WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ]
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
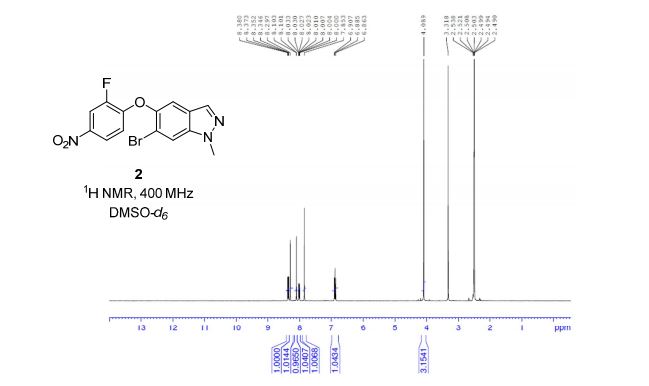
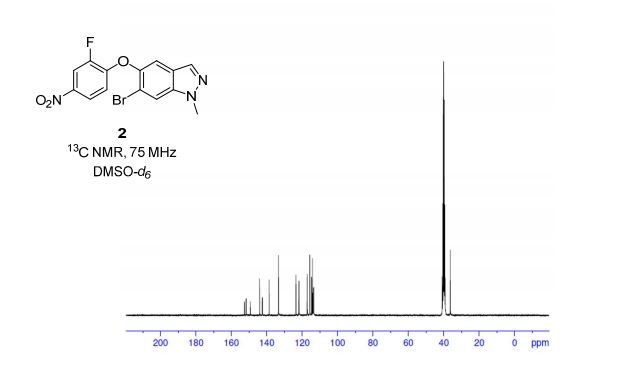
HRMS (EI, TOF) m/z calcd for C24H21F2NO3 [M] 409.1489 found 409.1478. Anal. Calcd for C24H21F2NO3: C 70.41, H 5.17, F 9.28, N 3.42. Found: C 70.46, H 5.23, F 9.24, N 3.34.
To a cooled (0 °C) solution of lactone 19 (2.0 g, 4 mmol) in 160 mL of dry diethyl ether was added 12 mL of 1 M solution of t-BuMgCl in diethyl ether. After 2 h, 30 mL of aq NH4Cl was added. The aqueous layer was extracted with ether (160 mL), the organic layer was washed with satd NaHCO3 (50 mL) and dried (MgSO4), and the solvent was removed under reduced pressure. Crude product 20 (1.64 g, 82%) obtained as a yellowish solid was used in the next step without further purification. An analytic sample was obtained by chromatography on silica gel (hexanes/ethyl acetate 7:3). Mp 130–133 °C [lit.(11) 132–134 °C]; [α]20D −42.2 (c 1.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.42–7.20 (11H, m), 7.02–6.90 (6H, m), 5,04 (2H, s), 4.72–4.68 (1H, m), 4.55 (1H, d J 2.2 Hz), 3.07 (1H, dt J 7.1, 2.2 Hz), 2.05–1.93 (3H, m) 1.89–1.82 (2H, m); 13C NMR (150 MHz, CDCl3) δ 167.6, 163.0, and 161.4 (d, JC–F 244.2 Hz), 159.8 and 158.1 (d, JC–F 241.8 Hz), 159.0, 140.0, 139.9, 136.6, 133.9, and 133.8 (d, JC–F 2.9 Hz), 129.6, 128.6, 128.1, 127.5, 127.4 and 127.4, (d, JC–F 8.0 Hz), 127.2, 118.4, 118.3, 115.8, 115.8, and 115.7 (d, JC–F 22.0 Hz), 115.5, 115.4, and 115.3 (d, JC–F 21.3 Hz), 73.3, 70.1, 61.1, 60.3, 36.5, 25.0; HRMS (ESI, TOF) m/z calcd for C31H27F2NO3Na [M + Na]+ 522.1851, found 522.1862; IR (KBr) v 3441, 1743, 1609, 1510 cm–1. Anal. Calcd for C31H27F2NO3: C 74.53, H 5.45, N 2.80, F 7.61. Found: C 74.40, H 5.53, N 2.74, F 7.56.
Ezetimibe has the chemical name 1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone (hereinafter referred to by its adopted name “ezetimibe”) and is structurally represented by Formula I.
Ezetimibe is in a class of lipid lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related phytosterols. It is commercially available in products sold using the trademark ZETIA as a tablet for oral administration containing 10 mg of ezetimibe, and in combination products with simvastatin using the trademark VYTORIN.
U.S. Pat. No. 6,096,883 discloses generically and specifically ezetimibe and its related compounds along with their pharmaceutical compositions. The patent also describes a process for the preparation of ezetimibe.
The process described in the patent involves the use of methyl-4-(chloroformyl) butyrate and also involves isolation of the compound (3R,4S)-1-(4-fluorophenyl)-3-[3-(chloroformyl)-3-oxo-propyl]-4-(4-benzyloxyphenyl)-2-azetidinone as an intermediate. Chlorinated compounds are unstable and difficult to handle in large scale productions. The process described in the patent also involves the purification of intermediates using column chromatography, thus making the process difficult to be scaled up.
Processes for preparation of ezetimibe and its intermediates have also been described in U.S. Pat. Nos. 6,207,822, 5,856,473, 5,739,321, and 5,886,171, International Application Publication No. WO 2006/050634, and in Journal of Medicinal Chemistry 1998, 41, 973-980, Journal of Organic Chemistry 1999, 64, 3714-3718, and Tetrahedron Letters, 44(4), 801-804.
EXAMPLE 10 PREPARATION OF 1-(4-FLUOROPHENYL)-3(R)-[3-(4-FLUOROPHENYL)-3(S)-HYDROXYPROPYL]-4(S)-(4-HYDROXYPHENYL)-2-AZETIDINONE (FORMULA I)
50 g of (3R,4S)-1-(4-fluorophenyl)-3-[3-(4-fluorophenyl)-3(s)-hydroxypropyl]-4-(4-benzyloxyphenyl)-2-azetidinone and 475 ml of methanol were taken into a round bottom flask. A mixture of 15 g of 5% palladium on carbon and 25 ml of water was added to it. The reaction mass was flushed with hydrogen gas and a hydrogen pressure of 3 to 5 kg/cm2 was applied. The reaction mass was stirred for 3 hours. Reaction completion was checked using thin layer chromatography. After the reaction was completed, the pressure was released and the reaction mass was filtered through perlite. The filter bed was washed with 100 ml of methanol. The filtrate was distilled completely at 70° C., and 400 ml of isopropanol was added to it. The reaction mass was heated to 45° C. and maintained for 10 minutes. The reaction mass was then allowed to cool to 28° C. 400 ml of water was added to the reaction mass and stirred for 1 hour, 20 minutes. The separated compound was filtered and washed with 100 ml of water. The wet cake was taken into another round bottom flask and 500 ml of chlorobenzene and 40 ml of methanol were added to it. The reaction mass was heated to 65° C. and maintained for 15 minutes. 25 ml of water was added to the reaction mass and stirred for 2 hours. The separated compound was filtered and washed with 100 ml of chlorobenzene. The wet cake was taken into another round bottom flask and 375 ml of chlorobenzene, and 30 ml of methanol were added to it. The reaction mass was heated to 62° C. and maintained for 10 minutes. The reaction mass was then cooled to 28° C. and 20 ml of water was added to it. The reaction mass was stirred for 20 minutes and then filtered and washed with 100 ml of chlorobenzene. The wet cake was taken into another round bottom flask and 400 ml of isopropanol was added to it. The reaction mass was heated to 46° C. and maintained for 15 minutes. 800 ml of water was added to the reaction mass at 45 to 46° C. and stirred for one hour. The separated solid was filtered and washed with water. The process of recrystallization in a combination of isopropanol and water was repeated and the obtained compound was dried at 70° C. for 5 hours to get 19.8 g of the title compound. (Yield 49.2%)
Purity by HPLC: 99.68%.
EXAMPLE 11 PURIFICATION OF 1-(4-FLUOROPHENYL)-3(R)-[3-(4-FLUOROPHENYL)-3(S)-HYDROXYPROPYL]-4(S)-(4-HYDROXYPHENYL)-2-AZETIDINONE (FORMULA I)
15.0 g of ezetimibe obtained above and 120 ml of isopropanol were taken into a round bottom flask and the reaction mass was heated to 48° C. The reaction mass was filtered through a perlite bed in the hot condition to make the solution particle free. The filtrate was taken into another round bottom flask and heated to 47° C. 240 ml of water was added at 47° C. After completion of the addition, the reaction mass was maintained at 47° C. for 1 hour. The separated solid was filtered and washed with 30 ml of water. The wet compound was dried at 70° C. for 8 hours to get 13.4 g of the title compound. (Yield 89%)
Purity by HPLC: 99.92.
benzyl ezetimibe impurity: less than 0.0003 area-%,
N-{[4-((2R,3R)-1-(4-fluorophenyl)-3-{[(2R or S)-2-(4-fluorophenyl)-2-hydroxyethyl]thio}-4-oxoazetidin-2-yl)phenoxy]acetyl}glycyl-D-lysine, used as anticholesterol agents
Taking a look at the Warning Letters the FDA issued after inspections of activesubstance manufacturers in the 2015 fiscal year, which ended on 30 September 2015, it is first of all striking that only non-American companies are among the addressees. Almost half of them are Indian companies. Overall the numbers look like this: India (3 WLs); China (2 WLs); Canada (1 WL); Thailand (1 WL); Czech Republic (1 WL).
The top issue in the Warning Letters is the non-GMP compliant handling of electronic data or missing data integrity. Each of the 8 warning letters contains the following comment in the same wording:
“Failure to prevent unauthorized access or changes to data and to provide adequate controls to prevent omission of data.”
The lack of access control on electronic (raw) data is an issue the FDA investigators have been observing for a long time, especially during inspections in pharmaceutical companies. In this as well as in the last fiscal year there were significant deficiencies in several companies – medicinal product as well as API manufacturers – as the comments in the appropriate Warning Letters show. For more information also see the GMP news Another FDA Warning Letter with Focus on “Data Integrity” and FDA Warning Letter on Data Integrity.
Ultimately these deficiencies can be traced back to a failure of the quality assurance unit which also affects other areas. In the Warning Letters, the following examples can be found for this:
“Failure of your quality unit to ensure that materials are appropriately tested and the results are reported.”
“Failure of your quality unit to exercise its responsibility to ensure the APIs manufactured at your facility are in compliance with CGMP, and meet established specifications for quality and purity.” Data were manipulated by laboratory staff (change of the file name), to fake results from identity tests in batches which in reality were not performed. Quality assurance was not able to uncover this manipulation.
Despite an unknown peak in the examination for residual solvents the relevant batches were released. Upon receipt of a complaint regarding this peak an examination was conducted with the result that the contamination originated in the production process itself. Preventive control measures to avoid this contamination were not established.
“Failure to adequately investigate complaints and extend the investigations to other batches that may have been affected.” As a result of a complaint (bad smell), a cause study was initiated which was completed prior to implementation of the preventive measures again. The CAPA measures subsequently carried out were obviously not associated with the reason for the complaint.
“Failure to have appropriate controls for issuance of batch records”.
The use of document templates for batch records is out of control. These can be printed out from the production staff’s personal computers. Although there is an SOP for the control of batch records there are no appropriate training records.
“Failure to have appropriate documentation and record controls.” Data for tracing raw materials are not available. Log entries are without date/visa and partly corrected with Tippex. There is an SOP prohibiting the use of correction fluid, however this was not trained.
“Failure to record activities at the time they are performed and destruction of original records.” Original records of critical process data on uncontrolled memos were transferred subsequently in new report templates (after batch approvals) and then destroyed.
This selection of examples shows the lack of fundamental GMP principles which leads to a blatant misconduct of staff and ultimately to quality defects in the final product. The main responsibility usually has the quality unit, which task it actually would be to ensure a thorough training in production and quality control and to monitor compliance with the appropriate regulations. These examples of non-GMP-compliant behavior are not limited to active ingredient manufacturers; there are very similar findings in Warning Letters issued to medicinal product manufacturers. An analysis of these Warning Letters issued in the fiscal year 2015 will be part of one the coming newsletters.
/////Warning Letters, FDA, active ingredient manufacturers
Pharmaceutical Research Institute (IF), Rydygiera 8, Warszawa 01-793, Poland
Ezetimibe, (3R,4S)-1-(4-fluorophenyl)-3-((3S)-3-(4-fluorophenyl)- 3-hydroxypropyl)-4-(4-hydroxyphenyl)-2-azetidinone, is an anti-hyperlipidemic drug which is used to lower cholesterol level. It acts by decreasing cholesterol absorption in the intestine.
The three chiral centers in the ezetimibe molecule give rise to eight stereoisomers and the synthesis of stereochemical pure ezetimibe is a significant challenge. The synthesis of ezetymibe is described in many patents and patent applications, however the problem of stereochemical purity of the final product and its intermediates is almost completely omitted.
The synthesis of ezetimibe was realized by a procedure shown below, according to Schering Co. patents No US 6,207,822, EP 1137634:
We have investigated the sterochemical course of all steps of this process and found that for the preparation of optical pure ezetimibe the providing of pure (S,R,S,S) – EZ-6 is cru-cial. This diastereomer (product of anti-condensation of EZ-4 + EZ-5) is usually contaminated with (S,R,R,S) – EZ-6 isomer (syn-condensation), and also with (R,R,S,S) – EZ-6 isomer derived from small amount of (R,S)-alcohol EZ-4 which is usually occurring in required (S,S)-alcohol. The presence of (R,R,S,S) – EZ-6 diastereomer leads to (R,R,S) -“iso-ezetimibe” which is very difficult to remove from ezetimibe.
The synthesis of ezetimibe was optimized, all chemical and sterochemical impurities were isolated and/or synthesized and characterized by NMR, MS and HPLC techniques. The method for the purification of desired key intermediate (S,R,S,S)-6 was elaborated. These al-lowed us to develop the large scale efficient synthesis of pharmaceutical pure Ezetimibe (HPLC > 99,5 %, (R,R,S)-isomer < 0,1 %, single unknown impurity < 0,1 %, total impurities < 0,6 % ).
Ezetimibe has the chemical name 1-(4-fluorophenyl)-3(R)-[3-(4-fluorophenyl)-3(S)-hydroxypropyl]-4(S)-(4-hydroxyphenyl)-2-azetidinone (hereinafter referred to by its adopted name “ezetimibe”) and is structurally represented by Formula I.
Ezetimibe is in a class of lipid lowering compounds that selectively inhibit the intestinal absorption of cholesterol and related phytosterols. It is commercially available in products sold using the trademark ZETIA as a tablet for oral administration containing 10 mg of ezetimibe, and in combination products with simvastatin using the trademark VYTORIN.
U.S. Pat. No. 6,096,883 discloses generically and specifically ezetimibe and its related compounds along with their pharmaceutical compositions
The preparation of ezetimibe ezetimibe first disclosed in U.S. Patent US 5767115.
Hydrogen Debenzylation get ezetimibe, the method disclosed in this patent require the use of several key intermediates purified by column chromatography, increasing the difficulty and cost of industrial production.
US patent US5767115 to improve the synthesis process have also been reported.For example: W02006137080 US5767115 on the basis of synthesis of intermediate compound 3 were improved optimization, using pivaloyl chloride and the formation of a mixed anhydride intermediate compound 2, and then with the chiral auxiliary (S) -4- phenyl-2- oxazolidinone reaction intermediate compound 3; US Patent US6133001 discloses a microbial catalytic asymmetric reduction of carbonyl to give chiral hydroxy, instead US5767115 Synthesis of (R) -CBS catalyst;
W02008089984 reported the use of a rhodium catalyst [(S, S) -N- (piperidyl-N-sulfonyl) -I, 2-diphenyl ethylenediamine] (η 6-mesitylene) Ruthenium right of intermediate compound 9Said reduction.
W02008032338 reports by reacting the intermediate compound 8 with a salt of an aliphatic amine which was purified manner, although effectively improve the purity, but adds steps, and the yield was significantly reduced.
In addition to the synthetic route based on open Pu Xi US5767115, US Patent US6207822, US Patent US5856473, US patent US5886171, W02005066120, W02005113496, W02006050634, W02007017705 also disclose the ezetimibe different preparation methods.
Patent W02007072088 discloses another synthetic route for preparing ezetimibe ezetimibe, which is a small step synthesis reaction, the specific synthetic route is as follows:
Another US: 5739321; US: 1 5886171 reported the route: the (4S) – hydroxytetrahydrofuran _2_ one and N- (4- fluorophenyl) -4-benzyloxy-benzylidene methylamine as starting Preparation of raw ezetimibe, the reaction scheme is as follows:
Since the first report since the synthesis method, there are already several ezetimibe ezetimibe synthetic route reports, such as document US 5856473, US 5739321, EP 1137634, EP 720599, WO 1995/08532 EP 0720599, provides ezetimibe ezetimibe synthetic route.
Example 9 Preparation of Compound 8 embodiment.
