













| Patent | Submitted | Granted |
|---|---|---|
| COMPOUNDS AND METHODS FOR TREATING INFLAMMATORY AND FIBROTIC DISORDERS [US2009318455] | 2009-12-24 | |
| COMPOSITION AND METHOD FOR TREATING PROTEINURIA [US2010099719] | 2010-04-22 | |
| COMPOSITION AND METHOD FOR TREATING FIBROTIC DISEASES [US2009258911] | 2009-10-15 | |
| Composition and Method for Treating Fibrotic Diseases [US2008319027] | 2008-12-25 | |
| METHODS FOR TREATING ACUTE MYOCARDIAL INFARCTIONS AND ASSOCIATED DISORDERS [US2010190731] | 2010-07-29 | |
| Methods for Treating Acute Myocardial Infarctions and Associated Disorders [US2011218515] | 2011-09-08 | |
| METHODS OF TREATING HIV PATIENTS WITH ANTI-FIBROTICS [US2012014917] | 2012-01-19 | |
| Composition and Method for Treating Fibrotic Diseases Composition and Method for Treating Fibrotic Diseases [US2009005424] | 2007-08-30 | |
| Crystalline 1-(3-fluorophenyl)-5-methyl-2-(1H)pyridone, the preparation methods, compositions and applications thereof [US8232408] | 2009-03-10 | 2012-07-31 |
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Dofequidar fumarate

Dofequidar fumarate
Phase III
A P-glycoprotein inhibitor potentially for the treatment of breast cancer and non-small lung cancer (NSCLC).
![]()
MS-209; Dofequidar fumarate
CAS No. 129716-58-1 (Dofequidar FREE )
CAS No 153653-30-6 (Dofequidar fumarate 1;1)…..C34H35N3O7, 597.66
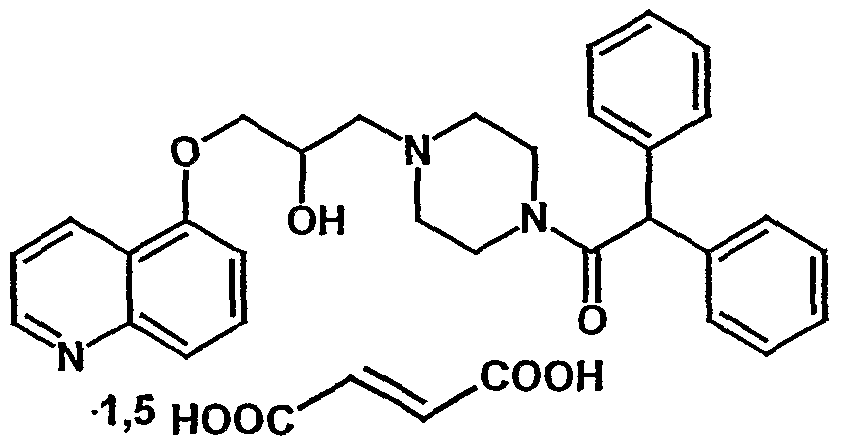
5-[3-[4-(2,2-Diphenylacetyl)piperazin-1-yl]-2-hydroxypropoxy]quinoline sesquifumarate
1-[4-(2,2-Diphenylacetyl)piperazin-1-yl]-3-(quinoliln-5-yloxy)-2-propanol sesquifumarate
1-(Diphenylacetyl)-4-[(2RS)-2-hydroxy-3-(5-quinolyloxy)propyl]piperazine sesquifumarate
CAS Number 158681-49-3, C30H31N3O3 · 1.5 C4H4O4, Molecular Weight 655.69
4-(Diphenylacetyl)-a-[(5-quinolinyloxy)methyl]-1-Piperazineethanol (E)-2-butenedioate fumarate (1:1.5), C30 H31 N3 O3 . 3/2 C4 H4 O4
1-Piperazineethanol, 4-(diphenylacetyl)-α-[(5-quinolinyloxy)methyl]-, (E)-2-butenedioate (2:3)
1-Piperazineethanol, 4-(diphenylacetyl)-α-[(5-quinolinyloxy)methyl]-, (E)-2-butenedioate (2:3)

Dofequidar fumarate(MS-209 fumarate), an orally active quinoline compound, has been reported to overcome MDR by inhibiting ABCB1/P-gp, ABCC1/MDR-associated protein 1, or both.
Dofequidar fumarate(MS-209 fumarate), an orally active quinoline compound, has been reported to overcome MDR by inhibitingABCB1/P-gp, ABCC1/MDR-associated protein 1, or both.
IC50 value:
Target: P-gp
in vitro: MS-209 at 3 microM effectively overcame docetaxel resistance in MDR cancer cells, and this concentration was achieved in blood plasma for > 7 h without serious toxicity [1]. MS-209 restored chemosensitivity of SBC-3 / ADM cells to VP-16, ADM, and VCR in a dose-dependent manner in vitro [2]. dofequidar inhibits the efflux of chemotherapeutic drugs and increases the sensitivity to anticancer drugs in CSC-like side population (SP) cells isolated from various cancer cell lines. Dofequidar treatment greatly reduced the cell number in the SP fraction [3]. In 4-1St cells, which are extremely resistant to ADM and VCR, MS-209 at a concentration of 3 microM enhanced the cytotoxicity of ADM and VCR, 88- and 350-fold, respectively [4].
in vivo: Treatment with docetaxel alone at the maximal tolerated dose (MTD) showed an apparent antitumor activity to an intrinsically resistant HCT-15 tumor xenograft, and MS-209 additionally potentiated the antitumor activity of docetaxel. Against a MCF-7/ADM tumor xenograft expressing larger amounts of P-gp, docetaxel alone at the MTD showed no antitumor activity, whereas the MTD of docetaxel combined with MS-209 greatly reduced MCF-7/ADM tumor growth [1]. Intravenous injection with SBC-3 or SBC-3 / ADM cells produced metastatic colonies in the liver, kidneys and lymph nodes in natural killer (NK) cell-depleted severe combined immunodeficiency (SCID) mice, though SBC-3 / ADM cells more rapidly produced metastases than did SBC-3 cells. Treatment with VP-16 and ADM reduced metastasis formation by SBC-3 cells, whereas the same treatment did not affect metastasis by SBC-3 / ADM cells. Although MS-209 alone had no effect on metastasis by SBC-3 or SBC-3 / ADM cells, combined use of MS-209 with VP-16 or ADM resulted in marked inhibition of metastasis formation by SBC-3 / ADM cells to multiple organs [2].
Dofequidar fumarate is a multidrug resistance (MDR)-reversing quinoline derivative that interacts directly with P-glycoprotein and inhibits the efflux of antitumor agents. The agent had been in phase III clinical development by Nihon Schering (now Bayer) for the treatment of advanced and recurrent breast cancer and non-small lung cancer (NSCLC) and at the National Cancer Institute in combination with docetaxel for the treatment of solid tumors. In 2000, Schering AG obtained dofequidar fumarate when Nihon Schering acquired Mitsui Pharmaceuticals, originator of the compound.
PAPER
Structure-activity relationship of newly synthesized quinoline derivatives for reversal of multidrug resistance in cancer
J Med Chem 1997, 40(13): 2047
5-[3-{4-(2,2-Diphenylacetyl)piperazin-1-yl}-2-hydroxypropoxy]quinoline 1.5Fumarate (16, MS-209)
free form of 16 (7.37 g, 70%): mp 161−162 °C; 1H-NMR (CDCl3) δ 2.2−2.8 (m, 6 H), 3.5−3.6 (m, 2H), 3.7−3.9 (m, 2H), 4.1−4.3 (m, 3H), 5.20 (s, 1H), 6.86 (d, 1H, J = 7.3 Hz), 7.2−7.4 (m, 11H), 7.59 (t, 1H, J = 8.1 Hz), 7.71 (d, 1H, J = 8.1 Hz), 8.54 (d, 1H, J = 7.3 Hz), 8.91 (dd, 1H, J = 2, 4 Hz); IR (KBr) 2954, 1630, 1587, 1268, 1091, 802, 748, 703 cm-1.
16 1.5Fumarate(1.0 g, 60%): mp 210 °C dec; 1H-NMR (DMSO-d6) δ 2.2−2.6 (m, 6H), 3.4−3.6 (m, 4H), 4.0−4.2 (m, 3H), 5.53 (s, 1H), 6.63 (s, 3H), 7.03 (d, 1H, J = 8.1 Hz), 7.2−7.4 (m, 10H), 7.5−7.7 (m, 3H), 8.61 (d, 1H, J = 8.1 Hz), 8.89 (dd, 1H, J = 1.5, 4.4 Hz); IR (KBr) 3424, 1644, 1592, 1277, 1180, 1110, 799 cm-1.
Patent
WO 2004099151
https://www.google.co.in/patents/WO2004099151A1?cl=en
A method for producing the purest rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyI] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate and the purest rac-1 – {4- [2-hydroxy-3- (5-quinoly loxy) propylene l] piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate
The invention relates to a method for producing the purest rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate as well as rac -1- {4- [2- hydroxy-3- (5-quinolyloxy) propyl] piperazin-1-yl} -2,2-diphenylethan-1-one fumarate with a purity of at least 99.55%
The multidrug resistance modulator rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] – piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate, its preparation and use as carcinostatic drug is described as well as other derivatives of this compound in EP 575,890.
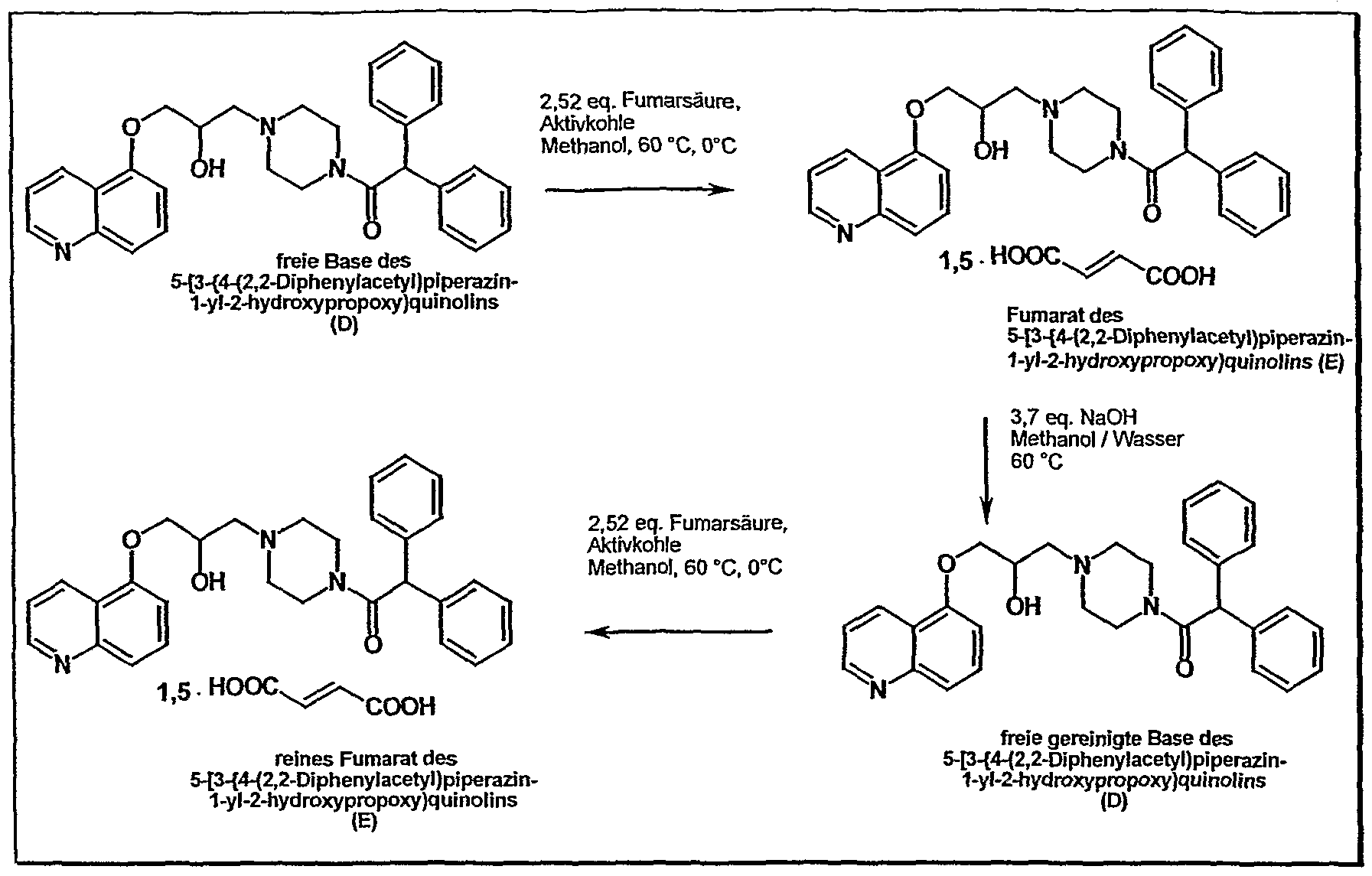
According to the process described in EP 575 890 A process for the preparation of pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-dϊphenylethan-1-one fumarate is first by coupling the two modules epoxiline (B) (5- (2,3-epoxypropoxy) – quinoline) and Diphenpiperazid (C) (N- (2,2-Diphenylacetyl) piperazine), the free base 5- [3- {4- (2,2-diphenylacetyl) piperazin-1-yl} -2-hydroxypropoxy] quinoline isolated as a crude product. This implementation includes two sub-stages. First, the Epoxylat with hydroxyquinoline (A) is reacted. In the second step the epoxiline (B) (5- (2,3-epoxypropoxy) -quinolin) by Diphenpiperazid (C) (N- (2,2-Diphenylacetyl) piperazine) is opened, it gives the secondary alcohol (D). This reaction takes place in ethanol, water catalyzes the conversion. The workup / isolation is then carried out by precipitation from acetone / water and drying under vacuum at 60 ° C.
The overall reaction results from the following scheme:

On the isolation of the free base, the many impurities (purity of the crude product is typically about 80%), joins in the next step a very expensive cleaning procedures. After charcoal treatment of the free base and the formation of the fumarate in methanol, the free base is again prepared by treatment with dilute sodium hydroxide solution for purification. Subsequently, as the last step, repeated fumarate formation. The two fumarate formations are procedurally identical and differ only in the batch size (T. Suzuki et al., J. Med. Chem. (1997) 40, 2047) (JP 2000281653). Starting from the crude free base, the typical yield for this laboratory cleaning sequence 45% of theory.
A disadvantage of this method is not only the low yield (about 50% loss in the final stage), but also the complex technical implementation, which binds many operational capacities and thus caused increased costs. A particular disadvantage is the extremely poor filterability of the free base, the filter must be dried partially over several weeks.
Despite the high procedural expenses according to this known method, the extremely high purity requirements of rac-1 – {4- [2-hydroxy-3- (5- quinolyloxy) propyl] piperazine-1-yl} -2,2-diphenylethane-1 -one fumarate not always be achieved completely satisfactory.
. Furthermore provides the method described in EP 575 890 any reasonable results during scale-up an overview of the individual reactions are the following scheme:
It has now been found that these known disadvantages can be overcome with the process of this invention. In the process of this invention also the epoxiline (B) and Diphenpiperazid (C) is first coupled by opening of the epoxide. But is not the free base (D) but after the addition of solid fumaric acid directly the fumarate salt (E) is then isolated as a crude product.
The present application thus provides a process for the preparation of pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1 -one fumarate , which is characterized in that firstly
a) a Epoxytosylat of structure I
OTs
(0 with

b) 5-hydroxyquinoline (II)
(II) and cesium carbonate in a suitable solvent and at a suitable temperature to 5- (2,3-epoxypropoxy) -quinolin of formula III

allowed to react, and then the 5- (2,3-epoxypropoxy) -quinolin of formula III
c) with N- (2,2-Diphenylacefyl) piperazine of the formula IV

in a suitable solvent and at a suitable temperature followed by the addition of solid fumaric acid to the crude rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] – piperazin-1-yl} -2,2-diphenylethane 1-one fumarate of the formula V
And subsequently reacting (V)
d) the thus formed crude rac-1 – fumarate {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1 -one (V) is isolated and is dissolved in a solvent mixture of methanol and methylene chloride, is treated with activated carbon and subsequently filtered through a pressure filter having silica gel as column material, and the thus obtained pure rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate (V) is crystallized from a suitable alcohol.
Preparation Example
Preparation of rac-1 – 4- [2-Hy droxy-3- (5-quinolyloxy) propylene l] -piperazin-1 -yl> -2,2-diphenylethan-1-one fumarate
A) Under nitrogen, 44.2 g of 5-hydroxy-quinoline and 151.9 g of cesium carbonate with 560 ml acetone will give at room temperature together and stirred for 30 minutes at 60 ° C bath temperature. At 50 ° C internal temperature 73.0 g of 5- (2,3-epoxypropoxy) -quinolin dissolved in
153.3 g of dichloromethane, admit. The mixture is stirred at 50 ° C for two hours. The mixture is filtered at 50 ° C. The filter residue (inorganic salts) is washed with 560 ml of 50 ° C warmed acetone. 85.4 g are then N- (2,2-diphenyl-acetyl) piperazine admit and concentrated at a bath temperature of 40 ° C under vacuum to 374 g final weight. It will then add 374 g of demineralized water and 2
Stirred at 40 ° C hours. Then 255 g of acetone and 201 g of demineralized water will admit. The mixture is cooled to room temperature and 89.1 g of fumaric acid are in solid form to Gege-ben. It is stirred for 60 minutes at 60 ° C bath temperature and then stirred at 0 ° C for 2 hours. The solid is suction filtered and washed with 150 ml of ice-cold methanol. The filter residue is dried at 60 ° C under vacuum.
Yield: 65 – 85% of theory
B) 56.0 g of the thus prepared rac-1 – {4- [2-hydroxy-3- (5-quinolyIoxy) propyl] -piperazin-1-yl} – 2,2-diphenylethan-1-one fumarate were nitrogen and treated at room temperature with 5.6 g of activated carbon, Norit SX plus, 672 ml of methanol and 1008 ml of dichloromethane. The resulting suspension is stirred at a bath temperature of 75 ° C to warm to reflux temperature and refluxed for 30 min. At an internal temperature of 40 ° C is rac-1 – {4- [2-hydroxy-3- (5-quinolyloxy) propyl] -piperazin-1-yl} -2,2-diphenylethan-1-one fumarate in solution. The mixture is then filtered hot through 300% silica gel and the silica gel with 560 ml of a mixture of 168 ml of methanol and 392 ml of dichloromethane at room temperature RT. The solution is concentrated at a bath temperature of 40 ° C and an initial vacuum of 400 mbar to a final volume of 517 ml. The ultimate vacuum of 350 mbar. The distilled volume is about the difference in volume (about 1, 7 I). There are 404 ml of methanol was added so that a final volume of 921 ml is achieved. The solution is cooled to 0 ° C, whereupon the product precipitates. The resulting suspension is stirred for 2 hours at 0 ° C and then filtered through a paper filter. The filter residue is washed with 56.0 ml of ice-cold methanol. The filter residue is dried at 60 ° C and under vacuum at 100 mbar for 10 hours.
Yield (. Uncorr): 47.29 g (84.45% FS)
Purity: 99.65% (HPLC, 100% method)
References on Dofequidar fumarate
http://jco.ascopubs.org/content/25/4/411.full.pdf
SEE………http://apisynthesisint.blogspot.in/2016/01/ms-209-dofequidar-fumarate.html
///////////MS-209, Dofequidar fumarate, PHASE 3
What are “complex manufacturing processes”? A recent reply from the EMA
The Variations Regulation (EC) no. 1234/2008 of the European Commission defines the procedure for variations of existing marketing authorisations. The “detailed guidelines for the various categories of variations“, which were published in the consolidated version in August 2013 in the European Official Journal, explain the interpretation and application of this Variations Regulation.
Although the “detailed guidelines” describe a number of scenarios of possible variations in some detail, there are formulations in the Guideline text which require clarification due to their blur. The EMA adopted such a case in a recent update of itsquestions and answers collection “Quality of Medicines Questions and Answers: Part 1” to concretise the case through a statement.
It is about the term “complex manufacturing processes”, which is used in two scenarios associated with type II variations (found in the “detailed guidelines” p 40ff):
- Replacement or addition of a manufacturing site for part or all of the manufacturing process of the finished product (Guideline change code B.II.b.1)
…
c) Site where any manufacturing operation(s) take place, except batch release, batch control, and secondary packaging, for biological/immunological medicinal products, or for pharmaceutical forms manufactured by complex manufacturing processes. - Change in the batch size (including batch size ranges) of the finished product (Guideline change code B.II.b.4)
…
d) The change relates to all other pharmaceutical forms manufactured by complex manufacturing processes .
The EMA now clarified this term as follows:
- Guideline Change Code B.II.b.1: Complex manufacturing processes are given when the understanding of the relation between quality characteristics of the product and its in vivo efficacy is lacking. This is often the case in innovative medicines such as products of nanomedicine.
- Guideline Change Code B.II.b.4: Complex manufacturing processes are those which contain one or more sub-steps, where a scale-up can lead to problems.
In both scenarios, the approving authority will decide on a case by case basis. If the applicant submits the variation as a Type IB, he must provide a valid justification that the production process is not “complex”. However, in doubt the authority may upgrade the variation to a Type II. Therefore, the EMA recommends that the applicant clarifies the situation with the authority before submitting the variation.
What are “complex manufacturing processes”? A recent reply from the EMA………..http://www.gmp-compliance.org/enews_05072_What-are-%22complex-manufacturing-processes%22-A-recent-reply-from-the-EMA.html
Statistics and Process Validation: current Findings of the FDA

