
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

For description see at synfacts
https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0033-1340505
Contributor: Philip Kocienski
 Philip Kocienski, Professor of Organic Chemistry.
Philip Kocienski, Professor of Organic Chemistry.
https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0033-1340505

Bao H, Bayeh L, Tambar UK * The University of Texas Southwestern Medical Center at Dallas, USA
Catalytic Enantioselective Allylic Amination of Olefins for the Synthesis of ent-Sitagliptin.
Synlett 2013;
24: 2459-2463

P. J. Kocienski
School of Chemistry
University of Leeds
Leeds LS2 9JT, UK
p.kocienski@chem.leeds.ac.uk
http://www.chem.leeds.ac.uk
Philip J. Kocienski was born in Troy, New York, in 1946. His love for organic chemsitry, amply stimulated by Alfred Viola whilst an undergraduate at Northeastern University, was further developed at Brown University, where he obtained his PhD degree in 1971 under Joseph Ciabattoni. Postdoctoral study with George Büchi at MIT and later with Basil Lythgoe at Leeds University, England, confirmed his interest in the synthesis of natural products. He was appointed Brotherton Research lecturer at Leeds in 1979 and Professor of Chemistry at Southampton University in 1985. In 1990 he was appointed Glaxo Professor of Chemistry at Southampton University. He moved to the University of Glasgow in 1997, where he was Regius Professor of Chemistry and now he is a Professor of Chemistry at Leeds University.
In addition to Prof. Kocienski’s work as an author he is also a member of the SYNTHESIS Editorial Board and contributes greatly to the development of Thieme Chemistry’s journals
Furthermore, Prof. Kocienski has also contributed to the Science of Synthesis project where he was an author for Volume 4, Compounds of Group 15 (As, Sb, Bi) and Silicon Compounds.
Prof. Kocienski is also responsible for compiling a database called Synthesis Reviews. This resource is free and contains 16,257 English review articles (from journals and books) of interest to synthetic organic chemists. It covers literature from 1970 to 2002.
////////////////////////////////////////
SITAGLIPTIN……………..



GREENING UP DRUGS Merck process chemists redesigned and significantly shortened the original synthesis of type 2 diabetes drug candidate sitagliptin (Januvia) to include an unprecedented efficient hydrogenation of an unprotected enamine.
MERCK was selected for the award in the greener synthetic pathways category for revising the synthesis for sitagliptin, a chiral β-amino acid derivative that is the active ingredient in Januvia, the company’s pending new treatment for type 2 diabetes. The breakthrough leading to the new synthesis was the discovery that the amino group of the key enamine intermediate doesn’t need to be protected prior to enantioselective catalytic hydrogenation of the double bond.
This development has solved a long-standing problem in the synthesis of β-amino acid derivatives, which are known for their pharmacological properties and are commonly used as chiral building blocks, noted Karl B. Hansen, a Merck process chemist involved with the synthetic effort. The outcome has been to slash the number of reaction steps in the sitagliptin synthesis from eight to three, leading to an equally dramatic reduction in the amount of chemicals and solvent needed and the amount of waste generated.
Merck’s first-generation synthesis of sitagliptin involved preparing a β-hydroxy carboxylic acid, which was converted to a protected β-lactam and then coupled to a triazole building block. Deprotecting the resulting intermediate provided the β-amino acid moiety, and sitagliptin was isolated as a phosphoric acid salt.
This synthesis involved a roundabout route involving four steps to introduce the pivotal chiral amino group of sitagliptin. The synthesis worked well to prepare more than 100 kg of the compound for clinical trials, and with modifications it was deemed to be a viable though not very green manufacturing process, Hansen pointed out. For example, the original synthesis required a number of distillations and aqueous extractions to isolate intermediates, leading to a large volume of waste to treat.
“Being environmentally friendly and economically savvy can, and does, go hand-in-hand.”
Merck process chemists recognized that a much more efficient process was possible by synthesizing the β-amino acid portion of the molecule directly from an enamine. But the protection-deprotection of the amine nitrogen with an acyl group during the hydrogenation is difficult on a large scale, and unprotected reactions generally result in lower yields and lower enantiomeric excesses, Hansen said.
Undaunted, the Merck scientists working on the revised synthesis discovered that the amino group could be efficiently introduced by an unprotected hydrogenation using a rhodium catalyst with a ferrocenyl phosphine ligand named Josiphos (C&EN, Sept. 5, 2005, page 40). Merck turned to Solvias, a Swiss company with experience in asymmetric hydrogenations that manufactures Josiphos, as a partner to help speed up the process development.
The new synthesis involves first coupling trifluorophenyl acetic acid and triazole building blocks to form a diketoamide, which in turn is converted to the enamine. This sequence is carried out without isolating intermediates. The enamine is then hydrogenated, sitagliptin is isolated and recrystallized as the phosphoric acid salt, and the rhodium Josiphos catalyst is recovered.
In sum, the revised synthesis increases the overall yield of sitagliptin by nearly 50% and reduces the amount of waste by more than 80%. A key difference is that the original synthesis produced more than 60 L of aqueous waste per kg of product, while the new synthesis completely eliminates aqueous waste. When tallied up, Merck expects these savings will prevent formation of 150,000 metric tons of solid and aqueous process waste over the lifetime of Januvia. Industry analysts speculate that regulatory approval of the drug will come by early next year and that it’s destined to become a top-selling drug.
The novel enamine hydrogenation “is arguably the most efficient means to prepare β-amino acid derivatives,” noted R. P. (Skip) Volante, Merck’s vice president of process research. The company currently is using the procedure to make several other exploratory drug candidates, he added. Overall, the redesigned synthesis of sitagliptin “is a green chemistry solution to the preparation of a challenging synthetic target and is an excellent example of a scientific innovation resulting in benefits to the environment,” Volante said.

