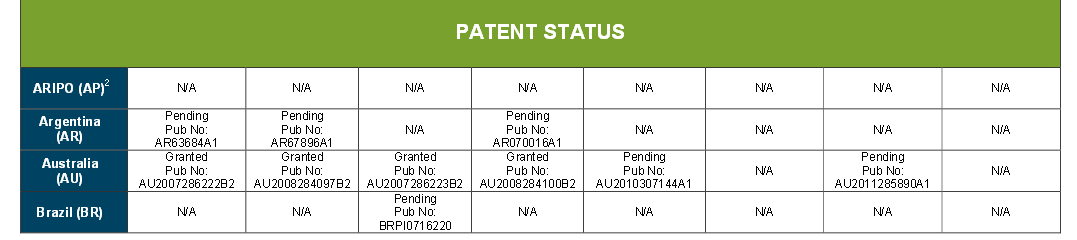
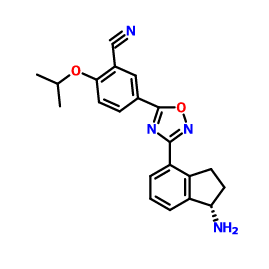
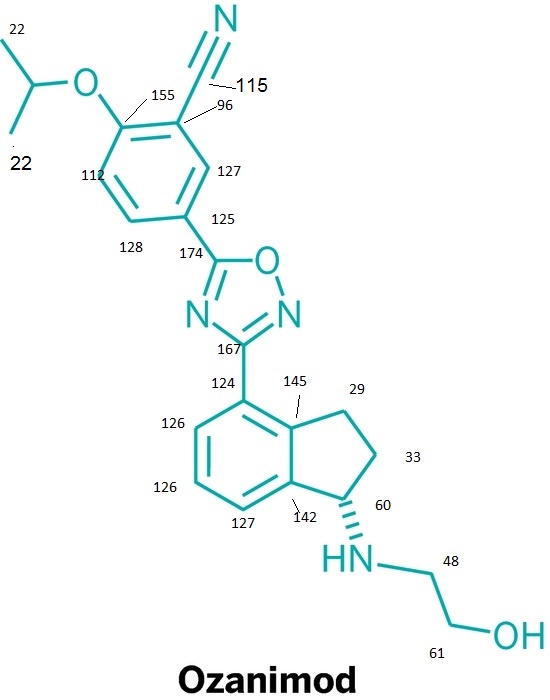
![ChemSpider 2D Image | 5-(3-{(1S)-1-[(2-Hydroxyethyl)amino]-2,3-dihydro-1H-inden-4-yl}-1,2,4-oxadiazol-5-yl)-2-isopropoxybenzonitrile | C23H24N4O3](http://www.chemspider.com/ImagesHandler.ashx?id=34979946&w=250&h=250)
cas 1306760-87-1
Ozanimod, RPC1063
Receptos, Inc. INNOVATOR
IUPAC/Chemical name: (S)-5-(3-(1-((2-hydroxyethyl)amino)-2,3-dihydro-1H-inden-4-yl)-1,2,4-oxadiazol-5-yl)-2-isopropoxybenzonitrile

SMILES: N#CC1=CC(C2=NC(C3=CC=CC4=C3CC[C@@H]4NCCO)=NO2)=CC=C1OC(C)C
C23H24N4O3
Molecular Weight: 404.46
Elemental Analysis: C, 68.30; H, 5.98; N, 13.85; O, 11.87
Ozanimod is a selective sphingosine 1 phosphate receptor modulators and methods which may be useful in the treatment of S1P1-associated diseases. ozanimod, a sphingosine-1-phosphate receptor 1 (S1P1) agonist in Phase III studies as a treatment for ulcerative colitis and multiple sclerosis (MS). Although Novartis’s S1P1 modulator Gilenya has been available to treat MS since 2010,
Relapsing multiple sclerosis (RMS) is a chronic autoimmune disorder of the central nervous system (CNS), characterized by recurrent acute exacerbations (relapses) of neurological dysfunction followed by variable degrees of recovery with clinical stability between relapses (remission). The CNS destruction caused by autoreactive lymphocytes can lead to the clinical symptoms, such as numbness, difficulty walking, visual loss, lack of coordination and muscle weakness, experienced by patients. The disease invariably results in progressive and permanent accumulation of disability and impairment, affecting adults during their most productive years. RMS disproportionately affects women, with its peak onset around age 30. In the past, the treatments for RMS were generally injectable agents with significant side effects. There is a substantial market opportunity for effective oral RMS therapies with improved safety and tolerability profiles.
RPC1063 is a novel, orally administered, once daily, specific and potent modulator of the sphingosine 1-phosphate 1 receptor (S1P1R) pathway. The S1P1R is expressed on white blood cells (lymphocytes), including those responsible for the development of disease. S1P1R modulation causes selective and reversible retention, or sequestration, of circulating lymphocytes in peripheral lymphoid tissue. This sequestration is achieved by modulating cell migration patterns (known as “lymphocyte trafficking”), specifically preventing migration of autoreactive lymphocytes to areas of disease inflammation, which is a major contributor to autoimmune disease. S1P1R modulation may also involve the reduction of lymphocyte migration into the central nervous system (CNS), where certain disease processes take place. This therapeutic approach diminishes the activity of autoreactive lymphocytes that are the underlying cause of many types of autoimmune disease.


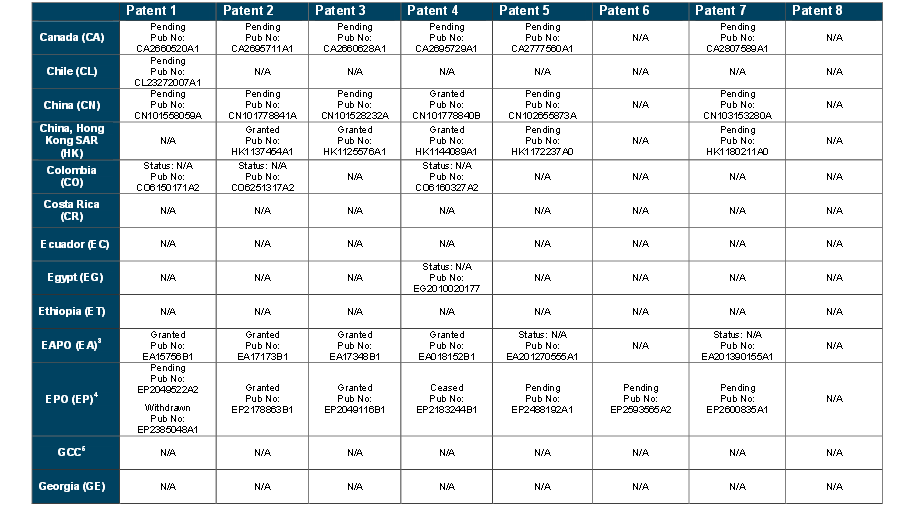
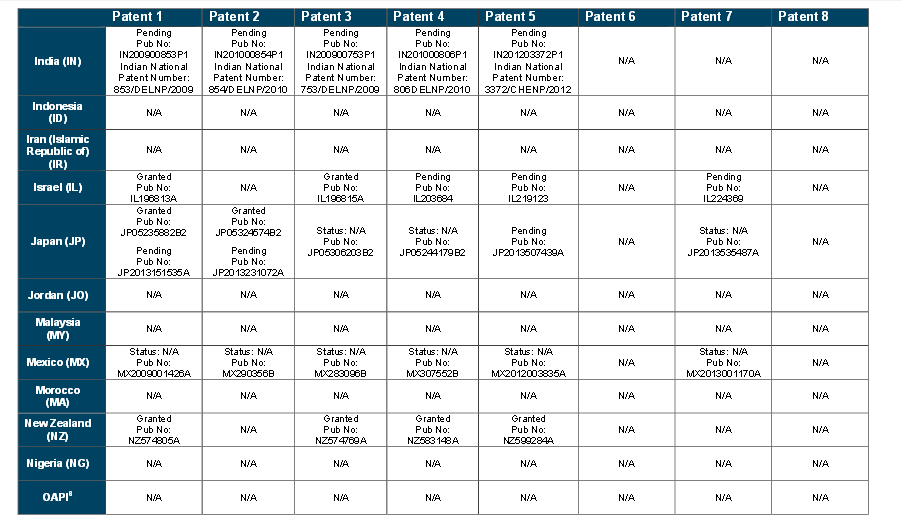
WO 2015066515
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015066515&recNum=1&maxRec=&office=&prevFilter=&sortOption=&queryString=&tab=PCTDescription
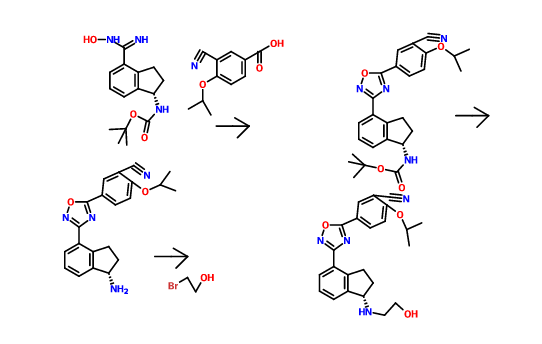
Scheme 3:

Reagents: (i) (a) MsCl, pyridine; (b) TsCl, pyridine; (c) NsCl, pyridine; (d) SOCl2, DCM; (e) SOCl2, pyridine, DCM; (f) NaN3, PPh3, CBr4; (ii) (a) DIEA, DMA, HNR’R”; (b) DIEA, NaBr or Nal, DMA, HNR’R”.
Enantiomerically enriched material can be prepared in the same manner outlined in Scheme 3 using the (R)- or (5)-indanols.
Scheme 4:

Reagents: (i) Zn(CN)2, Pd(PPh3)4, NMP; (ii) (i?)-2-methylpropane-2-sulfmamide, Ti(OEt)4, toluene; (iii) NaBH4, THF; (iv) 4M HCl in dioxane, MeOH; (v) Boc20, TEA, DCM; (vi) NH2OH HCl, TEA, EtOH; (vii) HOBt, EDC, substituted benzoic acid, DMF (viii) 4M HCl in dioxane; (ix) (a) R’-LG or R”-LG, where LG represents a leaving group, K2C03, CH3CN; (b) R -C02H or R2-C02H, HOBt, EDC, DMF or R -COCl or R2-COCl, TEA, DCM; (c) R -S02C1 or R3-S02C1, TEA, DCM (d) R2-CHO, HO Ac, NaBH4 or NaCNBH3 or Na(OAc)3BH, MeOH; (e) R -OCOCl or R2-OCOCl, DIEA, DMF; (f) HN(R5R5), CDI, TEA, DCM; (g) H2NS02NH2, Δ, dioxane; (h)
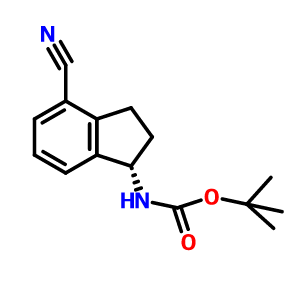
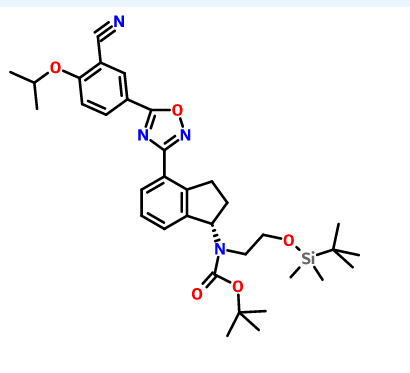
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2 ,3-dihydro- lH-inden- 1-yl)carbamate INT-16)

Prepared using General Procedure 9. To a flame-dried flask under N2 was added {R)-tert- vXy\ 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 (8.3 g, 32.1 mmol) in anhydrous DMF (240 mL). The reaction mixture was cooled to 0°C and sodium hydride (3.8 g, 60% in oil, 160.6 mmol) was added portionwise. After stirring at 0°C for 2.75 h, (2-bromoethoxy)(tert-butyl)dimethylsilane (16.9 mL, 70.7 mmol) was added. The ice bath was removed after 5 mins and the reaction mixture was allowed to warm to room temperature. After 1.5 h, the reaction mixture was quenched by the slow addition of sat. NaHC03 at 0°C. Once gas evolution was complete the reaction was extracted with EA. The organic layers were washed with water and brine, dried over MgS04 and concentrated. The product was purified by chromatography (EA / hexanes) to provide 10.76 g (80%) of {R)-tert-bvXy\ 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 as a colorless oil. LCMS-ESI (m/z) calculated for C23H36N203Si: 416.6; found 317.2 [M-Boc]+ and 439.0 [M+Na]+, tR = 4.04 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.46 (d, J = 7.6, 1H), 7.38- 7.32 (m, 1H), 7.33 – 7.18 (m, 1H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 3.70 (ddd, J = 48.8, 26.6, 22.9, 1.5 H), 3.50 – 3.37 (m, 1H), 3.17 (ddd, J = 16.7, 9.4, 2.2, 2H), 2.93 (m, 1.5 H), 2.45 (s, 1H), 2.21 (dd, J = 24.5, 14.5, 1H), 1.56 – 1.37 (bs, 4.5H), 1.22 (bs, 4.5H), 0.87 – 0.74 (m, 9H), -0.04 (dd, J = 26.6, 8.2, 6H). 13C NMR (101 MHz, CDC13) δ 155.03, 146.55, 145.54, 131.16, 130.76, [128.11, 127.03], 117.58, 109.20, 79.88, [63.93, 61.88], [61.44, 60.34], [49.73, 46.76], 30.30, 29.70, 28.44, 28.12, [25.87, 25.62], -5.43. (5)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro- 1 H-inden- 1 -yl)carbamate INT- 17 is prepared in an analogous fashion using INT-9.
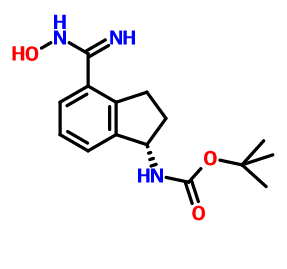
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate (INT-18)

Prepared using General Procedure 3. To a solution of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 (12.0 g, 28.9 mmol) in EtOH (120 mL), under an atmosphere of N2 was added hydroxylamine-HCl (6.0 g, 86.5 mmol) and triethylamine (13.4 mL, 9.7 g, 86.5 mmol). The reaction mixture was refluxed at 80°C for 4 h. The reaction mixture was cooled to room temperature and concentrated to dryness and then diluted with DCM (500 mL). The organic layer was washed with NaHC03, water, and brine. The combined organic layers were dried over MgSC^ and concentrated to produce 11.8 g of {R)-tert- vXy\ 2-(tert-butyldimethylsilyloxy) ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 as a white foamy solid, which was used without purification in the next experiment. LCMS-ESI (m/z) calculated for C23H39N304Si: 449.7; found 350.2 [M-Boc]+ and 472.2 [M+Na]+, tR = 1.79 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.32 (t, J= 7.3 Hz, 1H), 7.21 – 7.07 (m, 2H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 4.89 (s, 2H), 3.85 – 3.50 (m, 2H), 3.31 (ddd, J = 12.2, 9.2, 2.5 Hz, 2H), 3.28 – 3.03 (m, 2H), 3.03 – 2.70 (m, 1H), 2.29 (t, J= 23.6 Hz, 1H), 1.43 (bs, 4.5H), 1.28 (bs, 4.5H), 1.16 – 1.04 (m, 1H), 0.90 – 0.71 (m, 9H), 0.08 – -0.14 (m, 6H). 13C NMR (101 MHz, CDC13) δ 170.99, [156.20, 155.62], 152.38, [144.53, 143.57], [141.82, 141.21], 129.61, 126.78, [126.59, 126.25], [125.02, 124.77], [79.91, 79.68], 64.04, 61.88, [61.57, 61.23], [46.03, 45.76], 30.76, 30.21, [28.53, 28.28], 25.95, [25.66, 25.29], 25.13, [18.28, 17.94], 3.72, -5.34. (S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate INT-19 is prepared in an analogous fashion using INT- 17.
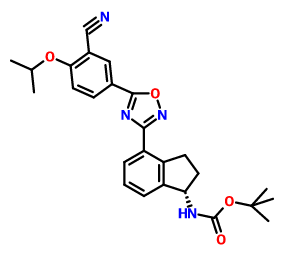
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl)carbamate and (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl) (2-hydroxethyl) carbamate

Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (4.5 g, 21.9 mmol) in anhydrous DMF (100 mL) was added HOBt (5.4 g, 40.0 mmol) and EDC (5.6 g, 29.6 mmol). After 1 h, (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT- 18 (11.8 g, 26.3 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. LCMS analysis showed complete conversion to the intermediate, (R)-tert-butyl 2-(tert-butyldimethylsilyloxy) ethyl (4-(N-(3-cyano-4-isopropoxybenzoyloxy) carbamimidoyl)-2,3-dihydro-7H-inden-l-yl)carbamate INT-20. The reaction mixture was then heated to 80°C for 12 h. The reaction mixture was cooled to room temperature and diluted with EA (250 mL). NaHC03 (250 mL) and water (350 mL) were added until all the solids dissolved. The mixture was extracted with EA and the organic layers washed successively with water and brine. The organic layers were dried over MgS04 and concentrated to produce 15.3 g of a mixture of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5 -(3 -cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)- 2,3-dihydro-iH-inden-l-yl) carbamate INT-21, and the corresponding material without the TBS protecting group, {R)-tert-bvXy\ 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxy ethyl) carbamate INT-22. The mixture was a brown oil, which could used directly without further purification or purified by chromatography (EA/hexane). INT-21: LCMS-ESI (m/z) calculated for C34H46N405Si: 618.8; found 519.2 [M-Boc]+ and 641.3 [M+Na]+, tR = 7.30 min (Method 1). 1H NMR (400 MHz, CDC13) δ 8.43 (d, J =
2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.07 (d, J= 8.1, 1H), 7.46 – 7.26 (m, 2H), 7.12 (d, J = 9.0, 1H), 5.85 (s, 0.5H), 5.37 (s, 0.5H), 4.80 (dt, J = 12.2, 6.1, 1H), 3.92 – 3.32 (m, 3.5 H), 3.17 (s, 2H), 2.95 (s, 0.5 H), 2.62 – 2.39 (m, 1H), 2.38 – 2.05 (m, 1H), 1.53 (s, 4.5H), 1.48 (d, J = 6.1, 6H), 1.33 – 1.27 (m, 4.5H), 0.94 – 0.77 (m, 9H), 0.01 (d, J = 20.9, 6H). 13C NMR (101 MHz, DMSO) δ 173.02, 169.00, 162.75, [156.22, 155.52], [145.18, 144.12], [143.39, 142.76], 134.16, 133.89, 128.20, [128.01, 127.85], [127.04, 126.90], 126.43, 123.31, 116.93, 115.30, 113.55, 103.96, [79.95, 79.68], 72.73, 67.61, 63.42, [61.91, 61.77], 60.99, 46.11, 31.78, [30.47, 29.87], [28.55, 28.26], 25.93, 21.75, 18.30, 0.00, -5.37. INT-22: LCMS-ESI calculated for C28H32N405: 504.6; found 527.2 [M+Na]+, tR = 2.65 min (Method 1). 1H NMR (400 MHz, CDC13) δ 8.36 (d, J = 2.1, 1H), 8.27 (dd, J = 8.9, 2.2, 1H), 8.03 (d, J = 7.2, 1H), 7.35 – 7.26 (m, 2H), 7.06 (d, J = 9.0, 1H), 5.44 (s, 1H), 4.73 (dt, J= 12.2, 6.1, 1H), 3.64 (s, 2H), 3.44 (ddd, J= 17.5, 9.5,
3.2, 2H), 3.11 (dt, J = 17.4, 8.6, 3H), 2.54 – 2.38 (m, 1H), 2.04 (td, J = 17.6, 8.8, 1H), 1.50 – 1.24 (m, 15H).
(S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-23 and {S)-tert- vXy\ 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT-24 were made in an analogous fashion.
(S) IS DESIRED CONFIGURATION
……………………………………
(S)-tert-Butanesulfinamide
 CAS 343338-28-3
CAS 343338-28-3
 3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile
3-CYANO-4-ISOPROPOXYBENZOIC ACID;3-cyano-4-(propan-2-yloxy)benzoic acid;5-(1-hydroxyvinyl)-2-isopropoxybenzonitrile
cas 258273-31-3
 (S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride
(S)-1-Amino-2,3-dihydro-1H-indene-4-carbonitrile hydrochloride
cas 1306763-57-4 HCl, 1213099-69-4 FREE BASE
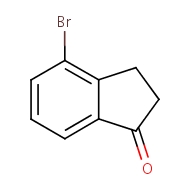
4-bromo-2,3-dihydro-1H-inden-1-one
cas 15115-60-3
 S CONFIGURATION
S CONFIGURATION
Carbamic acid, N-[(1S)-4-cyano-2,3-dihydro-1H-inden-1-yl]-, 1,1-dimethylethyl ester, cas 1306763-31-4
(S) IS DESIRED CONFIGURATION
……………….