The “new” FDA’s process validation guideline has been effective since January 2011. One considerable change was made to the original validation guideline from 1987 to put a significantly greater emphasis on statistics in the context of process validation. So far, relatively few inspection deficiencies had been observed by the FDA with regard to statistics. At a conference in September 2015 co-sponsored by the FDA, Grace McNally – Senior FDA official – reported about current “findings” in the 483s deficiency reports and in Establishment Inspection Reports (EIR). Now, deficiencies regarding statistical problematics can also be found here.
For example, it has been criticised that a (statistical) sampling plan had be misinterpreted. Wrong AQL values with regard to the number of samples have been noted based on MIL-STD-105D. Moreover, it has been criticised that the company didn’t know the operation characteristics of its sampling plan.
Another criticised “finding” was that PPQ batches had been considered as “accepted” when all in-process controls and release specifications were met. It has also been criticised that no intra-batch variabilities have been examined. In addition, it has been noticed that there was no information available in the validation plan concerning the assessment of the process itself. There was also no indication about the objective of the determination of inter-batch variabilities.
Although OOS results had been found in 2 out of 4 PPQ batches, reduced IPC tests have been recommended in the PPQ report giving the justification that this was a standard procedure. Regarding this point, the FDA criticises the lack of scientific rationales for reduced sampling and monitoring. Interestingly, Grace McNally mentions possibilities for rationales of IPC sampling plans and the adaptation to a reduced size. In this context, she refers to the ANSI/ASQ Z1.4 norm and ISO 2859 whereby it is expressly pointed out that the ANSI norm recommends the production of at least 10 successful batches before reducing testing. According to the ISO norm even 15 successful batches are necessary.
The FDA notified a tablet process, criticising the fact that no rationales for warning and action limits were available. Furthermore, it has been criticised that no analyses on variabilities were available although they had been required internally and no capacity indices had been determined. There have been no analyses on the distribution of data, neither planned nor performed. The FDA also remarked that the calculation of variabilities is necessary to be able to make statements about process capacities.
Conclusion: Reinforcing the emphasis on statistics in the US FDA Process Validation Guideline from 2011 hasn’t been really often addressed in the official deficiencies reports. This seems to be changing.


////////////Statistics, Process Validation, current Findings, FDA
FDA gives Advice on the Use of Control Charts

In our News entitled “Statistics and Process Validation: current Findings of the FDA“, we pointed out that the FDA has been observing more and more deficiencies in the area of statistics with regard to process validation. One of the “statistical methods” – also stated in FDA’s Process Validation Guidance – is the use of control charts in the context of a statistical process control (SPC). A SPC can be a valuable resource, particularly in stage 3 of the process validation lifecycle (continued/ongoing process verification).
During an event with the Product Quality Research Institute (PQRI), Dr Daniel Peng from CDER’s Office of Processes and Facilities (OPF) presented his views on the use of SPC under the heading “Using Control Charts to Evaluate Process Variability”. He began with the history of control charts and their fields of application. He furthermore considered the most important rules to create and manage control charts, including types of control charts, sampling and evaluation rules according to the Western Electric Rules.
He concluded his presentation with examples of control charts for evaluating intra and inter-batch variabilities and site performance monitoring.
The slides of the presentation “Using Control Charts to Evaluate Process Variability” are very easy to read and available for free.
Are you interested in SPC and control charts? On 18/19 February, the ECA organises the Education Course “Statistical Process Control – A key tool for process understanding in the process validation life cycle” in Heidelberg, Germany.
Preclinical characterization of substituted 6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one P2X7 receptor antagonists
MW 422.79, MF C18 H14 Cl F3 N6 O
cas 1627748-32-6
1,2,4-Triazolo[4,3-a]pyrazin-8(5H)-one, 7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6,7-dihydro-6-methyl-3-(2-pyrazinyl)-, (6S)-
(6S)-7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6-methyl-3-pyrazin-2-yl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8-one
(6S)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
Janssen Pharmaceutica Nv INNOVATOR
Michael K. Ameriks, Jason C. Rech, Brad M. Savall

(6S)-7-[[2-chloro-3-(trifluoromethyl)phenyl]methyl]-6-methyl-3-pyrazin-2-yl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8-one
PAPER

The synthesis, SAR, and preclinical characterization of a series of substituted 6,7-dihydro[1,2,4]triazolo[4,3]pyrazin-8(5H)-one P2X7 receptor antagonists are described. Optimized leads from this series comprise some of the most potent human P2X7R antagonists reported to date (IC50s < 1 nM). They also exhibit sufficient potency and oral bioavailability in rat to enable extensive in vivo profiling. Although many of the disclosed compounds are peripherally restricted, compound 11d is brain penetrant and upon oral administration demonstrated dose-dependent target engagement in rat hippocampus as determined by ex vivo receptor occupancy with radiotracer 5 (ED50 = 0.8 mg/kg).
Volume 26, Issue 2, 15 January 2016, Pages 257–261

Preclinical characterization of substituted 6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one P2X7 receptor antagonists
- Janssen Pharmaceutical Research & Development L.L.C., 3210 Merryfield Row, San Diego, CA 92121, United States
![]()
http://www.sciencedirect.com/science/article/pii/S0960894X15303656

Scheme 3.
Synthesis of compounds 11d and 11l–t. Reagents and conditions: (a) Boc2O, NaOH, H2O/MeOH, 0 °C→rt (42%); (b) 2-chloro-3-trifluoromethylbenzaldehyde, Na(OAc)3BH, DCE, rt (85%); (c) methyl chlorooxoacetate, Et3N, CH2Cl2, 0 °C→rt (97%); (d) 4 N HCl/dioxane, rt, then Et3N, CH2Cl2, rt (100%); (e) Et3O+BF4−, DCM, rt, or Lawesson’s reagent, THF, 55 °C (67–99%); (f) RCONHNH2, 1-butanol, 130 °C (27–90%).
PATENT
US 20140275096
http://www.google.com/patents/US20140275096
- Intermediate 1. 3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
Step A. tert-butyl 3-ethoxy-5,6-dihydropyrazine-1(2H)-carboxylate
-
To a solution of tert-butyl 3-oxopiperazine-1-carboxylate (1 g, 5 mmol) in DCM (15 mL) was added triethyloxonium tetrafluoroborate (2.9 g, 15 mmol). Stirred for 2 h and neutralized with sat. aq NaHCO3. Layers separated and aqueous layer extracted with DCM. Combined organic layers dried over Na2SO4, filtered, and concentrated to give the title compound, which was used directly without further purification.
Step B. tert-butyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a solution of tert-butyl 3-ethoxy-5,6-dihydropyrazine-1(2H)-carboxylate (1.14 g, 5 mmol) in 1-butanol (30 mL) was added pyrazine-2-carbohydrazide (685 mg, 5 mmol). The reaction mixture was heated at reflux for 16 h. After cooling to rt, the reaction mixture was concentrated and purified by chromatography (SiO2; 2.5% MeOH in DCM) to afford the desired product as a white solid (700 mg, 50% over 2 steps). MS (ESI): mass calcd. for C14H18N6O2, 302.2; m/z found, 303.2 [M+H]+.
-
1H NMR (500 MHz, CDCl3) d 9.57 (d, J=1.4 Hz, 1H), 8.62 (d, J=2.5 Hz, 1H), 8.59-8.54 (m, 1H), 4.94 (s, 2H), 4.63-4.50 (m, 2H), 3.89 (t, J=5.4 Hz, 2H), 1.51 (s, 9H).
Step C. 3-(pyrazin-2-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine
-
To a solution of tert-butyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (9.3 g, 30 mmol) in DCM (100 mL) was added 1.25M HCl in EtOH (30 mL, 37.5 mmol). After 3 h, the reaction mixture was concentrated, and the resulting solid was purified by chromatography (SiO2; 10% MeOH in DCM) to provide the desired product as a white solid (3.7 g, 61%). MS (ESI): mass calcd. for C9H10N6, 202.1; m/z found, 203.1 [M+H]+. 1H NMR (400 MHz, CD3OD) δ 9.35 (d, J=1.4 Hz, 1H), 8.72 (dd, J=2.5, 1.6 Hz, 1H), 8.66 (d, J=2.6 Hz, 1H), 4.50 (t, J=5.6 Hz, 2H), 4.22 (s, 2H), 3.24 (t, J=5.6 Hz, 2H).
Step D. 2-(trimethylsilyl)ethyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a solution of 3-(pyrazin-2-yl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine (1.0 g, 5.0 mmol) and N,N-diisopropylethylamine (1.7 mL, 9.9 mmol) in DMF (15 mL) was added 1-[2-(trimethylsilyl)ethoxycarbonyloxy]pyrrolidin-2,5-dione (1.5 g, 5.9 mmol). Stirred for 18 h and poured into ice cold brine (150 mL). Precipitate filtered and washed successively with water and ether to afford the desired product as a white solid (1.5 g, 89%). MS (ESI): mass calcd. for C15H22N6O2Si, 346.2; m/z found, 347.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.50 (d, J=1.4 Hz, 1H), 8.56 (d, J=2.5 Hz, 1H), 8.52-8.48 (m, 1H), 4.91 (s, 2H), 4.60-4.45 (m, 2H), 4.25-4.14 (m, 2H), 3.87 (t, J=5.3 Hz, 2H), 1.07-0.92 (m, 2H), 0.01-0.04 (m, 9H).
Step E. 2-(trimethylsilyl)ethyl 8-oxo-3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate
-
To a vigorously stirred solution of 2-(trimethylsilyl)ethyl 3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (172 mg, 0.5 mmol) in 1:1 CHCl3:MeCN (3.8 mL) was added a solution of ruthenium (IV) oxide hydrate (9.8 mg, 0.07 mmol) and sodium metaperiodate (504 mg, 2.3 mmol) in water (4.7 mL). After 4 h, the reaction mixture was diluted with water and extracted with CHCl3 (×3). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford a green oil. Purification by chromatography (SiO2; EtOAc—10% IPA/EtOAc) provided the desired product as a white solid (663 mg, 63%).
-
[0140]MS (ESI): mass calcd. for C15H20H6O3Si, 360.1; m/z found, 361.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.59 (d, J=1.5 Hz, 1H), 8.63 (d, J=2.5 Hz, 1H), 8.55 (dd, J=2.5, 1.6 Hz, 1H), 4.88-4.75 (m, 2H), 4.47-4.33 (m, 2H), 4.33-4.24 (m, 2H), 1.18-1.04 (m, 2H), 0.04-(−0.02) (m, 9H).
Step F. 3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
-
To a solution of 2-(trimethylsilyl)ethyl 8-oxo-3-(pyrazin-2-yl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxylate (1.0 g, 2.9 mmol) in DCM (29 mL) was added TFA (5.7 mL, 75 mmol). After 1 h, the reaction mixture was concentrated. The crude residue was diluted with EtOAc, sonicated, and filtered to provide the desired product as a white solid (1.2 g, 95%). MS (ESI): mass calcd. for C9H8N6O, 216.1; m/z found, 217.1 [M+H]+. 1H NMR (500 MHz, DMSO-d6) δ 9.39 (d, J=1.1 Hz, 1H), 8.77 (q, J=2.6 Hz, 2H), 8.56 (s, 1H), 4.73-4.60 (m, 2H), 3.67-3.55 (m, 2H).
-

