
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
TAVABOROLE, AN 2690, 他伐硼罗 Таваборол تافابورول
TAVABOROLE
- AN 2690
- AN-2690
- AN2690
- UNII-K124A4EUQ3
5-Fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole
5-Fluoro-2,1-benzoxaborol-1(3H)-ol;
1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole
MOLECULAR FORMULA C7H6BFO2
MOLECULAR WEIGHT 151.9
SPONSOR Anacor Pharmaceuticals, Inc.
CAS REGISTRY NUMBER 174671-46-6
Mp 118-120° C…..US20070265226
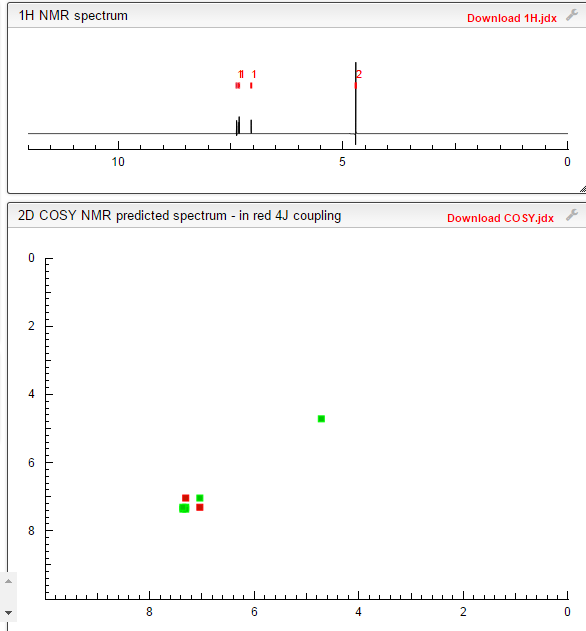
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H)
FDA APPROVED JULY 2 2014………..“FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
Has antifungal activity.
The US Food and Drug Administration (FDA) 2014 JULY 8 ratified the Anacor’s Kerydin (5% Tavaborole solution) for the topical treatment of nail fungal infections. Tavaboroleindications of toenail fungus Trichophyton rubrum or Trichophyton rubrum infections.Instructions recommended once a day for toenail infections, treatment for 48 weeks, on the recommendation of Anacor, and do not need to nail debridement.
I tis an oxaborole antifungal used topically, as a 5% w/w solution, for the treatment of onychomycosis of the toenails due to Trichophyton rubrumor T. mentagrophytes. It is applied to the affected toenail once daily for 48 weeks.
Ingrowing toenails and application site reactions including exfoliation, erythema, and dermatitis have been reported during use.
1H NMR FROM NET

CLICK ON IMAGE FOR CLEAR VIEW
COSY NMR PREDICT
Tavaborole (AN2690, trade name Kerydin) is a topical antifungal medication for the treatment of onychomycosis, a fungal infectionof the nail and nail bed. Tavaborole began its Phase 3 trials in December 2010[1] and was approved in July 2014.[2] Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection. No treatment-related systemic side effects were observed in any of its clinical trials.
Tavaborole is the first oxygen boron used to treat toenail infections dioxolane (oxaborole) antifungal agents, located in Palo Alto, Anacor focuses on boron-based drug development and production, according to the latest news, Tavaborole future also be used to infect fingernails. Wedbush Securities analyst predicts that next year the drug sales in the United States for $ 16 million, by 2021 will reach peak sales of $ 347 million.
Gram-negative bacteria cause approximately 70% of the infections in intensive care units. A growing number of bacterial isolates responsible for these infections are resistant to currently available antibiotics and to many in development. Most agents under development are modifications of existing drug classes, which only partially overcome existing resistance mechanisms. Therefore, new classes of Gram-negative antibacterials with truly novel modes of action are needed to circumvent these existing resistance mechanisms. We have previously identified a new a way to inhibit an aminoacyl-tRNA synthetase, leucyl-tRNA synthetase (LeuRS), in fungi via the oxaborole tRNA trapping (OBORT) mechanism.
Herein, we show how we have modified the OBORT mechanism using a structure-guided approach to develop a new boron-based antibiotic class, the benzoxaboroles, which inhibit bacterial leucyl-tRNA synthetase and have activity against Gram-negative bacteria by largely evading the main efflux mechanisms in Escherichia coli and Pseudomonas aeruginosa. The lead analogue, is active against Gram-negative bacteria, including Enterobacteriaceaebearing NDM-1 and KPC carbapenemases, as well as P. aeruginosa. This novel boron-based antibacterial, has good mouse pharmacokinetics and was efficacious against E. coli and P. aeruginosa in murine thigh infection models, which suggest that this novel class of antibacterials has the potential to address this unmet medical need.
Anacor continued development on that drug, tavaborole, and filed for FDA approval in July. The FDA will review the phase 3 trial data and issue a decision on July 29, 2014.
If approved, Anacor hopes tavaborole’s ability to clear onychomycosis in 10% of treated patients will be enough to win market share away from generic Lamisil and generic topical Pentac. While Lamisil cleared the fungus in 38% of patients, it’s been associated with rare cases of liver failure. And Pentac requires frequent debridement of the nail and only clears the fungus in 5.5% to 8.5% of patients.
Tavaborole is a novel, topical antifungal medication being developed for the topical treatment of onychomycosis, a nail fungus infection, which affects seven to ten percent of the U.S. population. Early studies show AN-2690 penetrates the nail effectively and has robust activity against dermatophytes, which cause onychomycosis.

1H NMR PREDICT

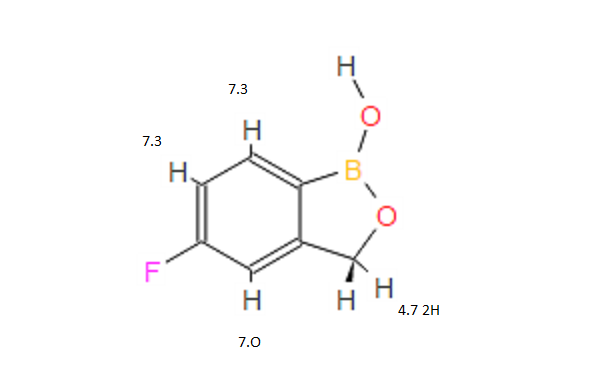
……………………………………………………………………………
13 C NMR PREDICT

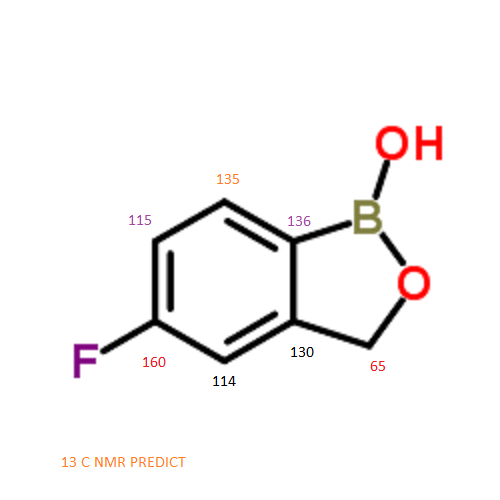
ARTICLE
Anacor Pharmaceuticals to Present Pivotal Phase 3 Data of Tavaborole for the Topical Treatment of Toenail Onychomycosis
Abstract Accepted for Oral Presentation at the 2013 American Podiatric Medical Association Annual Scientific Meeting
PALO ALTO, Calif.–(BUSINESS WIRE)– Anacor Pharmaceuticals (NASDAQ:ANAC) announced today that its abstract “Pivotal Phase 3 Safety and Efficacy Results of Tavaborole (Formerly AN2690), a Novel Boron-Based Molecule for the Topical Treatment of Toenail Onychomycosis” was accepted for oral presentation at the 2013 APMA Annual Scientific Meeting (The National) to be held in Las Vegas, Nevada. Max Weisfeld, DPM, will present the data from tavaborole’s Phase 3 studies on Monday, July 22, 2013 during the Evidence-Based Medicine and Oral Abstracts session.
As announced earlier this year, tavaborole achieved statistically significant and clinically meaningful results on all primary and secondary endpoints in two Phase 3 pivotal studies without concomitant debridement. Anacor is seeking approval for tavaborole from the Food and Drug Administration (FDA) and will file a New Drug Application imminently. Currently, there is only one FDA-approved topical treatment for onychomycosis, a fungal infection of the nail and nail bed, which affects approximately 35 million people in the United States.
“I’m impressed with tavaborole’s safety and efficacy data. There is no FDA-approved topical treatment for onychomycosis with tavaborole’s range of efficacy and ability to penetrate the nail to reach the site of the infection,” said Dr. Weisfeld. “Tavaborole’s Phase 3 results demonstrate its ability to clear the nail and eliminate the infection which is important to both patients and the physicians who treat them. In addition, tavaborole is easy to apply and dries quickly which makes it convenient for patients to use.”
“We are pleased to present these positive data at the APMA’s Annual Scientific Meeting, the leading annual meeting of podiatrists. As we seek FDAapproval for tavaborole, we look forward to developing relationships with podiatrists to potentially offer them a new treatment option for the large number of patients who seek treatment for onychomycosis,” said David Perry, Chief Executive Officer of Anacor Pharmaceuticals.
About the Studies
Anacor conducted two separate Phase 3 studies of tavaborole on patients with distal subungual onychomycosis affecting 20 to 60 percent of the target great toenail. Approximately 600 patients aged 18 years and older with no upper age limit (the oldest subject was 88 years old) were enrolled in each study and randomized two-to-one to receive either tavaborole or the vehicle control. Patients were instructed to apply tavaborole solution or the vehicle to the toenail once daily for 48 weeks.
A copy of the presentation will be available on Anacor’s website following the oral session.
About Anacor Pharmaceuticals
Anacor is a biopharmaceutical company focused on discovering, developing and commercializing novel small-molecule therapeutics derived from its boron chemistry platform. Anacor has discovered eight compounds that are currently in development. Its two lead product candidates are topically administered dermatologic compounds — tavaborole, a topical antifungal for the treatment of onychomycosis, and AN2728, a topical anti-inflammatory PDE-4 inhibitor for the treatment of atopic dermatitis and psoriasis. In addition to its two lead programs, Anacor has discovered three other wholly-owned clinical product candidates — AN2718 and AN2898, which are backup compounds to tavaborole and AN2728, respectively, and AN3365 an antibiotic for the treatment of infections caused by Gram-negative bacteria. We have discovered three other compounds that we have out-licensed for further development — two compounds for the treatment of animal health indications that are licensed to Eli Lilly and Company and AN5568, also referred to as SCYX-7158, for human African trypanosomiasis (HAT, or sleeping sickness), which is licensed to Drugs for Neglected Diseases initiative, or DNDi. We also have a pipeline of other internally discovered topical and systemic boron-based compounds in development. For more information, visit http://www.anacor.com.
Patents
WO 1995033754
WO 2004009578….
WO 2006089067
WO 2008025543
…………………………..
SYNTHESIS

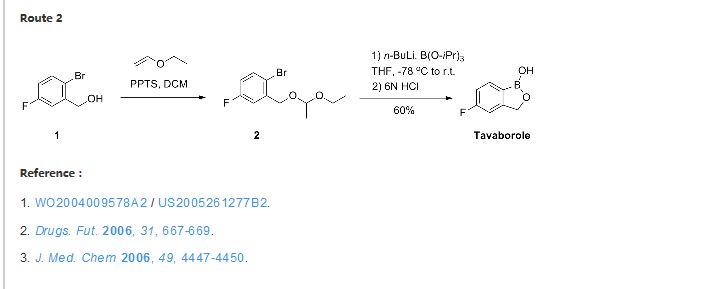
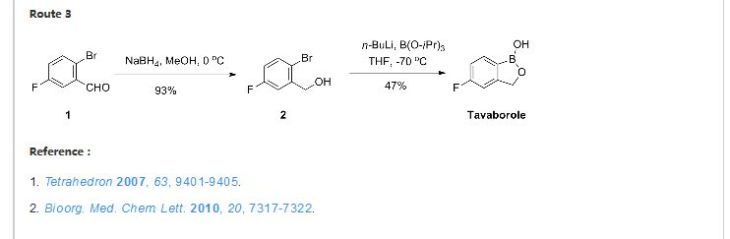
Reference:
ELI LILLY AND COMPANY Patent: WO2004/9578 A2, 2004 ; Location in patent: Page 36-37 ; WO 2004/009578 A2
PATENT
Anacor Pharmaceuticals Patent: US2007/265226 A1, 2007 ; Location in patent: Page/Page column 59 ;
http://www.google.com/patents/US20070265226

1,3-Dihydro-5-fluoro-1-hydroxy-2,1-benzoxaborole (19b)
To a solution of 5b (73.2 g, 293 mmol) in dry THF (400 mL) was added n-butyllithium (1.6 M in hexanes; 200 mL) over 45 min at −78° C. under nitrogen atmosphere. Anion precipitated. After 5 min, (i-PrO)3B (76.0 mL, 330 mmol) was added over 10 min, and the mixture was allowed to warm to room temperature over 1.5 h. Water and 6 N HCl (55 mL) were added, and the solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure. To a solution of the residue in tetrahydrofuran (360 mL) was added 6 N HCl (90 mL), and the mixture was stirred at 30° C. overnight. The solvent was removed under reduced pressure to about a half volume. The mixture was poured into ethyl acetate and water. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the residue was treated with i-Pr2O/hexane to give 19b (26.9 g, 60%) as a white powder:
mp 118-120° C.;
1H NMR (300 MHz, DMSO-d6) δ (ppm) 4.95 (s, 2H), 7.15 (m, 1H), 7.24 (dd, J=9.7, 1.8 Hz, 1H), 7.74 (dd, J=8.2, 6.2 Hz, 1H), 9.22 (s, 1H);
ESI-MS m/z 151 (M−H)−;
HPLC purity 97.8%; Anal (C7H6BFO2) C, H.
…………………
Gunasekera, Dinara S.; Gerold, Dennis J.; Aalderks, Nathan S.; Chandra, J. Subash; Maanu, Christiana A.; Kiprof, Paul; Zhdankin, Viktor V.; Reddy, M. Venkat Ram Tetrahedron, 2007 , vol. 63, # 38 p. 9401 – 9405
…………………….
Baker, Stephen J.; Zhang, Yong-Kang; Akama, Tsutomu; Lau, Agnes; Zhou, Huchen; Hernandez, Vincent; Mao, Weimin; Alley; Sanders, Virginia; Plattner, Jacob J. Journal of Medicinal Chemistry, 2006 , vol. 49, # 15 p. 4447 – 4450
………………..
Ding, Charles Z.; Zhang, Yong-Kang; Li, Xianfeng; Liu, Yang; Zhang, Suoming; Zhou, Yasheen; Plattner, Jacob J.; Baker, Stephen J.; Liu, Liang; Duan, Maosheng; Jarvest, Richard L.; Ji, Jingjing; Kazmierski, Wieslaw M.; Tallant, Matthew D.; Wright, Lois L.; Smith, Gary K.; Crosby, Renae M.; Wang, Amy A.; Ni, Zhi-Jie; Zou, Wuxin; Wright, Jon Bioorganic and Medicinal Chemistry Letters, 2010 , vol. 20, # 24 p. 7317 – 7322
…………..
PATENT
PREPARATION 13 5-Fluoro-3H-benzo[c][1,2)oxaborol-1-ol