CAS 1306763-70-1, Carbamic acid, N-[(1S)-2,3-dihydro-4-[(hydroxyamino)iminomethyl]-1H-inden-1-yl]-, 1,1-dimethylethyl ester
…………………

CAS 1306763-71-2, Carbamic acid, N-[(1S)-4-[5-[3-cyano-4-(1-methylethoxy)phenyl]-1,2,4-oxadiazol-3-yl]-2,3-dihydro-1H-inden-1-yl]-, 1,1-dimethylethyl ester

1306760-73-5, Benzonitrile, 5-[3-[(1S)-1-amino-2,3-dihydro-1H-inden-4-yl]-1,2,4-oxadiazol-5-yl]-2-(1-methylethoxy)-
………………………..

1306763-63-2,
………………….

86864-60-0, (2-Bromoethoxy)dimethyl-tert-butylsilane
Synthesis
……………………………………
WO 2011060392
http://www.google.com/patents/WO2011060392A1?cl=en
(R)-N-(4-cyano-2,3-dihydro-lH-indene-l-ylidene)-2-methylpropane-^
(INT-4
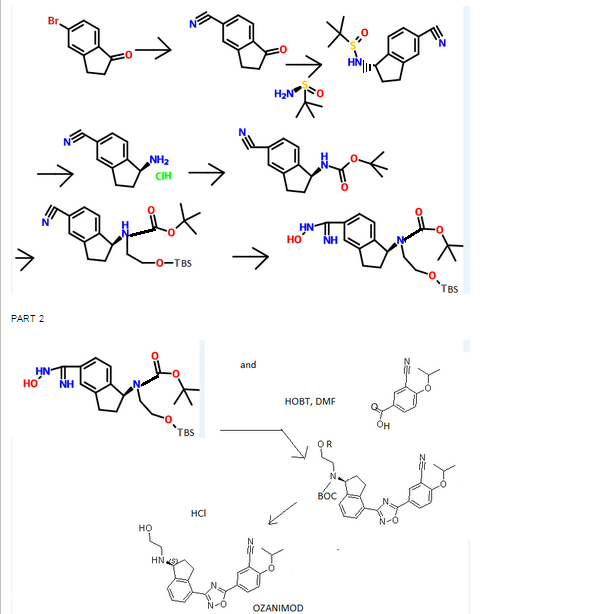
[0304] To l-oxo-2,3-dihydro-/H-indene-4-carbonitrile INT-1 (42.5 g, 0.27 mol) and (R)-2- methylpropane-2-sulfmamide (36.0 g, 0.30 mol) in toluene (530 mL) was added titanium tetraethoxide (84.1 mL, 92.5 g, 0.40 mol) and the reaction mixture was heated at 60°C for 12 h under N2. The crude (R)-N-(4-cyano-2,3-dihydro-lH-indene-l-ylidene)-2-methylpropane- 2-sulfinamide INT-4 was used directly in the next experiment. LCMS-ESI (m/z) calculated for C14Hi6N2OS: 260.3; found 261.1 [M+H]+, tR= 3.19 min.
[0305] (R)-N'((R)-4-cyano-2,3-dihydro-lH nden-l-yl)-2-n thylprop ne-2-sulfirmmide
(INT-5)

[0306] To a flask containing the crude suspension of (R)-N-(4-cyano-2,3-dihydro-iH-indene- l-ylidene)-2-methylpropane-2-sulfrnaniide INT -4 under N2 was added THF (1.0 L) and the reaction mixture cooled to -78°C. Sodium borohydride (40.9 g, 1.08 mol) was added portion- wise over 30 mins. (The internal temperature did not rise during the addition). The reaction mixture was stirred at -78°C for 30 mins, half out of the bath for 30 mins, then warmed to 0°C over 1 h. The 0°C reaction mixture was placed in an ice bath and quenched with brine (100 mL) followed by saturated sodium potassium tartrate (420 mL) and the Ti salts precipitated. The reaction mixture was diluted with EA (1.5 L) and stirred at room temperature overnight. The organic layers were decanted and washed successively with saturated NH4CI, water, and brine. The organic layers were dried over MgS04 and filtered through a pad of MgS04. The filtrate was concentrated to produce 52.9 g of crude (R)-N-((/?)-4-cyano-2,3-dihydro-lH- inden-l-yl)-2-methylpropane-2-sulfmamide INT-5 as a brown oil, which was used directly in the next step. LCMS-ESI (m/z) calculated for C14H18 2OS: 262.3; found 263.1 [M+H]+, tR = 2.99 min. 1H NMR (400 MHz, CDC13) δ 7.89 (d, J = 7.7, 1H), 7.56 (t, J = 6.8, 1H), 7.36 (t, J = 7.7, 1H), 4.97 (q, J = 7.5, 1H), 3.50 (d, J = 7.6, 1H), 3.22 (ddd, J = 16.9, 8.8, 3.9, 1H), 3.01 (dt, J = 22.4, 6.9, 1H), 2.70 – 2.53 (m, 1H), 2.15 – 1.95 (m, 1H), 1.33 – 1.20 (m, 9H).
[0307] (R)-l-amino-2,3-dihydro-lH-indene-l-yl)-4-carbonitrile (T^T-6)

[0308] To crude (R)-N-((R)-4-cyano-2,3-dihydro-iH-inden-l-yl)-2-methylpropane-2- sulfinamide INT-5 (52.9 g, 0.20 mol) in MeOH (200 mL) was added 4N HC1 in dioxane (152.0 mL, 0.60 mol) and the resulting yellow suspension was stirred at room temperature for 1.5 h. The crude reaction mixture was diluted with MeOH (500 mL) and filtered to remove some Ti by-products. The filtrate was concentrated and the resulting solid refluxed in acetonitrile (500 mL). The resulting white solid was collected to produce 13.0 g (31% over 3 steps) of the HC1 salt of (R)-l-amino-2,3-dihydro-7H-indene-l-yl)-4-carbonitrile INT-6. LCMS-ESI (m/z) calculated for Ci0H10N2: 158.2; found 142.0 [M-NH2]+, fR = 0.84 min. Ή NMR (400 MHz, DMSO) δ 8.61 (s, 3H), 7.96 (d, J = 7.7, 1H), 7.83 (d, J = 7.5, 1H), 7.52 (t, J = 7.7, 1H), 4.80 (s, 1H), 3.23 (ddd, J = 16.6, 8.7, 5.2, 1H), 3.05 (ddd, J = 16.6, 8.6, 6.3, 1H), 2.62 – 2.51 (m, 1H), 2.15 – 2.01 (m, 1H). 13C NMR (101 MHz, DMSO) δ 148.09, 141.15, 132.48, 130.32, 127.89, 117.27, 108.05, 54.36, 39.08, 29.64. The free base can be prepared by extraction with IN NaHC03and DCM. LCMS-ESI (m/z) calculated for Ci0H10N2: 158.2; found 142.0 [M-NH2]+, tR = 0.83 min. 1H NMR (400 MHz, CDC13) δ 7.52 – 7.38 (m, 2H), 7.23 (dd, 7 = 17.4, 9.8, 1H), 4.35 (t, J = 7.6, 1H), 3.11 (ddd, 7 = 16.8, 8.7, 3.2, 1H), 2.89 (dt, J = 16.9, 8.5, 1H), 2.53 (dddd, J = 12.8, 8.1, 7.3, 3.2, 1H), 1.70 (dtd, J = 12.8, 8.8, 8.0, 1H). 13C NMR (101 MHz, DMSO) δ 150.16, 146.67, 130.19, 128.74, 127.38, 117.77, 107.42, 56.86, 38.86, 29.14. Chiral HPLC: (R)-l-amino-2,3-dihydro-7H-indene-l-yl)-4-carbonitrile was eluted using 5% EtOH in hexanes, plus 0.05% TEA: 95% ee, ¾ = 23.02 min. The (S)- enantiomer INT-7 was prepared in an analogous fashion using (5)-2-methylpropane-2- sulfinamide. tR for (S)-enantiomer = 20.17 min.
[0309] (R)-tert-butyl 4-cyano-2,3-dihydro-lH-inden-l-ylcarbamate (INT-8)