Hydrogenation bottle was added 7a (2.14 g, 4.3 mmol), methanol (30 mL), was added Pd / C (50 mg :), transferred into the autoclave, and replaced with hydrogen three times, filled with hydrogen 5 atm, room temperature and stirred for 6 hours, venting of hydrogen, filtered through Celite, with a small amount of methanol (10 mL), dried and concentrated, the residue was mixed solvent of methyl t-butyl ether, and recrystallized from n-hexane to give compound 8, 78% yield
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5 Lc (193 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (4.12 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol ). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated
(I?) – 2a, 85% yield, 93% ee.
Example 4 Compound Example (Preparation -2b of.
The reaction is as follows: Under an argon atmosphere, [Pd (C 3 H 5 ) Cl] 2 (54.8 mg, 0.15 mmol) and (&& 5) -La (165 mg, 0.25 mmol) were added to a Schlenk tube, was added anhydrous CH 2 C1 2 C50 mL), stirred at room temperature for 10 minutes, the substrate was added successively lb (3.78 g, 10 mmol), K 2 C0 3(1.0 M solution, 30 mL, 30 mmol) and p-fluoroaniline (3.33 g, 30 mmol). After stirring at room temperature for three hours, liquid separation, the aqueous phase was extracted with dichloromethane (3 x 50 mL), The combined organic phase was dried over anhydrous sodium sulfate, filtered and concentrated, purified by column chromatography to give asymmetric amination the product of (i?) – 2b. The reaction formula is as follows:
(I?) – 2a (3.44 g, 8.48 mmol) and nucleophiles 3a (2.82 g, 12.7 mmol) was added in an eggplant-shaped flask, tetrahydrofuran (100 mL), DBU (4.25 g, 16.96 mmol was stirred at room temperature for 12 hours, thin layer chromatography until starting material disappeared by TLC the reaction mixture was concentrated and purified by column chromatography, to obtain compound 4a, 82% yield (Note: Allyl allyl).
ESI-MS m / z: 628.4 [M + H + ]; HRMS (ESI) m / z:. calcd for C 37 H 36 N0 6 F 2 +1 : 628.2505, Found:
+ ]. After the reaction system may also not treated directly in the next step. The reaction formula is as follows:
: Example 6 Preparation of Compound 5a.
To the reaction system of Example 5 is continued morpholine (4.43 g, 50.88 mmol) and Pd (PPh 3 ) 4 , and stirring was continued at room temperature for 6 hours, concentrated purified by chromatography (98 mg, 0.0848 mmol) after column .
The total yield from the compound 2a to 5a rate of 71%. Compound 5a is composed of a pair of non-enantiomer at a ratio of 2 or 3: 1. No need to separate the non-enantiomer, can be used directly in the next step.
ESI-MS m / z: 544.2 [M + H +]; HRMS (ESI) m / z:. Calcd for C33H31NO4 F 2 Na +1 : 566.2113, Found: 566.2113 [M + Na + ].
Preparation Example 7 Compound 6a embodiment.
Compound 5a (3.5 g, 6.4 mmol) and anhydrous tetrahydrofuran (50 mL) was added an eggplant-shaped flask, and cooled to -20 ° C under slowly added dropwise amino lithium hexamethyldisilazide (LiHMDS) (1.0 M THF, 14 ml, 14 mmol). The reaction system was stirred at this temperature continued for 40 minutes, 5 mL of water was added to quench the reaction, and extracted with dichloromethane (3 x 100 mL), the organic phase was dried over anhydrous sodium sulfate
6a, 77% yield. [A] D 2Q = +1.9 (c 1.00, MeOH), 95% EE [by the high performance liquid chromatography, chiral OD-H column n is isopropanol = 70:30, 1.0 mL / min, 254 nm; t R (Major) = 19.60 min; t R . (minor) = 25.83 min] 1H NMR (400 MHz, CDCl 3 ) [delta] = 7.98-7.94 (m, 2H), 7.41-7.30 ( m, 5H), 7.25-7.23 (m, 4H), 7.09 (t, J = 8.8 Hz, 2H), 6.96-6.88 (m, 4H), 5.02 (s, 1H), 4.67 (d, J = 2.4 Hz , 1H), 3.31-3.23 (m, 1H), 3.17-3.08 (m, 2H), 2.42-2.20 (m, 2H) ppm; 13 C NMR (100 MHz, CDCl 3 ) [delta] = 197.2, 167.1, 165.6 ( d, J (F , C) = 253.9 Hz), 158.9, 158.8 (d, J (F , C) = 242.2 Hz), 136.5, 133.7 (d, J (F , C) = 2.7 Hz), 132.9 (d , J (F , C) = 2.8 Hz), 130.5 (d, J (F , C) = 9.4 Hz), 129.3, 128.5, 127.9, 127.3, 127.1, 118.2 (d, J (F , C) = 7.9 Hz ), 115.7 (d, J (F , C) = 8.4 Hz), 115.5 (d, J (F , c) = 8.3 Hz), 115.3, 69.9, 60.9, 59.6, 35.4, 23.0 ppm; 19 F NMR (376 MHz, CDCl 3 ) [delta] -104.8, -117.9 ppm.
Compound 6a is the same as reported in the literature specific rotation direction, the same NMR data reported in the literature. References:
(A) Wu, G; Wong, Y;. Chen, X .; Ding, ZJ Org Chem 1999, 64, 3714. (b) Sasikala, CHVA;. Padi, PR; Sunkara, V; Ramayya, P .; Dubey , PK; Uppala, VBR;… Praveen, C. Org Process Res Dev 2009, 13, 907. (c) Sova, M .; Mravljak, J .; Kovac, A .; Pecar, S .; Casar, Z .; Gobec, S .; Synthesis, 2010, 20, 3433.
Preparation Example 8 Compound 7a embodiment.
In dichloromethane (40 mL) and tetrahydrofuran (5 mL) were added to an eggplant-shaped flask, and cooled to 0 ° C, was added borane dimethyl sulfide complex (0.46 mL, 7.23 mmol) and – (+) – 2-methyl–CBS- oxazaborolidine (133 mg, 0.482 mmol). Compound 6 (; 2.4 § , 4.82 11 ^ 101) was dissolved in dichloromethane (2011 ^) in the join. Stirred at the same temperature for 5 hours. After completion of the reaction with methanol (10 mL) quenched the reaction was concentrated, added to 1 mol per liter of dilute hydrochloric acid, methylene-wan (X) was extracted, the organic phase was washed with saturated sodium chloride wash paint, concentrated, ethyl acetate – n-hexane to give the compound 7a, 90% yield,> 99%. Reaction
Another report line 2: (5S) – acetyl-5- (4-fluorophenyl) valeric acid as reaction intermediates for the preparation of ezetimibe, the synthesis process is as follows:
Seventh Embodiment
The 7 (20g, 0.04mol) was dissolved in methanol (25OmL) was added ammonium formate (25g, 0.4mol), 10% palladium / carbon (Ig) and formic acid (2mL, 0.04mol), stirred at room temperature 20min, filtered palladium / carbon, the filtrate was concentrated to dryness.The residue was dissolved in ethyl acetate, washed with saturated brine, and dried.The organic phase was concentrated to approximately 40mL, was slowly added thereto at room temperature, methyl tert-butyl ether, stirring lh, floc filtered and the filtrate was concentrated to dryness.The residue was dissolved in ethyl acetate, petroleum ether was added, stirred at room temperature 2h, filtered, and dried to give a white solid Ezetimibe 6.4g, yield 38.9%, [a] 2 ° D = _23.7.IH-NMR (DMS0-d6) δ: 9.51 (s, 1Η), 7.32-7.08 (m, 10Η), 6.75 (d, J = 8.4, 2Η), 5.27 (d, J = 4.5, 1Η), 4.80 ( d, J = 2.1, 1Η), 4.49 (m, 1Η), 3.08 (m, 1Η), 1.68-1.82 (m, 4Η).
ezetimibe ezetimibe synthesis and purification methods:
A Method: IOOml reactor was added to 60ml of ethanol, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 6g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 10 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake rinsed with an aqueous solution of an ice drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml ko alcohol, purified water was slowly added dropwise 160ml a large number of solid precipitation, filtration, alcohol use ko – after pumping out water rinse, 60 ° C and dried under vacuum to obtain the product 4. Og, yield: 81 · 4%.
B method: to IOOml reaction flask 60ml of methanol, acetic acid 2. 4g and 6g compound 10,
Stirred for 20 minutes, added I. 2g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen under a hydrogen atmosphere, the reaction for 18 hours at ambient temperature and pressure inch, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml isopropanol, filtered, and the mother liquor was slowly added dropwise 56ml of purified water, a large number of solid precipitation, filtration, filter cake washed with isopropanol – water rinse after pumping dried, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml isopropanol was slowly added dropwise 160ml of purified water, large amount of solid precipitated, suction filtered, washed with isopropanol – water rinse after draining, 60 ° C under vacuum drying products 3. 7g, yield: 75.3%.
C Method: To a IOOml 60ml of methanol was added to the reaction vessel, was added glacial acetic acid and 6g 2. 4g compound 10, followed by stirring for 20 minutes, O. 8g 20% Pd (OH) 2 / C, purged with nitrogen, purged with hydrogen , under hydrogen atmosphere, 30 ° C at atmospheric pressure for 18 hours the reaction inches, TLC analysis showed complete conversion of compound 10, suction filtered, the mother liquor was concentrated to dryness under reduced pressure to a pale yellow solid, the resulting solid was dissolved with 40ml ko alcohol, filtered, the mother liquor 56ml of purified water was slowly added dropwise, after the large amount of solid precipitated, suction filtered, the filter cake washed with methanol – water rinsing after drained, and dried in vacuo to give a white solid product, the resulting product was dissolved in 32ml of methanol, 160ml of purified water was slowly added dropwise a large number of solid precipitation, filtration, washed with methanol – after pumping out water rinse, 60 ° C under vacuum drying products 3. 5g, Yield: 71.3%.
Processes for preparation of ezetimibe and its intermediates have also been described in U.S. Pat. Nos. 6,207,822, 5,856,473, 5,739,321, and 5,886,171, International Application Publication No. WO 2006/050634, and in Journal of Medicinal Chemistry 1998, 41, 973-980, Journal of Organic Chemistry 1999, 64, 3714-3718, and Tetrahedron Letters, 44(4), 801-804.
EXAMPLE 1 DETERMINATION OF IMPURITIES IN EZETIMIBE
Determining the level of impurities in ezetimibe using HPLC. The HPLC analysis conditions are as described in Table 1.
TABLE 1
HPLC method for detecting the level of the impurities.
Column:
Zorbax SB-C18 150 × 4.6 mm, 3.5 μm
Flow:
1.0 ml/minute
Column oven
Ambient
temperature:
Wave length:
230 nm
Injection volume:
10 μl
Run time:
65 minutes
Elution:
Gradient
Diluent:
Acetonitrile
Gradient Program:
Time
% B
% A
(in minutes)
concentration.
concentration.
0.01
35
65
10.0
35
65
35.0
80
20
55.0
80
20
60.0
35
65
65.0
35
65
Mobile phase A = Buffer:Acetonitrile is 80:20 (v/v)
Mobile phase B = Buffer:Acetonitrile is 20:80 (v/v)
Buffer: 2.76 g of sodium dihydrogen phosphate monohydrate was
dissolved in 1000 ml of water and the pH was adjusted to 5.0 with
[0042] To a 500ml bottle of single oral Compound I (PG1 = PG2 = trimethylsilyl) (10g, 13.3mmol), BSA (lOml), TBAF (0. 2g, 0. 66mmol) and methyl tert-butyl ether 100ml.Stirred for 15 minutes at room temperature, the disappearance of the test compound 8, the reaction was terminated.The pre-mixed solution of isopropanol and 2N sulfuric acid was added to the above solution and stirred at room temperature for 1 hour.Crystallized from aqueous isopropanol final product, the product was filtered and washed with aqueous isopropanol, washed with water until the eluate pH is less than 5.Drying at 60 ° to give the final product 5g, yield: 91%, optical yield was 100%.ΐ NMR (400MHz, d6-D MS0): δ 1. 68 (m, 2H), 1 · 82 (m, 2H), 3. 07 (m, 1H), 4. 47 (d, 1H), 4. 79 (d, 1H), 5. 25 (d, 1H), 6. 75 (d, 2H), 7. 10 (m, 4H), 7. 21 (m, 4H), 7. 29 (m, 2H ), 9. 43 (s, 1H).
CN2006 / 10150638 discloses another improved synthetic process, the intermediate acid chloride (IV) first converted to the Weinreb amide, and then reacted with a Grignard reagent to give the key intermediate I.
The synthesis of four-membered azacycles is of importance because of the chemical and biological relevance of these compounds. Recent progress in copper-catalyzed reactions has been applicable to a variety of research fields, such as heterocyclic synthesis. The aim of the current review is to summarize the synthesis of strained four-membered ring taking advantage of copper catalyzed and mediated processes.
Taken toluene (250 ml) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. Borane DMS complex and (R)-tetrahydro-l-phenyl-3,3-diphenyl-l H,3H-pyrrol (l,2-c)(l,3,2) oxaza borolidine (R-phenyl CBS) is charged into the reaction mass at 0°C. 25 gm of Keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-5°C. Maintained the reaction mass for 3 hrs and quenched with methanol and followed by 1 N hydrochloric acid solution. Organic layer separated and washed with 5% hydrogen peroxide solution and 5% sodium sulfate solution and followed by with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 75°C. Product is isolated in diisopropyl ether and dried the product at 60-70°C for 6 hrs. (Yield: 15 gm). Example-2: Preparation of compound of hydroxy compound of formula-XL
Taken toluene (250 nil) into cleaned R.B.Flask under nitrogen atmosphere and cooled to 0-5°C. DIP Chloride (Mole ratio 1:1.5) into the reaction mass at O0C. 25 gm of keto compound of formula-X is dissolved in toluene(50 ml) and added to the reaction mass at 0-50C. Maintained the reaction mass for 3 hrs and quenched with ammonia solution. Organic layer separated and washed with 10% sodium chloride solution. Distilled the solvent completely under reduced pressure at below 750C. Residue is taken for next stage directly without any purification.
Step-h: Preparation of compound of formula-I (Ezetimibe).
Taken compound of formula-XII (10 gm) and isopropanol (100 ml) into a hydrogenation flask, added 5 % Pd/C ( 4gm) at 25°C and maintained at 45-500C for 3 hrs under hydrogen pressure, filtered through hyflow and washed the Pd/C with isopropanol(20 ml). Distilled the solvent completely under vacuum at below 7O0C, product is recrystallised in dichloromethane (Yield: 6 gm).
Purification of Ezetimibe (formula-1).
Ezetimibe (10 gm) is dissolved in 30 ml of methanol and filtered through hyflow and saturated with DM.Water(30 ml) and stirred for 1 hr at 20-250C. Product filtered and dried for 6-8 hrs at 80-850C (Yield:9 gm):
is an useful intermediate for the preparation of ezetimibe. The intermediates represented by the formula 2 can be prepared economically in good yields as represented by the scheme B.
wherein X is O or S; Y is O, S or N(lower alkyl); and R is alkyl, unsubstituted or substituted phenyl, unsubstituted or substituted naphthyl or lower alkoxy carbonyl, wherein substituents on phenyl and naphthyl are selected from the group consisting of lower alkyl and phenyl. The starting compounds of formula 3 are known or can be obtained from known methods. The reduction may be carried out in a neutral organic solvent or a combination of the neutral organic solvents. Neutral organic solvent means the solvent that is unreactive in the reduction reaction. The preferable organic solvents are chloroalkanes such as methylene dichloride, chloroform, carbon tetrachloride and ethylene dichloride; carbocyclic aromatics such as toluene and benzene; ethers such as methyl tert-butyl ether, diethylether and isopropyl ether; heterocyclic compound such as tetrahydrofuran; dimethylformamide; dimethylsulfoxide; alkanes such as pentane and hexane; and acetonitrile. More preferable solvents are toluene, diethyl ether, isopropyl ether, hexane, methylene dichloride and ethylene dichloride. The preferable reaction temperature is below the boiling temperature of the solvent used, more preferably between about -40°C and the boiling temperature of the solvent, still more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. Quantity of (-)-DIP chloride used is preferably at least about 0.3 mole, more preferably about 0.5 to 10 mole, most preferably about 0.8 to 5 mole per mole of the keto compound of formula 3. Yield of the hydroxy compound of formula 2 is usually above 80%, typically between 90 % to 100%. The compounds of formula 2 wherein X is O; Y is O; and R is alkyl, unsubstituted or substituted phenyl are the preferred. Preferable conditions for obtaining a hydroxy compound of formula 2 from the corresponding keto compound of formula is that the keto compound of the formula 3 is mixed with a neutral solvent, reduced with (-)-DIP chloride at a temperature between -40°C and the boiling temperature of the solvent, more preferably between about -20°C and 40°C and most preferably between about -10°C and 10°C. The reaction mass may be subjected to usual work up. The reaction mass may be used directly in the next step to produce finally ezetimibe, or the hydroxy compound may be isolated and used in the next step. The invention will now be further described by the following examples, which are illustrative rather than limiting. Example 1 3-[5-(4-fluorophenyl)-1 ,5-dioxopentyl]-4-phenyl-2-oxazolidinone (100 gm) is dissolved in toluene (750 ml), the mixture of (-)-β- chlorodiisopinocampheylborane ((-)-DIP chloride) in heptane (545 ml, 1.5M) and toluene (750 ml) is added at 0°C to 5°C for 1 hour. The reaction mixture is stirred for 15 hours at 25°C to 30°C and 340 ml of 10% sodium chloride is then added at the same temperature. The layers are separated and the organic layer is washed with 5% sodium bicarbonate (300 ml), 1 N sulfuric acid (300 ml), and 10% sodium chloride (300 ml). Then the organic layer is dried on sodium sulfate to give 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4-phenyI-2- oxazolidinone in 96% yield. Example 2 The organic layer of 3-[(5S)-5-(4-fluorophenyl)-5-hydroxy-1-oxopentyl]-4- phenyl-2-oxazolidinone from example 1 is mixed with 4-fluoro-N-(4- hydroxyphenyl)methylene-benzenamine (121 gm) and cooled to -10°C. Then diisopropylethylamine (260 ml) is added to the reaction mixture for 45 minutes at -10°C to -15°C, trimethylsilylchloride (135 ml) is added and stirred for 1 hour at -20°C to -25°C. The reaction mixture is cooled to -30°C, TiCI4 (35 ml) is slowly added to the reaction mixture at -30°C to -35°C and stirred for 3 hours at the same temperature. 5% Aq. tartaric acid solution (1700 ml) is added to the reaction mixture at 0°C, stirred for 1 hour and allowed the temperature to rise to 25°C. Then 20% Aq. NaHSO3 (350 ml) solution and stirred for 2 hours at 25°C to 30°C. The organic layer is separated and washed with 1000 ml water, concentrated to 250 ml volume and added 100 ml bistrimethylsilylacetamide. Then the reaction mixture is heated to reflux for 30 minutes. The organic layer is concentrated to remove methylene dichloride, crystallized from the mixture of ethyl acetate (250 ml) and n-heptane (250 ml), and filtered and dried to give 135 gm of compound 4 (prot = trimethylsilyl).