-
Intermediate 3 was made in a manner analogous to Intermediate 1 substituting (±)-tert-butyl 2-methyl-5-oxopiperazine-1-carboxylate for tert-butyl 3-oxopiperazine-1-carboxylate in Step A. MS (ESI): mass calcd. for C10H10N6O, 230.1; m/z found, 231.1 [M+H]+.
- Intermediate 3. (±)-6-methyl-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
Intermediate 4. (6S)-1-(2-chloro-3-(trifluoromethyl)benzyl)-6-methylpiperazine-2,3-dione
-
[0146]

Step A. (S)-tert-butyl(2-aminopropyl)carbamate
-
To a solution of (S)-1,2-diaminopropane dihydrochloride (16 g, 109 mmol) in MeOH (64 mL) and water (16 mL) was added di-tert-butyl dicarbonate (28.5 g, 131 mmol) in MeOH (16 mL). The resulting solution was cooled in an ice bath, and 4N NaOH (35 mL, 140 mL) was added dropwise over 2 h. The mixture was allowed to warm to rt and stirred for a total of 20 h. The reaction was filtered, and the filtrate concentrated to remove MeOH. 200 mL EtOAc, 200 mL water, and 16 mL 1M HCl were added sequentially. The layers were separated and the aqueous layer washed with EtOAc (200 mL). The combined organic extracts were washed with 0.04M HCl (208 mL). The organic phase was separated and discarded. The aqueous phases were combined, adjusted to pH=14 with 10N NaOH (20 mL), and extracted with DCM (400 mL×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a clear oil (8.0 g, 42%). MS (ESI): mass calcd. for C8H18N2O2, 174.1; m/z found, 175.2 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 5.01 (br s, 1H), 3.24-3.09 (m, 1H), 3.09-2.95 (m, 1H), 2.92-2.84 (m, 1H), 1.45 (s, 9H), 1.35-1.19 (m, 2H), 1.07 (d, J=6.4 Hz, 3H).
Step B. (6S)-tert-butyl(2-((2-chloro-3-(trifluoromethyl)benzyl)amino)propyl) carbamate
-
A solution of (S)-tert-butyl(2-aminopropyl)carbamate (4.0 g, 23 mmol) and 2-chloro-3-trifluoromethylbenzaldehyde (4.8 g, 23 mmol) in DCE (100 mL) was stirred at rt for 2 h. Sodium triacetoxyborohydride (7.3 g, 34 mmol) was added at once and stirring continued overnight. Saturated aqueous NaHCO3 was added, and the resulting mixture was extracted with DCM (×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford a clear oil. Purification by chromatography (SiO2; hex—60% EtOAc/hex) provided the desired product as a clear oil (7.2 g, 85%). MS (ESI): mass calcd. for C16H22ClF3N2O2, 366.1; m/z found, 367.2 [M+H]+. 1H NMR (400 MHz, CDCl3) δ 7.72-7.56 (m, 2H), 7.35 (t, J=7.7 Hz, 1H), 4.94 (s, 1H), 3.99 (d, J=14.1 Hz, 1H), 3.90 (d, J=14.1 Hz, 1H), 3.29-3.14 (m, 1H), 3.11-2.99 (m, 1H), 2.84 (dd, J=11.1, 6.2 Hz, 1H), 1.44 (s, 9H), 1.11 (d, J=6.4 Hz, 3H).
Step C. (6S)-methyl 2-((1-((tert-butoxycarbonyl)amino)propan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate
-
To an ice cold solution of (6S)-tert-butyl(2-((2-chloro-3-(trifluoromethyl)benzyl)amino)propyl) carbamate (7.2 g, 20 mmol) and triethylamine (2.8 mL, 21 mmol) in DCM (121 mL) was added methyl chlorooxoacetate (1.9 mL, 21 mmol) dropwise. The resulting mixture was warmed to rt and stirred overnight. After diluting with brine, the layers were separated, and the aqueous layer washed with DCM. The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a white solid (8.5 g, 97%). 1H NMR (400 MHz, CDCl3) δ 7.72-7.56 (m, 1H), 7.49-7.32 (m, 2H), 4.83 (d, J=17.1 Hz, 1H), 4.79-4.62 (m, 1H), 4.51 (d, J=17.1 Hz, 1H), 4.11-3.97 (m, 1H), 3.93 (s, 3H), 3.24-3.13 (m, 2H), 1.44 (s, 9H), 1.16-1.12 (m, 3H).
Step D. (6S)-methyl 2-((1-aminopropan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate hydrochloride
-
To a solution of 4M HCl in dioxane (75 mL) was added (6S)-methyl 2-((1-((tert-butoxycarbonyl)amino)propan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate (7.5 g, 16.7 mmol). After 30 minutes, the reaction mixture was concentrated and the product was used in the next step without further purification (6.5 g, 100%). MS (ESI): mass calcd. for C14H16ClF3N2O3, 352.1; m/z found, 353.1 [M+H]+.
Step E. (6S)-1-(2-chloro-3-(trifluoromethyl)benzyl)-6-methylpiperazine-2,3-dione
-
To a solution of (6S)-methyl 2-((1-aminopropan-2-yl)(2-chloro-3-(trifluoromethyl)benzyl)amino)-2-oxoacetate hydrochloride (7.3 g, 18.9 mmol) in DCM (90 mL) was added triethylamine (7.9 mL, 57 mmol) at once. After 2 h, 1N HCl was added and the layers were separated. The aqueous layer was extracted with DCM (×2). The combined organic extracts were dried (Na2SO4), filtered, and concentrated to afford the desired product as a white solid (5.9 g, 98%). MS (ESI): mass calcd. for C13H11ClF3N2O2, 320.1; m/z found, 321.1 [M+H]+. 1H NMR (600 MHz, CDCl3) δ 8.24 (d, J=3.6 Hz, 1H), 7.68 (dd, J=7.8, 1.1 Hz, 1H), 7.59 (d, J=7.7 Hz, 1H), 7.39 (t, J=7.8 Hz, 1H), 5.22 (d, J=15.7 Hz, 1H), 4.52 (d, J=15.7 Hz, 1H), 3.82-3.73 (m, 1H), 3.69-3.61 (m, 1H), 3.31 (ddd, J=13.2, 5.2, 2.3 Hz, 1H), 1.46-1.38 (m, 3H).
-
Example 14
- (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one………..
- ……………………(±) FORM
-

-
Example 14 was made in a manner analogous to Example 2 substituting Intermediate 3 for Intermediate 1 and 1-(bromomethyl)-2-chloro-3-(trifluoromethyl)benzene for 1-(bromomethyl)-2,3-dichlorobenzene to provide the desired compound as a white solid (102 mg, 63%). MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+. 1H NMR (500 MHz, DMSO-d6) 89.48 (d, J=1.2 Hz, 1H), 8.84-8.82 (m, 2H), 7.85-7.82 (m, 2H), 7.56 (t, J=7.8 Hz, 1H), 5.20 (d, J=16.5 Hz, 1H), 4.98 (dd, J=13.8, 2.2 Hz, 1H), 4.80 (dd, J=13.8, 4.6 Hz, 1H), 4.56 (d, J=16.6 Hz, 1H), 4.23-4.10 (m, 1H), 1.23 (d, J=6.7 Hz, 3H).
- Example 15
- (6R)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one
- ……………………UNDESIRED R CONFIGURATION
-

-
Chiral SFC separation of (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one on a CHIRALCEL OD-H column (5 μM, 250×20 mm) using 70% CO2/30% MeOH provided 39 mg of the title compound as the first eluting enantiomer. [α]=+40° (c 2.2, CHCl3).
-
MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+. 1H NMR (500 MHz, CDCl3) δ 9.66 (d, J=1.5 Hz, 1H), 8.68 (d, J=2.5 Hz, 1H), 8.59 (dd, J=2.5, 1.5 Hz, 1H), 7.76-7.72 (m, 1H), 7.69 (dd, J=7.9, 1.6 Hz, 1H), 7.41 (t, J=7.8 Hz, 1H), 5.44 (d, J=15.5 Hz, 1H), 5.17 (dd, J=13.9, 2.1 Hz, 1H), 4.62-4.54 (m, 2H), 4.08-4.02 (m, 1H), 1.36 (d, J=6.8 Hz, 3H).
- Example 16
- (6S)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one……………… DESIRED
-