[0310] To ( ?)-l-amino-2,3-dihydro-/H-indene-l-yl)-4-carbonitrile HC1 INT-6 (11.6 g, 59.6 mmol) in DCM (100 mL) at 0°C was added TEA (12.0 mL, 131.0 mmol). To the resulting solution was added a solution of Boc anhydride (14.3 g, 65.6 mmol) in DCM (30 mL) and the reaction mixture stirred at room temperature for 1.5 h. The reaction mixture was washed with brine, and the organic layers were dried over MgS04 and filtered. Additional DCM was added to a total volume of 250 mL and Norit (4.5 g) was added. The product was refluxed for 15 mins and the hot mixture filtered through a pad of celite / silica. The filtrate was concentrated and recrystallized from EA (50 mL) and hexane (150 mL) to produce 12.93 g (84%) of (/?)-tert-butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 as an off-white solid. LCMS-ESI (m/z) calculated for C15H18N202: 258.3; found 281.1 [M+Na]+, tR = 3.45 min. Elemental Analysis determined for C^H^^O^ C calculated = 69.74%; found = 69.98%. H calculated = 7.02%; found = 7.14%. N calculated = 10.84%; found = 10.89%. 1H NMR (400 MHz, CDC13) δ 7.64 – 7.49 (m, 2H), 7.34 (dt, / = 7.7, 3.8, 1H), 5.36 – 5.20 (m, 1H), 4.78 (d, J = 6.8, 1H), 3.20 (ddd, J = 16.9, 8.9, 3.3, 1H), 3.02 (dt, J = 25.4, 8.4, 1H), 2.82 – 2.53 (m, 1H), 1.88 (dq, J = 13.2, 8.6, 1H), 1.55 – 1.44 (m, 9H). 13C NMR (101 MHz, DMSO) δ 155.52, 146.68, 146.32, 130.89, 128.70, 127.63, 117.51, 107.76, 77.98, 55.09, 31.88, 29.11, 28.19. Chiral HPLC: (R)-tert-butyl 4-cyano-2,3-dihydro-lH-inden-l- ylcarbamate was eluted using 2.5% EtOH in hexanes: >99.9% ee, tR = 19.36 min. The (5)- enantiomer INT-9 was prepared in an analogous fashion using (S)-l-amino-2,3-dihydro-7H- indene-l-yl)-4-carbonitrile HC1. tR for (5)-enantiomer = 28.98 min.
General Procedure 3. Preparation oflndane Amide Oximes
[0311] To (R)- or (5)-tert-butyl 4-cyano-2,3-dihydro-7H-inden-l-ylcarbamate (1 eq) in EtOH
(0.56 M) was added hydroxylamine hydrochloride (3 eq) and TEA (3 eq) and the reaction mixture heated at 85°C for 1-2 h. The organic soluble amide oximes were isolated by removal of the solvent and partitioning between water and DCM. The water soluble amide oximes were chromatographed or used directly in the cyclization. Pure amide oximes can be obtained by recrystallization from alcoholic solvents.
[0312] (R)-tert-butyl 4-(N -hydroxy carbamimidoyl )-2, 3-dihydro-lH-inden-l -ylcarbamate
(INT-10)

[0313] Prepared using General Procedure 3. To (R)-tert-butyl 4-cyano-2,3-dihydro-iH- inden-1 -ylcarbamate INT-8 (15.0 g, 58.2 mmol) in EtOH (100 niL) was added hydroxylamine hydrochloride (12.1 g, 174.2 mmol) and TEA (17.6 mL, 174.2 mmol) and the reaction mixture heated at 85°C for 2 h. The solvents were removed and the resulting white solid was partitioned between water and DCM. The organic layers were dried over Na2S04, concentrated, and recrystallized from isopropanol (50 mL) to afford 14.4 g (85%) of (R)-tert- butyl 4-(N-hydroxycarbaniimidoyl)-2,3-dihydro-iH-inden-l-ylcarbamate INT-10 as white crystalline solid. LCMS-ESI (m/z) calculated for C15H21N303: 291.4; found 292.1 [M+H]+, ¾ = 2.04 min. 1H NMR (400 MHz, DMSO) δ 9.53 (s, 1H), 7.38 – 7.32 (m, 1H), 7.32 – 7.12 (m, 3H), 5.68 (s, 2H), 4.97 (q, J = 8.5, 1H), 3.07 (ddd, J = 16.6, 8.7, 2.6, 1H), 2.86 (dt, J = 16.8, 8.4, 1H), 2.30 (ddd, J = 12.6, 7.6, 3.6, 1H), 1.75 (dq, J = 12.3, 9.0, 1H), 1.44 (s, 9H). General Procedure 4. Cyclization to Indane Oxadiazole Amines
[0314] A solution of the appropriate acid (1 eq), HOBt (1.3 eq), and EDC (1.3 eq) in DMF
(0.08 M in acid) was stirred at room temperature under an atmosphere of N2. After the complete formation of the HOBt- acid complex (1-3 h), the (R)- or (5)-amide oxime (1.1 eq) was added to the mixture. After complete formation of the coupled intermediate (ca. 0.5- 2 h), the mixture was heated to 75-95°C until the cyclization was complete (8-12 h). The reaction mixture was diluted with saturated NaHC03 and extracted with EA. The combined organic extracts were dried, concentrated, and either purified by chromatography (EA/hexanes) or taken on directly. The oxadiazole was treated with HC1 (5N in dioxane, 5 eq) at 50-60°C for 0.5-6 h. The reaction mixture could be extracted (DCM /NaHC03), or the resulting HC1 salt concentrated, suspended in Et20, and collected. Pure indane amines can be obtained by recrystallization from alcoholic solvents or by chromatography.
( R)-tert-butyl 4-(5-( 3-cyano-4-isopropoxyphenyl)-l,2, 4-oxadiazol-3-yl )-2,3-dihydro-lH- inden-l-ylcarbamate (INT- 12)

[0315] Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (7.74 g, 37.7 mmol) in DMF (50 mL) was added HOBt (6.02 g, 44.6 mmol) and EDC (8.53 g, 44.6 mmol) at room temperature. The reaction was stirred for 2 h until complete formation of the HOBt-acid complex. (R)-tert-butyl 4-(N-hydroxycarbamimidoyl)-2,3- dihydro-iH-inden-l-ylcarbamate INT-10 (10.0 g, 34.3 mmol) was added and the reaction mixture stirred at room temperature for 2 h until the formation of INT-11, (R)-tert-butyl 4- (N-(3-cyano-4-isopropoxybenzolyloxy) carbamimidoyl)-2,3-dihydro-iH-inden-l- ylcarbamate. The mixture was partitioned between EA and NaHC03 and the organic layer was collected and dried over MgS04. INT-11 (16.3 g, 34.0 mmol) was re-dissolved in DMF (50 mL) and the mixture was heated to 95°C for 12 hrs. The reaction was diluted with NaHC03 (200 mL) and extracted with EA (3 X 50 mL). The organic layer was dried over Na2S04and concentrated under reduced pressure to produce 12.8 g (81%) of (R)-tert-butyl 4- (5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden- 1-ylcarbamate INT-12 as a light brown solid and used without further purification in the next step. LCMS- ESI (m/z) calculated for C26H28N404: 460.5; found 483.2 [M+Na]+, tR = 4.25 min. Ή NMR (400 MHz, CDCI3) δ 8.43 (d, J = 2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.09 (d, J = 7.6, 1H), 7.51 (d, / = 7.5, 1H), 7.39 (t, J = 7.6, 1H), 7.12 (d, J = 9.0, 1H), 5.28 (d, J = 8.2, 1H), 4.80 (hept, J = 6.0, 1H), 3.47 (ddd, J = 17.4, 8.9, 3.5, 1H), 3.27 – 3.03 (m, 1H), 2.68 (d, J = 8.7, 1H), 1.87 (td, J = 16.7, 8.5, 1H), 1.53 – 1.43 (m, 15H). 13C NMR (101 MHz, CDC13) δ 173.00, 168.82, 162.70, 155.68, 145.31, 142.96, 134.05, 133.83, 128.25, 127.21, 126.79, 123.09, 116.78, 115.24, 113.52, 103.87, 79.52, 72.70, 55.72, 33.86, 31.47, 28.39, 21.70. Chiral HPLC: (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-lH-inden-l-ylcarbamate was eluted using 20% /-PrOH in hexanes: >99.9% ee, ?R = 13.33 min. The (5)-enantiomer INT-13 was prepared in an analogous fashion using (S)-tert- butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate using General Procedures 3 and 4 (tR for (Syenantiomer = 16.31 min).
( R )-5-( 3-(l -amino-2,3-dihydro-lH-inden-4-yl)-l,2, 4-oxadiazol-5-yl)-2-isopropoxy- benzonitrile h drochloride (Compound 49)

[0317] To (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-iH-inden-l-ylcarbamate(12.8 g, 27.8 mmol) in dioxane (200 mL) was added 4N HCl in dioxane (69 mL). The solution was heated to 55°C for 1 h, and product precipitated. Dioxane was removed and the resulting solid suspended in ether and collected. The material was recrystallized from MeOH (200 mL) to produce 8.11 g (81%) of (R)-5-(3-(l-amino-2,3- dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxybenzonitrile 49 as the HCl salt. LCMS-ESI (m/z): calcd for: C21H20N4O2: 360.4; found 383.2 [M+Na]+, tR = 2.49 min. Elemental Analysis and NMR spectra determined for C21H21N402C1 * 0.5 H20; C calculated = 62.14%; found = 62.25%. H calculated = 5.46%; found = 5.30%. N calculated = 13.80%; found = 13.84%. CI calculated = 8.73%; found = 8.34%. 1H NMR (400 MHz, DMSO) δ 8.71 (s, 3H), 8.49 (d, J = 2.3, 1H), 8.39 (dd, J = 9.0, 2.3, 1H), 8.11 (d, J = 7.6, 1H), 7.91 (d, J = 7.6, 1H), 7.55 (t, J = 8.5, 2H), 4.97 (hept, J = 6.1, 1H), 4.80 (s, 1H), 3.47 (ddd, J = 17.4, 8.7, 5.3, 1H), 3.23 (ddd, 7 = 17.4, 8.6, 6.4, 1H), 2.55 (ddd, 7 = 13.7, 8.3, 3.2, 1H), 2.22 – 1.97 (m, 1H), 1.38 (d, J = 6.0, 6H). 13C NMR (101 MHz, CDC13) δ 173.28, 167.98, 162.53, 143.69, 141.29, 134.59, 133.80, 128.93, 128.11, 127.55, 122.72, 115.87, 115.24, 114.91, 102.46, 72.54, 54.38, 31.51, 29.91, 21.47. Chiral HPLC of the free base: (R)-5-(3-(l-amino-2,3- dihydro-lH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy benzonitrile was eluted using 15% i‘-PrOH in hexanes plus 0.3% DEA: > 99.9% ee, tR = 30.80 min.
(S)- 5-(3-(l-amino-2,3- dihydro-lH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy-benzonitrile 50 was prepared in an analogous fashion from (S)-tert-b tyl 4-cyano-2,3-dihydro-lH-inden-l-ylcarbamate: >99.9% ee, tR for (5)-enantiomer = 28.58 min.
(R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-lH-inden-l- yl)carbamate ( -16)

