-hydroxybenzimidoyl chloride (1.04 g, 6.68 mmol) and ethyl 4,4,4-trifluorobut-2-ynoate (1.238 g, 7.45 mmol) in diethyl ether (20 mL) at room temperature was added triethylamine (1.86 mL, 13.4 mmol) over 15 min., resulting in a precipitant. After the addition was complete, the pale yellow slurry was stirred at room temperature over a weekend. The heterogeneous reaction mixture was filtered under reduced pressure to remove the triethylamine hydrochloride salt, and the filtrate was concentrated to give the product mixture as a dark yellow, viscous oil (2.03 g). By HPLC, the reaction mixture was composed of a mixture of the desired regioisomer, ethyl 3-phenyl-4-(trifluoromethyl)isoxazole-5- carboxylate, and the undesired regioisomer, ethyl 3-phenyl-5- (trifluoromethyl)isoxazole-4-carboxylate, in an approximately 15:85 ratio. The compound mixture was dissolved in hexane and sonicated for 5 min. The hexane was decanted off, and the dark red, oily residue was found to have only trace product by HPLC. The hexane was removed under reduced pressure, and the residue (1.89 g) was purified by preparative HPLC. The desired fractions containing ethyl 3-phenyl- 4-(trifluoromethyl)isoxazole-5-carboxylate were concentrated, and the residue was diluted with dichloromethane, washed with a saturated aqueous solution of sodium bicarbonate, and dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded ethyl 3-phenyl-4-(trifluoromethyl) isoxazole-5-carboxylate (0.087 g, 0.305 mmol, 4.6 % yield) as a pale yellow solid. The compound had an HPLC ret. time = 2.88 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. 1H NMR (400 MHz, CDCl3) δ ppm 1.46 (t, J=7.15 Hz, 3H), 4.53 (q, J=7.03 Hz, 2H), 7.48 – 7.55 (m, 3H), and 7.58 (d, J=7.53 Hz, 2H).
An Alternative Preparation of 1-D-l : Ethyl 3-phenyl-4-(trifluoromethyl)isoxazole-5- carboxylic acid starting with ethyl 4,4,4-trifluorobut-2-enoate
1-D-l. Ethyl 2,3-dibromo-4,4,4-trifluorobutanoate
Br L /COOEt
Br (1-D-l) [00129] Bromine (18.4 mL, 357 mmol) was added dropwise over 30 minutes to a solution of (E)-ethyl 4,4,4-trifluorobut-2-enoate (50 g, 297 mmol) in carbon tetrachloride (50 mL) at room temperature under nitrogen. The resulting dark red solution was refluxed for 4 hours. Additional bromine (2ml) was added and heating was continued until the HPLC analysis showed that the starting material had been consumed. The reaction mixture was concentrated under reduced pressure to give light brown oil which used in the next step without purification. HPLC (XBridge 5μ Cl 8 4.6×50 mm, 4 mL/min, Solvent A: 10 % MeOH/water with 0.2 % H3PO4, Solvent B: 90 % MeOH/water with 0.2 % H3PO4, gradient with 0-100 % B over 4 minutes): 2.96 and 3.19 minutes.
l-D-2. (Z/E)-Ethyl 2-bromo-4,4,4-trifluorobut-2-enoate
,COOEt
F3C
Br (l-D-2)
To a solution of ethyl 2,3-dibromo-4,4,4-trifluorobutanoate (1-B-l) in hexane (200 mL) cooled to 00C was added triethylamine (49.7 ml, 357mmol) drop- wise over 35 minutes, during which time a white precipitate formed. The reaction mixture was stirred for an additional 2 hours until LC indicated complete conversion. The solid was filtered and rinsed with hexane (3 x 5OmL), and the filtrate was concentrated and passed through a short silica gel pad eluting with 10% ethyl acetate/hexane to give (Z/E)-ethyl 2-bromo-4,4,4-trifluorobut-2-enoate (65.5 g, 265mmol, 89 % yield for two steps) as a colorless oil. Alternatively, the crude product can be purified by distillation (85 0C / -60 mmHg). 1H NMR (CDCl3, 400 MHz) 5 7.41 (q, IH, J= 7.28 Hz), 4.35 (q, 2H, J= 7.11 Hz), 1.38 (t, 3H, J= 7.15 Hz); HPLC (XBridge 5μ Cl 8 4.6×50 mm, 4 mL/min, Solvent A: 10 % MeOH/water with 0.2 % H3PO4, Solvent B: 90 % MeOH/water with 0.2 % H3PO4, gradient with 0- 100 % B over 4 minutes): 3.09 minutes.