-
Chiral SFC separation of (±)-7-[2-Chloro-3-(trifluoromethyl)benzyl]-6-methyl-3-pyrazin-2-yl-6,7-dihydro[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one on a CHIRALCEL OD-H column (5 μM, 250×20 mm) using 70% CO2/30% MeOH provided 40 mg of the title compound as the second eluting enantiomer.
-
[α]=−44° (c 2.2, CHCl3).
-
MS (ESI): mass calcd. for C18H14ClF3N6O, 422.1; m/z found, 423.1 [M+H]+.
-
1H NMR (500 MHz, CDCl3) δ 9.66 (d, J=1.5 Hz, 1H), 8.68 (d, J=2.5 Hz, 1H), 8.59 (dd, J=2.5, 1.5 Hz, 1H), 7.76-7.72 (m, 1H), 7.69 (dd, J=7.9, 1.6 Hz, 1H), 7.41 (t, J=7.8 Hz, 1H), 5.44 (d, J=15.5 Hz, 1H), 5.17 (dd, J=13.9, 2.1 Hz, 1H), 4.62-4.54 (m, 2H), 4.08-4.02 (m, 1H), 1.36 (d, J=6.8 Hz, 3H).
| Patent | Submitted | Granted |
|---|---|---|
| P2X7 MODULATORS [US2014275096] | 2014-03-14 | 2014-09-18 |
see,,,,,,,,,http://worlddrugtracker.blogspot.in/2016/01/preclinical-characterization-of.html
//////////////P2X7, 6,7-Dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one, Autoradiography, Depression, CNS, Preclinical characterization, substituted 6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one, P2X7 receptor antagonists, Janssen Pharmaceutical Research & Development L.L.C, 1627748-32-6
FC(F)(F)c4cccc(CN1C(=O)c2nnc(n2C[C@@H]1C)c3cnccn3)c4Cl
CC1CN2C(=NN=C2C(=O)N1CC3=C(C(=CC=C3)C(F)(F)F)Cl)C4=NC=CN=C4
FK-3311
FK-3311
FK 3311; 116686-15-8; FK-3311; N-[4-acetyl-2-(2,4-difluorophenoxy)phenyl]methanesulfonamide; COX-2 Inhibitor V, FK3311; FK3311;
A prostaglandin receptor antagonist potentially for the treatment of rheumatoid arthritis.

cas 116686-15-8
| Molecular Formula: | C15H13F2NO4S |
|---|---|
| Molecular Weight: | 341.329826 g/mol |

This compound has been obtained by two different ways: 1) The oxidation of 4′-amino-3′-chloroacetophenone (I) with NaNO2 and HCl in water gives 3′-chloro-4-nitroacetophenone (II), which is condensed with 2,4-difluorophenol (III) by means of K2CO3 in xylene yielding 3′-(2,4-difluorophenyl)-4′-nitroacetophenone (IV). The reduction of (IV) with Fe and NH4Cl in ethanol affords the corresponding 4′-amino compound (V), which is finally treated with methanesulfonyl chloride and pyridine. 2) The reaction of 4′-aminoacetophenone (VI) with methanesulfonyl chloride as before gives the corresponding sulfonamide (VII), which is brominated with Br2 in acetic acid yielding N-(4-acetyl-3-bromophenyl)methanesulfonamide (VIII). Finally, this compound is condensed with 2,4-difluorophenol (III) by means of K2CO3 and CuCl as before.
Chem Pharm Bull 1992,40(9),2399
|
1 to 4 of 4
|
|---|
| Patent | Submitted | Granted |
|---|---|---|
| METHODS TO TREAT INFECTIONS [US2014329777] | 2014-04-22 | 2014-11-06 |
| NOVEL NIMESULIDE COMPOSITIONS [US2012063996] | 2011-07-28 | 2012-03-15 |
| NANOPARTICULATE MELOXICAM FORMULATIONS [US2014141083] | 2013-07-12 | 2014-05-22 |
| Alkanesulfonanilide derivatives, processes for preparation thereof and pharmaceutical composition comprising the same [US4866091] | 1989-09-12 | |
\\\\\\\\\\\\\CC(=O)C1=CC(=C(C=C1)NS(=O)(=O)C)OC2=C(C=C(C=C2)F)F
SEE………..http://apisynthesisint.blogspot.in/2016/01/fk-3311.html
Merck’s Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP)
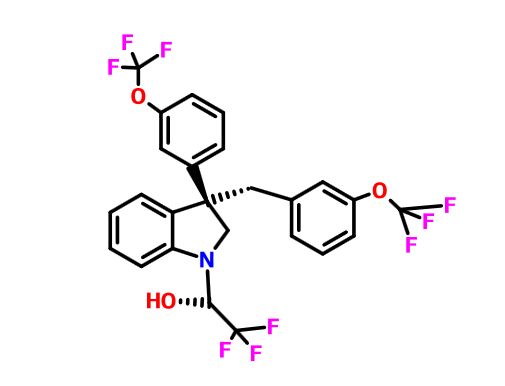
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3-(trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER
MF C26 H20 F9 N O3, MW 565.43
Merck Sharp & Dohme Corp. INNOVATOR

Using the collective body of known (CETP) inhibitors as inspiration for design, a structurally novel series of tetrahydroquinoxaline CETP inhibitors were discovered. An exemplar from this series, compound 5, displayed potent in vitro CETP inhibition and was efficacious in a transgenic cynomologus-CETP mouse HDL PD (pharmacodynamic) assay. However, an undesirable metabolic profile and chemical instability hampered further development of the series. A three-dimensional structure of tetrahydroquinoxaline inhibitor 6 was proposed from 1H NMR structural studies, and this model was then used in silico for the design of a new class of compounds based upon an indoline scaffold. This work resulted in the discovery of compound 7, which displayed potent in vitro CETP inhibition, a favorable PK–PD profile relative to tetrahydroquinoxaline 5, and dose-dependent efficacy in the transgenic cynomologus-CETP mouse HDL PD assay.
chemical compounds that inhibit cholesterol ester transfer protein (CETP) and are expected to have utility in raising HDL-C, lowering LDL-C, and in the treatment and prevention of atherosclerosis.
see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
Discovery of Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP) through a Structure-Guided Approach
†Department of Medicinal Chemistry and ‡Department of Structural Chemistry, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Rahway, New Jersey 07065, United States
§Department of Pharmacology, ∥Department of Drug Metabolism and Pharmacokinetics, and ⊥Department of Biology, Merck Research Laboratories, Merck & Co, Inc., P.O. Box 2000, Kenilworth, New Jersey 07033, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.5b00404
Publication Date (Web): January 4, 2016
Copyright © 2016 American Chemical Society
*E-mail: jonathan_wilson@merck.com.
PATENT
Atherosclerosis and its clinical consequences, including coronary heart disease
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
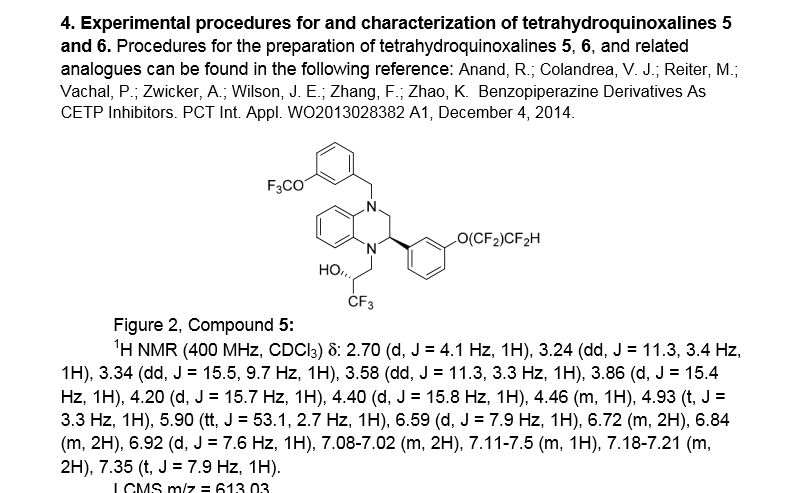
Example 18 as in patent

(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12

Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.

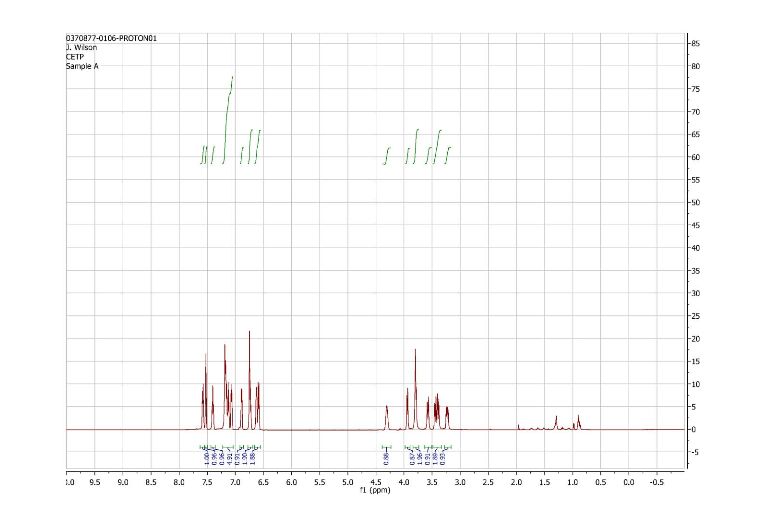
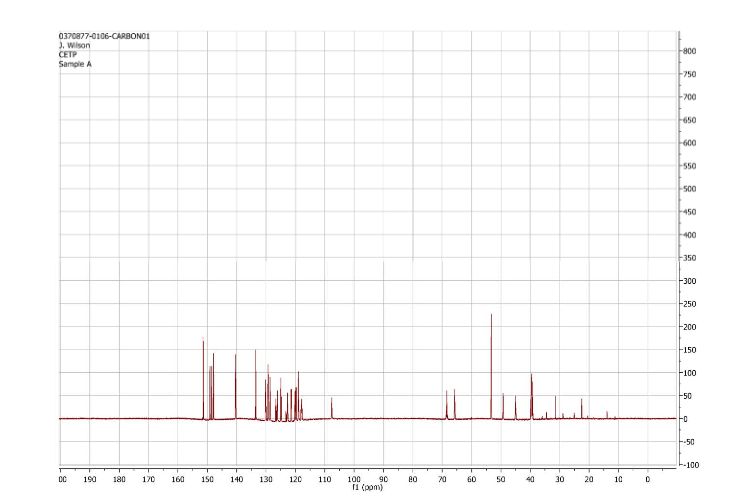
1H and 13C NMR spectra for compound 7
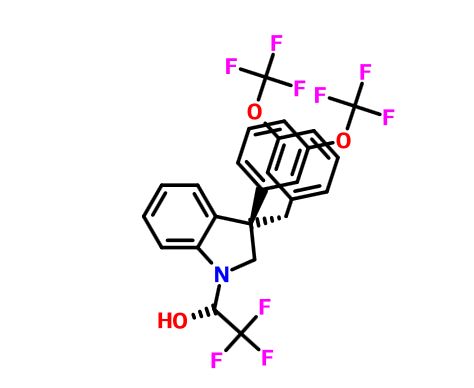
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al