[0366] Prepared using General Procedure 9. To a flame-dried flask under N2 was added (R)- tert-butyl 4-cyano-2,3-dihydro-iH-inden-l-ylcarbamate INT-8 (8.3 g, 32.1 mmol) in anhydrous DMF (240 mL). The reaction mixture was cooled to 0°C and sodium hydride (3.8 g, 60% in oil, 160.6 mmol) was added portionwise. After stirring at 0°C for 2.75 h, (2- bromoethoxy)(½rt-butyl)dimethylsilane (16.9 mL, 70.7 mmol) was added. The ice bath was removed after 5 mins and the reaction mixture was allowed to warm to room temperature. After 1.5 h, the reaction mixture was quenched by the slow addition of sat. NaHC03at 0°C. Once gas evolution was complete the reaction was extracted with EA. The organic layers were washed with water and brine, dried over MgS04 and concentrated. The product was purified by chromatography (EA / hexanes) to provide 10.76 g (80%) of (R)-teri-butyl 2-(tert- butyldimemylsilyloxy)emyl(4-cyano-2,3-dihydro-iH-inden-l-yl)carbamate INT-16 as a colorless oil. LCMS-ESI (m/z) calculated for C23H36N203Si: 416.6; found 317.2 [M-Boc]+ and 439.0 [M+Na]+, tR = 4.04 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.46 (d, J = 7.6, 1H), 7.38- 7.32 (m, 1H), 7.33 – 7.18 (m, 1H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 3.70 (ddd, J = 48.8, 26.6, 22.9, 1.5 H), 3.50 – 3.37 (m, 1H), 3.17 (ddd, J = 16.7, 9.4, 2.2, 2H), 2.93 (m, 1.5 H), 2.45 (s, 1H), 2.21 (dd, J = 24.5, 14.5, 1H), 1.56 – 1.37 (bs, 4.5H), 1.22 (bs, 4.5H), 0.87 – 0.74 (m, 9H), -0.04 (dd, J = 26.6, 8.2, 6H).13C NMR (101 MHz, CDC13) δ 155.03, 146.55, 145.54, 131.16, 130.76, [128.11, 127.03], 117.58, 109.20, 79.88, [63.93, 61.88], [61.44, 60.34], [49.73, 46.76], 30.30, 29.70, 28.44, 28.12, [25.87, 25.62], -5.43. (5)-tert-butyl 2-(tert- butyldimemylsilyloxy)emyl(4-cyano-2,3-dihydro-lH-inden-l-yl)carbamate INT-17 is prepared in an analogous fashion using INT -9. [0367] (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N-hydroxycarbamimidoyl)-2,3- dihydro-1 H-inden-1 -yl)carbamate (INT-18)

[0368] Prepared using General Procedure 3. To a solution of (R)-iert-butyl 2-(tert- butyldimemylsilyloxy)ethyl(4-cyano-2,3-dmydro-/H-inden-l-yl)carbamate INT-16 (12.0 g, 28.9 mmol) in EtOH (120 mL), under an atmosphere of N2 was added hydroxylamine-HCl (6.0 g, 86.5 mmol) and triemylamine (13.4 mL, 9.7 g, 86.5 mmol). The reaction mixture was refluxed at 80°C for 4 h. The reaction mixture was cooled to room temperature and concentrated to dryness and then diluted with DCM (500 mL). The organic layer was washed with NaHC03, water, and brine. The combined organic layers were dried over MgS04 and concentrated to produce 11.8 g of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy) ethyl (4-(N- hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 as a white foamy solid, which was used without purification in the next experiment. LCMS-ESI (m/z) calculated for C23H39N304Si: 449.7; found 350.2 [M-Boc]+ and 472.2 [M+Na]+, ¾ = 1.79 min (Method 1). 1H NMR (400 MHz, CDC13) δ 7.32 (t, / = 7.3 Hz, 1H), 7.21 – 7.07 (m, 2H), 5.69 (s, 0.5 H), 5.19 (s, 0.5 H), 4.89 (s, 2H), 3.85 – 3.50 (m, 2H), 3.31 (ddd, / = 12.2, 9.2, 2.5 Hz, 2H), 3.28 – 3.03 (m, 2H), 3.03 – 2.70 (m, 1H), 2.29 (t, J = 23.6 Hz, 1H), 1.43 (bs, 4.5H), 1.28 (bs, 4.5H), 1.16 – 1.04 (m, 1H), 0.90 – 0.71 (m, 9H), 0.08 – -0.14 (m, 6H). 13C NMR (101 MHz, CDC13) 6 170.99, [156.20, 155.62], 152.38, [144.53, 143.57], [141.82, 141.21], 129.61, 126.78, [126.59, 126.25], [125.02, 124.77], [79.91, 79.68], 64.04, 61.88, [61.57, 61.23], [46.03, 45.76], 30.76, 30.21, [28.53, 28.28], 25.95, [25.66, 25.29], 25.13, [18.28, 17.94], 3.72, -5.34. ^-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl (4-(N- hydroxycarbamimidoyl)-2,3-dihydro-lH-inden-l-yl)carbamate INT-19 is prepared in an analogous fashion using INT-17. [0369] (R)-tert-butyl 2-( tert-butyldimethylsilyloxy)ethyl( 4-( 5-( 3-cyano-4-isopropoxyphenyl)- l,2,4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl)carbamate and (R)-tert-butyl 4-(5-(3-cyano- 4-isopropoxyphenyl )-l,2, 4-oxadiazol-3-yl)-2,3-dihydro-lH-inden-l-yl) (2-hydroxethyl) carbamate

[0370] Prepared using General Procedure 4. To a solution of 3-cyano-4-isopropoxybenzoic acid (4.5 g, 21.9 mmol) in anhydrous DMF (100 mL) was added HOBt (5.4 g, 40.0 mmol) and EDC (5.6 g, 29.6 mmol). After 1 h, {R)-tert-buiy\ 2-(tert-butyldimethylsilyloxy)ethyl (4- (N-hydroxycarbamimidoyl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-18 (11.8 g, 26.3 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. LCMS analysis showed complete conversion to the intermediate, (R)-tert-b\xty\ 2-(tert- butyldimethylsilyloxy) ethyl (4-(N-(3-cyano-4-isopropoxybenzoyloxy) carbamimidoyl)-2,3- dihydro-7H-inden-l-yl)carbamate INT-20. The reaction mixture was then heated to 80°C for 12 h. The reaction mixture was cooled to room temperature and diluted with EA (250 mL). NaHC03 (250 mL) and water (350 mL) were added until all the solids dissolved. The mixture was extracted with EA and the organic layers washed successively with water and brine. The organic layers were dried over MgS04 and concentrated to produce 15.3 g of a mixture of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4- oxadiazol-3-yl)- 2,3-dihydro-iH-inden-l-yl) carbamate INT-21, and the corresponding material without the TBS protecting group, (R)-tert-butyl 4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT -22. The mixture was a brown oil, which could used directly without further purification or purified by chromatography (EA hexane). INT-21: LCMS-ESI (m/z) calculated for C34H46N4O5S1: 618.8; found 519.2 [M-Boc]+ and 641.3 [M+Na]+, tR = 7.30 min (Method 1). Ή NMR (400 MHz, CDC13) δ 8.43 (d, J = 2.1, 1H), 8.34 (dd, J = 8.9, 2.2, 1H), 8.07 (d, J = 8.1, 1H), 7.46 – 7.26 (m, 2H), 7.12 (d, / = 9.0, 1H), 5.85 (s, 0.5H), 5.37 (s, 0.5H), 4.80 (dt, J = 12.2, 6.1, 1H), 3.92 – 3.32 (m, 3.5 H), 3.17 (s, 2H), 2.95 (s, 0.5 H), 2.62 – 2.39 (m, 1H), 2.38 – 2.05 (m, 1H), 1.53 (s, 4.5H), 1.48 (d, J = 6.1, 6H), 1.33 – 1.27 (m, 4.5H), 0.94 – 0.77 (m, 9H), 0.01 (d, J = 20.9, 6H). 1C NMR (101 MHz, DMSO) δ 173.02, 169.00, 162.75, [156.22, 155.52], [145.18, 144.12], [143.39, 142.76], 134.16, 133.89, 128.20, [128.01, 127.85], [127.04, 126.90], 126.43, 123.31, 116.93, 115.30, 113.55, 103.96, [79.95, 79.68], 72.73, 67.61, 63.42, [61.91, 61.77], 60.99, 46.11, 31.78, [30.47, 29.87], [28.55, 28.26], 25.93, 21.75, 18.30, 0.00, -5.37. INT-22: LCMS-ESI calculated for C28H32N4Os: 504.6; found 527.2 [M+Na]+, tR = 2.65 min (Method 1). Ή NMR (400 MHz, CDC13) δ 8.36 (d, J = 2.1, 1H), 8.27 (dd, / = 8.9, 2.2, 1H), 8.03 (d, / = 7.2, 1H), 7.35 – 7.26 (m, 2H), 7.06 (d, / = 9.0, 1H), 5.44 (s, 1H), 4.73 (dt, J = 12.2, 6.1, 1H), 3.64 (s, 2H), 3.44 (ddd, / = 17.5, 9.5, 3.2, 2H), 3.11 (dt, J = 17.4, 8.6, 3H), 2.54 – 2.38 (m, 1H), 2.04 (td, J = 17.6, 8.8, 1H), 1.50 – 1.24 (m, 15H). (5 -teri-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH-inden-l-yl)carbamate INT-23 and (S)-terf-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-iH- inden-l-yl) (2-hydroxyethyl) carbamate INT -24 were made in an analogous fashion.
[0371] (R)-5-(3-(l-(2-hydroxyethylamino)-2,3-dihydro-lH-inden-4-yl)-l,2,4-oxadi zol-^ 2-isopropoxybenzonitrile (Compound 85)