Resolution of trans-2-(alkoxycarbonylethyl)-laitams useful in synthesis of 1-(4-fluorophenyl)-3(R)-[3(s)-hydroxy-3-(flurophenyl)-propyl)]-4(s)-(4-hydroxyphenyl)-2-azetidinone
A method of manufacturing (3r,4s)-l-(4-fluorophenyl)-3-[(3s)-3-(4-fluorophenyl)-3- hydroxypropyl)]-4-(4-hydroxyphenyl)-2-azetidinone and its intermediates
Processes for the preparation of (3r,4s)-4-((4-benzyloxy)phenyl)-1-(4-fluorophenyl)-3-((s)-3-(4-fluorophenyl)-3-hydroxypropyl)-2-azetidinone, an intermediate for the synthesis of ezetimibe
On October 5, the U.S. Food and Drug Administration approved Aristada (aripiprazole lauroxil) extended release injection to treat adults with schizophrenia. Aristada is administered by a health care professional every four to six weeks using an injection in the arm or buttocks.
Schizophrenia is a chronic, severe and disabling brain disorder affecting about one percent of Americans. Typically, symptoms are first seen in adults younger than 30 years of age and include hearing voices, believing other people are reading their minds or controlling their thoughts, and being suspicious or withdrawn.
“Long-acting medications to treat schizophrenia can improve the lives of patients,” said Mitchell Mathis, M.D., director of the Division of Psychiatry Products in the FDA’s Center for Drug Evaluation and Research. “Having a variety of treatment options and dosage forms available for patients with mental illness is important so that a treatment plan can be tailored to meet the patient’s needs.”
The efficacy of Aristada was demonstrated in part by a 12-week clinical trial in 622 participants. In participants with acute schizophrenia who had been stabilized with oral aripiprazole, Aristada was found to maintain the treatment effect compared to a placebo.
Aristada and other atypical antipsychotic drugs used to treat schizophrenia have a Boxed Warning alerting health care professionals about an increased risk of death associated with the off-label use of these drugs to treat behavioral problems in older people with dementia-related psychosis. No drug in this class is approved to treat patients with dementia-related psychosis. Aristada must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The most common side effect reported by participants receiving Aristada in clinical trials was feeling the urge to move constantly (akathisia).
Aristada is manufactured by Alkermes, Inc. of Waltham, Massachusetts.
Chemical process research and development is recognised as a key function during the commercialisation of a new product particularly in the generic and contract manufacturing arms of the chemical, agrochemical and pharmaceutical industries.
The synthesis and individual processes must be economic, safe and must generate product that meets the necessary quality requirements.
This 2-day course presented by highly experienced process chemists will concentrate on the development and optimisation of efficient processes to target molecules with an emphasis on raw material cost, solvent choice, yield improvement, process efficiency and work up, and waste minimisation.
Process robustness testing and reaction optimisation via stastical methods will also be covered.
A discussion of patent issues and areas where engineering and technology can help reduce operating costs.
The use of engineering and technology solutions to reduce costs will be discussed and throughout the course the emphasis will be on minimising costs and maximising returns.
Young Chemists who have just started work in industry as development chemists
Organic Chemists/Medicinal Chemists in Research and Development who would like to gain an appreciation of development and scale up and who are perhaps contemplating moving into chemical development.
Development and Production Chemists in industry who would like to improve their efficiency and gain an insight into alternative approaches to chemical development.
Chemical Engineers who wish to understand a chemist’s approach to chemical development of batch processes. (Engineers would, however, need a good grounding in organic chemistry)
Students who are about to enter the industry and can obtain company sponsorship.
Experienced Chemists looking to refresh and/or augment their knowledge of chemical development
Analytical Chemists who wish to gain a broader appreciation of process chemistry
Managers who might benefit from a comprehensive and up to date overview of chemical development
Introduction Route selection, raw material choice
• Choosing the best route
• Using the cheapest raw materials and reagents, back integration of raw material supply
• Reducing the number of steps vs. reagent choice / yield and cost
Solvent selection
• Solvent cost, recyclability
• Solvent reactivity and solvent swapping
• Solvent choice for reaction and work up
Process optimisation
• Reaction quench
• Work up
• Product isolation (crystallisation, filtration and drying)
Statistical methods of optimisation
• Design of experiments
• Factorial and fractional factorial design
• Response surface analysis
• Robustness testing
Regulatory and Quality issues
• Impurity control and tracking
• Process validation and QbD
• Vessel cleaning
Patent issues
• Patents basics
• Patent definition
• Where patents are in force
• How to work around patents
Use of technology and engineering
• Flow chemistry
• SMB chromatography
• Separation technologies
At the end of the course participants will have gained:
• A logical investigative approach to chemical development and optimisation
• An insight into the factors involved in development and scaleup
• A preliminary knowledge of statistical methods of optimisation
• Improved ability to decide which parts of the chemical process to examine in detail.
• Ideas for efficient resource allocation
• Improved troubleshooting and problem solving ability
• A basic outline of the patent system
• An appreciation of how to assess the main cost contributors in a process
C22H48N12O2
Mw: 512.40232
Mechanism of Action: an intravenously administered anticoagulant Reversal Agent
Blood coagulation factor modulators; Factor Xa inhibitors
Indication: Anticoagulant Reversal
Development Stage: Phase II
Developer:Perosphere, Inc..Perosphere Inc.
Highest Development Phases
Phase IIHaemorrhage
Most Recent Events
02 Apr 2015Ciraparantag receives Fast Track designation for Haemorrhage [IV] (In volunteers) in USA
05 Nov 2014Efficacy and adverse events data from a phase I/II trial in Haemorrhage released by Perosphere
Ciraparantag, also known as PER977, is a A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. PER977 is water-soluble, cationic molecule that is designed to bind specifically to unfractionated heparin and low-molecular-weight heparin through noncovalent hydrogen bonding and charge–charge interactions.
PER-977 is an intravenous heparin neutralizer in phase II clinical trials at Perosphere to reverse edoxaban’s induced anticoagulation.
In April 2015, fast track designation was assigned in the U.S. as an investigational anticoagulant reversal agent.
is synthesized by reacting excess equivalents (e.g., at least about two equivalents) of compound 1
with one equivalent of compound 2
in the presence of a peptide coupling reagent, to obtain a compound 3
wherein PI is a protecting group and P2 is a protecting group or is a hydrogen.
the coupling involved reacting compound 1, wherein PI was Boc and P2 was a hydrogen (depicted as Boc-Arg-OH HCl below), with compound 2 as depicted below:
The resultant crude product was more than 95% pure by thin layer
chromatography (TLC).
Subsequently, the deprotection step was carried out as depicted below:
The deprotected product was purified by preparative HPLC using 1% acetic acid buffer. Product purity of >98% was observed. Residual TFA was removed by low quantity of DOWEX resin. The molecular weight of DAP (the compound of Formula V) is 512.4, and the compound synthesized according to the above scheme exhibited the following primary peak by mass spectroscopy: [M+H]+=513.4.
References
1: Dzik WH. Reversal of oral factor Xa inhibitors by prothrombin complex concentrates: a re-appraisal. J Thromb Haemost. 2015 Jun;13 Suppl 1:S187-94. doi: 10.1111/jth.12949. PubMed PMID: 26149022.
2: Crowther M, Crowther MA. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler Thromb Vasc Biol. 2015 Aug;35(8):1736-45. doi: 10.1161/ATVBAHA.114.303402. Epub 2015 Jun 18. PubMed PMID: 26088576.
3: Sullivan DW Jr, Gad SC, Laulicht B, Bakhru S, Steiner S. Nonclinical Safety Assessment of PER977: A Small Molecule Reversal Agent for New Oral Anticoagulants and Heparins. Int J Toxicol. 2015 Jun 15. pii: 1091581815590667. [Epub ahead of print] PubMed PMID: 26079256.
4: Mo Y, Yam FK. Recent advances in the development of specific antidotes for target-specific oral anticoagulants. Pharmacotherapy. 2015 Feb;35(2):198-207. doi: 10.1002/phar.1532. Epub 2015 Feb 3. PubMed PMID: 25644580.
5: Yates SW. Interrupting anticoagulation in patients with nonvalvular atrial fibrillation. P T. 2014 Dec;39(12):858-80. PubMed PMID: 25516695; PubMed Central PMCID: PMC4264672.
6: Vanden Daelen S, Peetermans M, Vanassche T, Verhamme P, Vandermeulen E. Monitoring and reversal strategies for new oral anticoagulants. Expert Rev Cardiovasc Ther. 2015 Jan;13(1):95-103. doi: 10.1586/14779072.2015.987126. Epub 2014 Nov 28. PubMed PMID: 25431993.
7: Costin J, Ansell J, Laulicht B, Bakhru S, Steiner S. Reversal agents in development for the new oral anticoagulants. Postgrad Med. 2014 Nov;126(7):19-24. doi: 10.3810/pgm.2014.11.2829. Review. PubMed PMID: 25387210.
8: Ansell JE, Bakhru SH, Laulicht BE, Steiner SS, Grosso M, Brown K, Dishy V, Noveck RJ, Costin JC. Use of PER977 to reverse the anticoagulant effect of edoxaban. N Engl J Med. 2014 Nov 27;371(22):2141-2. doi: 10.1056/NEJMc1411800. Epub 2014 Nov 5. PubMed PMID: 25371966.
9: Hankey GJ. Intracranial hemorrhage and novel anticoagulants for atrial fibrillation: what have we learned? Curr Cardiol Rep. 2014 May;16(5):480. doi: 10.1007/s11886-014-0480-9. Review. PubMed PMID: 24643903.
To improve glycemic control in adults with type 2 diabetes mellitus as an adjunct to diet and exercise Drug Trials Snapshot Phase II Developer:Theracos, Inc.
Conditions
Phases
Recruitment
Interventions
Sponsor/Collaborators
Diabetes Mellitus Type 2
Phase 2
Completed
Drug: EGT0001442|Drug: Placebo capsules to match EGT0001442
L-Proline, compd. with (1S)-1,5-anhydro-1-C-(4-chloro-3-((4-(2-(cyclopropyloxy)ethoxy)phenyl)methyl)phenyl)-D-glucitol (2:1)
Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.
Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys.
Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).
The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)
To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.
Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)
To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)
A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)
To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)
To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.
Preparation of (2-chloro-5-iodophenyl)methan-1-ol
A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.
Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex
Example 1A
Preparation of 2-cyclopropoxyethanol (1)
To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)
To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)
A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)
To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)
To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.
To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7
[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8
A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References: 1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
Bexagliflozin (Brenzavvy). Bexagliflozin (3) was discoveredanddevelopedbyTheracosBioforthetreatmentof type2diabetesmellitus.28Bexagliflozinisasodium-dependent glucose cotransporter 2 (SGLT2) inhibitor. Inhibition of SGLT2 reduces blood sugar without stimulating insulin release.29 Bexagliflozin shows >2000-fold selectivity forSGLT2 over SGLT1 and demonstrated improvement inglycemiccontrolwithaoncedaily,20mgdose.28Since 2011, there have been 11 therapeutics targeting SGLT2.30Thesedrugsexhibit commonstructural features(abiarylmethaneandglycoside)andlikelyfacesimilarsynthetic challenges.31 The medicinal chemistry efforts to identifybexagliflozinweredisclosedintheprimaryliterature.32Apatent fromTheracos, Inc. in2013describedasyntheticapproachto bexagliflozinonmultikilogramscale.33Slightvariations inthe reactionconditions,yieldandisolationstrategyofintermediates wereincludedinthepatent.Theimplementationoftelescoping intheprocessislikelyduetopoorcrystallinityofintermediates, whichmaybeacommonchallengetootherSGLT2inhibitors.31 Anotherpatent disclosedbyPiramal Enterprises suggesteda similarbondformationstrategybut includedanacetylationof bexagliflozinprior tothefinal isolation inorder toprovidea crystallinesolid.34 Bexagliflozinwas assembled by cryogenicmetal halogen exchangeof aryl iodide3.1with turboGrignard(i-PrMgCl·LiCl)andsubsequentadditiontoprotectedgluconolactone3.2 whichwaspreparedbytreatmentofD-(+)-glucono-1,4-lactonewithTMSClandNMMinTHFin94%yield(Scheme4).WhentheGrignardadditionwascomplete,thereactionwasquenchedand a solution of the product inEtOAcwas treatedwith activated carbon, filtered, concentrated, and diluted with methanol.ThissolutionwastreatedwithconcentratedHCl to remove thesilyl protectinggroupsandprovidecrudemethyl ketal3.3inyields rangingfrom79to95%.Themethyl ketal functionalitywasreducedusingtriethylsilaneandBF3·Et2Oin DCMandMeCNatcryogenictemperaturestoprovidecrude bexagliflozin (3) as a solid after concentrating the reaction mixture. Alternatively, a larger-scale demonstration of this processinthepatenttelescopedasolutionofcrudebexagliflozin toformabis-L-prolinecomplexinethanol,water,andheptane, whichwasisolatedasacrystallinesolidin81%yield.Thiswas convertedto the free formin82%yieldbycrystallization in methanolandwater.Arecrystallizationofbexagliflozin(3)was reported in 92% yield. Details on stereoselectivity of this approachwerenotdisclosed. Amilligram-togram-scaleconstructionofthearyliodide3.1 wasalsodisclosedintheTheracospatent from2013(Scheme 5).33First,carboxylicacid3.5wasreducedtoprimaryalcohol 3.6using sodiumborohydride and iodine. Next, the diaryl methanecorewas assembledbyFriedel−Crafts alkylationof phenol with3.6 after activationwithHBr andZnCl2. This reactionwasdemonstratedonmilligramscaleandachieved65% conversion, with 52% isolated yield after chromatographic purification.Analternativeapproachtoabromovariantofaryl iodide3.7waspresentedina2009patentfromTheracos,where Friedel−Craftsacylationprovidedtheanalogousbenzophenone intermediatewhichwas thensubsequentlyreduced.35Finally,alkylationofthephenolwasconductedusingthetosylatedether 3.8toprovidearyl iodide3.1in75%yieldonkilogramscale.A syntheticapproachtothetosylatedetherwasprovidedinthe earlyTheracospatent,35wherecyclopropylether formationin 3.10wasgeneratedviaGrignardformationandrearrangement of 2-(2-bromoethyl)-1,3-dioxolane 3.9 (Scheme 6). The primary alcohol 3.10was protectedas the tosylate3.8and employedinthealkylationstepwithoutpurification.Noyields wereprovided.