First generation route to sitagliptin. BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; EDC = N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride; DIAD = di-isopropyl azodicarboxylate; NMM = N-methylmorpholine……..http://www.technology.matthey.com/article/55/2/135-139/

http://pubs.rsc.org/en/content/articlelanding/2011/cc/c1cc11592h#!divAbstract

http://www.nature.com/nature/journal/v485/n7397/fig_tab/nature11117_F4.html
PAPER
First Generation Process for the Preparation of the DPP-IV Inhibitor Sitagliptin

A new synthesis of sitagliptin (MK-0431), a DPP-IV inhibitor and potential new treatment for type II diabetes, suitable for the preparation of multi-kilogram quantities is presented. The triazolopyrazine fragment of sitagliptin was prepared in 26% yield over four chemical steps using a synthetic strategy similar to the medicinal chemistry synthesis. Key process developments were made in the first step of this sequence, the addition of hydrazine to chloropyrazine, to ensure its safe operation on a large scale. The beta-amino acid fragment of sitagliptin was prepared by asymmetric reduction of the corresponding beta-ketoester followed by a two-step elaboration to an N-benzyloxy beta-lactam. Hydrolysis of the lactam followed by direct coupling to the triazolopiperazine afforded sitagliptin after cleavage of the N-benzyloxy group and salt formation. The overall yield was 52% over eight steps.