Dissolve 1-bromo-2-(1-ethoxy-ethoxymethyl)-4-fluoro-benzene(5.4 g, 19.5 mmol) in dry THF (100 mL) and cool to −78° C. under nitrogen. Add butyl lithium (2.5M in Hexanes, 10.2 mL, 25.4 mmol) dropwise at −78° C. Upon complete addition, stir the reaction at −78° C. for 10 minutes and then add trimethyl borate (4.4 mL, 39 mmol) and warm the reaction to room temperature. Pour the reaction into 1N HCl (100 mL) and stir for 1 hour. Extract the biphasic mixture with ether three times. Dry the combined organic layers with sodium sulfate, filter and concentrate in vacuo. Triturate the oily residue with cold hexanes to yield 2.1 g (70%) of the title compoud as a white solid.
1H NMR (d6-DMSO)
9.18 (s, 1H),
7.70 (dd, J=8.2, 5.8 Hz, 1H),
7.20 (dd, J=9.5, 2.7 Hz, 1H),
7.11 (m, 1H), 4.92 (s, 1H).
…………………
SEE
http://jpet.aspetjournals.org/content/early/2012/11/28/jpet.112.200030.full.pdf
………………………………..
SEE
Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ.
J Med Chem. 2006 Jul 27;49(15):4447-50.
Boron-containing inhibitors of synthetases.
Baker SJ, Tomsho JW, Benkovic SJ.
Chem Soc Rev. 2011 Aug;40(8):4279-85. doi: 10.1039/c0cs00131g. Epub 2011 Feb 7. Review.
- Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles.
Zhang YK, Plattner JJ, Freund YR, Easom EE, Zhou Y, Ye L, Zhou H, Waterson D, Gamo FJ, Sanz LM, Ge M, Li Z, Li L, Wang H, Cui H.
Bioorg Med Chem Lett. 2012 Feb 1;22(3):1299-307. doi: 10.1016/j.bmcl.2011.12.096. Epub 2011 Dec 28.
Tavaborole Market Opportunity
Anacor is developing tavaborole specifically to address the current limitations of existing treatment options for onychomycosis. This includes designed leaps forward in both the potential safety and efficacy profile aimed to make the drug a best-in-class therapy. Additionally, management has used the company’s expertise in medicinal chemistry to improve delivery of the compound through the nail plate to the nail bed, the site of onychomycosis infection. For example, preclinical studies indicate that tavaborole is able to penetrate the nail plate 250 times more effectively than ciclopirox.
Tavaborole novel mechanism of action inhibits an essential fungal enzyme, leucyl transfer RNA synthetase, or LeuRS required for protein synthesis. The inhibition of protein synthesis leads to termination of cell growth and cell death, eliminating the fungal infection.
Likewise, the topical dosing was designed to eliminate systemic absorption. Previous preclinical and clinical data shows topical treatment with tavaborole resulted in little or no detectable levels of drug in the blood or urine. No treatment related systemic side effects have been observed in any clinical trials to date. Safety data from the company’s studies to date was recently presented at the 100th National APMA meeting in Washington, DC.
Anacor’s topical solution currently in two phase III trials for onychomycosis. Phase II data with tavaborole suggests efficacy superior to ciclopirox with little to no systemic exposure.
Data from an open-label phase 2 program with tavaborole showed 50% patients using a 7.5% solution saw 2 mm clear nail growth and negative fungal cultures after six months. Roughly 25% of the patients saw 5 mm clear nail growth and negative fungal cultures after six months.
Anacor and partner Merck (NYSE:MRK) met with the U.S. FDA in 2009 to discuss the phase II data. Merck has since returned the rights to tavaborole to Anacor. The original deal was with Schering-Plough in 2007. Merck most likely felt as though tavaborole clashed with existing products or did not have peak sales potential large enough to continue the partnership with Anacor. We see tavaborole as a specialty promoted product, into podiatrists and dermatologists. For a company like Anacor, it’s an attractive first product.
Anacor’s first phase III trial completed enrollment in November 2011. The second phase III trial completed enrollment in December 2011. Data from these trials are expected around the middle of January 2013. Data from the second study is expected six weeks later. Given the positive phase II data noted above, we think odds favor a positive outcome. A benchmark for the trial is the efficacy of Lamisil, which is a complete cure rate of around 35% to 40%, and a mycological cure of around 70% after a typical course of treatment.
I note that on Anacor’s third quarter conference call management noted that they are pleased with the conduct of the trial to date. Specifically, the compliance rate appears to better than management had expected. The trial was designed with a 20% drop-out rate. It looks as though the drop-out rate is only around 13%, at a minimum suggestive of good safety and tolerability, but potentially also a sign that the drug is working.
I see onychomycosis as a significant market opportunity for Anacor. An estimated 35 million Americans have nail fungus, with about 95% of the infections in the toenail. With efficacy similar to Lamisil, we think Anacor can capture 20% of the market. With a price per course of treatment at around $1,200, I think peak sales of tavaborole are $500 million.
Conclusion
I’ll note two more important pieces of information for investors. Firstly, besides optimism for tavaborole, Anacor has apipeline of anti-infectant drugs. For this article I discussed only tavaborole. A second article can be dedicated entirely to AN2728 for the treatment of psoriasis and atopic dermatitis. Anacor also has an animal health collaboration with Eli Lilly (NYSE:LLY).
The second important thing to note is Anacor’s cash position. The company reported financial results on November 7, 2012. The company held $36.6 million in cash on the balance sheet as of September 30, 2012. However, in October 2012, the company completed an underwritten public offering of 4.0 million shares of common stock at $6.00 per share to raise net proceeds of $22.7 million. I view the current cash position as sufficient to report data from both phase 3 trials and, if positive, file the new drug application (NDA) around the middle of 2013.
With phase 3 data expected in less than two months, good prior evidence of both safety and efficacy, and a solid cash position, I think Anacor could be an attractive investment at today’s price. The stock is down meaningfully over the past month and investors can buy sizably below the October offering.


SYNTHESIS

References
- Clinical trial number NCT01270971 at ClinicalTrials.gov
- “FDA Approves Anacor Pharmaceuticals’ KERYDIN™ (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails”. Market Watch. July 8, 2014.
- http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204427s000lbl.pdf
- http://www.molbase.com/en/hnmr_174671-46-6-moldata-1568017.html#tabs
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
5-Fluoro-2,1-benzoxaborol-1(3H)-ol
|
|
| Clinical data | |
| Trade names | Kerydin |
| Legal status |
|
| Routes of administration |
Topical use only |
| Identifiers | |
| CAS Registry Number | 174671-46-6 |
| ATC code | None |
| PubChem | CID: 11499245 |
| ChemSpider | 9674047 |
| Synonyms | AN2690 |
| Chemical data | |
| Formula | C7H6BFO2 |
| Molecular mass | 151.93 g/mol |
NMR PREDICT
 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole H-NMR spectral analysis |

 CAS NO. 174671-46-6, 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole C-NMR spectral analysis |

more

 Anacor
Anacor
Anacor Pharmaceuticals is out to change that. The Palo Alto, Calif.-based biotechnology company is developing a family of boron-containing small-molecule drugs. And with the assistance of Naeja Pharmaceutical, a Canadian contract research organization, Anacor has licensed one of those molecules to GlaxoSmithKline and taken another one into Phase III clinical trials.
Anacor was founded in 2002 to develop technology created by Lucy Shapiro, a Stanford University bacterial geneticist, and Stephen J. Benkovic, a Pennsylvania State University organic chemist. Through a long-standing scientific collaboration, the two researchers had discovered boron-containing compounds that inhibited specific bacterial targets………..https://pubs.acs.org/cen/coverstory/89/8912cover3.html
mp 118-120….http://www.syninnova.com/catalog/product/SL-264
antifugal AN2690 by Anacor
Tavaborole inhibits an essential fungal enzyme, Leucyl-tRNA synthetase, or LeuRS, required for protein synthesis.
Minimum Inhibitory Concentration: 1, 1, 0.5, 0.25, and 0.25 μg/mL for T.rubrum, T.mentagrophytes, C.albicans, C.neoformans, A.fumigatus, respectivley.
AN2690 is a new boron-containing antifungal agent for the potential treatment of onychomycosis. Onychomycosis is caused mainly by dermatophytes, a class of fungus that dwells on skin, hair, and nails and is the cause of other cutaneous fungal infections such as athlete’s foot.
In vitro: AN2690 showed the most active against fungi and especially against the dermatophytes T. rubrum and T. mentagrophytes, the primary fungal pathogens causing onychomycosis. In addition, AN2690 was identified as having a unique profile of in vitro antidermatophyte activity, maintenance of this activity in the presence of keratin, and exceedingly good penetration of human nails [1].
Ex vivo: AN2690 was found to have superior penetration compared to ciclopirox, and achieves levels within and under the nail plate that suggest it has the potential to be an effective topical treatment for onychomycosis [2].
Clinical trial: The efficacy of tavaborole as a topical treatment for onychomycosis has been evaluated in two identical randomised, double-blind phase III studies, NCT01270971 (301) and NCT01302119 (302), enrolling 593 and 601 patients, respectively. Completely or almost clear nail and negative mycology was achieved in 15.3 and 17.9 % of tavaborole recipients compared with 1.5 and 3.9 % of vehicle recipients [3]
References:
[1] Baker SJ, Zhang YK, Akama T, Lau A, Zhou H, Hernandez V, Mao W, Alley MR, Sanders V, Plattner JJ. Discovery of a new boron-containing antifungal agent, 5-fluoro-1,3-dihydro-1-hydroxy-2,1- benzoxaborole (AN2690), for the potential treatment of onychomycosis. J Med Chem. 2006;49(15):4447-50.
[2] Hui X, Baker SJ, Wester RC, Barbadillo S, Cashmore AK, Sanders V, Hold KM, Akama T, Zhang YK, Plattner JJ, Maibach HI. In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci. 2007;96(10):2622-31.
[3] Markham A. Tavaborole: first global approval. Drugs. 2014;74(13):1555-8.
UPDATE
http://www.google.im/patents/EP1976536A2?cl=en
EXAMPLE 23
Alternative Preparation of 4 from 3
A 22.0 L 3-neck flask was equipped with a stir motor, N2 inlet, addition funnel, heating mantle, and condenser. The flask was charged with 3500 g (17.1 moi) of 2-bromo-5-fluorobenzyl alcohol followed by the addition of 3556 g of tetrahydrofuran and 16.4 g (0.17 mol) of methanesulfonic acid. Next, 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 100C. This step is exothermic so no additional charges should be made until exotherm subsides. The temperature was increased to 27°C, stirred for 15 min and then charged with 400 g (4.7 mol) of 3,4-dihydro-2H- pyran at 240C. Again the temperature increased (24°C to 380C). The mixture was stirred for 15 min. Once the exotherm subsided, the flask was again charged with 40Og (4.7 mol) of 3,4-dihydro-2H-pyran at 350C. The temperature again increased to 470C over a 20 min period. Once the exotherm subsided, the mixture was stirred for 15 min. Finally the remaining 400 g (4.7 mol) of 3,4-dihydro-2H-pyran was added at 440C. The temperature increased to 510C. After stirring for one hour, a sample was removed to check for removal of starting material. Upon reaction completion, contents were cooled to 20 ± 5 0C.
EXAMPLE 24

Alternative Preparation of 5 from 4
To a 22.0 L 3-neck flask equipped with a stir motor, N2 inlet, addition funnel, cooling bath, and condenser was charged 436 g (17.96 mol) of magnesium turnings. 5334 g of tetrahydrofuran was then added followed by 291 g (0.51 mol) of diisobutylaluminum hydride (DIBAL) (25%wt) in toluene. The mixture was stirred for 60 min at 20 ± 5 0C. Some gas evolution was seen. Next, 260-430 g -3-5% (by weight if solution of 4 was dropped to drums) of 4 in THF was added. The mixture was stirred for 15-30 min at which time a slight exotherm should be seen (ΔT = 10- 150C). Once the exotherm was observed, the reaction mixture was cooled to 5 ± 5 0C. To this mixture, the remaining 8.22-8.39 kg of 4 in THF was added at a rate such that the temperature was kept below 300C (t = 3h). The reaction was stirred at 20-25 0C for 30 min, at which time an aliquot was removed, quench with 3 N HCl (10 mL), and analyzed.
Upon completion, the contents were cooled to -25 ± 5°C. A solution of trimethylborate in THF was prepared by mixing 2665 g (25.7 mol) of trimethyl borate and 6666 g of tetrahydrofuran. This solution can be prepared in a drum with stirring. [0618] Next, the 9331 g of trimethyl borate in THF was added at a rate such that the temperature was kept between -35 and -20 °C (t = 2.5h). The mixture became very thick so THF was added. After stirring at -25 ± 5°C for 10 min, 50 mL aliquot was removed, quenched with 25 mL of 3N HCl, and submitted for CoR. Stirring continued at -25 ± 50C for Ih, and then the mixture was allowed to warm to ambient temperature, where it was stirred for at least 12h. Pull two samples (one at 6h and the other at 12h).
Results:
1H-NMR (300 MHz, DMSO-d6) δ (ppm) 1.45-1.75 (m, 6H), 3.53 (s, 6H), 3.45 (m, IH), 3.75 (m, IH), 4.69 (t, J=3 Hz, IH), 4.97 (d, J=14.1 Hz, IH), 5.14 (d, J=14.1 Hz, IH), 7.03 ((td, J=8.4, 2.7 Hz, IH), 7.24 (dd, J=10.8, 2.1 Hz, IH), 7.89 (t, J=7.8 Hz, IH), 8.76 (s, IH).
EXAMPLE 25
Alternative Preparation of I from 5