(28) Hoy, S. M. Bexagliflozin: first approval. Drugs 2023, 83, 447− 453. (29) Hsia, D. S.; Grove, O.; Cefalu, W. T. An update on sodium glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 73−79. (30) Guo, Y.-Y.; Zhang, J.-Y.; Sun, J.-F.; Gao, H. A comprehensive review of small-molecule drugs for the treatment of type 2 diabetes mellitus: Synthetic approaches and clinical applications. Eur. J. Med. Chem. 2024, 267, No. 116185. (31) Aguillón, A. R.; Mascarello, A.; Segretti, N. D.; de Azevedo, H. F. Z.; Guimaraes, C. R. W.; Miranda, L. S. M.; de Souza, R. O. M. A. Synthetic strategies toward SGLT2 inhibitors. Org. Process Res. Dev. 2018, 22, 467−488. (32) Xu, B.; Feng, Y.; Cheng, H.; Song, Y.; Lv, B.; Wu, Y.; Wang, C.; Li, S.; Xu, M.; Du, J.; et al. C-aryl glucosides substituted at the 4′ position as potent and selective renal sodium-dependent glucose co transporter 2 (SGLT2) inhibitors for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett. 2011, 21, 4465−4470. (33) Xu, B.; Lv, B.; Xu, G.; Seed, B.; Roberge, J. Y. Process for the preparation of benzyl-benzene C-glycosides via coupling reaction as potential SGLT2 inhibitors. US 20130267694, 2013. (34) Gharpure, M.; Sharma, S. K.; Vishwasrao, S.; Vichare, P.; Varal, D. Aprocess for the preparation of SGLT2 inhibitor and intermediates thereof. WO 2018207113, 2018. (35) Song, Y.; Chen, Y.; Cheng, H.; Li, S.; Wu, Y.; Feng, Y.; Lv, B.; Xu, B.; Seed, B.; Hadd, M. J.; et al. Preparation of benzylbenzene glycoside derivatives as antidiabetic agents. WO 2009026537, 2009.
Bexagliflozin (Brenzavvy) On January 20, 2023, the FDA granted approval to Bexagliflozin, a medication developed by Theracos Inc, for the treatment of type 2 diabetes mellitus (T2DM) [104–106]. The SGLT2 inhibitor Bexagliflozin can increase energy expenditure, reduce fluid retention, and increase urinary glucose excretion by inhibiting SGLT2 in renal tubular epithelial cells [106]. SGLT2 inhibitors have significant advantages compared to other drugs: (1) they can lower both pre-meal and post-meal blood sugar levels (not all drugs can lower both); (2) they have a lower risk of hypoglycemia as they do not stimulate insulin secretion; (3) they have adiuretic effect due to their primary action on the renal tubules, which lowers systolic blood pressure; (4) research has shown that SGLT2 in hibitors have therapeutic effects on diabetic kidney disease [107,108]. The process of synthesizing Bexagliflozin started by conducting theFriedel-Crafts acylation of ethoxybenzene (BEXA-002) with 5-bromo-2-chlorobenzoic acid (BEXA-001) (Scheme 29) [109]. This reaction produced ketone BEXA-003. Subsequently, the carbonyl reduction of BEXA-003 was carried out using trifluoromethanesulfonic acid (TfOH),triethylsilane, and TFA. This step yielded BEXA-004. Next, n-butyllithium (n-BuLi) and pyrone BEXA-005 were combined with BEXA-004 at78◦C. This reaction produced an intermediate, which was thenreacted with triethylsilane and BF◦3⋅Et2O at 0C. The final product obtained from this reaction was BEXA-006, which contained a sugar ring. BEXA-006 underwent dealkylation upon treatment with boron tribromide, resulting in the formation of BEXA-007, which was a phenol. Subsequently, BEXA-007 was alkylated using 2-cyclopropoxyethyl4-methylbenzenesulfonate (BEXA-008) to yield Bexagliflozin.
[104] S.M. Hoy, Bexagliflozin: first approval, Drugs 83 (2023) 447–453. [105] W. Zhang, A. Welihinda, J. Mechanic, H. Ding, L. Zhu, Y. Lu, Z. Deng, Z. Sheng, B. Lv, Y. Chen, J.Y. Roberge, B. Seed, Y.X. Wang, EGT1442, a potent and selectiveSGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats, Pharmacol. Res. 63 (2011) 284–293. [106] O. Azzam, R. Carnagarin, L.M. Lugo-Gavidia, J. Nolde, V.B. Matthews, M. P. Schlaich, Bexagliflozin for type 2 diabetes: an overview of the data, Expet Opin. Pharmacother. 22 (2021) 2095–2103. [107] B.F. Palmer, D.J. Clegg, Kidney-protective effects of SGLT2 inhibitors, Clin. J. Am. Soc. Nephrol. 18 (2023) 279–289. [108] M. Singh, A. Kumar, Risks associated with SGLT2 inhibitors: an overview, Curr. Drug Saf. 13 (2018) 84–91. [109] Y. Song, Y. Chen, H. Cheng, S. Li, Y. Wu, Y. Feng, B. Lv, B. Xu, B. Seed, M.J. Hadd, J. Du, C. Wang, J.Y. Roberge, Preparation of Benzylbenzene Glycoside Derivatives as Antidiabetic Agents, 2009. WO2009026537A1.
.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
LY2801653, also known as Merestinib,is an orally available, small molecule inhibitor of the proto-oncogene c-Met (mesenchymal-epithelial transition, also known as hepatocyte growth factor receptor [HGFR]) with potential antineoplastic activity. c-Met inhibitor LY2801653 selectively binds to c-Met, thereby inhibiting c-Met phosphorylation and disrupting c-Met signal transduction pathways. This may induce cell death in tumor cells overexpressing c-Met protein or expressing constitutively activated c-Met protein. This agent has potent anti-tumor efficacy in mono- and combination therapy in a broad range of cancers. c-Met, a receptor tyrosine kinase overexpressed or mutated in many tumor cell types, plays key roles in tumor cell proliferation, survival, invasion, metastasis, and tumor angiogenesis.
LY2801653 was identified and developed as a novel, potent, and orally active small molecule inhibitor of human c-Met. It demonstrated dose dependent inhibition of c-Met phosphorylation in xenograft tumors with a long lasting PD effect. LY2801653 displayed potent anti-tumor efficacy in a number of non small cell lung, renal, pancreatic, and breast tumor models. Examination of c-Met expression in these tumors by immunohistochemistry (IHC) revealed a good correlation between response and c-Met expression in the tumor tissue. LY2801653 treatment led to increase in functional vessel areas, and decrease in tumor hypoxia. Enhanced anti-tumor efficacy was achieved when Erlotinib was combined with LY2801653. . (source: http://cancerres.aacrjournals.org/cgi/content/meeting_abstract/70/8_MeetingAbstracts/3611).
Example 1 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), 1-(4-flurorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g, 7.6 mmol) and HOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5×100 mL). The combined aqueous solution is extracted with EtOAc (1×100 mL) and then the combined organic solutions are dried over N2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+H), 675 (M+Na).
Example 2 N-(3-Fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide
A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. 1-(4-Fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxylic acid (259.46 g, 1.05 mol) and EDCI (228.63 g, 1.19 mol) are added and it is rinse in with DMF (500 mL). Then HOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3×3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2×500 mL). The light tan solid is dried overnight in the vacuum oven at 40° C. to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (424 g, 68%). MS (m/z): 651.0 (M−H).
tert-Butyl 4-(5-(2-fluoro-4-(1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamido)phenoxy)-1-methyl-1H-indazol-6-yl)-1H-pyrazole-1-carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30° C. for 1 hour. Then the reaction mixture is heated to 45° C. and DCM (1.5 L) is added. After two hours, the solution is heated to 50° C. and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60° C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about ⅓ volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10-15° C. and then filtered. The solids are washed with MeOH. The solids are placed in a 55° C. vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M−H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(1-methyl-6-(1H-pyrazol-4-yl)-1H-indazol-5-yloxy)phenyl)-1-(4-fluorophenyl)-6-methyl-2-oxo-1,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pH is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOH solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOH (600 mL). The solids are placed in a vacuum oven at 35° C. overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery). TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55° C. in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling.
The jet mill (Aljet™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 lb header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial. The sample is heated to 70° C. with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8° C. until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45° C. for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238° C. by TGA. The form’s onset of melting is 247.8° C.
An NH4Cl-catalyzed ethoxy ethyl deprotection was developed for the synthesis of merestinib, a MET inhibitor. Alternative reactor technologies using temperatures above the solvent boiling point are combined with this mild catalyst to promote the deprotection reaction. The reaction is optimized for flow and has been used to synthesize over 100 kg of the target compound. The generality of the reaction conditions is also demonstrated with other compounds and protecting groups.
To a 100 mL round bottom flask is added tert-butyl 4-(5-(4-amino-2- fluorophenoxy)-l -methyl- IH- indazol-6-y I)- lH-pyrazole-1-carboxylate (1.43 g, 3.38 mmol), l-(4-fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxylic acid (1.25 g, 5.07 mmol), EDCI (1.48 g , 7.6 mmol) and ΗOBt (776 mg, 5.07 mmol) followed by DMF (15 mL, 193.99 mmol) and then DIPEA (1.47 mL, 8.44 mmol). The mixture is allowed to stir at RT overnight. The reaction mixture is diluted into EtOAc (300 mL) and washed with saturated aqueous sodium chloride (5 x 100 mL). The combined aqueous solution is extracted with EtOAc (1 x 100 mL) and then the combined organic solutions are dried over Na2SO4, filtered, and concentrated to dryness. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in a DCM solution (B), gradient from 100% (A) to 80% (A):20% (B) over 70 min to give tert-butyl 4-(5-(2- fluoro-4-(l-(4-fluoroplienyl)-6-metliy 1-2-oxo- 1,2-dihy dropyridine-3- carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 -carboxylate as a gold solid (2.20 g, 87% yield). MS (m/z): 653. (M+Η), 675 (M+Na).
To a round bottom flask is added tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)- 6-methyl-2-oxo- 1 ,2-dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6- yl)-lΗ-pyrazole-l -carboxylate (1.92 g, 2.94 mmol) and DCM (50 mL) followed by triethylsilane (1.88 mL, 11.77 mmol) and TFA (17.8 mL, 235.35 mmol). The reaction mixture is allowed to stir at RT for 1.5 hours. The solvent is removed and diluted into DCM (150 mL) and washed with saturated aqueous NaHCθ3 solution (2 x 100 mL). The organic solution is dried with Na2SO4, and concentrated under reduced pressure to give a solid material. The solid is purified on a silica gel column eluting with DCM (A) and a 10% MeOH in DCM solution (B), gradient from 100% (A) to 75%(A):25%(B) over 70 min, held at this 75:25 ratio for 15 min to give the title compound as an off-white solid. The solid is dissolved in hot EtOH (50 mL) followed by a portion-wise addition of distilled water (250 mL) causing a white solid to precipitate. The solid is filtered over a Buchner funnel and washed with distilled water (3 x 15 mL), air dried, and vacuum dried at 60 0C for 15 hours to give the title compound as an off-white solid (1.27 g, 78% yield). MS (m/z): 552.8 (M+H).
A 12 L round bottom flask is equipped with overhead agitation, a thermocouple, and a N2 purge. tert-Butyl 4-(5-(4-amino-2-fluorophenoxy)-l -methyl- lH-indazol-6-yl)- lH-pyrazole-1-carboxylate (404 g, 954.08 mmol) is dissolved in DMF (2 L) and charged to the flask. DMF (1 L) is used to rinse the flask. l-(4-Fluorophenyl)-6-methyl-2-oxo- l,2-dihydropyridine-3-carboxylic acid (259.46 g ,1.05 mol) and EDCI (228.63 g , 1.19 mol) are added and it is rinse in with DMF (500 mL). Then ΗOBt (189.94 g, 1.24 mol) is added and it is again rinsed in with DMF (500 mL). Finally, DIPEA is slowly added (184.97 g, 1.43 mol). The dark solution is then stirred at RT over the weekend. To a 20 L bottom outlet flask is added DI water (3 L) and DCM (5 L). The reaction mixture is poured in and it is rinsed in with DCM (1 L). The organic layer is separated, washed with DI water (3 X 3 L), dried over Na2SO4, filtered, rinsed solids with DCM and concentrated the filtrate. EtOAc (2 L) is added to the residue and the solution is stirred for 1 hour. The product crystallizes out. The mixture is concentrated. Another portion of EtOAc (2 L) is added and concentrated to remove all of the DCM. EtOAc (650 mL) and MTBE (3 L) are added to the residue and the solution is stirred in an ice bath for 1 hour. The tan slurry is filtered using a polypropylene pad. The cake is rinsed with MTBE (2 x 500 mL). The light tan solid is dried overnight in the vacuum oven at 40 0C to give the crude product (553 g). The crude product is purified by silica gel column chromatography eluting with (50% EtOAc (50%):35% DCM (35%): n-heptane (15%)) to give the pure desired product tert-butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (424 g, 68%). MS (m/z): 651.0 (M-H). tert-Butyl 4-(5-(2-fluoro-4-(l-(4-fluorophenyl)-6-methyl-2-oxo-l,2- dihydropyridine-3 -carboxamido)phenoxy)- 1 -methyl- lH-indazol-6-yl)- lH-pyrazole- 1 – carboxylate (423.9 g, 649.50 mmol) is dissolved in DCM (4.24 L). HCl in MeOH (5.74 N, 799.99 mL, 4.59 mol) is added and the solution is heated at 30 0C for 1 hour. Then the reaction mixture is heated to 45 0C and DCM (1.5 L) is added. After two hours, the solution is heated to 50 0C and DCM (2 L) is added. After 3 hours, DCM (2 L) is added followed by HCl in MeOH (4.5 N, 721.67 mL, 3.25 mol). After another 45 min, DCM (1 L), HCl in MeOH (4.5 N, 288.67 mL, 1.30 mol), and MeOH (1.5 L) are added. The reaction solution is then heated to 60 0C. After 4 hours, MeOH (2 L) is added and 10 min later DCM (1 L) is added followed by HCl in MeOH (4.5 N, 200 mL). After 5 hours, the reaction is complete. The reaction mixture is concentrated to about 1/3 volume. MeOH (2 L) is added and the solution is concentrated to a thick slurry. Again, MeOH (2 L) is added and the mixture is concentrated to a thick slurry. The slurry is cooled to about 10- 15 0C and then filtered. The solids are washed with MeOH. The solids are placed in a 55 0C vacuum oven for 2 days to give the desired product N-(3-fluoro-4-(l-methyl-6-(lH- pyrazol-4-yl)- lH-indazol-5-yloxy)phenyl)- 1 -(4-fluorophenyl)-6-methyl-2-oxo- 1,2- dihydropyridine-3-carboxamide hydrochloride (377 g, 92.8%). MS (m/z): 551.0 (M-H).
To a 22 L round bottom flask equipped with mechanical stirring under nitrogen is added N-(3-fluoro-4-(l-methyl-6-(lH-pyrazol-4-yl)-lH-indazol-5-yloxy)phenyl)-l-(4- fluorophenyl)-6-methyl-2-oxo-l,2-dihydropyridine-3-carboxamide hydrochloride (367 g, 0.62 mol) followed by DCM (7.34 L) and water (7.34 L). Na2CO3 (181.6 g, 1.71 mol) is added and the mixture is stirred at RT for 30 min. The pΗ is checked and found to be about 9.4. The mixture is filtered over polypropylene. The solids are collected and placed into a 5 L round bottom flask. A 20% water/MeOΗ solution (2.6 L) is added and the slurry is stirred for 30 min. The slurry is filtered and the solids are washed with 20% water/MeOΗ (600 mL). The solids are placed in a vacuum oven at 35 0C overnight. The first weighing indicates 394 g (theoretical yield 324.8 g, about 121% mass recovery).
TGA (Thermogravimetric analysis)/DSC (differential scanning calorimetry) shows about 17 wt % free water and 10-11 wt% volatile loss at the melt. The solids are dried at 55 0C in a vacuum oven with a N2 sweep for 3.5 hours (354.7 g, about 109% mass recovery, NMR shows about 9.3 wt % DCM). No free water is present according to TGA/DSC. The material is sent for milling. The jet mill (AIj et™ 0101) in a glove bag is assembled inside a walk in hood and hooked up to N2 to a 100 Ib header. The inlet pusher nozzle is adjusted for maximum draw and max nitrogen flow is introduced into the mill. Pressure readings are noted as 90 psi on pusher nozzle and 85 psi on both grind nozzles. The starting material (353.4 g) is slowly fed to the mill inlet, stopping to empty the receiver sock as needed. The total milling time is 22 min and 25 second. The calculated feed rate is 15.8 g/min (353.4 grams divided by 22.42 min). The milled material (335.7 g, 95%) is obtained with 17.7 g loss. Particle size analysis result of the milled material is d90 of 4.6 microns.
Anhydrous Crystal Form Preparation To 10 mL of EtOH is added 120 mg of the above compound into a 20 mL vial.
The sample is heated to 70 0C with stirring. Initially the solids start to dissolve and then a suspension forms followed by a white precipitate. The sample is cooled to RT while being stirred. A small sample of the slurry is taken by pipette and allowed to air dry. This material is highly crystalline and proves to be an ethanol solvate by TGA. To the remaining suspension, 10 mL of heptane is added and then heated to boiling. The measured temperature is monitored at 70.8 0C until the volume has been reduced to 10 mL. When the temperature starts to rise, the heat is removed and the slurry stirred at RT overnight. The solid is isolated by vacuum filtration and dried in a vacuum oven at 45 0C for 3 hours, resulting in 77% recovery. The crystalline form shows a weight loss of 0.17% from 25-238 0C by TGA. The form’s onset of melting is 247.80C.
References
1: Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, Shih C, Stewart JA, Stewart TR, Stout SL, Uhlik MT, Um SL, Wang Y, Wu W, Yan L, Yang WJ, Zhong B, Walgren RA. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2012 Dec 29. [Epub ahead of print] PubMed PMID: 23275061.