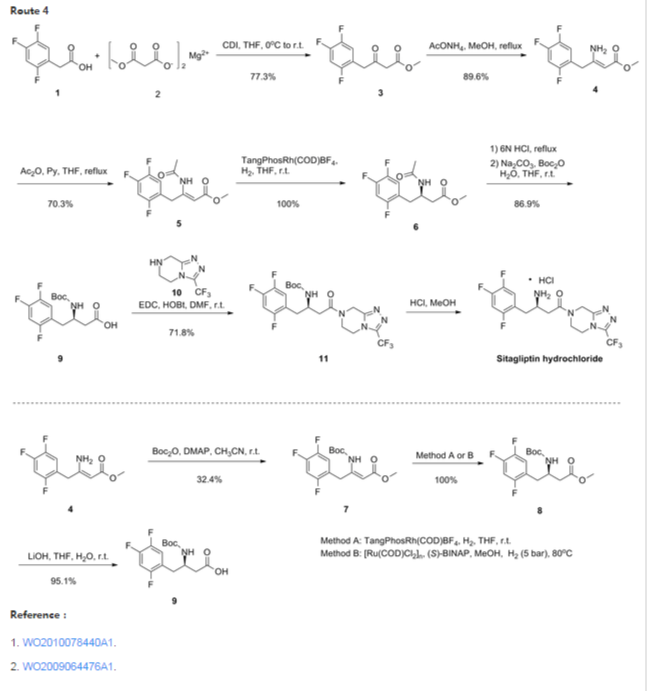
The synthesis of 1 was completed using a four-step through-process (Scheme 4). Lactam 5 or ester 13 was hydrolyzed to amino acid 2b with LiOH18 in THF/water by either stirring at room temperature or, in the case of 13, heating to 40 °C. While the benzyloxy group of 2b could be cleaved by hydrogenation and then protected with Boc2O to prevent side reactions during the coupling to triazole 3, the benzyloxy group of 2b was found to sufficiently protect the amino group to allow the desired amide to be formed. Thus, triazole 3 was coupled to2b at 0 °C using EDC−HCl and N-methylmorpholine (NMM) as base to afford 14in >99% assay yield. Following an aqueous workup, the organic extracts were distilled into ethanol and the solution was subjected to hydrogenation with 10% Pd on carbon. The presence of water in the hydrogenation was found to be crucial to the reaction success; anhydrous solutions of 14 hydrogenated with dry Pd on carbon proceeded only to low levels of conversion to 1, and addition of water to these reductions resulted in restored performance of the catalyst. Following hydrogenation, the catalyst was removed by filtration to provide an ethanol solution of 1. Sitagliptin was isolated in >99.5% purity as its anhydrous phosphoric acid salt by crystallizing from aqueous ethanol.
PATENT
Scott D Edmondson, Michael H Fisher,Dooseop Kim, Malcolm Maccoss, Emma R Parmee, Ann E Weber, Jinyou Xu
MORE INFO………
Sitagliptin phosphate monohydrate, a dipeptidyl-peptidase IV inhibitor, is marketed by Merck & Co. for the once-daily oral treatment of type 2 diabetes. The product was first launched in Mexico followed by commercialization in the U.S. The compound has also been filed for approval in the U.S. as adjunct to diet and exercise and in combination with other therapies to improve glycemic control in the treatment of diabetes. In 2007, the product was approved by the European Medicines Evaluation Agency (EMEA) and is currently available in the U.K., Germany and Spain. In 2009, sitagliptin phosphate monohydrate was approved and launched in Japan. The product is also available in Japan for the treatment of type 2 diabetes in combination with alpha-glucosidase inhibitors and in combination therapy with insulin. In 2012, the company filed for approval in Japan for the treatment of type 2 diabetes in patients with severe renal dysfunction, and in 2013 obtained the approval.
Sitagliptin phosphate monohydrate boasts a much lower risk of hypoglycemia than currently available insulin-inducing products due to its novel mechanism of action. MSD KK (formed in 2010 following the merger of Banyu and Schering-Plough KK) and Ono are developing the drug candidate in Japan. In 2008, the compound was licensed to Almirall by Merck Sharp & Dohme for comarketing in Spain for the treatment of type 2 diabetes. In 2010, FAES obtained a comarketing and commercialization license from Merck Sharp & Dohme in Spain for the treatment of type 2 diabetes.
Januvia (sitagliptin phosphate) is an antihyperglycaemic drug containing an orally active inhibitor of the dipeptidyl peptidase-IV (DPP-IV) enzyme. Developed by Merck Sharp & Dohme (MSD), a UK subsidiary of Merck & Co, sitagliptin is used for treating type 2 diabetes mellitus. The drug has proved effective in lowering blood sugar levels of diabetes patients when taken alone or in combination with other oral diabetes medications such as metformin and thiazolidinedione.
Sitagliptin was approved by the US Food and Drug Administration (FDA) in October 2006 and is marketed under the brand name Januvia in the US. Sitagliptin in combination with metformin was approved by the FDA in March 2007 and is marketed as Janumet in the US. In the EU, Januvia was approved in April 2007 and Janumet was approved in July 2008.
Sitagliptin is a triazolopiperazine based inhibitor of DPP-IV, which was discovered by
Merck. It is a potent (IC50= 18 nM) and highly selective over DPP-8 (48000 nM), DPP-9
(>100000 nM) and other isozymes.[16] It enhances the pancreatic β-cell functions, fasting and
post-prandial glycemic control in type 2 diabetic patients. In the crystal structure with DPP-IV,
unlike other substrate-based DPP-IV inhibitors, the binding orientation of the amide carbonyl of
sitagliptin is reversed, i.e. the aromatic trifluorophenyl moiety occupies S1 pocket and the β-
amino amide moiety fits into S2 pockets. The amino group forms a salt bridge and hydrogen
bonding interactions with Glu205 and Glu206, and Tyr662, respectively.The triazolopiperazinemoiety occupies the S2 extended pocket and stacks against Phe357. The exhibited binding
interactions of the trifluoromethyl group with the Arg358 and Ser209 are responsible for its high
selectivity profile. The presence of the trifluoromethyl group in the triazole ring also improves
the oral bioavailability in animal models. Sitagliptin inhibited the plasma DPP-IV up to 80% and
47% at 2 and 24 h, respectively, after a single dose of 25.0 mg in a dose-dependent manner. In a
24-week study, sitagliptin significantly decreased fasting glucose levels and HbA1c levels
(0.8%) at doses of 100 mg q.d. Thus, sitagliptin is well tolerated and body weight neutral. It is
the first DPP-IV inhibitor in the class approved by USFDA in 2006 and is used as either a
monotherapy or in combination with metformin


