
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA OKs Teva’s Injectable Treanda

FDA OKs Teva’s Injectable Treanda
FDA Approves Teva’s Injectable Treanda

bendamustine
Sept. 17, 2013 (GLOBES)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA; TASE: TEVA) has announced that the US Food and Drug Administration (FDA) has approved a new injectable version Treanda for treatment of indolent B-cell non-Hodgkin lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen, and chronic lymphocytic leukemia. read all at
http://www.pharmalive.com/fda-oks-tevas-injectable-treanda
Bendamustine (INN, trade names Treakisym, Ribomustin, Levact and Treanda; also known as SDX-105) is a nitrogen mustard used in the treatment of chronic lymphocytic leukemia[1] and lymphomas. It belongs to the family of drugs called alkylating agents. It is also being studied for the treatment of sarcoma.[2]
History
Bendamustine was first synthesized in 1963 by Ozegowski and Krebs in East Germany(the former German Democratic Republic). Until 1990 it was available only in East Germany. East German investigators found that it was useful for treating chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myelomaand lung cancer.
Bendamustine received its first marketing approval in Germany, where it is marketed under the tradename Ribomustin, by Astellas Pharma GmbH’s licensee, Mundipharma International Corporation Limited. It is indicated as a single-agent or in combination with other anti-cancer agents for indolent non-Hodgkin’s lymphoma, multiple myeloma, and chronic lymphocytic leukemia. SymBio Pharmaceuticals Ltd holds exclusive rights to develop and market bendamustine HCl in Japan and selected Asia Pacific Rim countries.
In March 2008, Cephalon received approval from the United States Food and Drug Administration to market bendamustine in the US, where it is sold under the tradename Treanda, for treatment of chronic lymphocytic leukemia.[3]
In October 2008, the FDA granted further approval to market Treanda for the treatment of indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen. [4]
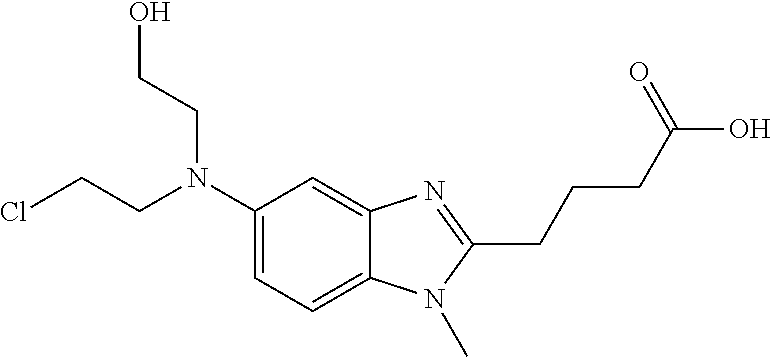
Bendamustine, 4-{5-[Bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric acid:

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there under the tradename Cytostasan®. See, e.g., W. Ozegowski and D. Krebs, IMET 3393 γ-[1-methyl-5-bis-(β-chloroethyl)-aminobenzimidazolo-(2)]-butyryl chloride, a new cytostatic agent of the group of benzimidazole nitrogen mustards. Zbl. Pharm. 110, (1971) Heft 10, 1013-1019, describing the synthesis of bendamustine hydrochloride monohydrate. Since that time, it has been marketed in Germany under the tradename Ribomustin®. Bendamustine is an alkylating agent that has been shown to have therapeutic utility in treating diseases such as chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to technical difficulties in its preparation and administration. Researchers, therefore, have investigated methods of improving the preparation and stability of bendamustine and its formulations. For example, German (GDR) Patent No. 159877 discloses a method for preparing bendamustine free base by reaction of the bis-hydroxyl precursor with thionyl chloride followed by recrystallization from water.
German (GDR) Patent No. 34727 discloses a method of preparing derivatives of bendamustine. The described derivatives differ from bendamustine in the substitution at the 1-position.
German (GDR) Patent No. 80967 discloses an injectable preparation of bendamustine hydrochloride monohydrate, ascorbic acid, and water. GDR 80967 describes that lyophilization of compounds such as bendamustine is only possible if the compound is of sufficient stability that it can withstand the processing conditions. The preparation described in GDR 80967 is not lyophilized.
German (GDR) Patent No. 159289 discloses a ready-to use, injectable solution of bendamustine hydrochloride that avoids lyophilization. GDR 159289 describes an anhydrous solution of bendamustine hydrochloride in 1,2-propylene glycol or ethanol.
U.S. application Ser. No. 11/330,868, filed Jan. 12, 2006, assigned to Cephalon, Inc., Frazer, P A, discloses methods of preparing lyophilized pharmaceutical compositions comprising bendamustine hydrochloride.
Chemotherapeutic uses
Bendamustine has been used both as sole therapy and in combination with other agents including etoposide, fludarabine, mitoxantrone,methotrexate, prednisone, rituximab, vincristine and 90Y-ibritumomab tiuxetan.
One combination for stage III/IV relapsed or refractory indolent lymphomas and mantle cell lymphoma (MCL), with or without prior rituximab-containing chemoimmunotherapy treatment, is bendamustine with mitoxantrone and rituximab.[5] In Germany in 2012 it has become the first line treatment of choice for indolent lymphoma.[6] after Trial results released in June 2012 showed that it more than doubled disease progression-free survival when given along with rituximab. The combination also left patients with fewer side effects than the older R-CHOP treatment.[7]
Common adverse reactions are typical for the class of nitrogen mustards, and include nausea, fatigue, vomiting, diarrhea, fever, constipation, loss of appetite, cough, headache, unintentional weight loss, difficulty breathing, rashes, and stomatitis, as well as immunosuppression, anemia, and low platelet counts. Notably, this drug has a low incidence of hair loss (alopecia) unlike most other chemotherapy drugs.[8]
References
- Kath R, Blumenstengel K, Fricke HJ, Höffken K (January 2001). “Bendamustine monotherapy in advanced and refractory chronic lymphocytic leukemia”. J. Cancer Res. Clin. Oncol. 127 (1): 48–54. doi:10.1007/s004320000180. PMID 11206271.
- Bagchi S (August 2007). “Bendamustine for advanced sarcoma”. Lancet Oncol. 8 (8): 674. doi:10.1016/S1470-2045(07)70225-5.PMID 17726779.
- “Cephalon press release – Cephalon Receives FDA Approval for TREANDA, a Novel Chemotherapy for Chronic Lymphocytic Leukemia”. Retrieved 2008-03-23.
- “Cephalon press release -Cephalon Receives FDA Approval for TREANDA to Treat Patients with Relapsed Indolent Non-Hodgkin’s Lymphoma”. Retrieved 2008-11-03.
- Weide R, Hess G, Köppler H, et al. (2007). “High anti–lymphoma activity of bendamustine/mitoxantrone/rituximab in rituximab pretreated relapsed or refractory indolent lymphomas and mantle cell lymphomas. A muticenter phase II study of the German Low Grade Lymphoma Study Group (GLSG)”. Leuk. Lymphoma. 48 (7): 1299–1306. doi:10.1080/10428190701361828. PMID 17613757.
- New Combo Replaces CHOP for Lymphoma. Dec 2012
- “‘Rediscovered’ Lymphoma Drug Helps Double Survival: Study”. June 3, 2012.
- Tageja, Nishant; Nagi, Jasdeepa; “Bendamustine: something old, something new”; Cancer Chemotherapy and Pharmacology, 2010 Aug;66(3):413-23. doi: 10.1007/s00280-010-1317-x.
External links
- Manufacturer’s official website intended for US patients
more info
Bendamustine hydrochloride, 4-{5-[Bis(2-chloroethyl) amino]- l-methyl-2- benzimidazolyl} butyric acid hydrochloride, of the formula (VI) :