To the reaction mixture above was added 5.3 kg of USP water. After stirring for 30 min, the mixture was charged 5.3 kg of acetic acid. Gas evolution was seen. After stirring for 30 min, an aliquot was removed for analysis. Mixture was then heated to reflux for 36-48 hours. During the reflux period, 12-13 L of THF were removed.
When the reaction was complete, the contents were cooled by the reactor to <40°C by setting jacket and by charging 10.5 kg of USP water. THF was removed until distillate did not remain. Contents of the reactor were transferred to Rosenmund filter dryer and allowed to cool to 20 ± 5°C. Reactor was rinsed with water, filtered, and then washed again with 10.5 kg of USP water. The flask was charged with 10.5 kg of 10% ACN in water (v/v) and agitated for Ih. After filtering, the cake was washed with 10.5 kg of 10% ACN in water (v/v), and then charged with 10.5 kg 10% ACN in water (v/v). The contents were agitated for Ih. The contents were subsequently washed with 10.5 kg of USP water, charged with 7.0 L of 5% Methyl t- Butyl Ether (MTBE)/Heptane (v/v), agitated for Ih, filtered, charged with 7.0 L of 5% MTBE/Heptanes (v/v) and again agitated for Ih. After filtering, the contents were charged again with 7.0 L of heptane and filtered. Solids were dried at <45°C to constant weight. Solids were recrystallized from toluene :heptane 75:25.
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////
Vedroprevir
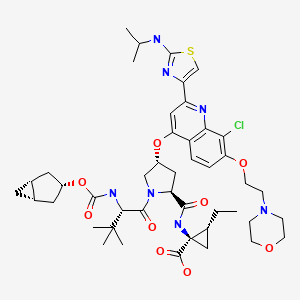
GS 9451, GS-9451, 1098189-15-1 USAN ZZ-81
VEDROPREVIR THERAPEUTIC CLAIM Treatment of hepatitis C
CHEMICAL NAMES 1. Cyclopropanecarboxylic acid, N-[[(1α,3β,5α)-bicyclo[3.1.0]hex-3- yloxy]carbonyl]-3-methyl-L-valyl-(4R)-4-[[8-chloro-2-[2-[(1-methylethyl)amino]- 4-thiazolyl]-7-[2-(4-morpholinyl)ethoxy]-4-quinolinyl]oxy]-L-prolyl-1-amino-2- ethyl-, (1R,2R)-
2. N-{[(1R,3r,5S)-bicyclo[3.1.0]hex-3-yloxy]carbonyl}-3-methyl-L-valyl-(4R)-4-[(8- chloro-2-{2-[(1-methylethyl)amino]thiazol-4-yl}-7-[2-(morpholin-4- yl)ethoxy]quinolin-4-yl)oxy]-L-prolyl-(1R,2R)-1-amino-2- ethylcyclopropanecarboxylic acid
MOLECULAR FORMULA C45H60ClN7O9S
MOLECULAR WEIGHT 910.5 daltons
SPONSOR Gilead Sciences, Inc.
CODE DESIGNATION GS-9451
CAS REGISTRY NUMBER1098189-15-1
WHO NUMBER9745
GS-9451 is a NS3 protease inhibitor in phase II clinical trials at Gilead for the oral treatment of hepatitis C.
…………………………………………………………………………
Discovery of GS-9451: An acid inhibitor of the hepatitis C virus NS3/4A protease Bioorg Med Chem Lett 2012, 22(7): 2629 ……………………………………………………………
PATENTS WO 2012087596 WO 2009005676 WO 2013106631 WO2013101550 ……………………….
WO2012087596A1 Compound 3 can be prepared using synthetic methods and intermediates like those described in USSN 12/215,605 (US 20090257978 A1). Compound 3 can also be prepared described in the following Example. Example 3: Preparation of Compound 3

Compound 315 (12 g, 13 mmol) was dissolved in THF (200 ml), LiOH (11g, 260 mmol) in H20 (200 ml) was added, followed by MeOH (200 ml). The mixture was kept stirring at room temperature for 20 hours. Upon completion of the reaction, 4 N HCI in H20 was added to adjust pH to 7 at 0 °C. The mixture was extracted with EtOAc (2 x 400 ml). The combined organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo to give compound 3 as a yellow solid (11 g, 93%). LC/MS = 911.52(M++1 ). 1H NMR (300MHz, CD3OD)57.95 (d, 1H), 7.90 (s, 1H), 7.48 (s, 1H), 7.31 (d, 1H), 5.42 (s, 1H), 4.37 (dd, 1H), 4.20 (m, 2H), 3.83-3.56 (m, 7H), 3.50 (m, 2H), 3.39 (m, 2H), 2.45 (m, 1H), 2.27(m, 1H), 1.62 (m, 2H), 1.50 (m, 1H), 1.33 (m, 2H), 1.18 (m, 1H), 1.05 (m, 8H), 0.90 (m, 3H), 0.76 (m, 11H), 0.14-0.04 (m, 2H) The intermediate compound 315 was prepared as follows.

301 302 a. Preparation of compound 301. To a dry, argon purged three-neck round bottom flask (1000 mL) were added anhydrous dichloromethane (100 mL) and Et2Zn (28 mL, 273 mmol) at 0 °C. (CAUTION: Source of argon can not be from needle. Use appropriate glass adapter only. A second bubbler can also be attached to the flask to prevent excessive pressure build up.) Cyclopenten-3-ol (10.0 mL, 119 mmol) was then added dropwise (large quantity of ethane gas was produced) to the flask and the reaction mixture was allowed to stir until the evolution of gas had ceased. Diiodomethane (22 mL, 242 mmol) was then added dropwise over a period of 30 minutes. The reaction was allowed to warm to room temperature and continued to stir overnight under a positive flow of argon, at which point TLC analysis had indicated complete disappearance of the starting alcohol. The reaction was then diluted with CH2CI2 and quenched with 2M HCI (white precipitate should be completely dissolved). The biphasic mixture was poured into a separatory funnel and the organic layer was collected. The solvent was removed under reduced pressure until 100 mL of material containing compound 301 remained. b. Preparation of compound 302. Anhydrous dichloromethane (525 mL) was added to the flask followed by the dropwise addition of triethylamine (34 mL, 245 mmol). The reaction continued to stir at room temperature under a positive flow of nitrogen at which point, disuccinimidylcarbonate (40.7 g, 159 mmol) was added to the flask portion wise. The reaction was allowed to stir until TLC analysis indicated complete disappearance of the starting material (2-3 days). Upon completion, the reaction mixture was quenched with 1 M HCI (200 mL x 2) and washed with H20 (200 mL x 2). The desired material was extracted using CH2CI2and the combined organic layers were dried using anhydrous MgS0 and passed through a silica plug. The solvent was removed under reduced pressure and the crude material was purified using flash chromatography (Rf = 0.33, 1 :1 Hex/EtOAc) to provide compound 302 (22 g, 75%): 1H NMR (300 MHz, CDCI3): δ 5. 24 (t, 1 H), 3.82 (s, 4H), 2.24 (m, 2H), 2.03 (d, 2H), 1.38 (m, 2H), 0.48 (m, 1 H), 0.40 (m, 1 H).


c. Preparation of compound 304. N-i-Boc-cis-4-Hydroxy-L-Proline methyl ester 303 (100.0 g, 407.7 mmol) and DABCO (1.5eq, 68.6g, 61 1.6 mmol) were dissolved in anhydrous toluene (200 mL) in a 2 L three necked round bottom flask with a mechanical stirrer and an addition funnel. After cooling the solution to 0 °C under N2, A solution of 4-Bromo-benzenesulfonyl chloride (1.3eq, 135.6g, 530.0 mmol) in 300 mL of toluene was added through addition funnel over 60 minutes. The reaction mixture was stirred and warmed to room temperature overnight (16 hours). The mixture was slowly poured into 2L 1 M Na2C03 (aq.), and the product was extracted with EtOAc (2L). After the organic phase was washed by 0.5 N HCI (2L), H20 (1 L), and brine (1 L), it was dried (MgS04), concentrated to give 195.45 g of a yellow oily brosylate product. To a solution of the above brosylate (407.7 mmol) in dichloromethane (300 mL) was slowly added 4.0 M HCI in dioxane (500 mL, 5eq) and the resulting solution was allowed to stir at room temperature for 2 hours. After ether (500mL) was added to the reaction mixture, the mixture was stirred for 15 minutes and the white precipitate was collected by filtration. The solid was washed with ether and hexane and then dried under vacuum overnight to obtain 153.0 g of the HCI amine salt of compound 304, 381.8 mmol, in 94% yield for two steps. d. Preparation of compound 305. To a solution of Boc-fert-butyl-glycine (97.0g, 420.0 mmol) in DMF (200mL) and DCM (200mL) were added HATU (217.76g, 572.7 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of the previous HCI salt (153.0 g, 381.8 mmol) and Hunig’s base (126 mL, 1 145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3h, with monitoring by LCMS. The reaction mixture was concentrated to remove dichloromethane under reduced pressure and the white solid that formed was filtered off. The remaining DMF solution was diluted with ethyl acetate (1 L), washed successively with 3% LiCI (aq) (3x650mL), sat’d NH4CI (2x500mL), 0.5N HCI (aq) (2x600ml_), brine (500ml_), sat’d NaHC03 (3x500mL), and brine (500mL). The resulting organic fraction was dried (MgS04) and concentrated to afford compound 305 (111g). e. Preparation of compound 306. To a solution of the methyl ester 305 (120 g, 207.8 mmol) in THF (300 ml_), MeOH (75 mL) was added a solution of LiOH (26.18 g, 623.4 mmol) in H20 (150 ml_). The solution was allowed to stir at room temperature for 4 hours. The mixture was cooled in an ice-bath while acidifying with 3N HCI to pH about 5.5, stirred for 10minut.es, and the resulting white solids were collected by filtration. The solids were washed with more water, ether and hexane. The solids were dried under vacuum at 40°C overnight to give 95.78g (82%) of the acid 306. f. Preparation of compound 307. To a solution of the carboxylic acid 306 (81.4 g, 144.27 mmol) in DMF (200ml_) and dichloromethane (200mL) was added HATU (82.3g, 216.4 mmol) and Hunig’s base (47.5 mL, 432.8 mmol) at room temperature. After the mixture was stirred for 20 minutes at room temperature, a solution of amine (158.7 mmol) and Hunig’s base (47.5 mL, 1145.4 mmol) in DMF (200mL) and dichloromethane (200mL) was added to the above acid mixture in one portion. The reaction mixture was stirred at room temperature for 3 hours and monitored by LCMS. After the mixture was concentrated under reduced pressure to remove dichloromethane, the white solids that formed were filtered off. The remaining DMF solution was diluted with ethyl acetate (600mL) and successively washed with 3% LiCI (aq) (2x550mL), sat’d NH4CI (500mL), 1 N HCI (aq) (500mL), sat’d NaHC03(500mL), and brine (300mL). The resulting organic fraction was dried (Na2S04) and concentrated to afford compound 307 (111 g). g. Preparation of compound 308. Compound 307 was dissolved in 4N HCI in dioxane (300 mL) at room temperature and stirred for 2 hours. It was then concentrated under vacuum, and co-evaporated with dichloromethane (2 x 200mL) to dryness. The residue was dissolved in EtOAc (600mL) and sat’d aq. NaHC03 (1 L). It was stirred vigorously. After 10 minutes, carbonic acid bicyclo[3.1.0]hex-3-yl ester 2,5-dioxo-pyrrolidin-1-yl ester 302 (41.4 g, 173.1 mmol) was added in one portion. After the resulting mixture was stirred for another 30 minutes, the organic layer was collected and washed with brine (500mL), dried (Na2S04), and concentrated. The crude product was purified by flash chromatography on silica gel with ethyl acetate/hexane to afford 94.44 g (92%) of compound 308.