Scheme A2

Scheme A3

R = Ar, NR2l C02R, CN, S02Me
es
es
SEE EXAMPLE ………SIMILAR BUT NOT SAME

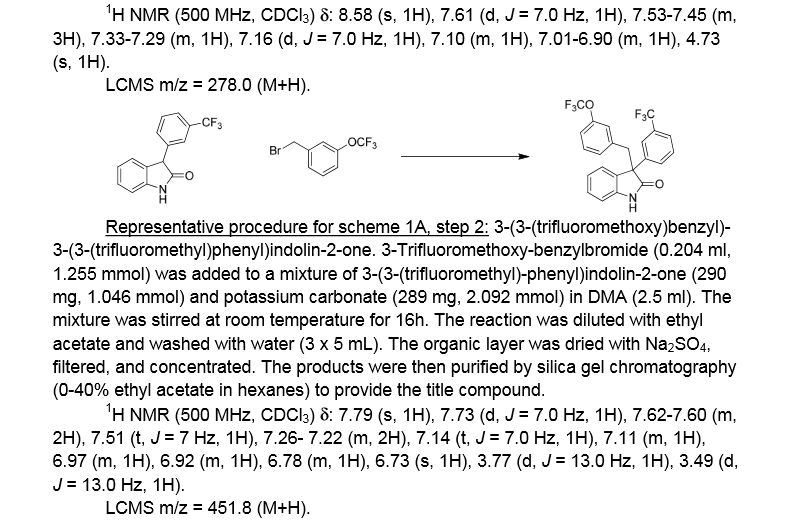
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.

3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).

3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)

3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)

(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a
SEE EG 10…….(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.

ABOUT AUTHOR
Jonathan Wilson
Associate Principal Scientist at Merck

https://www.linkedin.com/in/jonathan-wilson-23206523
Experience
Associate Principal Scientist
Merck
October 2013 – Present (2 years 4 months)
Senior scientist
Merck
May 2009 – October 2013 (4 years 6 months)
Postdoctoral researcher
Princeton University
October 2007 – May 2009 (1 year 8 months)
Associate Medicinal Chemist
Merck
2000 – 2002 (2 years)
Education
///////CETP inhibition, cholesterol ester transfer protein, HDL, indoline, tetrahydroquinoxaline, merck, discovery
c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
see…………http://worlddrugtracker.blogspot.in/2016/01/mercks-novel-indoline-cholesterol-ester.html
Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products

Recently, application of the flow technologies for the preparation of fine chemicals, such as natural products or Active Pharmaceutical Ingredients (APIs), has become very popular, especially in academia. Although pharma industry still relies on multipurpose batch or semibatch reactors, it is evident that interest is arising toward continuous flow manufacturing of organic molecules, including highly functionalized and chiral compounds. Continuous flow synthetic methodologies can also be easily combined to other enabling technologies, such as microwave irradiation, supported reagents or catalysts, photochemistry, inductive heating, electrochemistry, new solvent systems, 3D printing, or microreactor technology. This combination could allow the development of fully automated process with an increased efficiency and, in many cases, improved sustainability. It has been also demonstrated that a safer manufacturing of organic intermediates and APIs could be obtained under continuous flow conditions, where some synthetic steps that were not permitted for safety reasons can be performed with minimum risk. In this review we focused our attention only on very recent advances in the continuous flow multistep synthesis of organic molecules which found application as APIs, especially highlighting the contributions described in the literature from 2013 to 2015, including very recent examples not reported in any published review. Without claiming to be complete, we will give a general overview of different approaches, technologies, and synthetic strategies used so far, thus hoping to contribute to minimize the gap between academic research and pharmaceutical manufacturing. A general outlook about a quite young and relatively unexplored field of research, like stereoselective organocatalysis under flow conditions, will be also presented, and most significant examples will be described; our purpose is to illustrate all of the potentialities of continuous flow organocatalysis and offer a starting point to develop new methodologies for the synthesis of chiral drugs. Finally, some considerations on the perspectives and the possible, expected developments in the field are briefly discussed.
Two examples out of several in the publication discussed below……………
1 Diphenhydramine Hydrochloride

Scheme 1. Continuous Flow Synthesis of Diphenhydramine Hydrochloride
Diphenhydramine hydrochloride is the active pharmaceutical ingredient in several widely used medications (e.g., Benadryl, Zzzquil, Tylenol PM, Unisom), and its worldwide demand is higher than 100 tons/year.
In 2013, Jamison and co-workers developed a continuous flow process for the synthesis of 3minimizing waste and reducing purification steps and production time with respect to existing batch synthetic routes (Scheme 1). In the optimized process, chlorodiphenylmethane 1 and dimethylethanolamine 2 were mixed neat and pumped into a 720 μL PFA tube reactor (i.d. = 0.5 mm) at 175 °C with a residence time of 16 min. Running the reaction above the boiling point of 2and without any solvent resulted in high reaction rate. Product 3, obtained in the form of molten salt (i.e., above the melting point of the salt), could be easily transported in the flow system, a procedure not feasible on the same scale under batch conditions.
The reactor outcome was then combined with preheated NaOH 3 M to neutralize ammonium salts. After quenching, neutralized tertiary amine was extracted with hexanes into an inline membrane separator. The organic layer was then treated with HCl (5 M solution in iPrOH) in order to precipitate diphenhydramine hydrochloride 3 with an overall yield of 90% and an output of 2.4 g/h.
2 Olanzapine

Scheme 2. Continuous Flow Synthesis of Olanzapine
Atypical antipsychotic drugs differ from classical antipsychotics because of less side effects caused (e.g., involuntary tremors, body rigidity, and extrapyramidal effects). Among atypical ones, olanzapine 10, marketed with the name of Zyprexa, is used for the treatment of schizophrenia and bipolar disorders.
In 2013 Kirschning and co-workers developed the multistep continuous flow synthesis of olanzapine 10 using inductive heating (IH) as enabling technology to dramatically reduce reaction times and to increase process efficiency.(16) Inductive heating is a nonconventional heating technology based on the induction of an electromagnetic field (at medium or high frequency depending on nanoparticle sizes) to magnetic nanoparticles which result in a very rapid increase of temperature.As depicted in Scheme 2 the first synthetic step consisted of coupling aryl iodide 4 and aminothiazole 5 using Pd2dba3 as catalyst and Xantphos as ligand. Buchwald–Hartwig coupling took place inside a PEEK reactor filled with steel beads (0.8 mm) and heated inductively at 50 °C (15 kHz). AcOEt was chosen as solvent since it was compatible with following reaction steps. After quenching with distilled H2O and upon in-line extraction in a glass column, crude mixture was passed through a silica cartridge in order to remove Pd catalyst. Nitroaromatic compound 6 was then subjected to reduction with Et3SiH into a fixed bed reactor containing Pd/C at 40 °C. Aniline 7 was obtained in nearly quantitative yield, and the catalyst could be used for more than 250 h without loss of activity. The reactor outcome was then mixed with HCl (0.6 M methanol solution) and heated under high frequency (800 kHz) at 140 °C. Acid catalyzed cyclization afforded product 8 with an overall yield of 88%. Remarkably, the three step sequence did not require any solvent switch, and the total reactor volume is about 8 mL only.
The final substitution of compound 8 with piperazine 9 was carried out using a 3 mL of PEEK reactor containing MAGSILICA as inductive material and silica-supported Ti(OiPr)4 as Lewis acid. Heating inductively the reactor at 85 °C with a medium frequency (25 kHz) gave Olanzapine 10 in 83% yield.
SEE MORE IN THE PUBLICATION…………..
Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products
Dipartimento di Chimica, Università degli Studi di Milano Via Golgi 19, I-20133 Milano, Italy
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.5b00325
Publication Date (Web): November 26, 2015
Copyright © 2015 American Chemical Society
*E-mail: maurizio.benaglia@unimi.it., *E-mail: alessandra.puglisi@unimi.it.
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes.

Riccardo Porta
PhD Student

Dipartimento di Chimica, Università degli Studi di Milano Via Golgi 19, I-20133 Milano, Italy












![]()