was initially synthesized in 1963 in the German Democratic Republic (GDR) and was available from 1971 to 1992 there, as the hydrochloride salt, under the trade name Cytostasan®. Since that time, it has been marketed in Germany under the trade name Ribomustin®. Bendamustine Hydrochloride as injection is available in the United States under the tradename Treanda®. Bendamustine hydrochloride is an alkylating agent that is approved for the treatment of non-Hodgkin’s lymphoma, multiple myeloma and chronic lymphocytic leukemia.
Bendamustine hydrochloride is a benzimidazole analog. While bendamustine has been demonstrated as efficacious, it is known to be unstable, especially in aqueous solutions, leading to formation of non-bendamustine products (i.e. “degradation impurities”) which leads to technical difficulties in its preparation and administration. In light of its instability in aqueous solution, bendamustine is supplied as a lyophilized cake of bendamustine hydrochloride salt. US2006/159713, US 2006/128777 and WO2010/036702 disclose various impurities of Bendamustine hydrochloride which are as follows:

PC-1 PC-2
Jena et al. were the first to disclose the synthesis of Bendamustine hydrochloride in German (GDR) Patent No. 34727. Krueger et al. in German (GDR) Patent No. 159877 recite a method as summarized in scheme-1, for the synthesis of bendamustine hydrochloride comprising the reaction of the 4-[l-methyl-5-bis-(2- hydroxyethyl)-benzimidazolyl-2]butyric acid ethyl ester (4) (or the corresponding methyl, propyl or butyl ester) with thionyl chloride in chloroform at 0-5°C to form 4-[l- methyl-5-bis-(2-chloroethyl)-benzimidazolyl-2]butyric acid ethyl ester (5). Excess of thionyl chloride is destroyed by stirring the reaction mixture in aqueous HCl. Finally chloroform is distilled off and stirred at 95°C for 3 hours. The reaction mixture is partially concentrated and the residue is diluted with water and stirred upto crystallization. Further purification is done by recrystallization from water.
Scheme-1: Method disclosed by Krueger et al. in DD159877 for the synthesis of Bendamustine hydrochloride

Bendamustine hydrochloride (6)
Ozegowski et al in Zentralblatt fuer Pharmazie, Pharmakotherapie und Laboratoriumsdiagnostik 1 10 (10), 1013-1019 (1971) discloses a process for the preparation of bendamustine hydrochloride monohydrate. The Chinese journal “Chinese journal of New Drugs “, 2007, No. 23, Vol. 16, 1960-61 and J. Prakt. Chem. 20, 178-186 (1963) disclose another method for the synthesis of Bendamustine hydrochloride monohydrate starting from 2,4-dinitrochlorobenzene as summarized in scheme-2.

The crucial conversions are reaction of l-methyl-2-(4′-ethyl butyrate)-5- amino]-lH-benzimidazole 6 with ethylene oxide in the presence of water, sodium acetate and acetic acid, by maintaining at 5°C for 5 hours and overnight at 20°C to give 4-{5-[bis-(2-hydroxy-ethyl)-amino]-l-methyl-lH-benzimidazol-2-yl}-butyric acid ethyl ester (dihydroxy ester) 7 as a jelly mass, which on chlorination using thionyl chloride in chloroform and subsequent in situ hydrolysis with concentrated HCI gave bendamustine hydrochloride. It also discloses a process for the recrystallization of bendamustine hydrochloride from water and the product obtained is a monohydrate with a melting point of 148-151°C.
IP.com Journal 2009, 9(7B), 21 discloses another process as shown below for the preparation of ethyl-4-[5-[bis(2-hydroxyethyl) amino]- l-methylbenzimidazol-2- yl]butanoate (III) wherein ethyl-4-(5 -amino- 1 -methyl- lH-benzo[d]imidazol-2-yl) butanoate (II) is reacted with 2-halo ethanol in the presence of an inorganic base selected from the group consisting potassium carbonate, potassium bicarbonate, sodium