h. Preparation of compound 310.1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 309 (70.7 g, 354 mmol) was stirred in 48% aq. HBr (500 mL) at 110 °C for 72 hours. After the mixture was cooled to 0 °C with stirring, the solids were filtered and washed with water. The resulting solids were triturated with a saturated NaHC03 solution (-350 mL), filtered, washed with water, and dried under vacuum to give – 40 g (61%) of crude 310 as a dark brown solid. LC/MS = 186 (M++1). i. Preparation of compound 311. 1-(2-Amino-3-chloro-4-hydroxy-phenyl)-ethanone 310 (40 g, 215 mmol) was dissolved in DMF (360 ml). Cesium carbonate (140 g, 430 mmol) was added, followed by bromoacetaldehyde dimethyl acetal (54.5 g, 323 mmol). The mixture was then vigorously stirred at 65 °C for 24 hours. Upon cooling to room temperature, EtOAc (1 L) and H20 (1 L) were added to the mixture. The organic layer was extracted with EtOAc (1 x 400 ml). The combined organic layer was washed with aqueous 3% LiCI solution (2 x 1 L), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give compound 311 as a white solid (39 g, 67%). j. Preparation of compound 312. To a mixture of 1-[2-Amino-3-chloro-4-(2,2-dimethoxy-ethoxy)-phenyl]-ethanone 311 ( 13 g, 47.5 mmol) and isopropylaminothiazole-4-carboxylic acid hydrobromide (12.64 g, 47.5 mmol) in pyridine (150 ml) was slowly added phosphorus oxychloride (9.47 g, 61.8 mmol) at -40 °C. The mixture was then stirred at 0 °C for 4 hours. Upon completion of the reaction, H20 (30 ml) was added dropwise to the mixture. The mixture was then stirred at 0 °C for another 15 minutes. The mixture was concentrated in vacuo. The residue was diluted with EtOAc, washed with a sat. NaHC03 aqueous solution. The organic layer was dried (Na2S04) and concentrated in vacuo. The residue was dissolved in CH2CI2, hexanes were added slowly to the solution, and a yellow solid started to crash out. More hexanes were added until not much product was left in the mother liquid to provide compound 312 (18 g, 85%). k. Preparation of compound 313. 2-lsopropylamino-thiazole-4-carboxylic acid [6-acetyl-2-chloro-3-(2,2-dimethoxy-ethoxy)- phenyl]-amide 312 (18 g, 40.7 mmol) was suspended in toluene (400 ml). NaH (2.4 g, 61 mmol) was added to the vigorously stirred mixture while monitoring H2evolution. The mixture became a clear solution during heating to reflux. The reaction was complete after refluxing for 3 hours. The mixture was cooled to room temperature. A solution of AcOH (69.2 mmol) in H20 (3 vol) was added to the mixture. After vigorous agitation for 1 hour at 0 °C, the solids were collected by filtration, rinsed forward with H20. The wet cake was dried under high vacuum to a constant weight to provide compound 313 ( 5 g, 86%). I. Preparation of compound 314. To a mixture of brosylate intermediate 303 (15 g, 35 mmol) and compound 313 (27.5 g, 38.5 mmol) in NMP (200 ml) was added cesium carbonate (25.1 g, 77 mmol). The mixture was stirred at 65 °C for 5 hours. The reaction was cooled to room temperature and EtOAc (600 ml) and an aqueous solution of 3% LiCI (600 ml) were added to the mixture. The organic layer was washed with aqueous 3% LiCI (1 x 600 ml), brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired methyl ester as a yellow solid (23.6 g, 75%). LC/MS = 900. 1 3(M++ 1 ) . m. Preparation of compound 315. Methyl ester 314 (23.6 g, 26 mmol) was dissolved in glacial acetic acid (200 ml), 1.4 N HCI in H20 (75 ml) was added to the solution. The mixture was stirred at 60 °C for 1 hour. Upon completion of the reaction, the mixture was concentrated to remove the solvents, coevaporated with toluene (x 2) to remove residual acetic acid. The residue was then dissolved in EtOAc (500 ml) and sat. NaHC03 aqueous solution (enough to neutralize the mixture) while monitoring C02 evolution. The organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was further dried under high vacuum for 1 h and used as is for the next step. The crude was dissolved in CH2CI2 (360 ml), morpholine (3.4 g, 39 mmol) and sodium triacetoxyborohydride (7.2 g, 34 mmol) were added to the mixture at 0 °C. Then glacial acetic acid (0.47 g, 7.8 mmol) was added dropwise to the mixture. The reaction was complete in 10 minutes at 0 °C. Sat. NaHC03 aqueous solution was added to quench the reaction. After stirring for another 20 minutes, the organic layer was washed with brine, dried (Na2S04) and concentrated in vacuo. The residue was purified by silica gel chromatography to give the desired amine product 315 as a yellow solid (12 g, 50%). LC/MS = 924.63(M++ 1 )
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
TOLVAPTAN

TOLVAPTAN
的合成
N-(4-{[(5R)-7-chloro-5-hydroxy-2,3,4,5-tetrahydro-1H-1-benzazepin-1-yl]carbonyl}-3-methylphenyl)-2-methylbenzamide
| Formula | C26H25ClN2O3 |
|---|---|
| Mol. mass | 448.941 g/mol |
150683-30-0 CAS NO
+ form 331947-66-1 Rform
OPC-41061
Otsuka…..innovator
UPDATE 2022
Tolvaptan sodium phosphate, Samtasu,
|
トルバプタンリン酸エステルナトリウム
|
| 2022/3/28 JAPAN PPROVED |
| Formula |
C26H24ClN2O6P. 2Na
|
|---|---|
| CAS |
|
| Mol weight |
572.8849
|
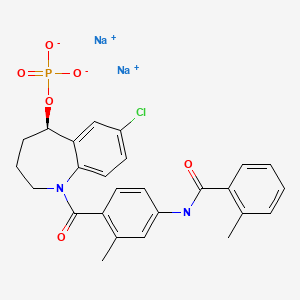
Tolvaptan sodium phosphate
disodium;[(5R)-7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-2,3,4,5-tetrahydro-1-benzazepin-5-yl] phosphate
European Medicines Agency (EMA) Accepts Otsuka’s Marketing Authorisation Application (MAA) for Tolvaptan, an Investigational Compound for Autosomal Dominant Polycystic Kidney Disease (ADPKD)
•Tolvaptan was discovered by Otsuka in Japan and, if approved by the EMA, would become the first pharmaceutical therapy in Europe for patients with ADPKD
•ADPKD is an inherited genetic disease that causes cyst growth in the kidneys, which gradually impairs their functioning. There is no current pharmaceutical treatment option
•Otsuka’s development of tolvaptan as a treatment for ADPKD illustrates the company’s commitment to address significant patient needs for diseases that traditionally have not been a priority for the pharmaceutical industry
TOKYO–(BUSINESS WIRE)–Otsuka Pharmaceutical Co., Ltd. announced today that the European Medicines Agency (EMA) has accepted the submission of a marketing authorisation application (MAA) for the potential approval of tolvaptan for the treatment of autosomal dominant polycystic kidney disease (ADPKD). Phase III clinical trial results that form the basis of the regulatory filing were published in the New England Journal of Medicine.
http://www.pharmalive.com/ema-accepts-otsukas-maa-for-tolvaptan
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP).
Tolvaptan is (±)-4′-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl) carbonyl]-otolu-m-toluidide. The empirical formula is C26H25ClN2O3. Molecular weight is 448.94. The chemical structure is:
 |
SAMSCA tablets for oral use contain 15 mg or 30 mg of tolvaptan. Inactive ingredients include corn starch, hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate and microcrystalline cellulose and FD&C Blue No. 2 Aluminum Lake as colorant.
SEE NEW UPDATE AT END OF PAGE
Tolvaptan (INN), also known as OPC-41061, is a selective, competitive vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated withcongestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone(SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca and in India is manufactured & sold by MSN laboratories Ltd. under the trade name Tolvat & Tolsama.
Tolvaptan is also in fast-track clinical trials[2] for polycystic kidney disease. In a 2004 trial, tolvaptan, when administered with traditional diuretics, was noted to increase excretion of excess fluids and improve blood sodium levels in patients with heart failure without producing side effects such as hypotension (low blood pressure) or hypokalemia(decreased blood levels of potassium) and without having an adverse effect on kidney function.[3] In a recently published trial (TEMPO 3:4 ClinicalTrials.gov number, NCT00428948) the study met its primary and secondary end points. Tolvaptan, when given at an average dose of 95 mg per day over a 3-year period, slowed the usual increase in kidney volume by 50% compared to placebo (2.80% per year versus 5.51% per year, respectively, p<0.001) and reduced the decline in kidney function when compared with that of placebo-treated patients by approximately 30% (reciprocal serum creatinine, -2.61 versus -3.81 (mg/mL)-1 per year, p <0.001)[4]
Tolvaptan was first approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, then approved by the European Medicines Agency (EMA) on August 3, 2009 and approved by Pharmaceuticals and Medical Devices Agency of Japan on Feb 4, 2013. It was developed and marketed as Samsca® by Otsuka in the US, DE and JP.
UPDATED
Tolvaptan is a selective vasopressin V2-receptor antagonist with an affinity for the V2-receptor that is 1.8 times that of native arginine vasopressin (AVP) and that is 29 times greater than for the V1a-receptor. When taken orally, 15 to 60 mg doses of tolvaptan antagonize the effect of vasopressin and cause an increase in urine water excretion that results in an increase in free water clearance (aquaresis), a decrease in urine osmolality, and a resulting increase in serum sodium concentrations. It is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia [serum sodium < 125 mEq/L or less marked hyponatremia that is symptomatic and has resisted correction with fluid restriction], including patients with heart failure, cirrhosis, and syndrome of inappropriate antidiuretic hormone (SIADH).
Samsca® is available as tablet for oral use, containing 7.5 mg/15 mg/30 mg of free Tolvaptan. The recommended starting dose is 15 mg once daily and it may be increased at intervals ≥ 24 hr to 30 mg once daily, and to a maximum of 60 mg once daily as needed to raise serum sodium.
| PATENT NUMBER | PEDIATRIC EXTENSION | APPROVED | EXPIRES (ESTIMATED) | |
|---|---|---|---|---|
| US5258510 | No | 1993-11-02 | 2010-11-02 |  |
| US5753677 | No | 1998-05-19 | 2020-05-19 | |
| US8501730 | No | 2013-08-06 | 2026-09-01 | |
| US5972882 | No | 1999-10-26 | 2018-12-14 | |
| US10905694 | No | 2021-02-02 | 2030-04-07 | |
Synthesis Reference
Bandi Parthasaradhi Reddy, “PROCESS FOR PREPARING TOLVAPTAN INTERMEDIATES.” U.S. Patent US20130190490, issued July 25, 2013.
US20130190490

Reference:1. US5258510A.

Reference:1. Bio. Med. Chemistry 2006, 14, 6165–6173.

Reference:1. Bio. Med. Chem. Lett. 2007, 17, 6455–6458.

Reference:1. CN102060769B.

Reference:1. Org. Lett. 2014, 16, 6041−6043.

Reference:1. Bio. Med. Chem. 1999, 7, 1743-1754.
2. WO2007026971A2.
SYN
SYN
Chemical synthesis:[5] 
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxy1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:
Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. Pat. No. 5,258,510.
Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine and 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro-1H-1-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-1-p-toluenesulfonyl-1H-1-benzazepine, and then reacted with polyphospholic acid. According to the journal, 7-chloro-1-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro-1-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine can be prepared by reacting 1-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro-1H-1-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation oftolvaptan can be prepared by the reduction of 7-chloro-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-1H-1-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
Biooganic and Medicinal Chemistry I (2007) 6455-6458, Biooganic andMedicinal Chemistry 14 (2000) 2493-2495 reported in the literature of the intermediate 2 – carboxylic acid -5 – (2 – methyl-benzoylamino) toluene synthesis method,


5-Chloro-2-nitrobenzoic acid (I) was converted into methyl ester (II) using dimethyl sulfate and K2CO3 in acetone. The nitro group of (II) was then reduced with SnCl2 to afford aniline (III), which was protected as the p-toluenesulfonamide (IV) with tosyl chloride in pyridine. Alkylation of (IV) with ethyl 4-bromobutyrate (V) yielded diester (VI). Subsequent Dieckmann cyclization of (VI) in the presence of potassium tert-butoxide provided benzazepinone (VIIa-b) as a mixture of ethyl and methyl esters, which was decarboxylated to (VIII) by heating with HCl in AcOH. Deprotection of the tosyl group of (VIII) was carried out in hot polyphosphoric acid. The resulting benzazepinone (IX) was condensed with 2-methyl-4-nitrobenzoyl chloride (X) to give amide (XI). After reduction of the nitro group of (XI) to the corresponding aniline (XII), condensation with 2-methylbenzoyl chloride (XIII) provided diamide (XIV). Finally, ketone reduction in (XIV) by means of NaBH4 led to the target compound.
……………………………………….
PATENT

……………………………………………..
PATENT

Synthesis of Intermediate III: 1.
Example
2-methyl-4-nitrobenzoic acid (available from Alfa Aesar Tianjin Chemical Co., purity> 99%, 25g,
0.14mol) was added to a 250ml reaction flask, is reacted with thionyl chloride under reflux conditions for 3h, thionyl chloride was distilled off under reduced pressure to give 2-methyl-4-nitrobenzoyl chloride (26.Sg, light yellow oily liquid), without purification, was used directly in the next step.
Intermediate II (20g, 0.1moI) and 2_ methyl _4_ nitrobenzoylchloride (22.4g, 0.llmol) was added to a 250ml reaction flask. Dichloromethane (50ml), cooled to ice bath with stirring to dissolve O~5 ° C, was slowly added dropwise N- methylmorpholine (11.2g, 0.llmol), Bi dropwise with stirring while, at room temperature the reaction 4h. TLC [developing solvent: ethyl acetate – petroleum ether (I: I), hereinafter] is displayed after completion of the reaction, saturated aqueous sodium bicarbonate (20ml), stirred for lOmin, filtered, the filter cake with dichloromethane (15ml X 2 ) washing. The filtrate and washings were combined, washed with saturated sodium chloride solution (30ml X 3), dried over anhydrous sodium sulfate and filtered. The filtrate under reduced pressure to recover the solvent, the residue was recrystallized from anhydrous methanol to give a white powder 111 (27.5g, 75.2%), mp 154.8 ~155.6 ° C. Purity 97.9% (HPLC normalization method).
Synthesis of Intermediate IV:
Intermediate III (10g, 28mmol) was added to a 250ml reaction flask, concentrated hydrochloric acid (40ml) and ethanol (50ml), with stirring, was slowly added dropwise stannous chloride (20g, 88mmol) in ethanol (40ml) . Bi room temperature drops 5h. After TLC showed completion of the reaction, ethanol was distilled off under reduced pressure to about 70ml, the residue was -10 ° C -0 ° C allowed to stand overnight to cool. Filtered, and the filter cake was washed with water poured into water (40ml) in. Plus 20% sodium hydroxide solution (approximately 60ml) was adjusted to pH 9. Filtered, washed with ethanol and recrystallized to give a pale yellow powdered solid IV (6.3g, 68.7%), mp 190.4~191.1 ° C. Purity 97.2% (HPLC normalization method).
Synthesis of intermediate V:
Intermediate IV (5g, 15mmol) and triethylamine (2.3g, 23mmol) was added followed by IOOml reaction flask was added dichloromethane (30ml), stir until dissolved. Solution of o-methylbenzoyl chloride (2.8g, 18mmol), dropwise at room temperature completion of the reaction Ih0 TLC showed the reaction was complete was poured into ice-water (about 40ml) in, (20ml X 3) and extracted with dichloromethane, the combined organic phases, and saturated sodium chloride solution successively (25ml X 3), dried over anhydrous sodium sulfate and filtered with 5% hydrochloric acid (25ml X 3). The filtrate under reduced pressure to recover the solvent (about 50ml), dried over anhydrous methanol residue – petroleum ether (2: 1) and recrystallized to give white crystals of Intermediate V (6.2g, 90.9%), mp 121.1 ~123.6 ° C. Purity 98.6% (HPLC normalization method).
Synthesis of tolvaptan: Example 4
Intermediate V (5g, Ilmmol) IOOml added to the reaction flask, was added anhydrous methanol (25ml), stirred and then added portionwise sodium borohydride (0.65g, 17mmol) to the reaction mixture, addition was complete the reaction at room temperature lh. After TLC showed the reaction was complete, the methanol recovered under reduced pressure (approximately 20ml), the residue was added methylene chloride (25ml), (25mlX3) and washed with saturated sodium chloride solution. Anhydrous sodium sulfate and filtered, and the filtrate under reduced pressure to recover the solvent, the residue with absolute methanol – petroleum ether (2: 1) and recrystallized tolvaptan white crystals (4.85g, 96.6%), mp 220.1~221.5 ° C. Purity 99.2% (HPLC normalization method). ES1-HRMS (C26H25C1N203, m / z) found (calc): 447.1476 (447.1481) [MH] – “
…………..
PATENT
http://www.google.com/patents/WO2012046244A1?cl=en
Tolvaptan is chemically, N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-hydroxylH-l- benzazepin- 1 -yl)carbonyl]-3-methylphenyl]-2-methylbenzamide. Tolvaptan is represented by the following structure:

Tolvaptan, also known as OPC-41061, is a selective, competitive arginine vasopressin receptor 2 antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.
Tolvaptan and its process for preparation were disclosed in U.S. patent no. 5,258,510. Processes for the preparation of 7-chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5- one, 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine and 7-chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine were reported in Bioorganic & medicinal chemistry 7 (1999), 1743-1754. According to the journal, 7-chloro-2,3,4,5-tetrahydro-lH-l- benzazepin-5-one can be prepared by reacting 7-chloro-4-ethoxycarbonyl-5-oxo-N-p- toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine with acetic acid in the presence of hydrochloric acid and water to obtain 7-chloro-5-oxo-2,3,4,5-tetrahydro-l-p- toluenesulfonyl-lH-l-benzazepine, and then reacted with polyphospholic acid.
According to the journal, 7-chloro- 1 -(2 -methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine can be prepared by reacting 7-chloro-5-oxo-2,3,4,5- tetrahydro-lH-l-benzazepine with 2-methyl-4-nitobenzoyl chloride in the presence of triethylamine.
According to the journal, 7-chloro- l-[2-methyl-4-[(2- methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine can be prepared by reacting l-(4-amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- lH-l-benzazepine with 2-methylbenzoylchloride in the presence of triethylamine.
PCT publication no. WO 2007/026971 disclosed a process for the preparation of tolvaptan can be prepared by the reduction of 7-chloro- l-[2-methyl-4-(2- methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one with sodium borohydride.
7-Chloro-2,3,4,5-tetrahydro-lH-l-benzazepin-5-one is a key intermediate for the preparation of tolvaptan.
SYNTHESIS CONSTRUCTION
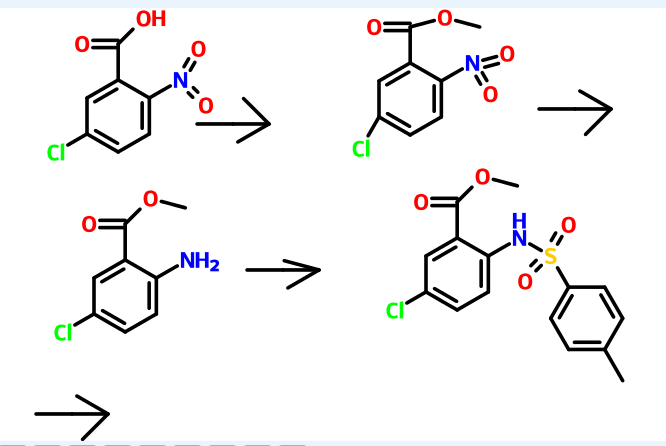



Reference example 1 :
Preparation of methyl 5-chloro-2-nitrobenzoate
Potassium carbonate (515 gm) was added to a solution of 5-chloro-2-nitro benzoic acid (500 gm) in acetone (2750 ml) at room temperature. Dimethyl sulphate (306.5 gm) was added to the reaction mixture slowly and heated to reflux for 30 minutes. The reaction mass was filtered and then concentrated to obtain a residual mass. The residual mass was poured to the ice water and extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual solid of methyl 5- chloro-2-nitrobenzoate (534 gm). Reference example 2:
Preparation of methyl 2-amino-5-chlorobenzoate
A mixture of methyl 5-chloro-2-nitrobenzoate (534 gm) as obtained in reference example 1 and concentrated hydrochloric acid (2250 ml) was added to ethyl acetate (1120 ml). To the reaction mixture was added a solution of tin chloride (1680 gm) in ethyl acetate (2250 ml). The reaction mass was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with aqueous sodium hydroxide solution (2650 ml). The separated aqueous layer was extracted with ethyl acetate and then concentrated to obtain a residual solid of methyl 2- amino-5-chlorobenzoate (345 gm). Reference example 3:
Preparation of methyl 5-chIoro-2-(N-p-toluenesulfonyl)aminobenzoate
To a solution of methyl-2-amino-5-chloro benzoate (345 gm) as obtained in reference example 2 in pyridine (1725 ml) was added p-toluenesulfonyl chloride (425 gm). The reaction mixture was stirred for 2 hours at room temperature and poured to the ice water. The separated solid was filtered and dried to obtain 585 gm of methyl 5- chloro-2-(N-p-toluenesulfonyl)aminobenzoate.
Reference example 4:
Preparation of methyl 5-chloro-2-[N-(3-ethoxycarbonyI)propyI-N-p- toluenesulfonyl] aminobenzoate
Methyl 5-chloro-2-(N-p-toluenesulfonyl)aminobenzoate (585 gm) as obtained in reference example 3, ethyl-4-bromo butyrate (369.6 gm) and potassium carbonate (664 gm) in dimethylformamide (4400 ml) were added at room temperature. The contents were heated to 120°C and maintained for 2 hours. The reaction mass was poured into water and filtered. The solid obtained was dried to give 726 gm of methyl 5-chloro-2-[N- (3 -ethoxycarbonyl)propyl-N-p-toluenesulfonyl] aminobenzoate.
Reference example 5:
Preparation of 7-chloro-4-ethoxycarbonyI-5-oxo-N-p-toluenesufonyl-2,3,4,5- tetrahydro-lH-l-benzazepine
To a heated mixture of potassium tetrabutoxide (363 gm) in toluene (1000 ml) at 70°C was added portion wise methyl 5-chloro-2-[N-(3-ethoxycarbonyl)propyl-N-p- toluenesulfonyl]aminobenzoate (726 gm) as obtained in reference example 4. The contents were heated to reflux and maintained for 30 minutes. The reaction mass was then cooled to room temperature and then poured to the ice water. The layers were separated and the aqueous layer was extracted with toluene. The solvent was distilled off under reduced pressure to obtain a residual solid of 7-chloro-4-ethoxycarbonyl-5-oxo-N- p-toluenesufonyl-2,3,4,5-tetrahydro-lH-l-benzazepine (455 gm).
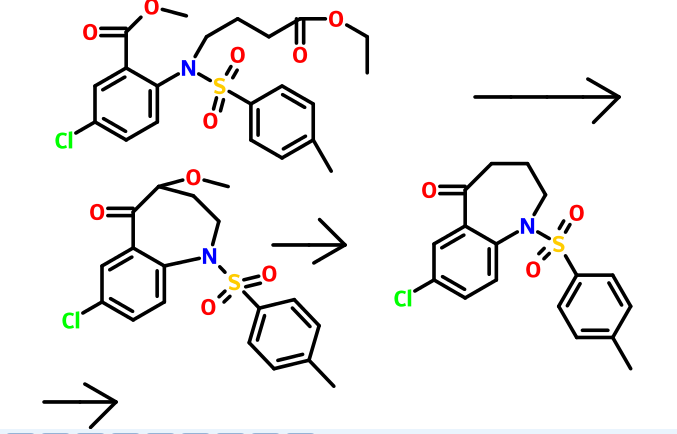
Example 1:
Preparation of 7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine
7-Chloro-4-ethoxycarbonyl-5-oxo-N-p-toluenesufonyl-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (455 gm) as obtained in reference example 5 was added to aqueous sulfuric acid (80%, 2275 ml). The contents heated to 75°C and maintained for 2 hours. The reaction mass was then cooled to room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 7.5 to 8.0 with sodium hydroxide solution (2575 ml). The solid obtained was collected by filtration and dried to give 160 gm of 7- chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 -benzazepine.
Example 2:
Preparation of 7-chIoro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l- benzazepine
7-Chloro-5-oxo-2,3,4,5-tetrahydro-lH-l -benzazepine (160 gm) as obtained in example 1 was dissolved in methylene dichloride (480 ml) and then added aqueous sodium bicarbonate solution (20%, 68.75 gm). The reaction mixture was then cooled to 0 to 5°C and then added 2-methyl-4-nitrobenzoylchloride (180 gm) slowly. The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (170 ml). The layers were separated and the aqueous layer was extracted with methylene chloride. The solvent was distilled off under reduced pressure to obtain a residual mass. To the residual mass was dissolved in isopropyl alcohol (7300 ml) and maintained for 2 hours at reflux temperature. The separated solid was filtered and dried to obtain 250 gm of 7-chloro-l-(2-methyl-4-nitrobenzoyl)-5-oxo-2,3,4,5-tetrahydro-lH-l-benzazepine. Example 3:
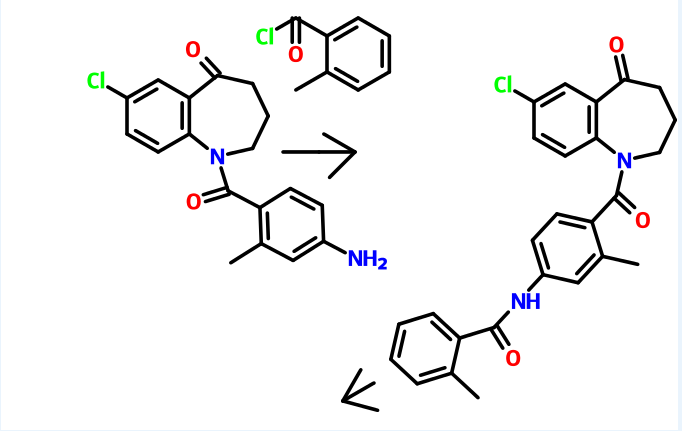
Preparation of l-(4-amino-2-methylbenzoyl)-7-chIoro-5-oxo-2,3,4,5-tetrahydro-lH- 1-benzazepine
7-Chloro- 1 -(2-methyl-4-nitrobenzoyl)-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (250 gm) as obtained in example 2 was dissolved in methanol (575 ml) and then added a solution of tin chloride (630 gm) in methanol (1130 ml). The reaction mixture was stirred for 16 hours at room temperature and then poured to the ice water. The pH of the reaction mass was adjusted to 8.0 to 9.0 with sodium hydroxide solution (1250 ml). The layers were separated and the aqueous layer was extracted with ethyl acetate. The solvent was distilled off under vacuum to obtain a residual solid of l-(4- amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 -benzazepine (185 gm).
Example 4:
Preparation of 7-chloro-l-[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-dxo- 2,3,4,5-tetrahydro-lH-l-benzazepine
1 -(4-Amino-2-methylbenzoyl)-7-chloro-5-oxo-2,3 ,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm) as obtained in example 3 was dissolved in methylene chloride (4000 ml) and then added sodium bicarbonate solution (10%, 47.3 gm). The reaction mass was cooled to 0 to 5°C and then added 2-methyl benzoyl chloride (95.7 gm) slowly. -The pH of the reaction mass was adjusted to 7.0 to 8.0 with aqueous sodium bicarbonate solution (120 ml). The separated aqueous layer was extracted with methylene chloride and then concentrated to obtain a residual solid of 7-chloro-l-[2- methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5-tetrahydro- 1 H- 1 – benzazepine (185 gm). Example 5:
Preparation of tolvaptan
7-Chloro- 1 -[2-methyl-4-[(2-methylbenzoyl)amino]benzoyl]-5-oxo-2,3,4,5- tetrahydro-lH-1 -benzazepine (63 gm) as obtained in example 4 was dissolved in methanol (570 ml) and then added sodium borohydride (2.07 gm) at room temperature. The reaction mass was stirred for 1 hour and pH of the reaction mass was adjusted to 6.0 to 7.0 with hydrochloric acid solution (1%, 630 ml). The separated solid was filtered and dried to obtain 57 gm of tolvaptan.
……………….

Process used to prepare Tolvaptan involves condensing 7-chloro-1, 2, 3, 4-tetrahydro-benzo[b]azepin-5-one with 2-methyl, 4-nitro benzoyl chloride, followed by reduction using SnCl2/HCl catalyst resulting in amine which is then condensed with o-toluoyl chloride followed by reduction with sodium borohydride to give Tolvaptan
///////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN 1
Synthetic Reference
Cordero-Vargas, Alejandro; Quiclet-Sire, Beatrice; Zard, Samir Z. A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan. Bioorganic & Medicinal Chemistry. Volume 14. Issue 18. Pages 6165-6173. 2006.