1-D-l. Ethyl 3 -phenyl -4-(trifluoromethyl)isoxazole-5-carboxylate

(Z/E)-Ethyl 2-bromo-4,4,4-trifluorobut-2-enoate, l-D-3, (39.7 g, 161 mmol) and N-hydroxybenzimidoyl chloride (30 g, 193mmol) were dissolved in ethyl acetate (15OmL). Indium (III) chloride (8.89 g, 40.2mmol) was added and the resulting mixture stirred for 60 minutes at RT under N2. Potassium hydrogen carbonate (32.2 g, 321mmol) was added to the reaction mixture which was allowed to stir overnight for 14 hours at RT. The solvent was removed in vacuo. The residue was re-suspended in 30OmL hexane and stirred for lOmiutes then filtered. The filter cake was washed with hexane (3X3 OmL) and the combined filtrate was concentrated in vacuo to give crude product, which was further purified with flash chromatography to generate 33g product (72%) as light yellowish oil as a mixture of the desired isomer 1-D-l and undesired isomer 1-D-la in a ratio of -30/1. MS m/e 286.06(M+H+); 1H NMR (CDCl3, 400 MHz) δ 7.56 (m, 5H), 4.53 (q, 2H, J= 7.3 Hz), 1.46 (t, 3H, J= 7.2 Hz); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10 % MeOH/water with 0.2 % H3PO4, Solvent B: 90 % MeOH/water with 0.2 % H3PO4, gradient with 0-100 % B over 4 minutes): 3.57 minutes.
Alt.1-D. 3-Phenyl-4-(trifluoromethyl)isoxazole-5-carboxylic acid, lithium salt

A mixture of ethyl 3-phenyl-4-(trifluoromethyl)isoxazole-5-carboxylate, 1-D-l, (0.085 g, 0.298 mmol) and lithium hydroxide hydrate (0.013 g, 0.298 mmol) in methanol (2.0 mL) and water (1.0 mL) was stirred at room temperature overnight. The reaction mixture was concentrated to dryness to give 3-phenyl-4- (trifluoromethyl)isoxazole-5-carboxylic acid, lithium salt (0.079 g, 0.299 mmol, 100 % yield) as a pale yellow solid. The compound had an HPLC ret. time = 1.72 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 258.0. 1H NMR (400 MHz, CDCl3) δ ppm 7.49 – 7.57 (m, 3H) and 7.58 – 7.62 (m, 2H).1-E. 3-Phenyl-4-(trifluoromethyl)isoxazole-5-carbonyl fluoride

To a mixture of 3-phenyl-4-(trifluoromethyl)isoxazole-5-carboxylic acid (3.00 g, 11.7 mmol) and pyridine (1.132 mL, 14.0 mmol) in dichloromethane (100 mL) at room temperature was added 2,4,6-trifluoro-l,3,5-triazine (cyanuric fluoride) (1.18 mL, 14.0 mmol). The reaction mixture was stirred at room temperature overnight, diluted with dichloromethane (300 mL), washed with an ice-cold solution of 0.5N aqueous hydrochloric acid (2 x 100 mL), and the organic layer was collected. The aqueous layer was back-extracted with dichloromethane (200 mL), and the combined organic layers were dried anhydrous sodium sulfate and concentrated to afford 3-phenyl-4-(trifluoromethyl)isoxazole-5-carbonyl fluoride (2.91 g, 11.2 mmol, 96 % yield) as a yellow, viscous oil. The product was found to react readily with methanol and on analysis was characterized as the methyl ester, which had an HPLC ret. time = 2.56 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 272.3 (methyl ester).1-F. tert-Butyl l-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol- 3-yl)-benzyl)azetidine-3-carboxylate

A suspension of 3-phenyl-4-(trifluoromethyl)isoxazole-5-carbonyl fluoride (2.91 g, 11.2 mmol), (Z)-tert-butyl 1-(4-(N’- hydroxycarbamimidoyl)benzyl)azetidine-3-carboxylate (Int. l, 3.43 g, 11.2 mmol), and Hunig’s Base (2.55 mL, 14.6 mmol) in acetonitrile (20 mL) was stirred at room temperature over the weekend. The reaction mixture had completely solidified (pinkish-tan in color), but was judged complete by HPLC and LCMS. The reaction mixture was partitioned between a saturated aqueous of sodium bicarbonate (150 mL) and dichloromethane (150 mL). The aqueous layer was extracted with dichloromethane (2 x 100 mL), and the