[0372] To a solution of (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3-cyano-4- isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3-dihydro-7H-inden-l-yl)carbamate INT-21 and (R)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2,3-dihydro-iH- inden-l-yl) (2-hydroxethyl) carbamate INT-22 (13.9 g, 27.5 mmol) in dioxane (70 mL) at 0°C was added 4N HCl in dioxane (68.8 g, 275.4 mmol). The reaction mixture was warmed to room temperature and then heated to 50°C for 1 h. The resulting suspension was cooled to room temperature and Et20 (75 mL) was added. The precipitate was collected by filtration, washed with Et20 and dried to produce 10.5 g of an off-white solid. The HCl salt was recrystallized from MeOH (165 mL) to produce 5.98 g (56% overall yield from (R)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-cyano-2,3-dihydro-iH-inden-l-yl) carbamate) of (R)-5- (3-(l-(2-hydroxyethylamino)-2,3-dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl)-2- isopropoxybenzonitrile 85 as a white solid. LCMS-ESI (m/z) calculated for C23H24N403: 404.5; found 405.4 [M+H]+, tR = 2.44 min. Ή NMR (400 MHz, DMSO) 5 9.25 (s, 2H), 8.53 (d, J = 2.3, 1H), 8.42 (dd, J = 9.0, 2.3, 1H), 8.17 (d, J = 7.7, 1H), 7.97 (d, J = 7.6, 1H), 7.63 – 7.50 (m, 2H), 5.28 (t, J = 5.0, 1H), 4.99 (hept, J = 6.1, 1H), 4.92 (s, 1H), 3.72 (q, J = 5.2, 2H), 3.57 – 3.43 (m, 1H), 3.27 (ddd, J = 17.6, 9.1, 5.0, 1H), 3.15-2.85 (m, J = 24.2, 2H), 2.53 (dtd, J = 9.0, 5.5, 5.3, 3.6, 1H), 2.30 (ddd, J = 13.4, 8.9, 4.6, 1H), 1.39 (d, J = 6.0, 6H). 13C NMR (101 MHz, DMSO) 6 173.25, 167.86, 162.47, 144.56, 139.13, 134.53, 133.77, 129.30, 128.93, 127.45, 122.83, 115.79, 115.15, 114.84, 102.40, 72.46, 61.04, 56.51, 46.38, 31.53, 27.74, 21.37. Elemental analysis for C23H25N403C1: C calc. = 62.65%; found = 62.73%; H calc. = 5.71%; found = 5.60%; N calc. = 12.71%; found = 12.64%; CI calc. = 8.04%; found = 8.16%. Chiral HRLC of the free base: (R)-5-(3-(l-(2-hydroxyemylamino)-2,3-dihydro-iH- inden-4-yl)-l,2,4-oxadiazol-5-yl)-2-isopropoxy – benzo-nitrile was eluted using 10% i‘-PrOH in hexanes plus 0.3% DEA: >99.9% ee, tR = 37.72 min.
(S)-5-(3-(l-(2-hydroxyethylamino)- 2,3-dihydro-iH-inden-4-yl)-l,2,4-oxadiazol-5-yl) -2-isopropoxy benzonitrile 86 was obtained in analogous fashion from (S)-tert-butyl 2-(tert-butyldimethylsilyloxy)ethyl(4-(5-(3- cyano-4-isopropoxyphenyl)- 1 ,2,4-oxadiazol-3-yl)-2, 3-dihydro-iH-inden- 1 -yl)carbamate INT-23 and (S)-tert-butyl 4-(5-(3-cyano-4-isopropoxyphenyl)-l,2,4-oxadiazol-3-yl)-2,3- dihydro-iH-inden-l-yl) (2-hydroxyethyl) carbamate INT-24: >99.9% ee, tR for (5)- enantiomer = 35.86 min.
(S) IS DESIRED CONFIGURATION
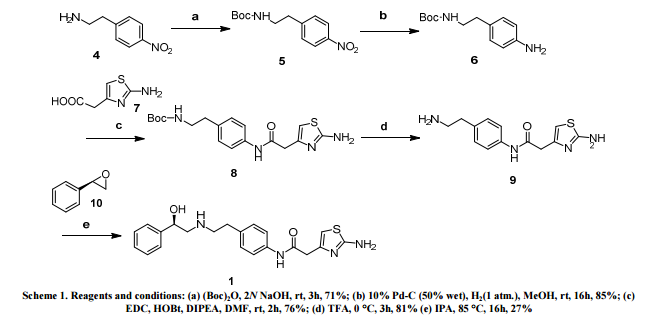
THE SYNTHESIS IS SUMMARISED BELOW

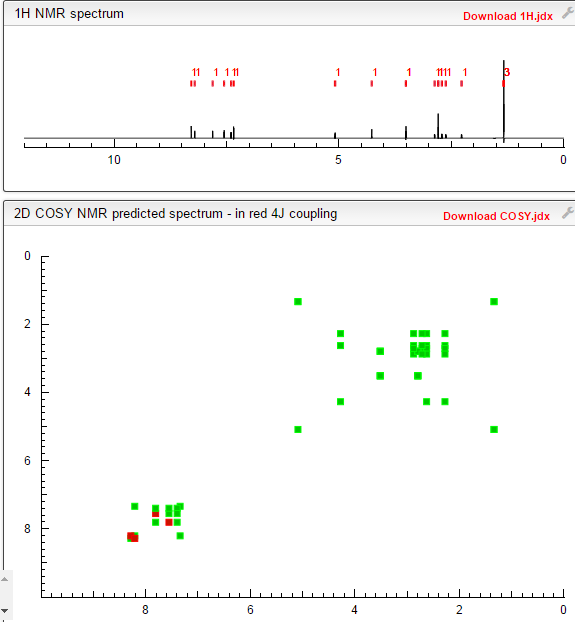
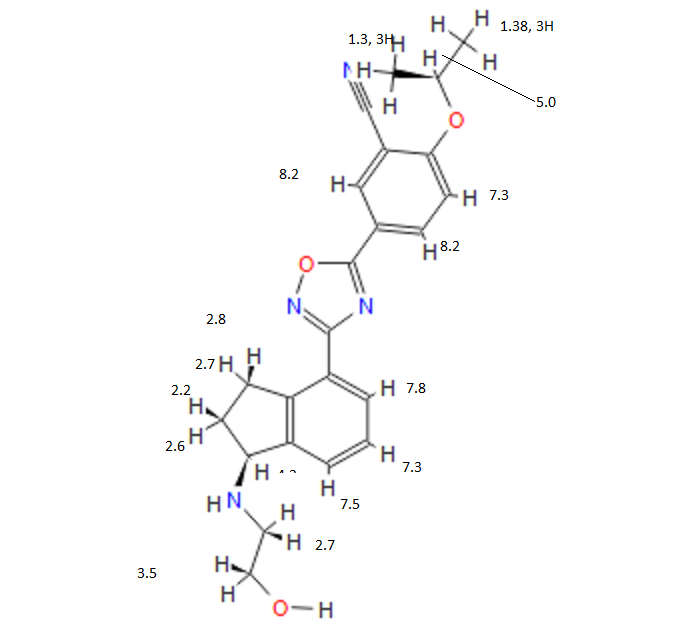
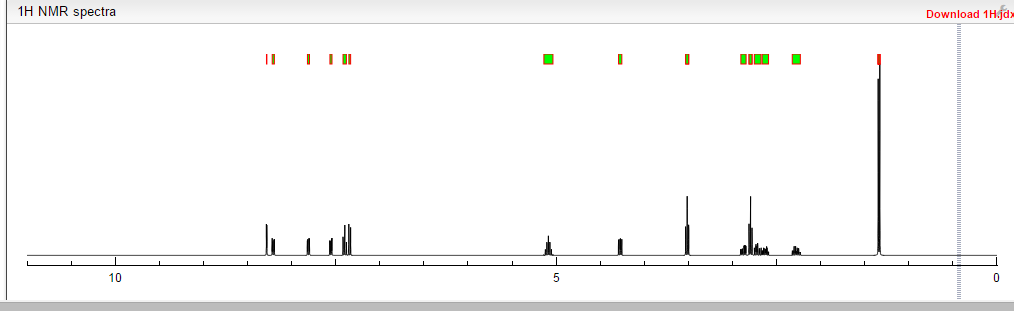
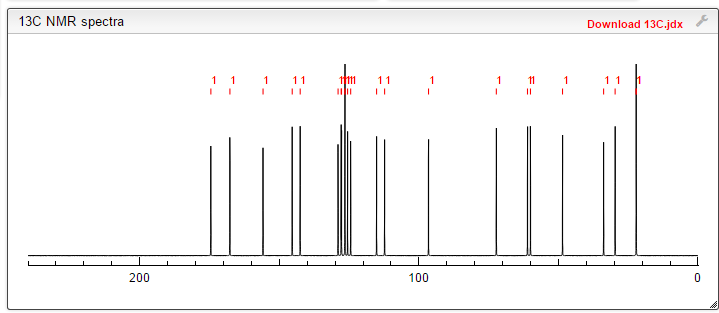
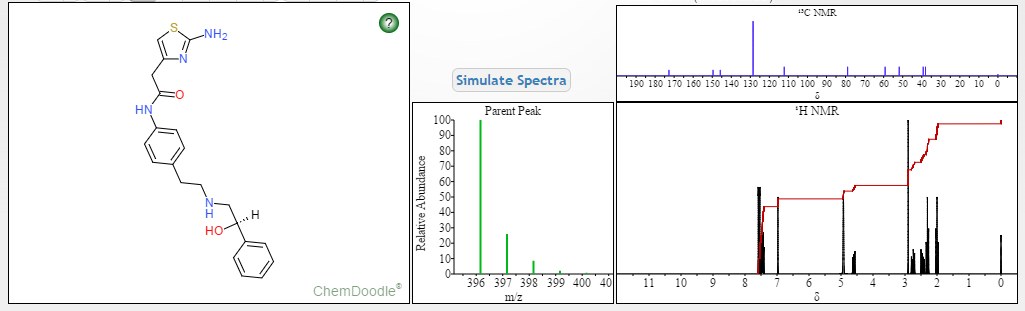
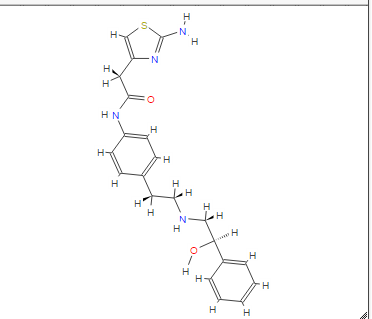
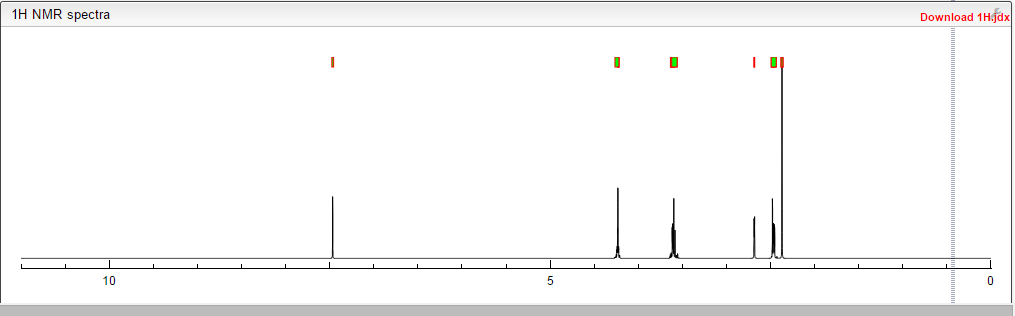
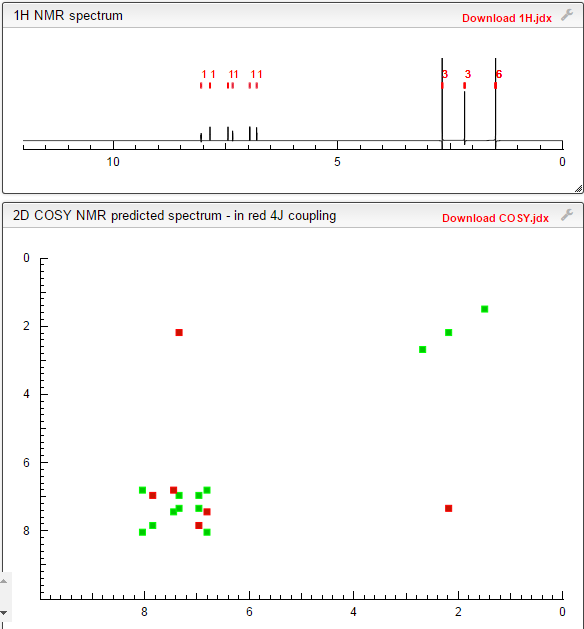
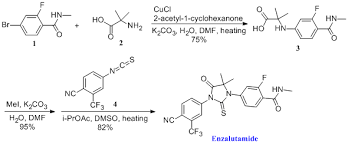
COSY PREDICT