Liu, X.; Newton, R. C.; Scherle, P. A.Expert Opin. Invest. Drugs2011, 20, 1225,DOI: 10.1517/13543784.2011.600687
Yan, S. B.; Peek, V. L.; Ajamie, R.; Buchanan, S. G.; Graff, J. R.; Heidler, S. A.; Hui, Y.;Huss, K. L.; Konicek, B. W.; Manro, J. R.; Shih, C.; Stewart, J. A.; Stewart, T. R.; Stout, S. L.; Uhlik, M. T.; Um, S. L.; Wang, Y.; Wu, W.; Yan, L.; Yang, W. J.; Zhong, B.; Walgren, R. A.Invest. New Drugs2013, 31, 833, DOI: 10.1007/s10637-012-9912-9
Kallman, N. J.; Liu, C.; Yates, M. H.; Linder, R. J.; Ruble, J. C.; Kogut, E. F.; Patterson, L. E.; Laird, D. L. T.; Hansen, M. M.Org. Process Res. Dev.2014, 18, 501,DOI: 10.1021/op400317z
Kallman, N.J.; Yates, M.H.; Linder, R.J.; Hansen, M.M.
Route design and development of c-Met inhibitor LY2801653
244th Am Chem Soc (ACS) Natl Meet (August 19-23, Philadelphia) 2012, Abst ORGN 212
////////
UPDATE…..
A new synthesis of a key indazole-containing building block for the MET kinase inhibitor merestinib was designed and demonstrated. Crucial to the successful construction of the challenging indazole is an SNAr reaction, which forges the heterocyclic ring. Continuous processing was applied to two of the five steps: nitration of a benzaldehyde and high-temperature hydrolysis of an aniline to phenol. Compared to a highly developed historical route, the new route shows clear benefits in terms of product quality and potentially manufacturability and robustness.
The chemical name of sildenafil is 5-[2-ethoxy-5-(4-methylpiperazin-1-ylsulfonyl)phenyl]-1- methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one and its formula is C22H30N6O4S. The melting point of sildenafil is 189-190oC. Its solubility is 3.5 mg/mL in water.
The 1H NMR data of sildenafil is given below. The abbreviations used are s for singlet, d for doublet, t for triplet and q for quartet. The chemical shifts are given in ppm (parts per million) and are followed by the number of Hydrogens the peaks account for:
Common side effects include headaches and heart burn, as well as flushed skin. Caution is advised in those who have cardiovascular disease. Rare but serious side effects include prolonged erections, which can lead to damage to the penis, and sudden-onset hearing loss. Sildenafil should not be taken by people who take nitrates such as nitroglycerin, as this may result in a severe and potentially fatal drop in blood pressure.[1]
It was originally discovered by Pfizerscientists Andrew Bell, David Brown, and Nicholas Terrett.[2][3] Since becoming available in 1998, sildenafil has been a common treatment for erectile dysfunction; its primary competitors are tadalafil (Cialis) and vardenafil (Levitra).
EP0463756A,US6469012,WO2008074512A1
Chemical synthesis
Dunn PJ (2005). “Synthesis of Commercial Phosphodiesterase(V) Inhibitors”. Org Process Res Dev2005 (1): 88–97. doi:10.1021/op040019c.
The preparation steps for synthesis of sildenafil are:[40]
Methylation of 3-propylpyrazole-5-carboxylic acid ethyl ester with hot dimethyl sulfate
The synthesis of sildenafil citrate was first reported in the Bioorganic & Medicinal Chemistry Letters, Vol 6, pp. 1819, 1824, 1996. The reaction scheme is reproduced below. Sildenafil was reported in this journal as “a potent and selective inhibitor of type 5 PDE with utility for the treatment of male erectile dysfunction”.
he first step of the synthesis is the reaction of a diketoester (1) and hydrazine to give the pyrazole ring. The regioselective N-methylation of the pyrazole and hydrolysis gives a carboxylic acid (3). Compound (3) is then reacted with HNO3 and H2SO4 to give a nitrated product.
This is then followed by a carboxamide formation and the reduction of the nitro group. The compound (4) is then acylated under basic conditions and this produces the pyrazolopyrimidinone (6). (6) is then chlorosulphonylated selectively on the 5′-position of the phenyl ring. This can then couple with an amine to give sildenafil (7).
The yield of each step is given on the reaction scheme.
This is the original synthesis which was reported in the literature when the molecule was first synthesised. A variant of the synthesis was published but the changes it involved only consisted in the change of a few reactants, and no major changes were reported. This synthesis appeared in the January 1999 issue of Chemistry in Britain. This journal only reported the original discovery synthesis and said that the synthesis used commercially had not been published.
The drug is commercially manufactured by an alternative route. The reaction scheme is described in the patent which was published on 17 decembre 1997. However, the synthesis used in the commercial manufacture could be different to this. The patent was filed by the Pfizer Research and Development Company. The scheme is reproduced below.
The synthesis was described in a lot of detail, including the solvents that were the best to use, however, these details have not been reproduced here. These and further details about the synthesis can be found on the original patent document.
The reaction pathway is explained in more detail below.
Compound 2 can be prepared by the chlorosulphonation of 2-ethoxybenzoic acid (1). The conversion of compound 2 to compound 4 is achieved by N-sulphonation of 1-methylpiperazine and may be conducted in a one or two step procedure. Coupling of compound 4 with compound 6 can be achieved by any of the known amide bond-forming reactions. The aminopyrazole (6) is obtainable by the conventional reduction of the corresponding nitropyrazole (5). The resulting solution of compound 6 may be used directly after filtration in the coupling reaction with compound 4.
The cyclisation of compound 7 to give sildenafil has been achieved in yields up to 95%. Thus the overall yield of sildenafil based on compound 1 as a starting material, depending on whether the one or two step sulphonylation procedure is used can be as high as 51.7% or 47.8% respectively. This compares favourably with the first synthesis in which the overall yield is 27.6%.
The cyclisation of compound 7 to sildenafil can be conducted under neutral or acidic conditions. Under neutral conditions, compound 7 is heated, optionally in the presence of a solvent and/or optionally in the presence of a dehydrating agent and/or mechanical water removal system. Under acidic conditions, the reaction is carried out with a prolic acid or Lewis acid optionally in the presence of a solvent.
The reagents employed in the reactions can vary, but the following are among the ones recommended by the submitters of the patent:
The first step is the chlorosulphonylation of 2-ethoxybenzoic acid. This can be achieved by reacting 1 equivalent mole of thionyl chloride with 4 equivalent mole of chlorosulphonic acid. Addition of 1-methylpiperazine to an aqueous suspension of compound 2 is a suitable reaction to obtain compound 4 in one step. The carboxylic function of compound 4 can be activated using a 5% excess of N,N’-carbonyldiimidazole in ethyl acetate. This intermediate can then be reacted with imidazolide and compound 6. Compound 6 is obtainable by reduction of the corresponding nitropyrazole 5 for example by using palladium catalysed hydrogenation in ethyl acetate. Compound 7 is then cyclised to complete the reaction scheme and give sildenafil.
Information about the synthesis used to manufatcure Viagra was not available, and the two presented above are only the ones which were published. It is not surprising that the commercial manufacture of the drug is by a pathway that is not published.
Preparation of 2- hydroxy-5-(4 methyl)-l-piperazinyl sulphonyl) benzoic acid Step-1: Preparation of 5-Chlorosulfonyl-2-hydroxy benzoic acid
To the chilled chlorosulfonic acid (1012 g), salicylic acid (200 g) was added at 0-50C over a period of 1 hour 40 min. The temperature of the reaction mixture was maintained at 20-250C for 2 hrs. Then thionyl chloride (172.4 g) was added over a period of 15 min and maintained for 12 hrs. The product formed was poured onto ice and maintained for lhr. The product was filtered and washed with DM water to get 5-Chlorosulfonyl-2- hydroxy benzoic acid.
Step-2: Preparation of 2-hydroxy-5-(4-methyI)-l-piperazinylsulphonyl)benzoic acid
5-Chlorosulfonyl-2-hydroxy benzoic acid (40Og) obtained in step 1 was dissolved in acetone (1200 ml) and cooled to 5-100C. To this clear solution N-methyl piperazine (254 g) was added and maintained for 2 hrs. The product formed was filtered, washed with water and purified in methanol to get 308 g of the titled compound.
Preparation of 4-[2-hydroxy-5-(4-methyI-l-piperazinyIsulphonyl)benzamido]-l- methyl-3-n-propyl-lH-pyrazole-5-carboxamide
2-Hydroxy-5-(4-methyl-l-piperazinylsulphonyl)benzoic acid (10Og) was dissolved in dichloromethane (500 ml) and triethylamine (50 ml) followed by distillation to get residual mass. The residual mass was dissolved in dichloromethane (1500ml) followed by the addition of 1,3-dicyclohexylcarbodiimide (75.6 g) and 1-hydroxybenzotriazole (45g). The reaction mixture was stirred at 27-280C and then 4-amino-l-methyl-3-n-propyl- pyrazole-5-carboxamide (60.6 g) was added. The reaction mixture was heated to reflux temperature and maintained for 3 hours. Filtered the undissolved material at hot and washed the cake with dichloromethane (200ml). The filtrate was distilled out completely to get residue. Dissloved the residue in methanol (300ml) at 4O0C and then cooled the mass to 27-280C and stirred overnight. Further, cooled the mass to 5-70C and stirred for lhr. Filtered the product and washed the cake with chilled methanol (100ml) and dried to get 130 g of title compound.
Preparation of 5-[2-hydroxy-5-(4-methylpiperazinyl-l-yl-sulphonyl)phenyl]-l- methyl -3-n- propyl-l,6-dihydro-7H-pyrazolo-[4,3-d]pyrimidin-7-one Sodium hydroxide (34 g) was added into diethylene glycol (780ml) and then heated to 110-1150C. 4-[2-hydroxy-5-(4-methyl-l-piperazinylsulphonyl)benzamido)-l-methyl-3-n- propyl-lH-pyrazole-5-carboxamide (130 g) obtained from example 2 was added to the above reaction mixture. The reaction mixture was maintained at 125-13O0C for 4-6 hrs. The reaction mixture was cooled to room temperature and then DM water (1300ml) was added slowly over 20min at 250C and maintained at this temperature for 1 hour. Filtered the mass and filtrate pH was adjusted to 6.5-7.5 with dilute hydrochloric acid at room temperature and stirred at room temperature for 2-3hrs. Product was filtered and slurried the cake with excess DM Water followed by purification in methanol to get 91 g of titled compound.
Preparation of 5-[2-ethoxycarbonyloxy-5-(4-methylpiperazin-l-yl-sulfonyI)phenyl]- l-methyI-3n-propyI-l,6-dihydro-7H-pyrazolo-[4,3-d]pyrimidin-7-one
5-[2-hydroxy-5-(4-methylpiperazinyl-l -yl-sulphonyl)phenyl]- 1 -methyl-3 -n- propyl- 1 ,6- dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (90 g) obtained from example 3 was dissolved in dichloromethane (360 ml) and added triethyl amine (41 ml) at room temperature and stirred for 10 min. The reaction mixture was cooled to 0-50C and followed by the addition of ethyl chloro formate (24ml) over 30 min under nitrogen atmosphere. The temperature of the reaction was raised slowly to 28-3O0C and maintained for 24 hrs. The reaction mixture was cooled to 0-50C and kept it for 1 hr. The product formed was filtered, washed with dichloromethane, dried and purified from methanol (270ml) to obtain 81 g of the title compound.
Preparation of 5-[2-ethoxy-5-(4-methyl piperazine-l-ylsulfonyl)phenyl]-l-methyl-3- n-propyl-1 ,6-dihydro-7H-pyrazolo [4,3-d] pyrimidin-7-one (Sildenafil base)
5-[2-Ethoxycarbonyloxy-5-(4-methylpiperazin-l-yl-sulfonyl)phenyl]-l-methyl-3-n- propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (50g) was dissolved in ethanol (750ml) in an autoclave and then added dicyclohexylcarbodimide (29.8g). The reaction temperature was raised to HO0C with internal pressure of 1.8-4.0 kg/cm and maintained for 6 hours followed by cooling to room temperature. The solvent was distilled off to get the crude Sildenafil base. The base thus obtained was dissolved in dichloromethane (380ml), filtered and filtrate was distilled out completely to get solid material, which is again dissolved in a mixture dichloromethane and isopropyl ether. The crude obtained was recrystallized from ethanol (260ml) to obtain 17.4gm of pure Sildenafil base.
Purity by HPLC: 99.77% Example 6
Preparation of 5-[2-Ethoxy-5-(4-methylpiperazine-l-yI-sulfonyl)phenyl]-l-methyl- 3-n-propyl-l,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one citrate (Sildenafil Citrate)
Sildenafil base (50 g) was dissolved in acetone (850 ml) at 550C and then slowly added citric acid solution (20 g in 100 ml acetone) over 45 min and maintain the reaction mixture for about 30 min. The reaction mixture was cooled, filtered and dried to get 65 g of Sildenafil citrate.
Purity by HPLC: 99.85%
Chemical synthesis
………………………………………………………………………………… SYNTHESIS
……………………………………………
…………………………………………………………………
SYNTHESIS
…………………………………………………………………………..
……………………………
PRECURSORS
………………………………….. SYNTHESIS
Patents
European Union
Pfizer’s patent on sildenafil citrate expired in some member countries of the EU, Austria, Denmark, France, Germany, Ireland, Italy, The Netherlands, Spain, Sweden, the United Kingdom and Switzerland on 21 June 2013.[53][54][55] A UK patent held by Pfizer on the use of PDE5 inhibitors (see below) as treatment of impotence was invalidated in 2000 because of obviousness; this decision was upheld on appeal in 2002.[56][57]
United States
In 1992, Pfizer filed a patent covering the substance sildenafil and its use to treat cardiovascular diseases.[58] This patent was published in 1993 and expired in 2012. In 1994, Pfizer filed a patent covering the use of sildenafil to treat erectile dysfunction.[59] This patent was published in 2002 and will expire in 2019. Teva sued to have the latter patent invalidated, but Pfizer prevailed in an August 2011 federal district court case.[60]
The patent on Revatio (indicated for pulmonary arterial hypertension rather than erectile dysfunction) expired in late 2012. Generic versions of this low-dose form of sildenafil have been available in the U.S. from a number of manufacturers, including Greenstone, Mylan, and Watson, since early 2013.[61] No legal barrier exists to doctors prescribing this form of sildenafil “off label” for erectile dysfunction, although the dosage typically required for treating ED requires patients to take multiple pills.
Canada
In Canada, Pfizer’s patent 2,324,324 for Revatio (sildenafil used to treat pulmonary hypertension) was found invalid by the Federal Court in June 2010, on an application by Ratiopharm Inc.[62][63]
On November 8, 2012, the Supreme Court of Canada ruled that Pfizer’s patent 2,163,446 on Viagra was invalid from the beginning because the company did not provide full disclosure in its application. The decision, Teva Canada Ltd. v. Pfizer Canada Inc., pointed to section 27(3)(b) of The Patent Act which requires that disclosure must include sufficient information “to enable any person skilled in the art or science to which it pertains” to produce it. It added further: “As a matter of policy and sound statutory interpretation, patentees cannot be allowed to ‘game’ the system in this way. This, in my view, is the key issue in this appeal.”[64]
Teva Canada launched Novo-Sildenafil, a generic version of Viagra, on the day the Supreme Court of Canada released its decision.[65][66][67] To remain competitive, Pfizer then reduced the price of Viagra in Canada.[68] However, on November 9, 2012, Pfizer filed a motion for a re-hearing of the appeal in the Supreme Court of Canada,[69] on the grounds that the court accidentally exceeded its jurisdiction by voiding the patent.[70] Finally, on April 22, 2013, the Supreme Court of Canada invalidated Pfizer’s patent altogether.[71]
India
Manufacture and sale of sildenafil citrate drugs known as “generic viagra” is common in India, where Pfizer’s patent claim does not apply. Trade names include Kamagra (Ajanta Pharma), Silagra (Cipla), Edegra (Sun Pharmaceutical), Penegra (Zydus Cadila), and Zenegra (Alkem Laboratories).
China
Manufacture and sale of sildenafil citrate drugs is common in China, where Pfizer’s patent claim is not widely enforced.
Other countries
Egypt approved Viagra for sale in 2002, but soon afterwards allowed local companies to produce generic versions of the drug, citing the interests of poor people who would not be able to afford Pfizer’s price.[72]
Pfizer’s patent on sildenafil citrate expired in Brazil in 2010.[73]
References
“Sildenafil Citrate”. The American Society of Health-System Pharmacists. Retrieved Dec 1, 2014.
Boolell M, Allen MJ, Ballard SA, Gepi-Attee S, Muirhead GJ, Naylor AM, Osterloh IH, Gingell C (June 1996). “Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction”. Int. J. Impot. Res.8 (2): 47–52. PMID8858389.
Vardi M, Nini A (2007). Vardi, Moshe, ed. “Phosphodiesterase inhibitors for erectile dysfunction in patients with diabetes mellitus”. Cochrane Database Syst Rev (1): CD002187. doi:10.1002/14651858.CD002187.pub3. PMID17253475.