SYN 2
Synthetic Reference
Torisawa, Yasuhiro; Abe, Kaoru; Muguruma, Yasuaki; Fujita, Shigekazu; Ogawa, Hidenori; Utsumi, Naoto; Miyake, Masahiro. Process for preparation of benzoylaminobenzoylbenzazepinones by reaction of benzazepinones with benzoylaminophenyl halides in the presence of carbonylating agents. Assignee Otsuka Pharmaceutical Co.,

SYN 3
Synthetic Reference
Zard, Samir; Cordero Vargas, Alejandro; Sire, Beatrice. Improved process for the preparation of benzazepines and their derivatives. Assignee Centre National de la Recherche Scientifique CNRS, Fr.; Ecole Polytechnique. FR 2867187. (2005).

SYN 4
Synthetic Reference
Gao, Junlong; Li, Peng; Liu, Kai; Guo, Dapeng. Method for preparing high-purity Tolvaptan intermediate. Assignee Jiangsu Hengrui Medicine Co., Ltd., Peop. Rep. China. CN 108503586. (2018).

SYN 5
Synthetic Reference
Han, Shin; Jeon, Seong Hyeon; Lee, Shin Yoon. Improved method for preparing synthetic intermediates for tolvaptan. Assignee Hexa Pharmatec Co., Ltd., S. Korea. JP 2018012690. (2018).

SYN 6
Synthetic Reference
Guo, Xinfu; Wang, Qiang; Liu, Zhaoguo; Wang, Zhipeng. Preparation method of tolvaptan. Assignee Tianjin Taipu Pharmaceutical Co., Ltd., Peop. Rep. China. CN 106883175. (2017).

SYN 7
Synthetic Reference
Lixin, Juanzi; Li, Jianzhi; Ma, Xilai; Chi, Wangzhou; Liu, Hai; Hu, Xuhua; Zheng, Xiaoli; Zhai, Zhijun; Li, Jianxun. Process for the preparation of tolvaptan. Assignee Shanghai Tianci International Pharmaceutical Co., Ltd., Peop. Rep. China. CN 105753735. (2016).

STR8
Synthetic Reference
Patel, Dhaval J.; Shah, Tejas C.; Singh, Manoj Kumar. A process for the preparation of tolvaptan. Assignee Cadila Healthcare Limited, India. IN 2012MU01559. (2014).

STR9
Synthetic Reference
Sethi, Madhuresh Kumar; Rawat, Vijendrasingh; Thirunavukarasu, Jayaprakash; Yerramala, Raja Krishna; Kumar, Anish. Improved process for the preparation of tolvaptan. Assignee Matrix Laboratories Ltd., India. IN 2011CH01303. (2013).

/////////////////////

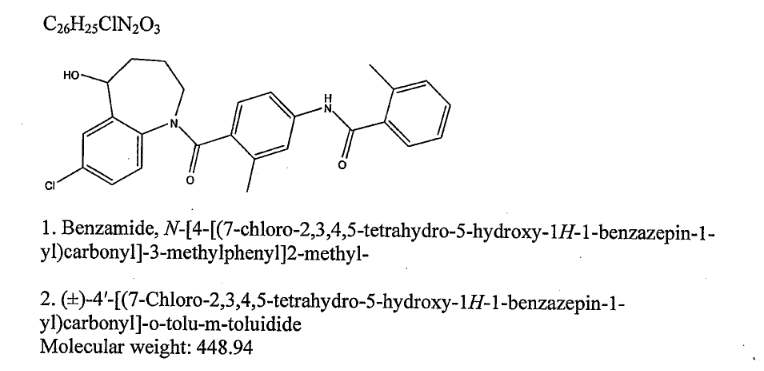

Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
- Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205.
- Otsuka Maryland Research Institute, Inc.
- Gheorghiade M, Gattis W, O’Connor C, et al. (2004). “Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: a randomized controlled trial”. JAMA 291 (16): 1963–71. doi:10.1001/jama.291.16.1963. PMID 15113814.
- (2012) Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease
- Kondo, K.; Ogawa, H.; Yamashita, H.; Miyamoto, H.; Tanaka, M.; Nakaya, K.; Kitano, K.; Yamamura, Y.; Nakamura, S.; Onogawa, T.; et al.; Bioor. Med. Chem. 1999, 7, 1743.
- http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm?source=govdelivery
- Gheorghiade M, Niazi I, Ouyang J et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04.PMID 12742979.
G. R. Belum, V. R. Belum, S. K. Chaitanya Arudra, and B. S. N. Reddy, “The Jarisch-Herxheimer reaction: revisited,” Travel Medicine and Infectious Disease, vol. 11, no. 4, pp. 231–237, 2013.
H. D. Zmily, S. Daifallah, and J. K. Ghali, “Tolvaptan, hyponatremia, and heart failure,” International Journal of Nephrology and Renovascular Disease, vol. 4, pp. 57–71, 2011.
M. N. Ferguson, “Novel agents for the treatment of hyponatremia: a review of conivaptan and tolvaptan,” Cardiology in Review, vol. 18, no. 6, pp. 313–321, 2010.
H. Ogawa, H. Miyamoto, K. Kondo, et al., US5258510, 1993.
K. Kondo, H. Ogawa, H. Yamashita et al., “7-Chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5- tetrahydro-1H-1-benzazepine (OPC-41061): a potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist,” Bioorganic and Medicinal Chemistry, vol. 7, no. 8, pp. 1743–1754, 1999.
| WO2012046244A1 * | Aug 23, 2011 | Apr 12, 2012 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| CN102060769A * | Dec 20, 2010 | May 18, 2011 | 天津药物研究院 | Preparation method of tolvaptan |
| CN102060769B | Dec 20, 2010 | Sep 18, 2013 | 天津药物研究院 | Preparation method of tolvaptan |
| US9024015 | Aug 23, 2011 | May 5, 2015 | Hetero Research Foundation | Process for preparing tolvaptan intermediates |
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| CN101817783A | May 12, 2010 | Sep 1, 2010 | 天津泰普药品科技发展有限公司 | Method for preparing tolvaptan intermediate |
| WO2007026971A2 | Sep 1, 2006 | Mar 8, 2007 | Otsuka Pharma Co Ltd | Process for preparing benzazepine compounds or salts thereof |
| Reference | ||
|---|---|---|
| 1 | Cordero-Vargas, Alejandro | |
| 2 | Kondo, Kazumi et al.7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoyl-amino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine (OPC-41061): A potent, orally active nonpeptide arginine vasopressin V2 receptor antagonist.《Bioorganic & Medicinal Chemistry》.1999,1743-1757. | |
| 3 | Quiclet-Sire, Beatrice | |
| 4 | Torisawa, Yasuhiro et al.Aminocarbonylation route to tolvaptan.《Bioorganic & Medicinal Chemistry Letters》.2007,6455-6458. | |
| 5 | Zard, Samir Z.A flexible approach for the preparation of substituted benzazepines: Application to the synthesis of tolvaptan.《Bioorganic & Medicinal Chemistry》.2006,6165-6173. | |
///////////////
CC1=CC=CC=C1C(=O)NC2=CC(=C(C=C2)C(=O)N3CCCC(C4=C3C=CC(=C4)Cl)OP(=O)([O-])[O-])C.[Na+].[Na+]
////////////UPDATE 2022
-Tolvaptan_Structural_Formula_V1.svg) |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Samsca, Jinarc, Jynarque, others |
| Other names | OPC-41061 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a609033 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown (40% absorbed) |
| Protein binding | 99% |
| Metabolism | Liver (CYP3A4-mediated)[7] |
| Elimination half-life | 12 hours (terminal) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.219.212 |
| Chemical and physical data | |
| Formula | C26H25ClN2O3 |
| Molar mass | 448.95 g·mol−1 |
| 3D model (JSmol) | |
| |
|
Tolvaptan, sold under the brand name Samsca among others, is an aquaretic drug that functions as a selective, competitive vasopressin receptor 2 (V2) antagonist used to treat hyponatremia (low blood sodium levels) associated with congestive heart failure, cirrhosis, and the syndrome of inappropriate antidiuretic hormone (SIADH). Tolvaptan was approved by the U.S. Food and Drug Administration (FDA) on May 19, 2009, and is sold by Otsuka Pharmaceutical Co. under the trade name Samsca.[8] Tolvaptan, as Jynarque, was granted approval for medical use in the United States in April 2018.[9]
The U.S. Food and Drug Administration (FDA) granted tolvaptan a fast track designation for clinical trials investigating its use for the treatment of polycystic kidney disease.[10] The FDA granted Jynarque an orphan drug designation in April 2012, for the treatment of autosomal dominant polycystic kidney disease.[11]
Tolvaptan is available as a generic medication.[12]
Medical uses
Tolvaptan (Samsca) is indicated for the treatment of clinically significant hypervolemic and euvolemic hyponatremia.[13]
Tolvaptan (Jynarque) is indicated to slow kidney function decline in adults at risk of rapidly progressing autosomal dominant polycystic kidney disease (ADPKD).[14]
Side effects
The FDA has determined that tolvaptan should not be used for longer than 30 days and should not be used in patients with underlying liver disease because it can cause liver injury, potentially leading to liver failure.[15] When using to treat hyponatremia, it may cause too rapid correction of hyponatremia resulting in fatal osmotic demyelination syndrome.[16]
Pharmacology
Tolvaptan is a selective vasopressin V2 receptor antagonist.[13][14]
Chemistry
Tolvaptan is a racemate, a 1:1 mixture of the following two enantiomers:[17]
| Enantiomers of tolvaptan | |
|---|---|
-Tolvaptan_Structural_Formula_V1.svg) (R)-Tolvaptan CAS number: 331947-66-1 |
-Tolvaptan_Structural_Formula_V1.svg) (S)-Tolvaptan CAS number: 331947-44-5 |
References
- ^ “Samsca 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 14 December 2020.
- ^ “Jinarc 15 mg tablets – Summary of Product Characteristics (SmPC)”. (emc). 21 April 2020. Retrieved 14 December 2020.
- ^ “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 14 December 2020.
- ^ “Samsca- tolvaptan tablet”. DailyMed. 26 October 2020. Retrieved 14 December 2020.
- ^ “Samsca EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ “Jinarc EPAR”. European Medicines Agency (EMA). Retrieved 14 December 2020.
- ^ Shoaf S, Elizari M, Wang Z, et al. (2005). “Tolvaptan administration does not affect steady state amiodarone concentrations in patients with cardiac arrhythmias”. J Cardiovasc Pharmacol Ther. 10 (3): 165–71. doi:10.1177/107424840501000304. PMID 16211205. S2CID 39158242.
- ^ “Drug Approval Package: Samsca (Tolvaptan) Tablets NDA #022275”. U.S. Food and Drug Administration (FDA). 21 July 2009. Retrieved 15 August 2020. Lay summary (PDF).
{{cite web}}: Cite uses deprecated parameter|lay-url=(help) - ^ “Drug Approval Package: Jynarque (tolvaptan)”. U.S. Food and Drug Administration (FDA). 8 June 2018. Retrieved 15 August 2020.
- ^ “Otsuka Maryland Research Institute, Inc. Granted Fast Track Designation For Tolvaptan In PKD”. Medical News Today. Healthline Media UK Ltd. Retrieved 6 December 2018.
- ^ “Tolvaptan Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 6 April 2012. Retrieved 15 August 2020.
- ^ “Drugs@FDA: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 15 August 2020.
- ^ Jump up to:a b “Samsca- tolvaptan tablet”. DailyMed. 28 May 2019. Retrieved 15 August 2020.
- ^ Jump up to:a b “Jynarque- tolvaptan kit Jynarque- tolvaptan tablet”. DailyMed. 31 March 2020. Retrieved 15 August 2020.
- ^ “U.S. Food and Drug Administration.” Samsca (Tolvaptan): Drug Safety Communication. N.p., 30 Apr. 2013. Web. 1 June 2014. <http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm350185.htm>[dead link]
- ^ Goodman & Gilman’s the pharmacological basis of therapeutics. Brunton, Laurence L, Knollmann, Björn C, Hilal-Dandan, Randa (Thirteenth ed.). New York. 5 December 2017. ISBN 9781259584732. OCLC 994570810.
- ^ Rote Liste Service GmbH (Hrsg.): Rote Liste 2017 – Arzneimittelverzeichnis für Deutschland (einschließlich EU-Zulassungen und bestimmter Medizinprodukte). Rote Liste Service GmbH, Frankfurt/Main, 2017, Aufl. 57, ISBN 978-3-946057-10-9, S. 222.
Further reading
- Gheorghiade M, Niazi I, Ouyang J, et al. (2003). “Vasopressin V2-receptor blockade with tolvaptan in patients with chronic heart failure: results from a double-blind, randomized trial”. Circulation. 107 (21): 2690–6. doi:10.1161/01.CIR.0000070422.41439.04. PMID 12742979.
External links
- “Tolvaptan”. Drug Information Portal. U.S. National Library of Medicine.
Synthesis
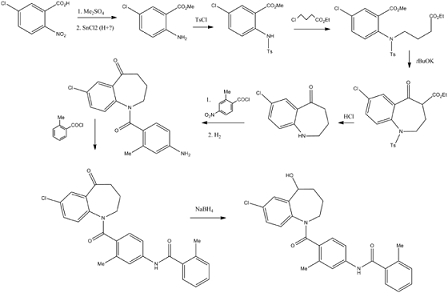
Fugure 1: Synthesis of Tolvaptan