//////////
Fluorofenidone
Fluorofenidone
1- (3-fluorophenyl) -5-methyl – 2 (1H) pyridone
2(1H)-Pyridinone, 1-(3-fluorophenyl)-5-methyl-
1- (3_ fluorophenyl) -5_ methylpyridine _2 (IH) – one
C12 H10 F N O, 203.2123
PRECLINICAL, IND Filing
An anti-inflammatory agent potentially for the treatment of organ fibrosis.
![]()
CAS No. 848353-85-5
Synthesis
PATENT
WO 2006108354
http://www.google.co.in/patents/WO2006108354A1?cl=en
PATENT
http://www.google.com/patents/CN102241625A?cl=zh
(Compound 1)
A. (3_ fluorophenyl) methyl pyridine _2 (IH) 1- -5_ – -one
9. 6gDMF, 45 0g (0 2mol.) Inter-fluoro-iodobenzene, 21 8g (0. 2mol) 5_ methylpyridine _2_ (IH) -.. -one, 28g of anhydrous potassium carbonate and 1. Og copper powder, 160 ° -170 °, the reaction was stirred at reflux for 20 hours, the natural cooling to 110~120 ° C, was slowly added to about 330ml 80~90 ° C hot water, cooled to 20 ° C. Suction filtered, the filter cake was washed with about 20ml of water, remove the cake, with about 300ml of ethyl acetate ultrasound 30min, suction filtered, the filter residue was washed with 20ml of ethyl acetate. The combined ethyl acetate, washed with water three times (50ml * 3), and the filtrate layers were separated and allowed to stand for 15min, ethyl acetate fraction was concentrated to a non-steamed, hot added under stirring for about 85ml of petroleum ether, cooling to 15~20 ° C insulation ~ 1.5 hours. Filtration, the filter cake was washed twice with petroleum ether (about 20ml * 2) used to give 34. 9g crude. Recrystallized from 20% ethanol to give the product 1- (3_ fluorophenyl) -5_ methylpyridine _2 (IH) – one as a white solid # 30. Ig0 Μ P.: 132 · 1 ~133 7 °.. C.
PATENT
http://www.google.co.in/patents/WO2009149188A1?cl=zh-CN
PATENT
CN 102241625
http://www.google.com/patents/CN102241625A?cl=zh
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009111947
PAPER
| CN1386737A * | Jun 11, 2002 | Dec 25, 2002 | 中南大学湘雅医学院 | Antifibrosis pyridinone medicine and its prepaing process |
| CN1846699A | Apr 13, 2005 | Oct 18, 2006 | 中南大学湘雅医院 | Application of 1-(substituted phenyl)-5-methyl-2-(1H)-pyridone compound in preparing medicine for anti-other organifibrosis and tissue fibrosis except renal interstitial fibrosis |
| CN101235013A* | Mar 10, 2008 | Aug 6, 2008 | 广东东阳光药业有限公司;张中能 | Crystallized 1-(3-fluorophenyl)-5-methyl-2-(1H)pyridine and its preparation method composition and application |
| US20070203203 | May 1, 2007 | Aug 30, 2007 | Tao Li J | Composition and Method for Treating Fibrotic Diseases |
/////////
CC1=CN(C(=O)C=C1)C2=CC(=CC=C2)F
PNQ 103 from Advinus for the potential treatment of COPD,; sickle cell disease (SCD)

Formula I and Formula II
OR

PNQ 103
STRUCTURE COMING…………
for the potential treatment of COPD & sickle cell disease (SCD)
Adenosine A2b receptor antagonist
Advinus Therapeutics Ltd
KEEP WATCHING THIS POST……….
PNQ-103 is a proprietary A2B Adenosine receptor (A2BAdoR antagonist), currently in the pre-clinical development stage for the potential treatment of COPD & sickle cell disease (SCD). Advinus is looking for partnering/co-development opportunities.
A2BAdenosine Receptor (A2BAdoR) Antagonist PNQ-103 for COPD and SCD
COPD
Chronic Obstructive Pulmonary Disease (COPD) is a disease that damages lung tissue or restricts airflow through the bronchioles and bronchi, and commonly leads to chronic bronchitis and emphysema. COPD, along with asthma, forms the third leading cause of death in both developed and developing countries and an annual direct and indirect cost of healthcare of more than $50 billion in the US alone. Current therapies suffer from lack of long term efficacy, patient compliance and a narrow therapeutic index.
Adenosine is a powerful bronchoconstrictor and pro-inflammatory agent in COPD and asthma. Adenosine regulates tissue function by activating its receptors: A1AdoR and A2AAdoR are high affinity receptors and A2BAdoR and A3AdoR are low affinity receptors. During pathological conditions in lung, local adenosine concentrations rise to high levels and activate A2BAdoR. A2BAdoR agonized by adenosine induces both bronchoconstriction and pro-inflammatory effects in lung by acting on multiple cell types that lead to airway hyperreactivity and chronic inflammation. Therefore, A2BAdoR antagonists are expected to be beneficial in COPD and asthma.
PNQ-103 is a proprietary A2BAdoR antagonist, currently in the pre-clinical development stage for the potential treatment of COPD. It is a potent, selective, orally bio-available agent with low clearance and small volume of distribution. PNQ-103 is efficacious in standard rodent asthma and lung fibrosis models. PNQ-103 was found to be safe in exploratory safety studies including a Drug Matrix Screen, mini-AMES test, and a test for cardiovascular liability in dog telemetry as well as a 30- day repeat dose study in rats.
SCD
Sickle Cell Disease (SCD) affects millions of people worldwide. It is caused by an autosomal mutation in the hemoglobin gene (substitution of amino-acid valine [Hb A] for glutamic acid [Hb S]. Hb S in low O2 condition polymerizes, leading to distortion of the cell membrane of red blood cells (RBC) into an elongated sickle shape. Sickled RBCs accumulate in capillaries causing occlusions, impair circulation and cause tissue damage and severe disabilities. Unfortunately, there is no targeted therapy for SCD.
Adenosine levels are elevated in SCD patients. Activation of the A2BAdoR by adenosine increases 2,3-DPG levels in RBCs, which reduces Hb S affinity to O2 and promotes its polymerization leading to RBC sickling. A recent study published in Nature Medicine (2011; 17:79-86) demonstrated potential utility of an A2BAdoR antagonist for the treatment of SCD, through selective inhibition of 2,3-DPG production in RBCs. Therefore, PNQ-103, a selective A2BAdoR antagonist, is expected to be useful for the treatment of SCD. In support, ex vivo PoC (selective inhibition of 2,3-DPG production) has been established for PNQ-103 in RBCs from normal and SCD patients.
EXAMPLES………
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012035548
Example 1: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyraz -4-yl]-l,6-dihydr»-purin-7-ylmethyl} ester

Step I: Synthesis of l-(3-Trifiuoroirethyl-ben:ijl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,354-tetrahydro-pyrimidin-5-yl)-amide
A mixture of 5,6-diamino-3-propyl-l H-pyrimidine-2,4-dione (4.25 g, 0.023 mol), l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6.23 g, 0.023 mol), prepared by conventional methods starting from pyrazole-4-carboxylic ester, in methanol (50 ml) were cooled to 0 °C and added EDCI.HC1 (8.82 g, 0.046 mol). The reaction mixture was stirred at 25 °C for 6 h and the organic volatiles were evaporated. To this residue water (50 ml) was added and the precipitate was filtered off, and washed with cold water (50 ml) to obtain l-(3-Trifluoromethyl-benzyl)- 1 H-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (7.2 g, 72 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.82 (t, J=7.6Hz, 3H); 1.46-1.51 (m, 2H); 3.64 (t, J=7.2Hz, 2H); 5.49 (s, 2H); 6.01 (s, 2H); 7.55-7.63 (m, 2H); 7.68-7.72 (m, 2H); 7.99 (s, 1H); 8.37 (s, 1H); 8.55 (s, 1H); 10.42 (s, 1H).
Step II: Preparation of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazoI-4-yl]-3,7-dihydro-purine-2,6-dione
A mixture of l-(3-Trifluoromethyl-benzyl)-lH-pyrazole-4-carboxylic acid (6-amino-2,4-dioxo-3-propyl-l,2,3,4-tetrahydro-pyrimidin-5-yl)-amide (30 g, 0.068 mol), P205(34.0g, 0.240.8 mol) and DMF (300ml) were heated at 100 °C for 30 minutes. The reaction mixture was cooled to 20-25 °C. The reaction mixture was slowly poured into water (1.5 L) with vigorous stirring. Solid material separated was filtered off, and washed with water (200ml) to obtain 1 -Propyl-8-[l -(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (25 g, 88 %) as a pale yellow solid.
‘HNMR(400MHz, DMSO d6): δ 0.87 (t, J=7.2Hz, 3H); 1.53-1.60 (m, 2H); 3.98 (t, J=7.2Hz, 2H); 5.53 (s, 2H); 7.57-7.64 (m, 2H); 7.69-7.71 (m, 2H); 8.08 (s, 1H); 8.47 (s, 1H); 1 1.83 (s, 1H); 13.39 (s, 1H)
Step III: Preparation of 2-ChIoro-l-propyI-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one
A mixture of l-Propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-3,7-dihydro-purine-2,6-dione (7.2 g, 0.017 mol), NH4C1 (4.54 g, 0.085 mol) and POCl3 (220 ml) were heated at 120-125 °C for 72 h. Reaction mixture was cooled to 20-25 °C. It was then concentrated under vacuum and quenched with cold water slowly and solid material was separated. It was filtered off and washed with water. The solid material was dried under vacuum. The crude product was purified by column chromatography using silica gel (230-400 mesh) and 0.5 to 4 % methanol in chloroform as an eluent to obtain 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)- lH-pyrazol-4-yl]-l,7-dihydro-purin-6-one (4.2 g, 58 %) as a pale yellow solid.
‘HNMR(400MHz, CD3OD): 6 1.02 (t, J=7.2Hz, 3H); 1.78-1.84 (m, 2H); 4.29 (t, J=7.6Hz,
2H); 5.52 (s, 2H); 7.56-7.57 (m, 2H); 7.63 (m, 2H); 8.12 (s, 1H); 8.35 (s, 1 H)
Step IV: Preparation of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile
A mixture of 2-Chloro-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-l H-pyrazol-4-yl]-l ,7-dihydro-purin-6-one (O. lg, 0.23 mmol), NaCN (0.016 g, 0.35 mmol), Nal (0.069g, 0.46 mmol) and DMF (2 ml) were stirred for 48 h at 65-70 °C. Reaction mixture was cooled to 20-25 °C and water was added. Solid material was separated. It was filtered off and washed with water. The product was dried under vacuum to obtain 6-Oxo-l-propyl-8-[l-(3-
trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-puriiAe-2-carbonitrile (0.075 g, 77 %) as an off white solid.
‘HNMR(400MHz, DMSO d6): δ 0.97 (t, J=7.6Hz, 3H); 1.71-1.77 (m, 2H); 4.12 (t, J=7.6Hz, 2H); 5.51 (s, 2H); 7.57-7.67 (m, 4H); 8.14 (s, 1H); 8.55 (s, 1H); 14.01 (bs, 1H)
Preparation of hosphoric acid di-tert-butyl ester chloromethyl ester:

Step I: Phosphoric acid di-tert-butyl ester
A mixture of di-tert-butylphosphite (5 g, 0.026 mol), NaHC03 (3.71 g, 0.044 mol) and water (50 ml) were taken and cooled to 0-(-5 , °C. KMn04 (6.18 g, 0.039 mol) was added to the reaction mixture in portion wise over ¾ period of 30 minutes at that temperature. The reaction mixture was allowed to warm to 20-25 °C ana stirred for 1.5 hours at that temperature. To this reaction mixture activated charcoal (25 g) was added and stirred at 55-60 °C for 1 hour. The reaction mixture was cooled to room temperature and filtered off and washed with water (200 ml). The filtrate was concentrated to half of its volume and cooled to 0 °C. It was then acidified with con. HC1 (pH~l-2) to obtain solid. The solid material was filtered off, washed with ice cold water and dried under vacuum to obtain Phosphoric acid di-tert-butyl ester as white solid (3.44 g, 63 %).
Step II. Phosphoric acid di-tert-butyl ester chloromethyl ester
A mixture of Phosphoric acid di-tert-butyl ester (1 g, 0.0048 mol), NaHC03 (0.806 g, 0.0096 mol), tetra butyl ammonium hydrogen sulphate (0.163 g, 0.00048 mol), water (40 ml) and DCM (25 ml) were taken. The mixture was cooled to 0 °C and stirred at that temperature for 20 minutes. Chloromethyl chlorosulphatc (0.943g, 0.0057 mol) in DCM (15 ml) was added to it at 0 °C. The reaction mixture allc ed to warm to room temperature and stirred for 18 hours. The organic layer was separated and aqueous layer was extracted with DCM (30 ml). The organic layer was washed with brine (60 ml) solution and dried over Na2SC>4. The organic layer was evaporated to obtain Phosphoric acid di-tert-butyl ester chloromethyl ester as colorless oil (0.79 g, 64%).