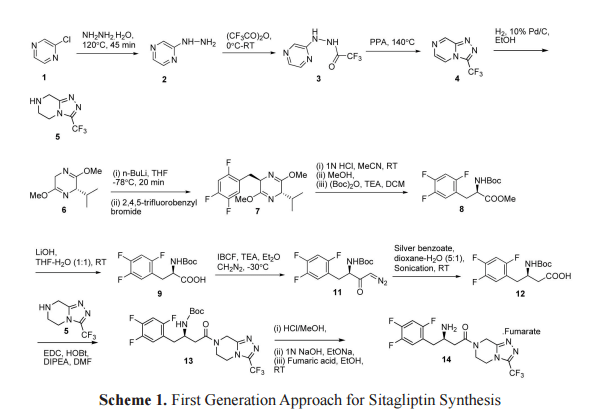
In the first synthetic approach, the synthesis of sitagliptin was started with the reaction of a Schollkopf reagent 6 with 2,4,5-trifluorobenzyl bromide to afford the compound 7, which was converted to compound 9 via hydrolysis of ester 8. The resulting Boc-protected amino acid 9 was converted to diazoketone 11 through mix anhydride protocol by using diazomethane. The intermediate 11 was converted to desired β-amino acid 12 by sonication in the presence of silver benzoate.[21] The sitagliptin (14) was synthesized by coupling of β-amino acid 12 with triazolopiperazine intermediate 5 followed by Boc deprotection of amino group of 13, and its corresponding hemi fumarate salt was then prepared (Scheme 1).[16]
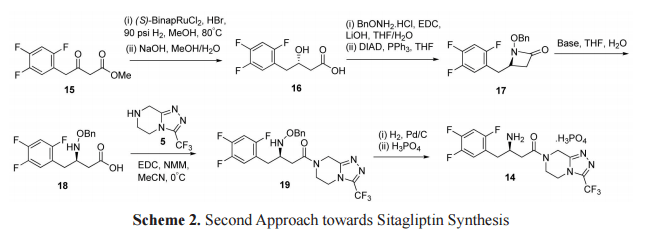
The second approach for synthesis of sitagliptinwas started from asymmetric reduction of β-ketoester 15 using the (S)-BinapRuCl2 complex with a catalytic amount of HBr in methanol followed by hydrolysis afforded the β-hydroxy acid 16. Lactam 17 was synthesized by coupling of 16 with BnONH2 •HCl using N-(3- dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC), followed by cyclization reaction with diisopropyl azodicarboxylate (DIAD) and PPh3 . [22] Treatment of a catalytic amount of 0.1% NaOH with lactam 17 hydrolyzed and directly afforded the β-amino acid 18. This wascoupled withtriazolopiperazine 5 using EDC•HCl and N-methylmorpholine to provide the N-benzyloxy protected compound 19, which after hydrogenation using Pd/C and by consequent treatment with phosphoric acid provided the phosphate salt of sitagliptin (14) (Scheme 2).
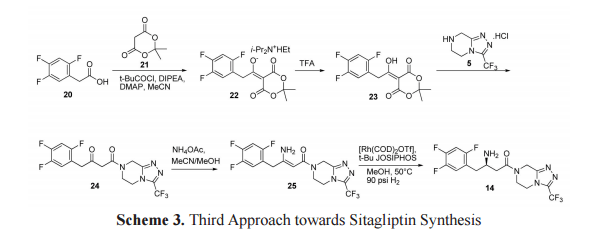
The third approach towards the synthesis of sitagliptin is outlined in scheme 3. Meldrum adduct 22 (Hunig’s base salt) was synthesized from trifluorophenylacetic acid 20 by the formation of a mixed anhydride with pivaloyl chloride in the presence of Meldrum’s acid 21, DIPEA and catalytic amount of dimethylamino pyridine (DMAP) in acetonitrile. Treatment of 22 with TFA resulted compound 23. β-keto amide 24 was formed on reaction of 23 with triazolopiperazine 5. β-keto amide 24 on treatment with ammonium acetate in methanol formed a key intermediate, dehydrositagliptin 25 (enamine amide). This intermediate contains the entire structure of sitagliptin 14 except two hydrogen atoms. Thus, sitagliptin 14 was synthesized by enantioselective hydrogenation of dehydrositagliptin 25 in the presence of [Rh(COD)2 OTf] 12,13 and t Bu JOSIPHOS in excellent yield with 95% ee.[23,24]
http://www.cbijournal.com/paper-archive/may-june-2014-vol-3/Review-Paper-1.pdf
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.




http://www.apiindia.org/medicine_update_2013/chap88.pdf
http://www.cbijournal.com/paper-archive/may-june-2014-vol-3/Review-Paper-1.pdf
////////////