combined organic layers were dried over anhydrous sodium sulfate. Concentration under reduced pressure afforded a tan solid which was purified by flash silica gel chromatography using a mixture of ethyl acetate in hexane (0-50%) to afford tert-butyl l-(4-(5-(3-phenyl-4-(trifluoromethyl) isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (4.60 g; 78%) as a white, crystalline solid. The material was suspended in methanol (-75 mL) and was sonicated for 5 minutes. The MeOH was removed under reduce pressure, and the residue was re-suspended in methanol (-50 mL) with sonication. Vacuum filtration and drying afforded tert-butyl l-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)- l,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (4.04 g, 7.67 mmol, 68 % yield) as a white, crystalline solid. The methanol filtrate was concentrated to afford additional tert-butyl l-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)- 1,2,4- oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (570 mg; 10%) as a slightly off- white solid. The compound had an HPLC retention time = 3.12 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 =527.1. 1H NMR (500 MHz, CDCl3) δ ppm 1.47 (s, 9H) 3.28 – 3.37 (m, 3H), 3.60 (br. s., 2H), 3.74 (br. s., 2H), 7.49 (d, J=7.70 Hz, 2H), 7.53 – 7.62 (m, 3H), 7.69 (d, J=7.15 Hz, 2H), and 8.16 (d, J=7.70 Hz, 2H). 1. Preparation of l-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4- oxadiazol-3-yl)benzyl)azetidine-3-carboxylic acid
A mixture of tert-butyl l-(4-(5-(3-phenyl-4-(trifluoromethyl)isoxazol-5- yl)-l,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (6.12 g, 11.6 mmol) and trifluoroacetic acid (50.1 mL, 651 mmol) was stirred at room temperature for 1.5 h. By HPLC, the deprotection appeared to be complete after 1 h. The TFA was removed under reduced pressure, and the oily residue was diluted with water (100 mL) and sonicated for 5 min. The resulting suspension was stirred for an additional 10 min until a consistent white suspension was observed. A IN aqueous solution of sodium hydroxide was added portion-wise until the pH was ~4.5 (42 mL of IN NaOH). Over time, the pH drifted back down to 3-4, and additional IN aqueous sodium hydroxide had to be added. The suspension was stirred overnight at room temperature. Several drops of IN aqueous sodium hydroxide were added to re-adjust the pH to 4.5, and after 60 min., the pH appeared to be stable. The solid was collected by vacuum filtration, washed with water several times, and dried under reduced pressure for 5 h. The solid was then suspended in methanol (110 mL) in a 150 mL round bottom flask and sonicated for 15 min. During the sonication, the solution became very thick. An additional 25 mL of methanol was added, and the suspension was stirred overnight. The product was collected by vacuum filtration, washed with methanol (-50 mL), and dried under reduced pressure. The solid was transferred to a 250 mL round bottom flask, re-suspended in methanol (115 mL), sonicated for 5 min., and stirred for 60 min. The solid was collected by vacuum filtration, washed with methanol (~50 mL), and dried over well under reduced pressure to give l-(4-(5-(3- phenyl-4-(trifluoromethyl)isoxazol-5-yl)-l,2,4-oxadiazol-3-yl)benzyl)azetidine-3- carboxylic acid (5.06 g, 10.7 mmol, 92 % yield) as a crystalline, white solid. The product had an HPLC ret. time = 2.79 min. – Column: CHROMOLITH® SpeedROD 4.6 x 50 mm (4 min.); Solvent A = 10% MeOH, 90% H2O, 0.1% TFA; Solvent B = 90% MeOH, 10% H2O, 0.1% TFA. LC/MS M+1 = 471.3. 1H NMR (500 MHz, DMSO-d6) δ ppm 3.20 – 3.46 (m, 5H), 3.66 (s, 2H), 7.53 (d, J=8.25 Hz, 2H), 7.60 – 7.70 (m, 5H), and 8.06 (d, J=7.70 Hz, 2H).