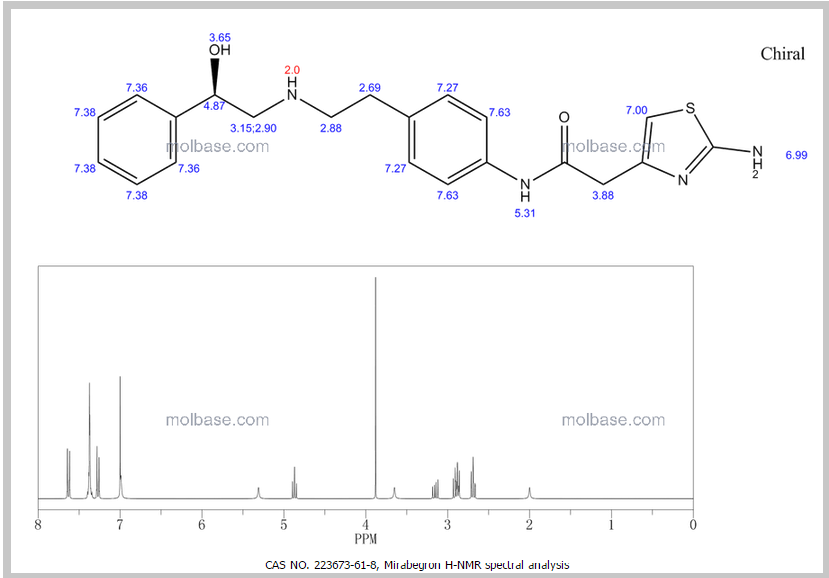
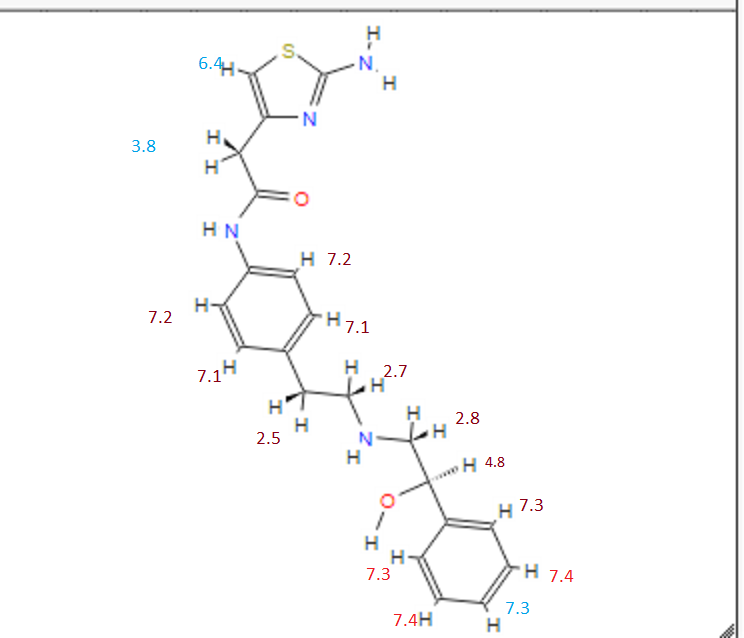
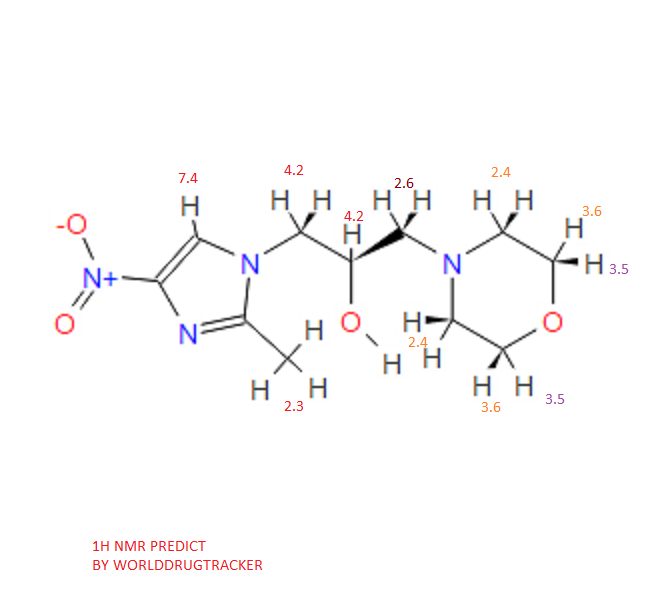
1H NMR PREDICT


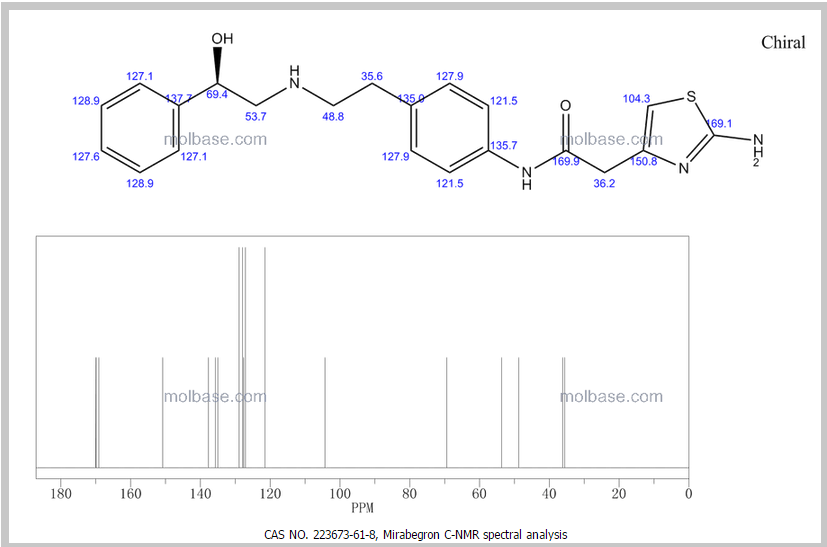
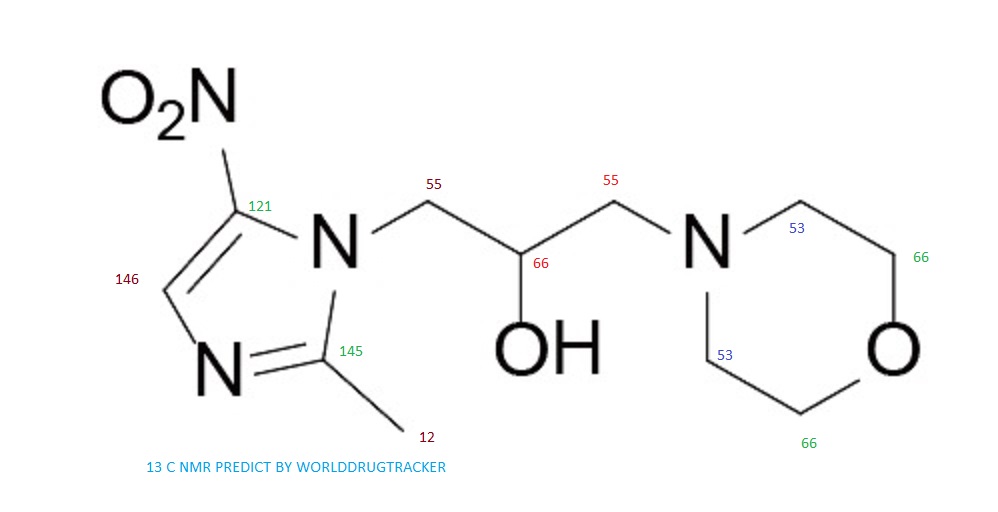
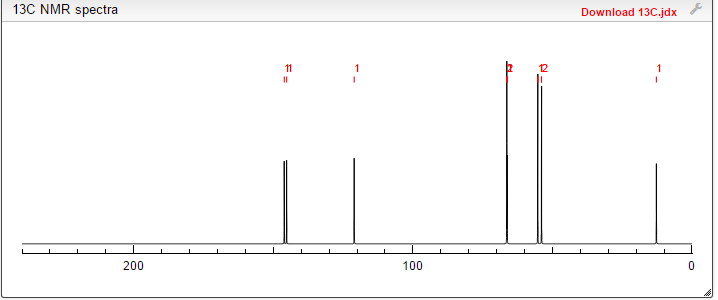
13C NMR PREDICT