NEW DRUG APPROVALS
HELP TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
Title: Tolvaptan
CAS Registry Number: 150683-30-0
CAS Name: N-[4-[(7-Chloro-2,3,4,5-tetrahydro-5-hydroxy-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide
Additional Names: 7-chloro-5-hydroxy-1-[2-methyl-4-(2-methylbenzoylamino)benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine
Manufacturers’ Codes: OPC-41061
Molecular Formula: C26H25ClN2O3
Molecular Weight: 448.94
Percent Composition: C 69.56%, H 5.61%, Cl 7.90%, N 6.24%, O 10.69%
Literature References: Nonpeptide arginine vasopressin V2 receptor antagonist. Prepn: H. Ogawa et al., WO 9105549; eidem, US 5258510 (1991, 1993 both to Otsuka); K. Kondo et al., Bioorg. Med. Chem. 7, 1743 (1999). Pharmacology: Y. Yamamura et al., J. Pharmacol. Exp. Ther. 287, 860 (1998). Clinical trial in heart failure: M. Gheorghiade et al., J. Am. Med. Assoc. 291, 1963 (2004).
Properties: Colorless prisms, mp 225.9°.
Melting point: mp 225.9°
Therap-Cat: In treatment of congestive heart failure.
Keywords: Vasopressin Receptor Antagonist.
Medicinal Chemistry International: DELDEPREVIR (NECEPREVIR)
Medicinal Chemistry International: DELDEPREVIR (NECEPREVIR)
WANT TO KNOW ABOUT VIR SERIES CLICK
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
DELDEPREVIR OR NECEPREVIR

CHEMICAL NAMES
1. Cyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-carboxamide, N-
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, (2R,6R,12Z,13aS,14aR,16aS)-
2. (2R,6R,12Z,13aS,14aR,16aS)-N-(cyclopropylsulfonyl)-6-[2-(3,3-difluoropiperidin-1-yl)-
2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-yl}oxy)-
5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16atetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecine-14a(5H)-
carboxamide
MOLECULAR WEIGHT 911.1
SPONSOR Achillion Pharmaceuticals, Inc.
CODE DESIGNATION ACH-0142684, ACH-2684
CAS REGISTRY NUMBER 1229626-28-1
WHO NUMBER 9600
NOTE: This adoption statement replaces adoption N12/17 and the name neceprevir is hereby rescinded.
(cyclopropylsulfonyl)-6-[2-(3,3-difluoro-1-piperidinyl)-2-oxoethyl]-
1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-tetradecahydro-2-[[7-methoxy-8-methyl-2-[4-(1-
methylethyl)-2-thiazolyl]-4-quinolinyl]oxy]-5,16-dioxo-, sodium salt (1:1),
(2R,6R,12Z,13aS,14aR,16aS)-
1-yl)-2-oxoethyl]-2-({7-methoxy-8-methyl-2-[4-(1-methylethyl)thiazol-2-yl]quinolin-4-
yl}oxy)-5,16-dioxo-1,2,3,6,7,8,9,10,11,13a,14,15,16,16a-
tetradecahydrocyclopropa[e]pyrrolo[1,2-a][1,4]diazacyclopentadecin-14a(5H)-
yl]formyl]azanide
| WO 2010068761 | ||
| US 2010152103 |

click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
Deleobuvir » All About Drugs
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
DELEOBUVIR

BI-207127 (free acid)
| Company | Boehringer Ingelheim GmbH |
| Description | Oral non-structural protein 5B (NS5B) RNA-dependent polymerase inhibitor |
| Molecular Target | HCV NS5B polymerase |
| Mechanism of Action | Viral polymerase inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase III |
| Indication | Hepatitis C virus (HCV) |
| Partner |
- Interferon-free hepatitis C treatment with faldaprevir proves safe and effective in people with cirrhosis. Alcorn, K. Aidsmap.com. 20 November 2012.
- S Zeuzem, J-F Dufour, M Buti, V Soriano, R Buynak, P Mantry, J Taunk, JO Stern, R Vinisko, J-P Gallivan, WO Bocher and FJ Mensa.“Interferon-free treatment with faldaprevir, deleobuvir (BI 207127) and ribavirin in SOUND-C3: 95% SVR12 in HCV GT-1b”. 23rd Conference of the Asian Pacific Association for the Study of the Liver (APASL) 6–9 June 2013. Retrieved 12 Sep 2013.
| WO 2013147749 |
| Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2010059667A1 | Nov 18, 2009 | May 27, 2010 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition of a potent hcv inhibitor for oral administration |
| WO2011005646A2 | Jul 1, 2010 | Jan 13, 2011 | Boehringer Ingelheim International Gmbh | Pharmaceutical composition for a hepatitis c viral protease inhibitor |
| WO2012041771A1 * | Sep 23, 2011 | Apr 5, 2012 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US4211771 | Feb 13, 1978 | Jul 8, 1980 | Robins Ronald K | Treatment of human viral diseases with 1-B-D-ribofuranosyl-1,2,4-triazole-3-carboxamide |
| US6063772 | Jun 15, 1998 | May 16, 2000 | Icn Pharmaceuticals, Inc. | Specific modulation of Th1/Th2 cytokine expression by ribavirin in activated T-lymphocytes |
| US6277830 | Jul 7, 1999 | Aug 21, 2001 | Schering Corporation | 5′-amino acid esters of ribavirin and the use of same to treat hepatitis C with interferon |
| US6323180 | Aug 5, 1999 | Nov 27, 2001 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| US6403564 | Oct 14, 1999 | Jun 11, 2002 | Schering Corporation | Ribavirin-interferon alfa combination therapy for eradicating detectable HCV-RNA in patients having chronic hepatitis C infection |
| US7141574 | Jul 18, 2002 | Nov 28, 2006 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| US7514557 | May 23, 2005 | Apr 7, 2009 | Boehringer Ingelheim International Gmbh | Process for preparing acyclic HCV protease inhibitors |
| US7582770 | Feb 18, 2005 | Sep 1, 2009 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7585845 | May 20, 2004 | Sep 8, 2009 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US7642352 | Feb 10, 2006 | Jan 5, 2010 | Boehringer Ingelheim International Gmbh | Process for preparing 2,3-disubstituted indoles |
| US20090087409 | Nov 26, 2008 | Apr 2, 2009 | Boehringer Ingelheim (Canada) Ltd. | Viral Polymerase Inhibitors |
| US20100068182 | Sep 16, 2009 | Mar 18, 2010 | Boehringer Ingelheim International Gmbh | Combination therapy for treating hcv infection |
| US20100093792 | Sep 15, 2009 | Apr 15, 2010 | Boehringer Ingelheim International Gmbh | Crystalline forms of a potent hcv inhibitor |
| Ref | ||
|---|---|---|
| 1 | BALAGOPAL GASTROENTEROLOGY vol. 139, 2010, pages 1865 – 1876 | |
| 2 | BERG ET AL. HEPATOL vol. 52, no. S1, 2010, | |
| 3 | * | DOMINIQUE LARREY ET AL: “Rapid and strong antiviral activity of the non-nucleosidic NS5B polymerase inhibitor BI 207127 in combination with peginterferon alfa 2a and ribavirin“, JOURNAL OF HEPATOLOGY, vol. 57, no. 1, 7 March 2012 (2012-03-07), pages 39-46, XP55040240, ISSN: 0168-8278, DOI: 10.1016/j.jhep.2012.02.015 |
| 4 | G. CAIRNS GENE VARIANT THAT HELPS HEPATITIS C TREATMENT MAY HINDER HIV TREATMENT, [Online] 2011, Retrieved from the Internet: <URL:http://www.bhiva.org/Ncws.aspx?NewsID=a7503829-94b9-4d2f-bd91-ld2fbaad6c8d> | |
| 5 | GE ET AL. NATURE vol. 461, 2009, pages 399 – 401 | |
| 6 | GHANY; MARC ET AL.: ‘An Update on Treatment of Genotype 1 Chronic Hepatitis C Virus Infection: 2011 Practice Guideline by the American Association for the Study of Liver Diseases‘ HEPATOLOGY vol. 54, no. 4, 2011, pages 1433 – 44 | |
| 7 | * | LIZ HIGHLEYMAN: “AASLD: All-Oral Combination of BI 201335, BI 207127 and Ribavirin Shows Good Efficacy at 12 Weeks“, INTERNET CITATION, [Online] 1 December 2011 (2011-12-01), pages 1-3, XP002684260, Retrieved from the Internet: URL:www.hivandhepatitis.com/hepatitis-c/he patitis-c-topics/hcv-treatment/3371-aasld- all-oral-combination-of-bi-201335-bi-20712 7-and-ribavirin-shows-good-efficacy-at-12- weeks> [retrieved on 2012-09-27] |
| 8 | * | POL S ET AL: “SVR AND PHARMACOKINETICS OF THE HCV PROTEASE INHIBITOR BI201335 WITH PEGIFN/RBV IN HCV GENOTYPE-1 PATIENTS WITH COMPENSATED LIVER CIRRHOSIS AND NON-RESPONSE TO PREVIOUS PEGIFN/RBV“, JOURNAL OF HEPATOLOGY, vol. 54, no. Suppl. 1, March 2011 (2011-03), page S486, XP55038942, & 46TH ANNUAL MEETING OF THE EUROPEAN-ASSOCIATION-FOR-THE-STUDY-OF-THE- LIVER (EASL); BERLIN, GERMANY; MARCH 30 -APRIL 03, 2011 ISSN: 0168-8278 |
| 9 | S. M. BIRGE ET AL. J. PHARM. SCI. vol. 66, 1977, pages 1 – 19 | |
| 10 | * | STEFAN ZEUZEM ET AL: “Efficacy of the Protease Inhibitor BI 201335, Polymerase Inhibitor BI 207127, and Ribavirin in Patients With Chronic HCV Infection“, GASTROENTEROLOGY, ELSEVIER, PHILADELPHIA, PA, vol. 141, no. 6, 1 December 2011 (2011-12-01), pages 2047-2055, XP002664706, ISSN: 0016-5085, DOI: 10.1053/J.GASTRO.2011.08.051 |
| 11 | SULKOWSKI MS ET AL. HEPATOL vol. 50, 2009, page 2A | |
| 12 | SULKOWSKI MS ET AL. J HEPATOL vol. 52, no. 1, 2010, pages S462 – S463 | |
| 13 | WHITE PW ET AL. ANTIMICROB AGENTS CHEMOTHER vol. 54, no. 11, 2010, pages 4611 – 4618 | |
| 14 | WHO COLLABORATIVE STUDY GROUP. VOX SANG vol. 76, 1999, pages 149 – 158 | |
| 15 | * | ZEUZEM STEFAN ET AL: “STRONG ANTIVIRAL ACTIVITY AND SAFETY OF IFN-SPARING TREATMENT WITH THE PROTEASE INHIBITOR BI 201335, THE HCV POLYMERASE INHIBITOR BI 207127 AND RIBAVIRIN IN PATIENTS WITH CHRONIC HEPATITIS C“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 52, no. Suppl, 1 October 2010 (2010-10-01), pages 876A-877A, XP009154421, ISSN: 0270-9139 |
| 16 | * | ZEUZEM STEFAN ET AL: “VIROLOGIC RESPONSE TO AN INTERFERON-FREE REGIMEN OF BI201335 AND BI207127, WITH AND WITHOUT RIBAVIRIN, IN TREATMENT-NAIVE PATIENTS WITH CHRONIC GENOTYPE-1 HCV INFECTION: WEEK 12 INTERIM RESULTS OF THE SOUND-C2 STUDY“, HEPATOLOGY, WILLIAMS AND WILKINS, BALTIMORE, MD, US, vol. 54, no. Suppl. 1, 1 November 2011 (2011-11-01), page 1436A, XP009163087, ISSN: 0270-9139, DOI: 10.1002/HEP.24666 [retrieved on 2011-09-30] |













click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
GRAZOPREVIR, MK 5172
- Grazoprevir hydrate
- UNII-4O2AB118LA
- MK 5172
MW804.99
quinoxalinyl)pentyl]cyclopropyl]oxy]carbonyl]-3-methyl-L-valyl-(4R)-4-hydroxy-L-prolyl-1-
amino-N-(cyclopropylsulfonyl)-2-ethenyl-, cyclic (1→2)-ether, hydrate (1 :1) (1R,2S)-
ethenylcyclopropyl}-5-(1,1-dimethylethyl)-14-methoxy-3,6-dioxo-
1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide hydrate
MOLECULAR WEIGHT 784.92
9857
ACS Med Chem Lett 2012, 3(4): 332DOI: 10.1021/ml300017p
Org Lett 2013, 15(16): 4174
[1]. Steven Harper , John A. McCauley , Michael T. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett., 2012, 3 (4), pp 332-336[2]. Summa V, Ludmerer SW, McCauley JA, MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012 Aug;56(8):4161-7.
IC50 Value: 7.4nM and 7nM for genotype1b and 1a respectively, in replicon system [1]
MK-5172 is a novel P2-P4 quinoxaline macrocyclic HCV NS3/4a protease inhibitor currently in clinical development.
in vitro: In biochemical assays, MK-5172 was effective against a panel of major genotypes and variants engineered with common resistant mutations observed in clinical studies with other NS3/4a protease inhibitors. In the replicon assay, MK-5172 demonstrated subnanomolar to low-nanomolar EC50s against genotypes 1a, 1b, and 2a [2].
in vivo: In rats, MK-5172 showed a plasma clearance of 28 ml/min/kg and plasma half-life of 1.4 hr. When dosed p.o. at 5 mg/kg, the plasma exposure of MK-5172 was good with an AUC of 0.7 uM.hr. The liver exposure of the compound was quite good (23 uM at 4 hr), and MK-5172 remained in liver 24 hr after a single p.o. 5 mg/kg dose. At 24 hr, the liver concentration of MK-5172 was 0.2 uM, which was over 25-fold higher than the IC50 in the replicon assay with 50% NHS. When dosed to dogs, MK-5172 showed low clearance of 5 ml/min/kg and a 3 hr half-life after i.v. 2 mg/kg dosing and had good plasma exposure (AUC=0.4 uM.hr) after a p.o. 1 mg/kg dose [1].
Clinical trial: Evaluation of Hepatic Pharmacokinetics for MK-5172 in Participants With Chronic Hepatitis C . Phase1