HPLC purity 100/99.8%, ret. time = 7.62 min. (A linear gradient using 5% acetonitrile, 95% water, and 0.05% TFA (Solvent A) and 95% acetonitrile, 5% water, and 0.05% TFA (Solvent B); t = 0 min., 10% B, t = l2 min., 100% B (15 min.) was employed on a SunFire C18 3.5u 4.6 x 150 mm column. Flow rate was 2 ml/min and UV detection was set to 220/254 nm.).
HPLC purity 100/99.9%, ret. time = 8.45 min. (A linear gradient using 5% acetonitrile, 95% water, and 0.05% TFA (Solvent A) and 95% acetonitrile, 5% water, and 0.05% TFA (Solvent B); t = 0 min., 10% B, t = l2 min., 100% B (15 min.) was employed on a XBridge Ph 3.5u 4.6 x 150 mm column. Flow rate was 2 ml/min and UV detection was set to 220/254 nm.).
CONSTRUCTION
![]()
Alt.1 -D- 1. Ethyl 3 -phenyl-4-(trifluoromethyl)isoxazole-5-carboxylate
ADDITIONAL INFORMATION
Sphingosine 1-phosphate (S1P) is the endogenous ligand for the sphingosine 1-phosphate receptors (S1P1–5) and evokes a variety of cellular responses through their stimulation. The interaction of S1P with the S1P receptors plays a fundamental physiological role in a number of processes including vascular development and stabilization, lymphocyte migration, and proliferation
REFERENCES
Watterson, S. H.; Guo, J.; Spergel, S. H.; Langevine, C. L.; Moquin, R. V.; Shen, D.
R.; Yarde, M.; Cvijic, M. E.; Banas, D.; Liu, R.; Suchard, S. J.; Gillooly, K.; Taylor,
T.; Rex-Rabe, S.; Shuster, D. J.; McIntyre, K. W.; Cornelius, G.; Darienzo, C.;
Marino, A.; Balimane, P.; Warrack, B.; Saltercid, L.; McKinnon, M.; Barrish, J. C.;
Carter, P. C.; Pitts, W. J.; Xie, J.; Dyckman, D. J. J. Med. Chem. 2016, 59, 2820.
Watterson, S.H.; Guo, J.; Spergel, S.H.; et al.
Potent and selective agonists of Sphingosine-1-Phosphate 1 (S1P1): The discovery and SAR of a novel isoxazole based series
241st Am Chem Soc (ACS) Natl Meet (March 27-30, Anaheim) 2011, Abst MEDI 96
/////Potent and Selective Isoxazole-Containing S1P1 Receptor Agonist, BMS 520, Sphingosine-1-Phosphate 1 (S1P1)
O=C(C1CN(CC2=CC=C(C3=NOC(C4=C(C(F)(F)F)C(C5=CC=CC=C5)=NO4)=N3)C=C2)C1)O