note——-(CH3 )2CH-O-AR appears at 72 ppm

////////

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











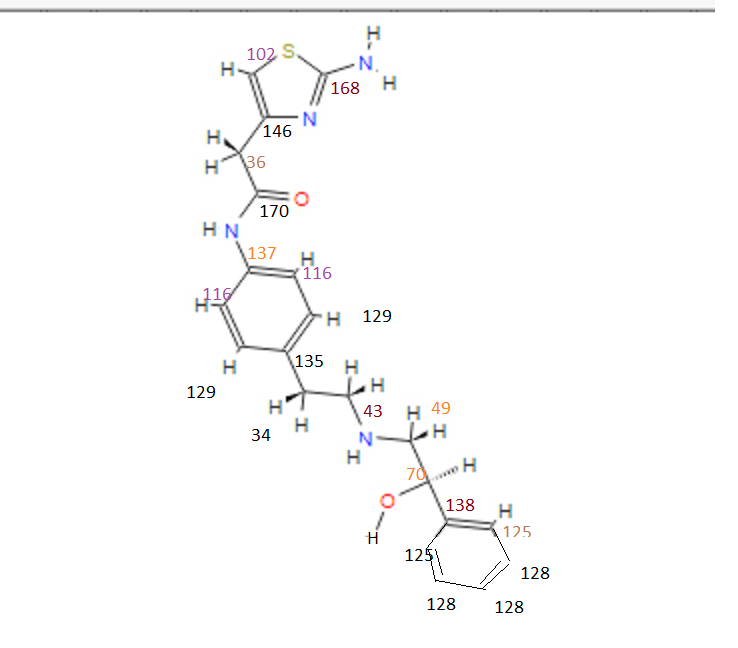
 CN 103896872
CN 103896872













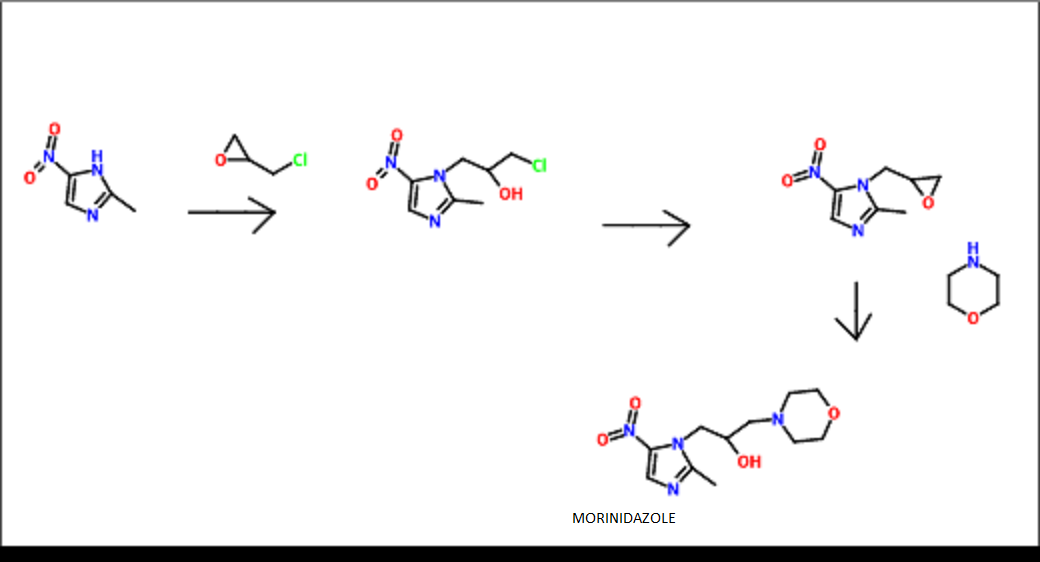

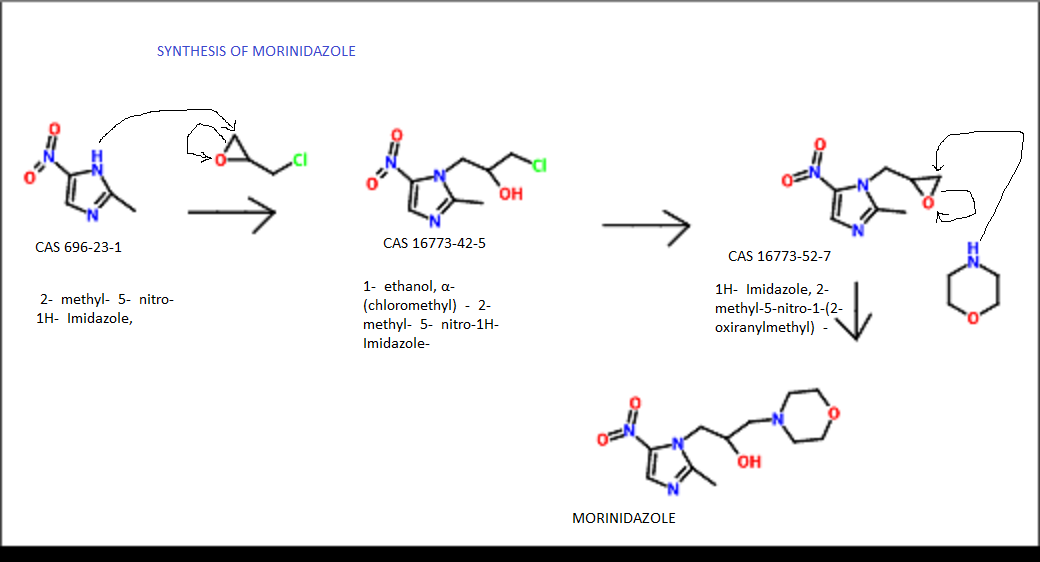
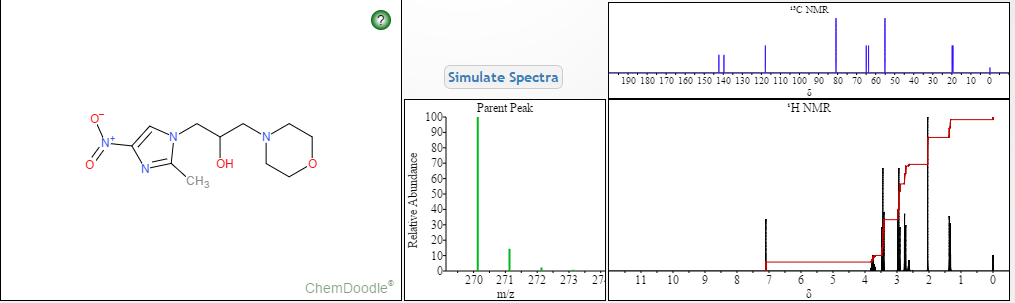


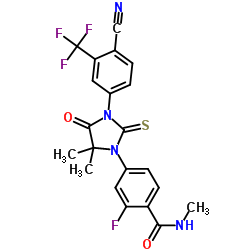
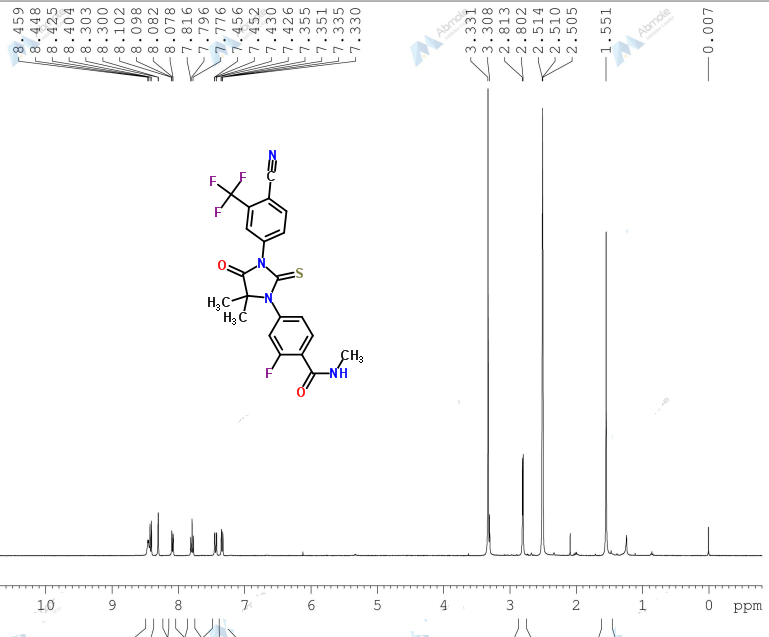

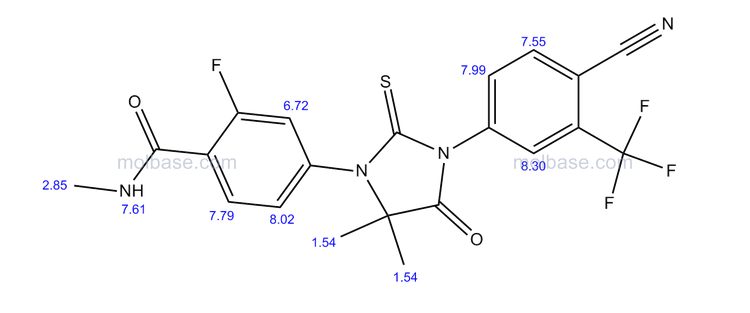

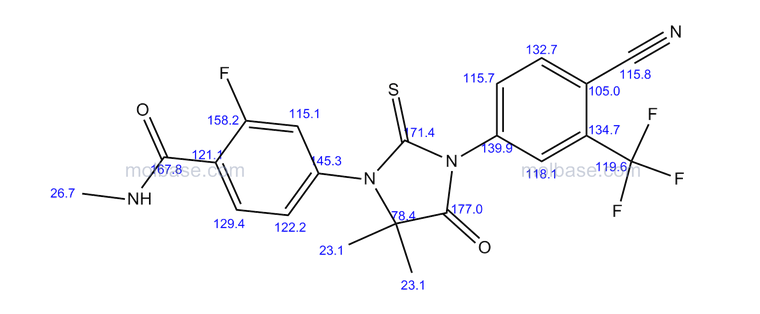
















 LIONEL MY SON
LIONEL MY SON
 Lenvatinib Mesilate
Lenvatinib Mesilate























 Click on images to view
Click on images to view