1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-
methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxalin-8-
yl]carbonyl}amino)-2-ethenylcyclopropyl]carbonyl}(cyclopropylsulfonyl)azanide (15 K-salt).
8.3 Hz, 1 H), 7.15 (m, 1 H), 7.04 (m, 1 H), 5.97 (m, 1 H), 5.73 (br s, 1 H), 4.96 (m, 1 H), 4.79 (apparent q, J = 9.3 Hz, 1 H), 4.26 (dd, J = 9.7, 7.7 Hz, 1 H), 4.20 (d, J = 11.3 Hz, 1 H), 4.14 (d, J = 8.8 Hz, 1 H), 3.90 (dd, J = 11.1, 3.2 Hz, 1 H), 3.86 (s, 3 H), 3.62 (m, 1 H), 2.86-2.60 (m, 3 H), 2.38 (m, 1 H), 2.21 (m, 1 H), 1.80-1.48 (m, 6 H), 1.42 (m, 5 H), 1.14 (m, 1 H), 0.95 (m, 10 H), 0.81 (m, 2 H), 0.72-0.50 (m, 3 H), 0.41 (m, 1 H) ppm.http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
vinylcyclopropyl)-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-
7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-
carboxamide (MK-5172, 15).
OD) δ 7.79 (dd, J = 9.6, 1.8 Hz, 1 H), 7.23 (s, 1 H), 7.22 (m, 1 H), 7.10 (d, J = 9.6 Hz, 1 H), 6.01 (apparent t, J = 3.6 Hz, 1 H), 5.74 (m, 1 H), 5.24 (dd, J = 17.0 Hz, 1.6 Hz, 1 H), 5.11 (dd, J = 10.4 Hz, 1.6 Hz, 1 H), 4.49 (d, J = 11.2 Hz, 1 H), 4.40 (m, 2 H), 4.13 (dd, J = 12.0 Hz, 4.0 Hz, 1 H), 3.92 (s, 3 H), 3.76 (m, 1 H), 2.92 (m, 2 H), 2.85 (m, 1 H), 2.55 (dd, J = 13.6 Hz, 6.4 Hz, 1 H), 2.28 (m, 1 H), 2.18 (apparent q, J =8.8 Hz, 1 H), 1.85 (dd, J = 8.0 Hz, 5.6 Hz, 1 H), 1.73 (m, 2 H), 1.5 (m, 2 H), 1.40 (dd, J = 9.6 Hz, 5.6 Hz, 1 H), 1.3 (m, 2 H), 1.23 (m, 4 H), 1.08 (s, 9 H), 0.99 (m, 2 H), 0.89 (m, 3 H), 0.73 (m, 1 H), 0.49 (m, 1 H) ppm; HRMS (ESI) m/z 767.3411 [(M+H)+; calcd for C38H51N6O9S: 767.3433].http://pubs.acs.org/doi/suppl/10.1021/ml300017p/suppl_file/ml300017p_si_001.pdf
| Intermediate # | Structure | Name | Lit. Reference |
| A1 |  |
(1R,2S)-1-Amino-N- (cyclopropylsulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride | Wang et al., U.S. Pat. No. 6,995,174 |

















Step 1: Quinoxaline Hydroxyproline Methyl Ester HCl Salt

A 250-ml RB, equipped with magnetic stirrer and N2 bubbler, was charged with chloroquinoxaline BOC hydroxyproline adduct in MeOH (100 ml), and the mixture was cooled in an ice bath. Acetyl chloride (17.9 g) was then added, and the mixture was stirred at RT for 2 h. The batch was diluted with IP Ac (80 ml). Solids were filtered off and washed with IPAc (20 ml). The washed solids were dried under vacuum for 3 d, to provide 48.9 g (100% yield ). Part of this material was used in the next step.
Example 17: Preparation of Compound A, Method A

Macrocyclic acid hemihydrate, the product of Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 mL to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 1.983 mmol) followed by DMAc (15 mL) was added at RT. The batch was cooled to 0°C to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HCl (4.49 g, 23.44 mmol) was added in portions or one portion at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. MeOAc (100 mL) followed by 15 wt% citric acid in 5% NaCl in water (50 mL) was added, while the internal temperature was maintained to < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 5% NaCl in water (50 mL) followed by 5% NaCl (50 mL). The organic phase was solvent-switched to acetone at a final volume of ~80 mL. Water (10 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~10 mg) and aged for 0.5 h tol h at 35°C to 40°C. Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (2x 40 mL). Suction drying at RT gave Compound A monohydrate form III as a white solid.
XH NMR (400 MHz, CDC13) δ 9.95 (s, br, 1 H), 7.81 (d, J = 9.1 Hz, 1 H), 7.18 (dd, J = 9.1, 2.7 Hz, 1 H), 7.16 (s, br, 1 H), 7.13 (d, J = 2.7 Hz, 1 H), 5.96 (t, J = 3.8 Hz, 1 H), 5.72 (m, 1 H), 5.68 (d, J = 10.1 Hz, 1 H), 5.19 (d, J = 17.1 Hz, 1 H), 5.07 (d, J = 10.1 Hz, 1 H), 4.52 (d, J = 11.4 Hz, 1 H), 4.45 (d, J = 9.8 Hz, 1 H), 4.36 (d, J = 10.5, 6.9 Hz, 1 H), 4.05 (dd, J = 11.5, 3.9 Hz, 1 H), 3.93 (s, 3 H), 3.78 (m, 1 H), 2.90 (m, 1 H), 2.82 (tt, J = 8.0, 4.8 Hz, 1 H), 2.74 (dt, J = 13.2, 4.8 Hz, 1 H), 2.59 (dd, J = 14.0, 6.7 Hz, 1 H), 2.40 (ddd, J = 14.0, 10.6, 4.0 Hz, 1 H), 2.10 (dd, J = 17.7, 8.7 Hz, 1 H), 1.98 (2 H, mono hydrate H20), 1.88 (dd, J 8.2, 5.9 Hz, 1 HO, 1.74 (m, 3 H), 1.61 (m, 1 H), 1.50 (m, 3 H), 1.42 (dd, J = 9.6, 5.8 Hz, 1 H), 1.22 (m, 2 H), 1.07 (s, 9 H), 0.95 (m, 4 H), 0.69 (m, 1 H), 0.47 (m, 1 H).
1 C NMR (100 MHz, CDC13) δ 173.5, 172.1, 169.1, 160.4, 157.7, 154.9, 148.4, 141.0, 134.3, 132.7, 129.1, 118.8, 118.7, 106.5, 74.4, 59.6, 59.4, 55.8, 55.5, 54.9, 41.8, 35.4, 35.3, 35.2, 34.3,. 31.2, 30.7, 29.5, 28.6, 28.2, 26.6, 22.6, 18.7, 11.2, 6.31, 6.17.
HPLC conditions: Ascentis Express Column, 10 cm x 4.6 mm, 2.7 μηι; Column temperature of 40°C; Flow rate of 1.8 mL/min; and Wavelength of 215 nm.
Gradiant: mm 0.1% ¾PO4
0 20 80
5 55 45
15 55 45
25 95 5
27 95 5
27.1 20 80
32 20 80
Retention time: mm.
Compound A 14.50
Example 18: Preparation of Compound A, Method B

To a 50-L flask equipped with overhead stirring was added macrocyclic acid (1.06 kg crude, 1.00 eq), amine-pTSA (862 g crude, 1.12 eq) and MeCN (7.42 L) at 19°C. The slurry was cooled in a water bath, pyridine (2.12 L, 13.8 eq) was added, aged 15 min, and then added EDC (586 g, 1.60 eq) in one portion, aged 1.5 h, while it turned into a clear homogeneous solution.
The solution cooled in a water bath, then quenched with 2 N HC1 (1.7 L), and seeded (9.2 g), aged 15 min, and the rest of the aqueous HC1 was added over 2.5 h. A yellow slurry was formed. The slurry was aged overnight at RT, filtered, washed with MeCN/water (1 : 1 v/v, 8 L), to obtain Compound A (Hydrate II).
Compound A was dissolved in acetone (4 L) at RT, filtered and transferred to a
12-L round-bottom flask with overhead stirring, rinsed with extra acetone (1 L), heated to 50°C, water (0.9 L) was added, seeded with Compound A monohydrate form III (-10 mg), and aged 15 min, and then added water (0.8 L) over 2.5 h, extra water 3.3 v over 2.5 h was added, stopped heating, cooled to RT, aged at RT overnight, filtered, washed with water/acetone (1 : 1 v/v, 4 L), and dried in air under vacuum. Compound A Hydrate III, 670 g, was obtained as an off-white solid.
Example 19: Preparation of Compound A, Method C

Macrocyclic acid hemihydrate from Example 15 (10.16 g, 18.03 mmol) was dissolved in THF (50 ml to 90 mL). The solution was azetropically dried at a final volume of 100 mL. Sulfonamide pTSA salt (7.98 g, 19.83 mmol) was added, followed by DMAc (15 mL), at RT. The batch was cooled to 0° to 10°C, and pyridine (10 mL) was added dropwise. Then, EDC HC1 (4.49 g, 23.44 mmol) was added (in portions or one portion) at 0°C to 10°C. The reaction mixture was aged at 0°C to 10°C for 1 h, and then warmed to 15°C to 20°C for 2 h to 4 h. THF (50 mL) was added, followed by 15 wt% citric acid in 15 wt% aq. NaCl (50 mL), while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was washed with 15 wt% citric acid in 1 % aq. NaCl (40 mL), followed by 15% NaCl (40 mL). The organic phase was solvent-switched to acetone at a final volume of ~75 mL Water (1 1 mL to 12 mL) was added dropwise at 35°C to 40°C. The batch was seeded with Compound A monohydrate form III (~20 mg) and aged for 0.5 h to 1 h at 35°C to 40°C.
Additional water (22 mL) was added dropwise over 2 h to 4 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 60% acetone in water (40 mL x 2). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 20: Preparation of Compound A, Method D

Macrocyclic acid hemihydrate from Example 12 (10 g, 98.4wt%, 17.74 mmol) was dissolved in THF (70 mL). The solution was azetropically dried at a final volume of ~60 mL. Sulfonamide pTSA salt (7.53 g, 18.7 mmol) was added at RT. The batch was cooled to 0°C to 5°C, and pyridine (1 1.4 mL) was added dropwise. Then, EDC HC1 (4.26 g, 22.2 mmol) was added in portions at 0°C to 15°C. The reaction mixture was aged at 10°C to 15°C for 2 h to 4 h. 35 wt% Citric acid in 10 wt% aq. NaCl (80 mL) was added, while the internal temperature was maintained at < 25°C with external cooling. The separated organic phase was solvent-switched to acetone at a final volume of ~75 mL. Water (12 mL) was added dropwise at 50°C. The batch was seeded with Compound A monohydrate form III (-300 mg) and aged for 0.5 h to 1 h at 50°C. Additional water (25 mL) was added dropwise over 6 h at 35°C to 40°C. The slurry was aged at 20°C for 2 h to 4 h before filtration. The wet cake was displacement washed with 65%) acetone in water (40 mL). Suction drying at RT or vacuum-oven drying at 45°C gave Compound A monohydrate form III as a white solid.
Example 24: Ring Closing Metathesis

To a 50 mL 2-neck RB flask with reflux condenser and needle for N2 bubbling was charged the product of Example 20 (1.034 g, 0.869 mmol, 1.0 eq), toluene (20.68 ml, 20X), and the resulting solution was degassed with N2. Hoveyda-Grubbs 2nd generation catalyst (10.90 mg, 0.017 mmol) was charged to the pot, and the system was heated to 80°C with constant sparge of N2, with color change from green to reddish. The reaction was sampled (5 h) and assay by HPLC to be approximately 80% converted. The system was removed from the heat and allowed to stir at RT overnight under N2. The reaction was again assayed and deemed complete by HPLC. Toluene was removed by concentration and the resulting red oil was purified by gradient silica gel chromatography (50 g BlOTAGE SNAP Si gel column; loaded with DCM; eluted with 0 to 10% EtOAc in DCM over 10 column volumes; then 10 to 20% EtOAc in DCM over 3 column volumes; then hold; detect by TLC-UV) to yield a yellow solid, which was further slurried in EtOAc (3 mL) and hexanes (6 mL). The resulting slurry was filtered and washed with 25% EtOAc in hexanes (6 mL) to yield the product (445 mg, 0.754 mmol, 87% yield) as a white solid.

Merck reported interim data from the Phase 2 C-WORTHY study in April 2014 at the International Liver Congress (ILC) in London that evaluated the efficacy and safety of its two-drug regimen based on NS3/4A protease inhibitor MK-5172 and NS5A replication complex inhibitor MK-8742, given with or without ribavirin, in GT1 HCV patients with cirrhosis. The once-daily single pill (without ribavirin) showed a 98% SVR12 (12-week sustained virologic response) in genotype-1, treatment-naive patients. Merck will start the phase III clinical trials (NCT02105688, NCT02105662, NCT02105467 andNCT02105701) for the combination in June 2014.
Development of a Practical, Asymmetric Synthesis of the Hepatitis C Virus Protease Inhibitor MK-5172.Org. Lett. 2013;
15: 4174-4177

MK-5172 is a hepatitis C virus protease inhibitor. Key steps in the synthesis depicted are (1) the regioselective SNAr reaction of dichloroquinoxaline A with prolinol derivative B and (2) construction of the 18-membered macrocycle using a macrolactamization (F → G).
Comment
The medicinal chemistry route to MK-5172 is based on a ring-closing metathesis strategy (S. Harper et al.ACS Med. Chem. Lett. 2012, 3, 332). The best regioselectivity (20:1) and minimization of double substitution in the SNAr reaction of A with B was achieved using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base in polar solvents such as DMSO, NMP, or DMAc.
SYNTHESIS, THESIS PROCEDURES, NMR see………..http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
/////////////http://www.allfordrugs.com/2015/07/31/mk-5172-grazoprevir/
Want to know everything on vir series
click
http://drugsynthesisint.blogspot.in/p/vir-series-hep-c-virus-22.html
AND
http://medcheminternational.blogspot.in/p/vir-series-hep-c-virus.html