Step I: Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester
A mixture of 6-Oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-6,7-dihydro-lH-purine-2-carbonitrile (0.5 g, 0.0012mol), K2C03 (0.485 g, 0.0036 mol ) and acetone ( 10 ml) were taken and stirred for 20 minutes at room temperature. Nal (0.702 g, 0.0047 mol) was added and then Phosphoric acid di-ten-butyl ester chloromethyl ester (0.619 g, 0.0024 mol in 2 ml acetone) was added to the reaction mixture drop wise. The reaction mixture was heated at 45 °C for 16 h. The reaction mixture was filtered through celite and washed with acetone. The organic layer was concentrated and the residue was taken in ethyl acetate (30 ml) and saturated NaHC03 solution (20 ml). The organic layer was separated and washed with saturated sodium thiosulphate solution (20 ml). The organic layer was washed with 0.5 N HC1 solution (20 ml) and brine solution (20 ml). The organic layer was dried over sodium sulphate and evaporated to obtain brown colored mass. The crude product, which is a mixture of N7 and N9 isomers was purified by column chromatography (230-400 mesh silica gel and it was first treated with 5% triethyl amine in hexane) using 5-20 % acetone in hexane (with 0.5 to 1% triethyl amine) as an eluent to obtain N7 isomer (0.34g, 45 % ) and N9 isomer ( 0.1 lg, 14 % )
Phosphoric acid di-tert-butyl ester 2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl ester (N7-isomer).
Ή NMR (400MHz, DMSO d6):6 0.95 (t J=8Hz, 3H); 125 (s, 18 H); 1.75-1.80 (m, 2H); 4.18 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.34 (d, ![]()
2H); 7.61-7.63 (m, 2H); 7.70-7.73 (m, 2H); 8.19 (s, 1H); 8.75 (s, 1H)
Phosphoric acid di-tert-butyl ester 2-cyano-8-[l-(3-trifluoromethyI-benzyl)-lH-pyrazol-4-yl]-6-oxo-l-propyl-l,6-dihydro-purin-9-ylmethyl ester (N9-isomer)
Ή NMR (400MHz, DMSO d6): δ 0.94 (t, J=8Hz, 3H); 125 (s, 18 H); 1.74-1.78 (m, 2H); 4.21 (t, J=7.2Hz, 2H); 5.59 (s, 2H); 6.05 (d, J=10.8Hz, 2H); 7.62-7.63 (m, 2H); 7.69-7.71 (m, 2H); 8.16 (s, 1H); 8.71 (s, 1H)
Step II: Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-ylmethyl} ester (N7-isomer).
The above product, N7 isomer (0.34 g, 0.52 mmol) was dissolved in DCM (20 ml) and TFA (0.29 ml, 4.2 mmol) was added to it. The reaction mixture was stirred at room temperature for 7 hours. The organic volatiles were evaporated and the residue was stirred with pentane: diethyl ether (3:1, 10 ml) and the solid material obtained was filtered off and washed with 10 % diethyl ether in pentane (10 ml) to obtain Phosphoric acid mono- {2-cyano-6-oxo-l -propyls’ [ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl } ester (0.239g, 85 %) as an off white solid.
(400MHz, DMSO d6): δ 0.96 (t, J=7.6Hz, 3H); 1.75-1.81 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.58 (s, 2H); 6.23 (d, J=6Hz, 2H); 7.61-7.63 (m, 2H); 7.69-7.75 (m, 2H); 8.22 (s, 1 H); 8.80 (s, 1H); (M+1): 538.2
Phosphoric acid mono-{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyl)-lH-pyrazol-4-yl]-l,6-dihydro-purin-9-ylmethyl} ester (N9-isomer, 28%)
(400MHz, DMSO d6): δ 0.93 (t, J=7.6Hz, 3H); 1.72-1.80 (m, 2H); 4.16 (t, J=7.2Hz, 2H); 5.54 (s, 2H); 5.95 (d, J=6Hz, 2H); 7.59-7.60 (m, 2H); 7.67-7.73 (m, 2H); 8.17 (s, 1H); 8.72 (s, 1H).
Step III: Phosphoric acid mon -{2-cyano-6-oxo-l-propyl-8-[l-(3-trifluoromethyl-benzyI)-lH-pyrazol-4-yl]-l,6-dihydro-purin-7-yimethyl} ester di sodium salt
The above product (0.239g, 0.44 mmol) and water (25 ml) were taken. To the suspension formed, NaHC03 solution (0.1 12g, 1.3 mmol in 20 ml water) was added. The reaction mixture was stirred at room temperature for 1.5 h and the solid material obtained was filtered off. The clear solution was passed through reverse phase column chromatography (LCMS). The fraction obtained was evaporated. It was lyophilized to obtain pure Phosphoric acid mono-{2-cyano-6-oxo- 1 -propyl-8-[ 1 -(3 -trifluoromethyl-benzyl)- 1 H-pyrazol-4-yl]- 1 ,6-dihydro-purin-7-ylmethyl} ester di sodium salt (0.208g; 80%) as an off white solid.
Ή NMR: (400MHz, D20): δ 0.97 (t, J=7.6Hz, 3H); 1.80-1.86 (m, 2H); 4.28 (t, J=7.6Hz, 2H); 5.53 (s, 2H); 6.04 (d, J=3.2Hz, 2H); 7.52-7.53 (m, 2H); 7.62-7.64 (m, 2H); 8.22 (s, 1H); 8.74 (s, 1H)
31P NMR: (400MHz, D20): δ 0.447
EXAMPLES…………..
Patent
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009118759
Example Al: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-purine-2, 6-dione

Step I: l-(3-p-ToIyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester
A mixture of l-prop-2-ynyl-lH-pyrazole-4-carboxylic acid ethyl ester obtained as given in example Bl (0.20Og, l.lmmol), 4-iodo toluene (0.254g, 1.1 mol), copper iodide (0.021g, O.l lmmol), dichlorobis (triphenylphosphine)-palladium (II) (39mg, O.Oόmmol), triethylamine (2ml), DMF (2ml) was degassed for lOmin. and stirred for 20hrs at 25-25 0C. Reaction mixture was diluted with water (10ml) and extracted with
• ethyl acetate. Organic layer was washed with brine solution and dried over Na2SO4.
The solvent was evaporated and crude product was purified by column chromatography
(Ethyl acetate: hexane-12:78) to obtain pure l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4- carboxylic acid ethyl ester compound (0.226g, 75%). 1HNMR^OOMHZ, CDCl3): δ 1.35 (t, J=6.8Hz, 3H); 2.37 (s, 3H); 4.31 (q, J=6.8Hz, 2H); 5.18 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 7.95 (s, IH); 8.21 (s, IH)
Step II: l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxy!ic acid l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid ethyl ester (0.226g, 0.84 mmol) was dissolved in a mixture of solvents THF: methanol: water (3:1:1, 10ml) and LiOH (0.07 Ig, 1.7mol) was added to the reaction mixture with stirring. The reaction mixture was then stirred at 20-25 0C for 2 hours. Solvents were evaporated and the residue was diluted with water (0.5 ml) and acidified with dil. HCl, filtered and dried to obtain off white precipitate, l-(3-p-Tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.182g, 90%).
1HNMR^OOMHZ, CDCl3): δ 2.37 (s, 3H); 5.2 (s, 2H); 7.16 (d, J=7.6Hz, 2H); 7.38 (d, J=8Hz, 2H); 8.01 (s, IH); 8.29 (s, IH) Step III: 1, 3-Dipropyl-8-[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yI]-3, 7-dihydro-‘ purine-2, 6-dione
A mixture of 5,6-diamino-l,3-dipropyl-lH-pyrimidine-2,4-dione (0.075g, 0.33 mmol), l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (0.080gm, 0.33mmol), methanol (5ml), EDCI (0.089g, 0.46mmol) were taken and stirred for 12 hours at 20-25 0C. The reaction mixture was concentrated to obtain intermediate l-(3-p-tolyl-prop-2-ynyl)-lH-pyrazole-4-carboxylic acid (6-amino-2, 4-dioxo-l, 3-dipropyl)-l, 2, 3, 4-tetrahydro-pyrimidine-5yl) amide (50mg, 34%) which was dissolved in hexamethyldisilazane (HMDS). To this reaction mixture ammonium sulphate (0.01 Og) was added. The reaction mixture was refluxed at 140 0C for 18hrs. The organic volatiles were evaporated and the residue was treated with crushed ice, the precipitate formed was filtered off. The product was then purified by column chromatography (l%MeOH in CHCl3) to obtain 1, 3-dipropyl-8~[l-(3-p-tolyl-prop-2ynyl)-lH-pyrazol-4-yl]-3, 7-dihydro-purine-2, 6-dione (0.035g, 92%). ‘HNMR(400MHz, DMSO d6): δ 0.76-0.87 (m, 6H); 1.51-1.57 (m, 2H); 1.68-1.74 (m, 2H); 2.29 (s, 3H); 3.82 (t, J=7.2Hz, 2H); 3.95 (t, J=7.2Hz, 2H); 5.36 (s, 2H); 7.18 (d, J=8Hz, 2H); 7.35 (d. J=8Hz, 2H); 8.08 (s, IH); 8.49 (s, IH); 13.9 (bs,lH)
Happy new year wishes 2016


/////////