Nurnberg HG, Hensley PL, Heiman JR, Croft HA, Debattista C, Paine S (2008). “Sildenafil Treatment of Women With Antidepressant-Associated Sexual Dysfunction”. JAMA300 (4): 395–404. doi:10.1001/jama.300.4.395. PMID18647982.
Richalet JP, Gratadour P, Robach P, et al. (2005). “Sildenafil inhibits altitude-induced hypoxemia and pulmonary hypertension”. Am. J. Respir. Crit. Care Med.171 (3): 275–81. doi:10.1164/rccm.200406-804OC. PMID15516532.
Perimenis P (2005). “Sildenafil for the treatment of altitude-induced hypoxaemia”. Expert Opin Pharmacother6 (5): 835–7. doi:10.1517/14656566.6.5.835. PMID15934909.
Fagenholz PJ, Gutman JA, Murray AF, Harris NS (2007). “Treatment of high altitude pulmonary edema at 4240 m in Nepal”. High Alt. Med. Biol.8 (2): 139–46. doi:10.1089/ham.2007.3055. PMID17584008.
Kloner RA (2005). “Pharmacology and drug interaction effects of the phosphodiesterase 5 inhibitors: focus on alpha-blocker interactions”. Am J Cardiol96 (12B): 42M–46M. doi:10.1016/j.amjcard.2005.07.011. PMID16387566.
Cheitlin MD, Hutter AM Jr, Brindis RG, Ganz P, Kaul S, Russell RO Jr, Zusman RM (1999). “ACC/AHA expert consensus document. Use of sildenafil (Viagra) in patients with cardiovascular disease. American College of Cardiology/American Heart Association”. Journal of the American College of Cardiology33 (1): 273–82. doi:10.1016/S0735-1097(98)00656-1. PMID9935041.
Smith KM, Romanelli F (2005). “Recreational use and misuse of phosphodiesterase 5 inhibitors”. J Am Pharm Assoc (2003)45 (1): 63–72; quiz 73–5. doi:10.1331/1544345052843165. PMID15730119.
Mondaini N, Ponchietti R, Muir GH, Montorsi F, Di Loro F, Lombardi G, Rizzo M (June 2003). “Sildenafil does not improve sexual function in men without erectile dysfunction but does reduce the postorgasmic refractory time”. Int. J. Impot. Res.15 (3): 225–8. doi:10.1038/sj.ijir.3901005. PMID12904810.
McCambridge J, Mitcheson L, Hunt N, Winstock A (March 2006). “The rise of Viagra among British illicit drug users: 5-year survey data”. Drug Alcohol Rev25 (2): 111–3. doi:10.1080/09595230500537167. PMID16627299.
Eloi-Stiven ML, Channaveeraiah N, Christos PJ, Finkel M, Reddy R (November 2007). “Does marijuana use play a role in the recreational use of sildenafil?”. J Fam Pract56 (11): E1–4. PMID17976333.
Venhuis BJ, de Kaste D. (2006–2012). “Towards a decade of detecting new analogues of sildenafil, tadalafil and vardenafil in food supplements: a history, analytical aspects and health risks”. Journal of Pharmaceutical and Biomedical Analysis69: 196–208. doi:10.1016/j.jpba.2012.02.014. PMID22464558.
Oh SS, Zou P, Low MY, Koh HL. Detection of sildenafil analogues in herbal products for erectile dysfunction (2006). “Detection of sildenafil analogues in herbal products for erectile dysfunction”. Journal of Toxicology and Environmental Health Part A69 (21): 1951–1958. doi:10.1080/15287390600751355. PMID16982533.
R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 9th edition, Biomedical Publications, Seal Beach, CA, 2011, pp. 1552–1553. http://www.biomedicalpublications.com/dt9.pdf
Sung, B. J.; Hwang, K.; Jeon, Y.; Lee, J. I.; Heo, Y. S.; Kim, J.; Moon, J.; Yoon, J.; Hyun, Y. L.; Kim, E.; Eum, S.; Park, S. Y.; Lee, J. O.; Lee, T.; Ro, S.; Cho, J. (2003). “Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules”. Nature425 (6953): 98–102. doi:10.1038/nature01914. PMID12955149.
Webb, D.J.; Freestone, S.; Allen, M.J.; Muirhead, G.J. (March 4, 1999). “Sildenafil citrate and blood-pressure-lowering drugs: results of drug interaction studies with an organic nitrate and a calcium antagonist”. Am. J. Cardiol.83 (5A): 21C–28C. doi:10.1016/S0002-9149(99)00044-2. PMID10078539.
Dunn PJ (2005). “Synthesis of Commercial Phosphodiesterase(V) Inhibitors”. Org Process Res Dev2005 (1): 88–97. doi:10.1021/op040019c.
“Research”. ABM. Abertawe Bro Morgannwg University Health Board. 4 July 2008. Archived from the original on 26 September 2008. Retrieved 6 August 2008. Our clinicians regularly offer patients the opportunity to take part in trials of new drugs and treatments. Morriston Hospital in Swansea, was the first in the world to trial Viagra!
Terrett NK, Bell AS, Brown D, Elllis P (1996). “Sildenafil (Viagra), a potent and selective inhibitor of Type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction”. Bioorg Med Chem Lett6 (15): 1819–1824. doi:10.1016/0960-894X(96)00323-X.
Kling J (1998). “From hypertension to angina to Viagra”. Mod. Drug Discov.1: 31–38. ISSN1532-4486. OCLC41105083.
Murray-, Rosie (23 January 2002). “Viagra ruling upsets Pfizer”. London: Telegraph Media Group Limited. Archived from the original on 22 August 2009. Retrieved 10 February 2009.
Defibrotide sodium is an oligonucleotide mixture with profibrinolytic properties. The chemical name of defibrotide sodium is polydeoxyribonucleotide, sodium salt. Defibrotide sodium is a polydisperse mixture of predominantly single-stranded (ss) polydeoxyribonucleotide sodium salts derived from porcine intestinal tissue having a mean weighted molecular weight of 13-20 kDa, and a potency of 27-39 and 28-38 biological units per mg as determined by two separate assays measuring the release of a product formed by contact between defibrotide sodium, plasmin and a plasmin substrate. The primary structure of defibrotide sodium is shown below.
DEFITELIO (defibrotide sodium) injection is a clear, light yellow to brown, sterile, preservative-free solution in a single-patient-use vial for intravenous use. Each milliliter of the injection contains 80 mg of defibrotide sodium and 10 mg of Sodium Citrate, USP, in Water for Injection, USP. Hydrochloric Acid, NF, and/or Sodium Hydroxide, NF, may have been used to adjust pH to 6.8-7.8.
Defibrotide is the sodium salt of a mixture of single-stranded oligodeoxyribonucleotides derived from porcine mucosal DNA. It has been shown to have antithrombotic, anti-inflammatory and anti-ischemic properties (but without associated significant systemic anticoagulant effects). It is marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries, but is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide is used to treat or prevent a failure of normal blood flow (occlusive venous disease, OVD) in the liver of patients who have had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others.
In 2012, an IND was filed in Japan seeking approval of the compound for the treatment of veno-occlusive disease.
Approved 3/30/3016 US FDA, defibrotide sodium, (NDA) 208114
To treat adults and children who develop hepatic veno-occlusive disease with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation
Polydeoxyribonucleotides from bovine lung or other mamalian organs with molecular weight between 15,000 and 30,000 Da
CAS 83712-60-1
Defibrotide is a polydisperse mixture of oligonucleotides produced by random, chemical cleavage (depolymerisation) of porcine DNA. It is predominantly single stranded, of varying base sequence, lengths and conformations; unfolded, folded or combined. The mean oligonucleotide length is 50 bases with a mean molecular weight of 17 ± 4 kDa. No individually defined component is at more than femtomolar concentration. The only meaningful scientific information that can be obtained about the biochemical nature of defibrotide (aside from determination of percentage of each nucleobase) is a measurement of its average length and its average percentage double stranded character. Therefore, it can be established that this active substance is of highly heterogenic nature.
Defibrotide (Defitelio, Gentium)[1] is a deoxyribonucleic acid derivative (single-stranded) derived from cow lung or porcine mucosa. It is an anticoagulant with a multiple mode of action (see below).
Jazz Pharmaceuticals plc announced that the FDA has accepted for filing with Priority Review its recently submitted New Drug Application (NDA) for defibrotide. AS ON OCT 2015
Defibrotide is an investigational agent proposed for the treatment of patients with hepatic veno-occlusive disease (VOD), also known as sinusoidal obstruction syndrome (SOS), with evidence of multi-organ dysfunction (MOD) following hematopoietic stem-cell transplantation (HSCT).
Priority Review status is designated for drugs that may offer major advances in treatment or provide a treatment where no adequate therapy exists. Based on timelines established by the Prescription Drug User Fee Act (PDUFA), FDA review of the NDA is expected to be completed by March 31, 2016.
“The FDA’s acceptance for filing and Priority Review status of the NDA for defibrotide is an important milestone for Jazz and reflects our commitment to bringing meaningful medicines to patients who have significant unmet needs,” said Karen Smith, M.D., Ph.D., Global Head of Research and Development and Chief Medical Officer of Jazz Pharmaceuticals. “We look forward to continuing to work closely with the FDA to obtain approval for defibrotide for patients with hepatic VOD with evidence of MOD in the U.S. as quickly as possible, as there are no other approved therapies for treating this rare, often fatal complication of HSCT.”
The NDA includes safety and efficacy data from three clinical studies of defibrotide for the treatment of hepatic VOD with MOD following HSCT, as well as a retrospective review of registry data from the Center for International Blood and Marrow Transplant Research. The safety database includes over 900 patients exposed to defibrotide in the clinical development program for the treatment of hepatic VOD.
The compound was originally developed under a collaboration between Sanofi and Gentium. In December 2001, Gentium entered into a license and supply agreement with Sigma-Tau Pharmaceuticals, pursuant to which the latter gained exclusive rights to distribute, market and sell the product for the treatment of VOD in the U.S. This agreement was expanded in 2005 to include all of North America, Central America and South America.
Defibrotide was granted orphan drug designations from the FDA in July 1985, May 2003 and January 2007 for the treatment of thrombotic thrombocytopenic purpura (TTP), for the treatment of VOD and for the prevention of VOD, respectively. Orphan drug was also received in the E.U. for the prevention and treatment of hepatic veno-occlusive disease (VOD) in 2004 and for the prevention of graft versus host disease (GvHD) in 2013.
Pharmacokinetics
Defibrotide is available as an oral, intravenous, and intramuscular formulation. Its oral bioavailability is in the range of 58-70% of theparenteral forms. T1/2 alpha is in the range of minutes while T1/2 beta is in the range of hours in studies with oral radiolabelleddefibrotide. These data suggest that defibrotide, in spite of its macromolecular nature, is absorbed well after oral administration. Due to the drug’s short half-life, it is necessary to give the daily dose divided in 2 to 4 doses (see below).
In 2014, Jazz Pharmaceuticals (parent of Gentium) acquired the rights of the product in U.S. and in the Americas
Mode of action
The drug appears to prevent the formation of blood clots and to help dissolve blood clots by increasing levels of prostaglandin I2, E2, and prostacyclin, altering platelet activity, increasing tissue plasminogen activator (tPA-)function, and decreasing activity of tissue plasminogen activator inhibitor. Prostaglandin I2 relaxes the smooth muscle of blood vessels and prevents platelets from adhering to each other. Prostaglandin E2 at certain concentrations also inhibits platelet aggregation. Moreover, the drug provides additional beneficial anti-inflammatory and antiischemic activities as recent studies have shown. It is yet unclear, if the latter effects can be utilized clinically (e.g., treatment of ischemic stroke).
Unlike heparin and warfarin, defibrotide appears to have a relatively mild anticoagulant activity, which may be beneficial in the treatment of patients at high risk of bleeding complications. Nevertheless, patients with known bleeding disorders (e.g., hemophilia A) or recent abnormal bleedings should be treated cautiously and under close medical supervision.
The drug was marketed under the brand names Dasovas (FM), Noravid, and Prociclide in a variety of countries. It is currently not approved in the USA. The manufacturer is Gentium.
Defibrotide also received fast track designation from the FDA for the treatment of severe VOD in recipients of stem cell transplants. In 2011, the compound was licensed to Medison Pharma by Gentium in Israel and Palestine. The license covers the management of named-patient sales program and local registration, authorization, marketing, reimbursement and medical affairs for the treatment of peripheral vascular disease.
Usual indications
Defibrotide is used to treat or prevent a failure of normal blood flow (Veno-occlusive disease, VOD) in the liver of patients having had bone marrow transplants or received certain drugs such as oral estrogens, mercaptopurine, and many others. Without intensive treatment, VOD is often a fatal condition, leading to multiorgan failure. It has repeatedly been reported that defibrotide was able to resolve the condition completely and was well tolerated.
Other indications are: peripheral obliterative arterial disease, thrombophlebitis, and Raynaud’s phenomenon. In very high doses, defibrotide is useful as treatment of acute myocardial infarction. The drug may also be used for the pre- and postoperative prophylaxis of deep venous thrombosis and can replace the heparin use during hemodialytic treatments.
Other recent preclinical studies have demonstrated that defibrotide used in conjunction with Granulocyte Colony-Stimulating Factor (rhG-CSF) significantly increases the number of Peripheral Blood Progenitor Cells (Stem cells). The benefit of this increase in stem cells may be crucial for a variety of clinical indications, including graft engineering procedures and gene therapy programs. This would expand the clinical usefulness of defibrotide to a complete distinct area.
Very recently (since early 2006) combination therapy trials (phase I/II) with defibrotide plus melphalan, prednisone, and thalidomide in patients with multiple myeloma have been conducted. The addition of defibrotide is expected to decrease the myelosuppressive toxicity of melphalan. However, is too early for any definitive results at that stage.
Cautions and contraindications
The efficacy of the drug has been reported to be poorer in patients with diabetes mellitus.
Pregnancy: The drug should not be used during pregnancy, because adequate and well controlled human studies do not exist.
Lactation: No human data is available. In order to avoid damage to the newborn, the nursing mother should discontinue either the drug or breastfeeding, taking into account the importance of treatment to the mother.
Known Bleeding Disorders or Bleeding Tendencies having occurred recently: Defibrotide should be used cautiously. Before initiation of treatment, the usual coagulation values should be obtained as baseline and regularly controlled under treatment. The patient should be observed regularly regarding local or systemic bleeding events.
Side-effects
Increased bleeding and bruising tendency, irritation at the injection site, nausea, vomiting, heartburn, low blood pressure. Serious allergic reactions have not been observed so far.
Drug interactions
Use of heparin with defibrotide may increase the aPTT, reflecting reduced ability of the body to form a clot. Nothing is known about the concomitant application of other anticoagulants than heparin and dextran containing plasma-expanders, but it can be anticipated that the risk of serious bleeding will be increased considerably.
PATENT
WO 2001078761
G-CSF (CAS registry number 143011-2-7/Merck Index, 1996, page 4558) is a haematopoietic growth factor which is indispensable in the proliferation and differentiation of the progenitor cells of granulocytes; it is a 18-22 kDa glycoprotein normally produced in response to specific stimulation by a variety of cells, including monocytes, fibroblasts and endothelial cells. The term defibrotide (CAS registry number 83712-60-1) normally identifies a polydeoxyribonucleotide obtained by extraction (US 3,770,720 and US 3,899,481) from animal and/or vegetable tissue; this polydeoxyribonucleotide is normally used in the form of a salt of an alkali metal, generally sodium. Defibrotide is used principally for its anti- thrombotic activity (US 3,829,567) although it may be used in different applications, such as, for example, the treatment of acute renal insufficiency (US 4,694,134) and the treatment of acute myocardial ischaemia (US 4,693,995). United States patents US 4,985,552 and US 5,223,609, finally, describe a process for the production of defibrotide which enables a product to be obtained which has constant and well defined physico-chemical characteristics and is also free from any undesired side-effects
References
“Jazz Pharma Acquiring Gentium for $1B”. Gen. Eng. Biotechnol. News (paper) 34 (2). January 15, 2014. p. 10.
Palmer KJ, Goa KL. Defibrotide: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in vascular disorders. Drugs 1993;45:259-94.
///////////Approved, 3/30/3016, US FDA, defibrotide sodium, NDA 208114, FDA 2016
Updates……….
FDA approves first treatment for rare disease in patients who receive stem cell transplant from blood or bone marrow
For Immediate Release
March 30, 2016
Release
The U.S. Food and Drug Administration today approved Defitelio (defibrotide sodium) to treat adults and children who develop hepatic veno-occlusive disease (VOD) with additional kidney or lung abnormalities after they receive a stem cell transplant from blood or bone marrow called hematopoietic stem cell transplantation (HSCT). This is the first FDA-approved therapy for treatment of severe hepatic VOD, a rare and life-threatening liver condition.
HSCT is a procedure performed in some patients to treat certain blood or bone marrow cancers. Immediately before an HSCT procedure, a patient receives chemotherapy. Hepatic VOD can occur in patients who receive chemotherapy and HSCT. Hepatic VOD is a condition in which some of the veins in the liver become blocked, causing swelling and a decrease in blood flow inside the liver, which may lead to liver damage. In the most severe form of hepatic VOD, the patient may also develop failure of the kidneys and lungs. Fewer than 2 percent of patients develop severe hepatic VOD after HSCT, but as many as 80 percent of patients who develop severe hepatic VOD do not survive.
“The approval of Defitelio fills a significant need in the transplantation community to treat this rare but frequently fatal complication in patients who receive chemotherapy and HSCT,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research.
The efficacy of Defitelio was investigated in 528 patients treated in three studies: two prospective clinical trials and an expanded access study. The patients enrolled in all three studies had a diagnosis of hepatic VOD with liver or kidney abnormalities after HSCT. The studies measured the percentage of patients who were still alive 100 days after HSCT (overall survival). In the three studies, 38 to 45 percent of patients treated with Defitelio were alive 100 days after HSCT. Based on published reports and analyses of patient-level data, the expected survival rates 100 days after HSCT would be 21 to 31 percent for patients with severe hepatic VOD who received only supportive care or interventions other than Defitelio.
The most common side effects of Defitelio include abnormally low blood pressure (hypotension), diarrhea, vomiting, nausea and nosebleeds (epistaxis). Serious potential side effects of Defitelio that were identified include bleeding (hemorrhage) and allergic reactions. Defitelio should not be used in patients who are having bleeding complications or who are taking blood thinners or other medicines that reduce the body’s ability to form clots.
The FDA granted the Defitelio application priority review status, which facilitates and expedites the development and review of certain drugs in light of their potential to benefit patients with serious or life-threatening conditions. Defitelio also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Defitelio is marketed by Jazz Pharmaceuticals based in Palo Alto, California

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












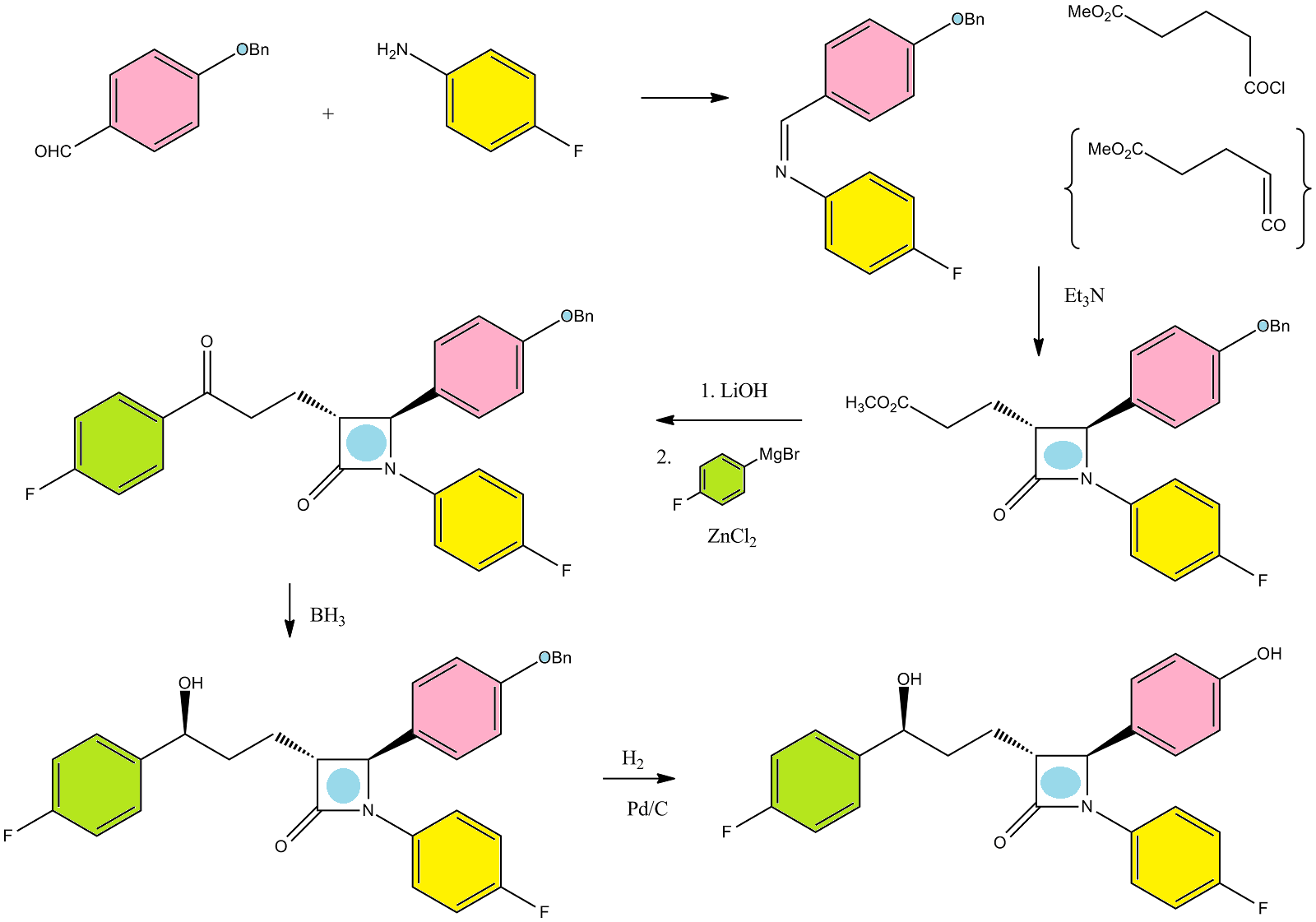

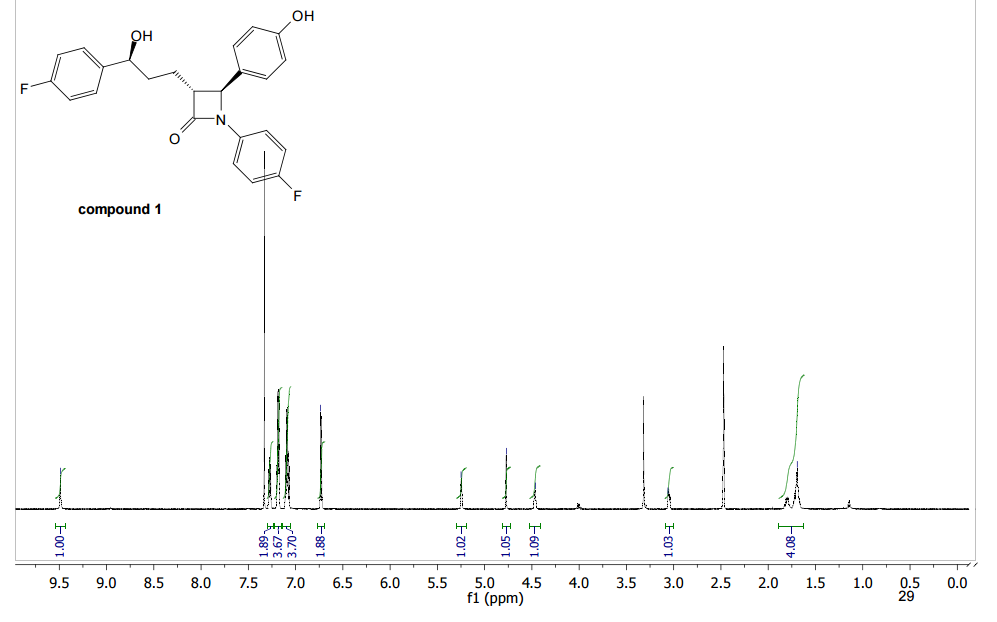
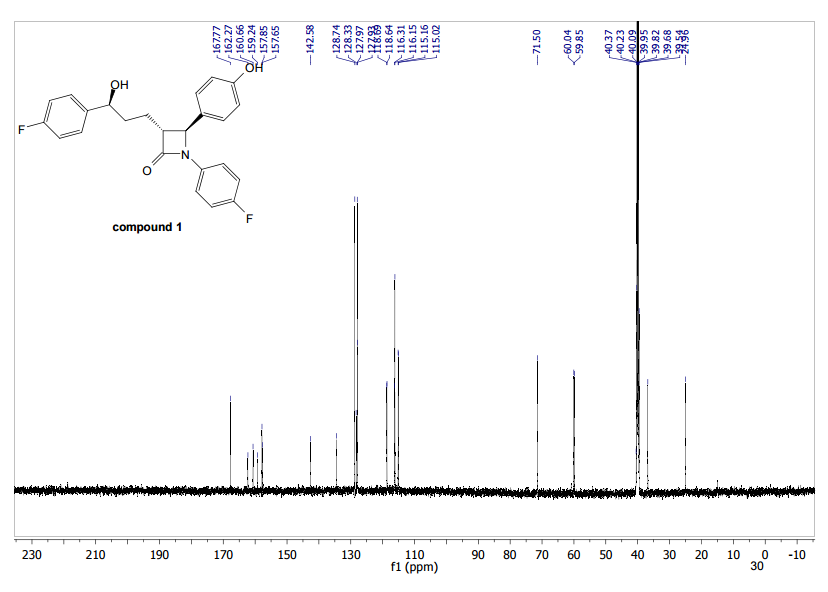

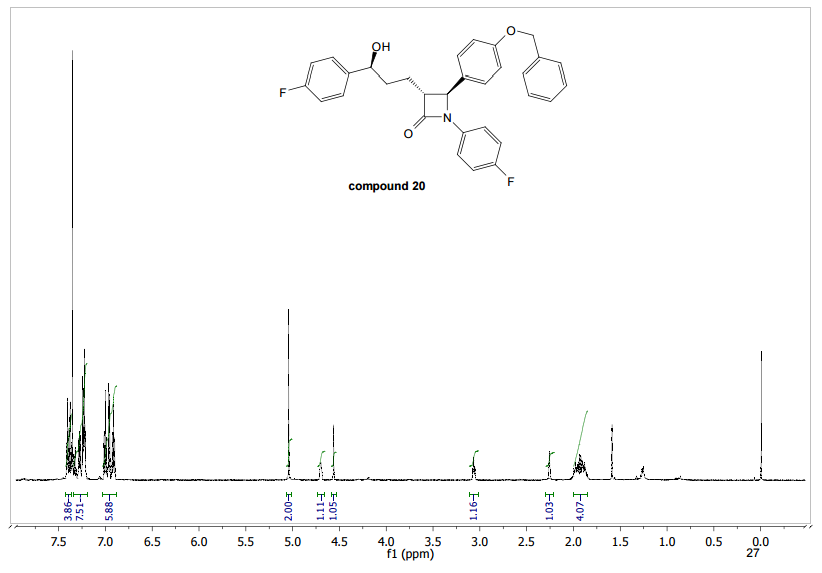






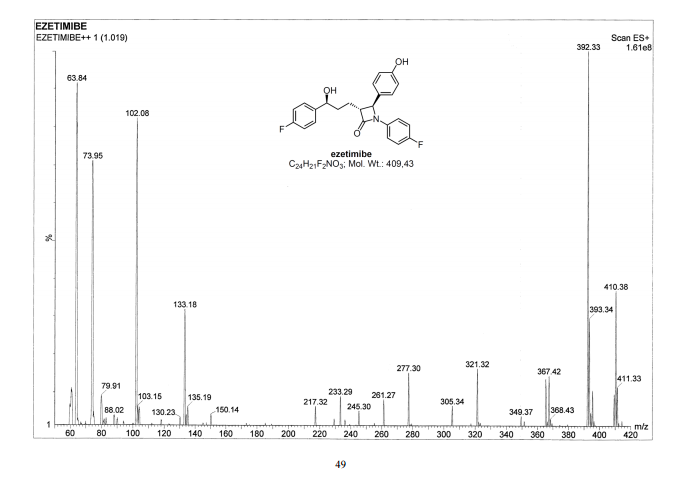
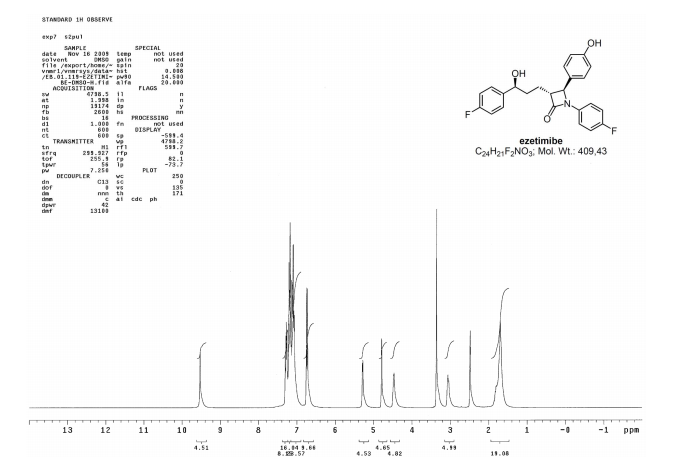


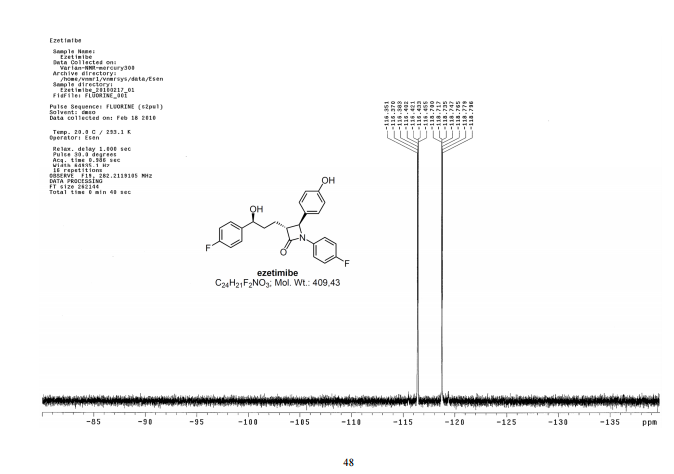







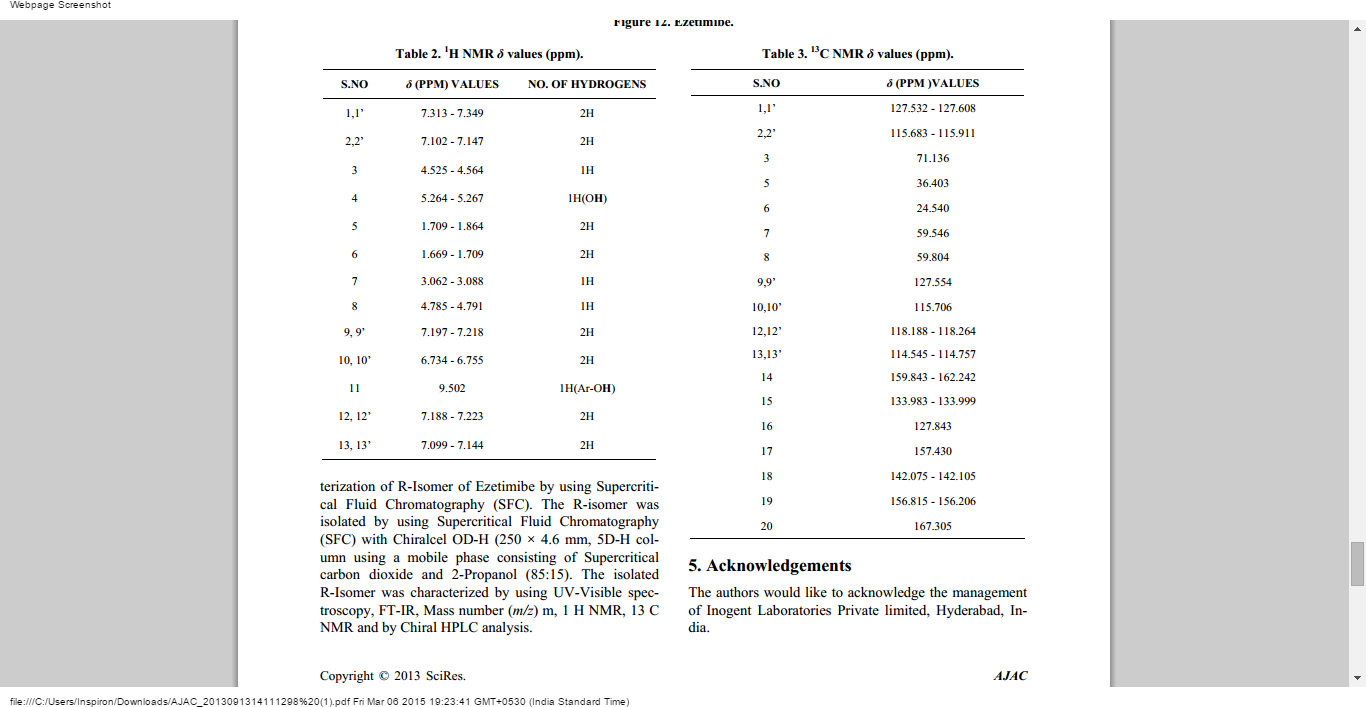






























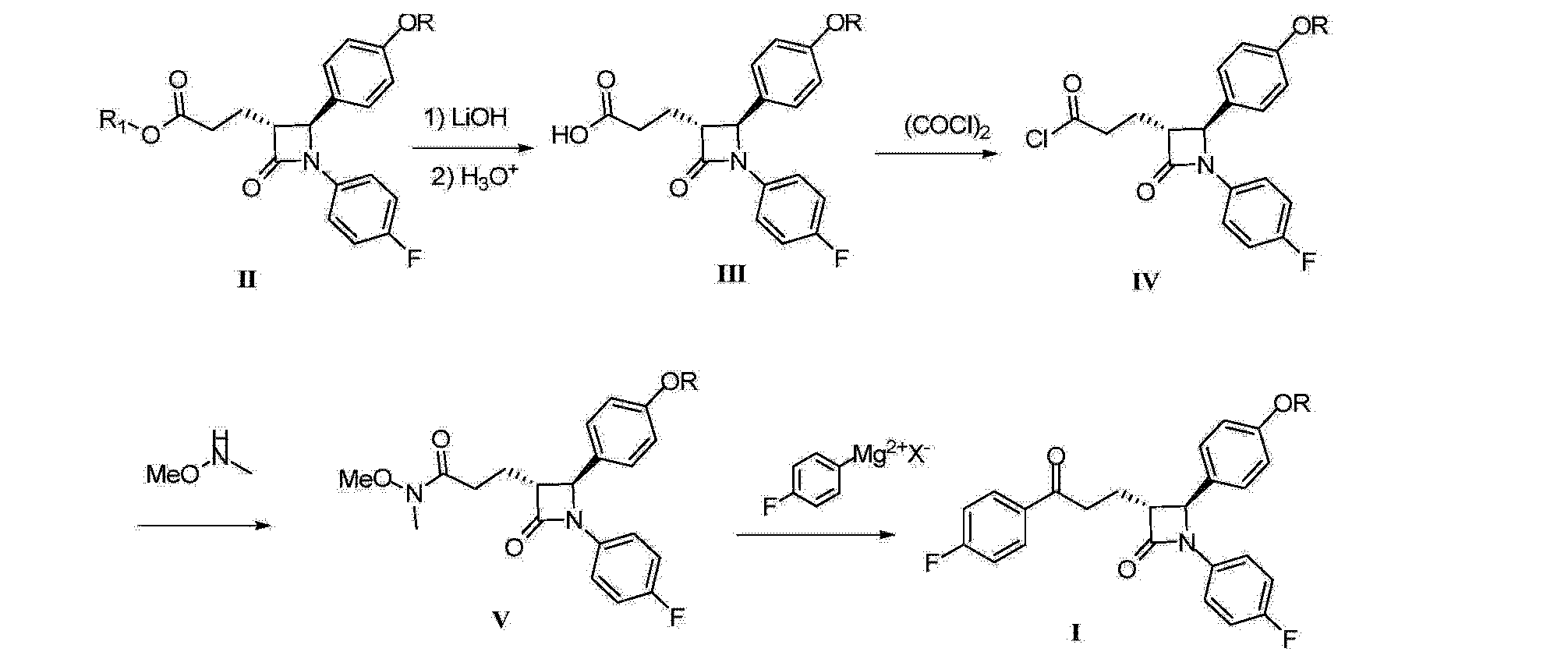




























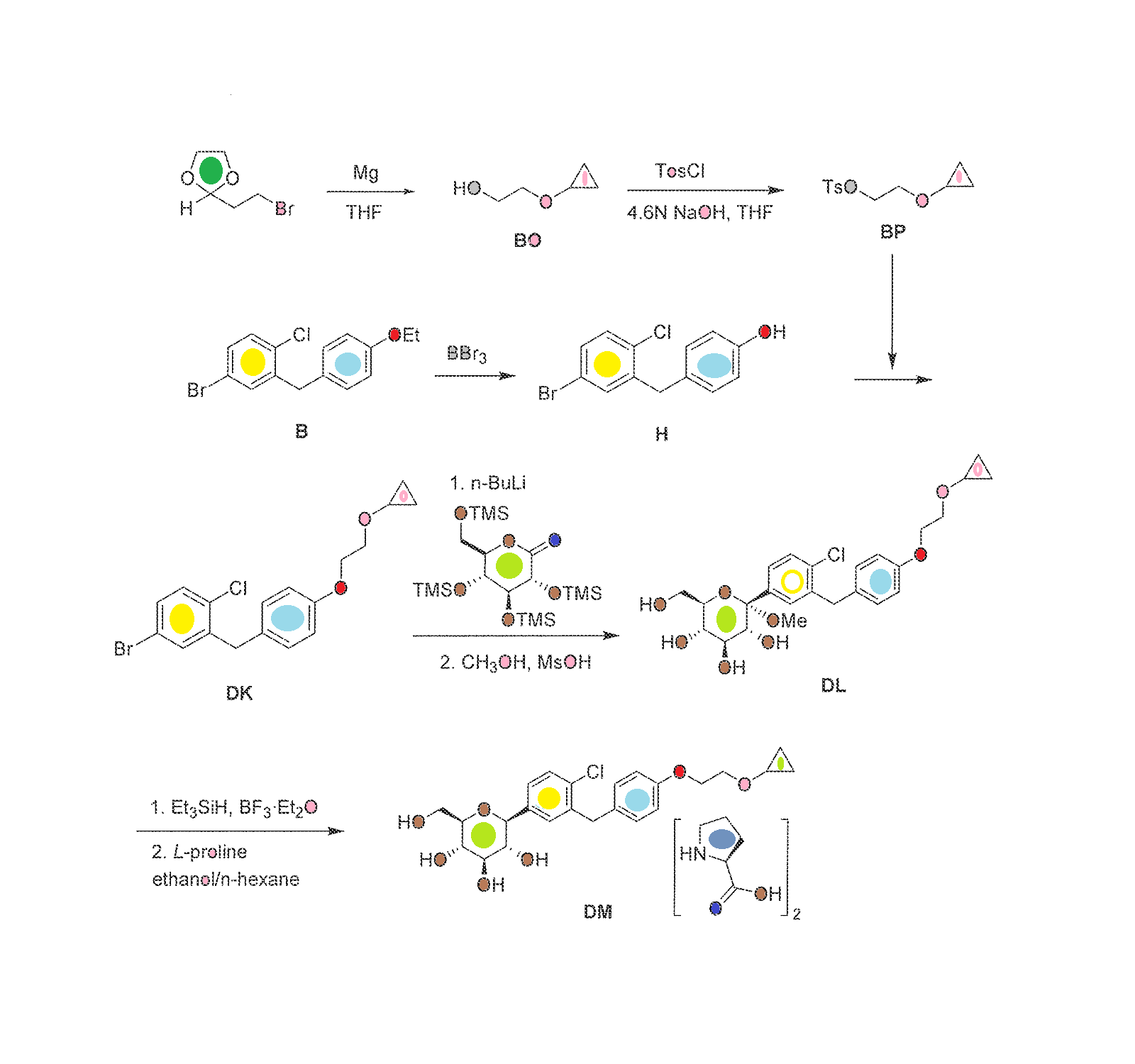











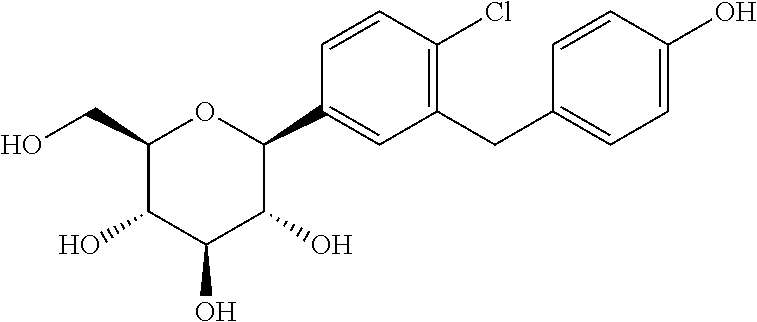



















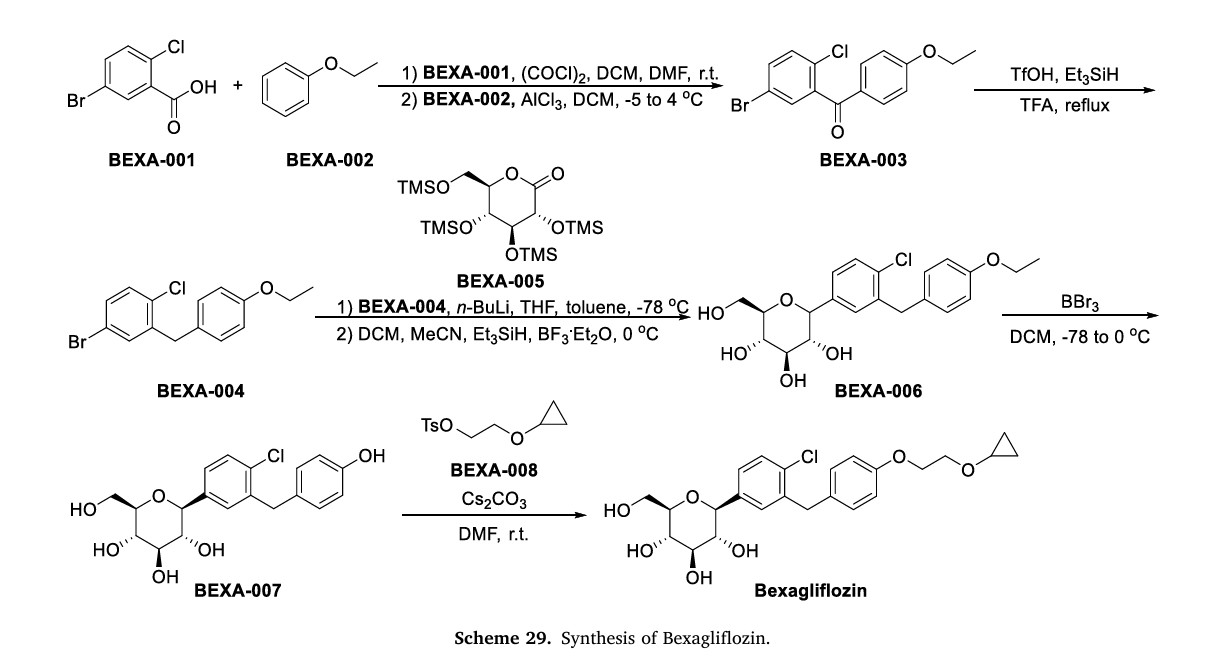







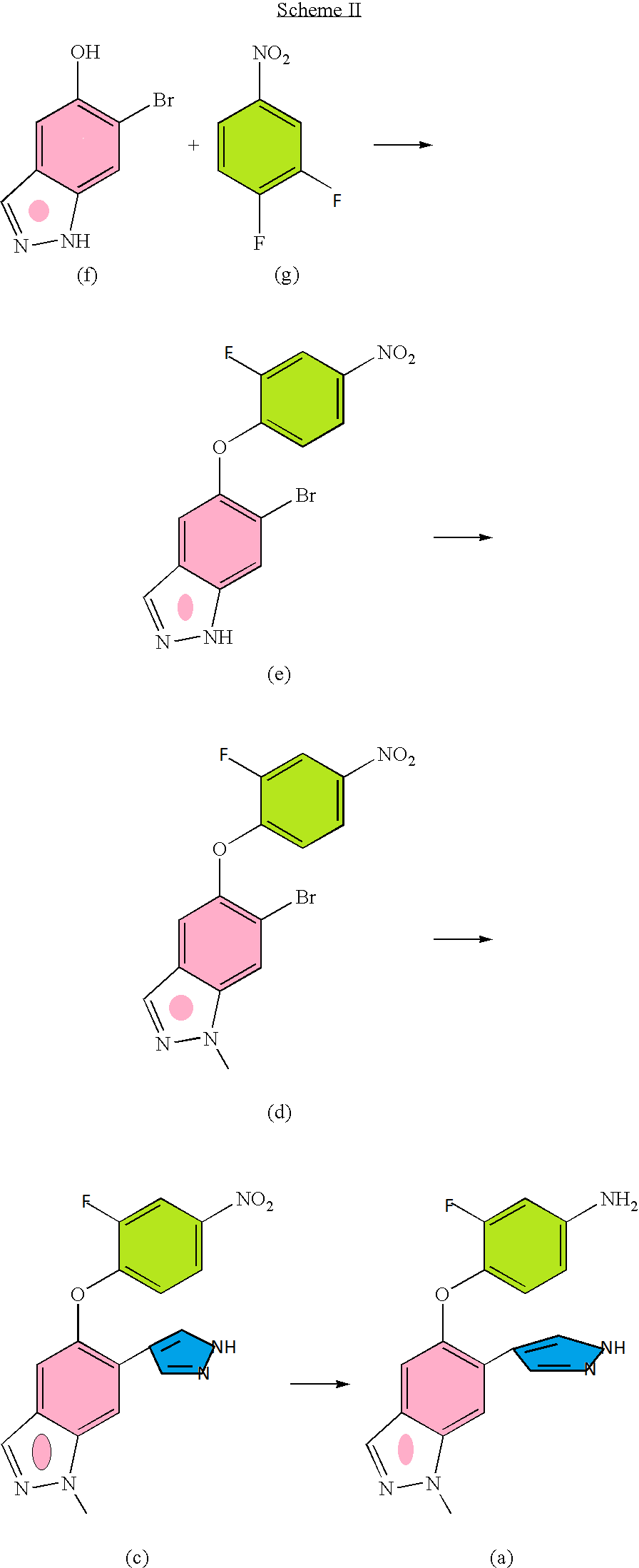
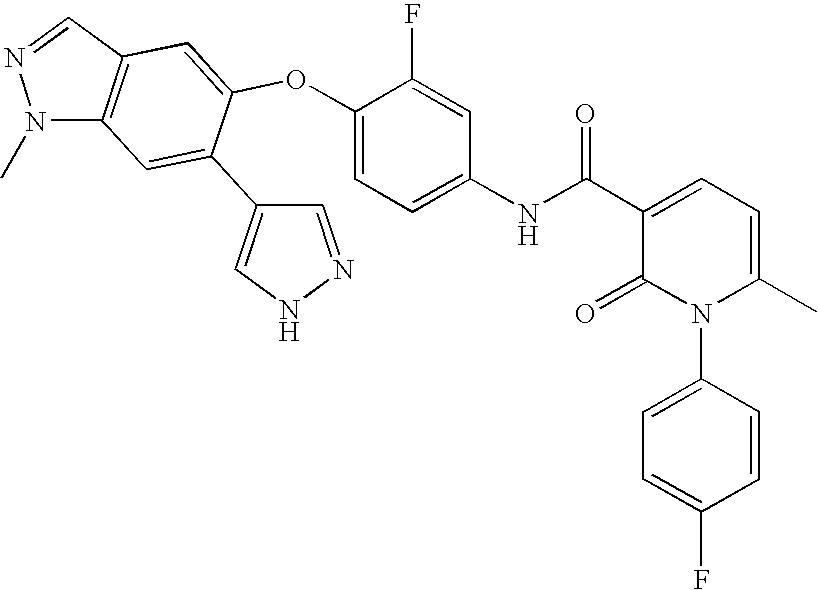

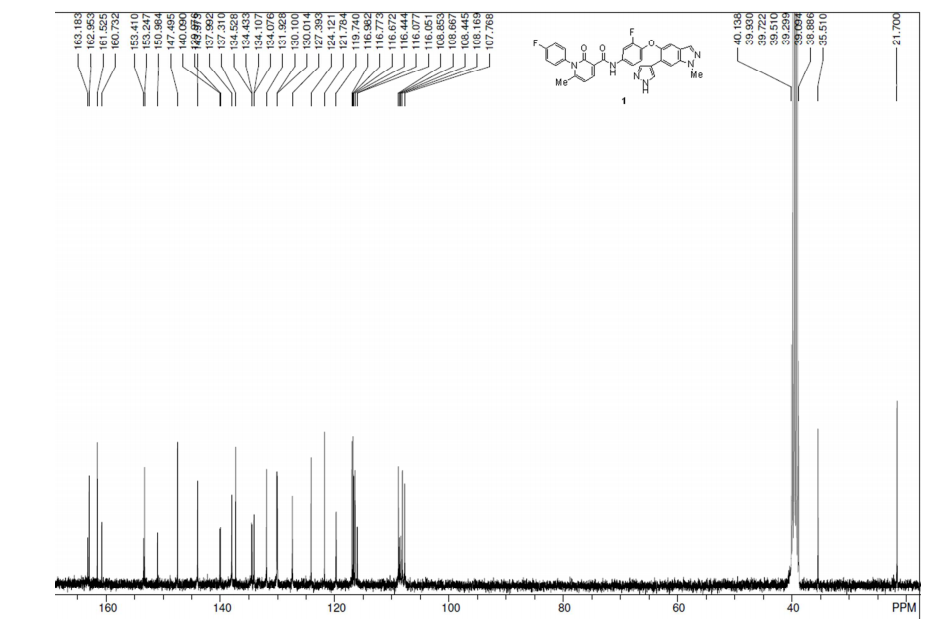

 1 MERESTENIB
1 MERESTENIB