The PCT application WO 2010/042568 assigned to Cephalon discloses the synthesis of Bendamustine hydrochloride as summarized in schem-3 starting from 2,4- dintroaniline in six steps. The crucial step is reductive alkylation of Il-a, using borane- tetrahydrofuran and chloroacetic acid at ambient temperature, producing compound of formula I-a. Acid mediated hydrolysis of I-a using concentrated hydrochloric acid at reflux produced bendamustine hydrochloride which has a purity of 99.1%. The above PCT Patent application also discloses a method of purification of Bendamustine hydrochloride by agitating the Bendamustine hydrochloride in a mixture of DMF and THF at 75°C for about 30 minutes followed by cooling to ambient temperature and isolating the solid by filtration.
Scheme-3:


iil-a


Bemdamuatine hydrochloride
The PCT application WO 2011/079193 assigned to Dr. Reddy’s Laboratories discloses the synthesis of Bendamustine hydrochloride as summarized in schem-4 starting from compound of formula (II). The crucial step is alkylation of compound of formula II with 2-haloethanol in the presence of an organic base to give a compound of formula (III) which on chlorination with a chlorinating agent affords a compound of formula (IV). Compound of formula (IV) on hydrolysis in acidic medium gives bendamustine hydrochloride. It further discloses purification of bendamustine hydrochloride using aqueous hydrochloric acid and acetonitrile.
Scheme-4:

Bendamustine hydrochloride (Pure)
The most of the prior art processes described above involve
• The use of ethylene oxide for the preparation of bendamustine hydrochloride, which is often not suitable for industrial scale processes due to difficulty in handling ethylene oxide, since it is shipped as a refrigerated liquid.
• Further, the known processes involve the use of strongly acidic conditions and high temperatures for the hydrolysis of ethyl ester of bendamustine and subsequent in-situ formation of bendamustine hydrochloride, thereby resulting in increased levels of various process-related impurities IMP. -A (RRT-0.46), IMP. -B (RRT-1.27) and IMP. -C (RRT-1.31) whose removal is quite difficult and make the process less economically viable.

IMP.-B
International Application Publication No. WO 2009/120386 describes various solid forms of bendamustine hydrochloride designated as bendamustine hydrochloride Form 1, bendamustine hydrochloride Form 2, bendamustine hydrochloride Form 3, bendamustine hydrochloride Form 4, amorphous bendamustine hydrochloride or a mixture thereof, processes for their preparation and lyophilized composition comprising the solid forms. According to the disclosure, monohydrate of bendamustine hydrochloride has been prepared previously. The monohydrate has a reported melting point of 152-156°C which is similar to that of the observed melting point of bendamustine hydrochloride Form 2.
It is known that synthetic compounds can contain extraneous compounds or impurities resulting from their synthesis or degradation. The impurities can be unreacted starting materials, by-products of the reaction, products of side reactions, or degradation products. Generally, impurities in an active pharmaceutical ingredient (API) may arise from degradation of the API itself, or during the preparation of the API. Impurities in Bendamustine hydrochloride or any active pharmaceutical ingredient (API) are undesirable and might be harmful.
Regulatory authorities worldwide require that drug manufacturers isolate, identify and characterize the impurities in their products. Furthermore, it is required to control the levels of these impurities in the final drug compound obtained by the manufacturing process and to ensure that the impurity is present in the lowest possible levels, even if structural determination is not possible. The product mixture of a chemical reaction is rarely a single compound with sufficient purity to comply with pharmaceutical standards. Side products and byproducts of the reaction and adjunct reagents used in the reaction will, in most cases, also be present in the product mixture. At certain stages during processing of the active pharmaceutical ingredient, the product is analyzed for purity, typically, by HPLC, TLC. or GC analysis, to determine if it is suitable for continued processing and, ultimately, for use in a pharmaceutical product. Purity standards are set with the intention of ensuring that an API is as free of impurities as possible, and, thus, are as safe as possible for clinical use. The United States Food and Drug Administration guidelines recommend that the amounts of some impurities are limited to less than 0.1 percent.
Generally, impurities are identified spectroscopically and by other physical methods, and then the impurities are associated with a peak position in a chromatogram (or a spot on a TLC plate). Thereafter, the impurity can be identified by its position in the chromatogram, which is conventionally measured in minutes between injection of the sample on the column and elution of the particular component through the detector, known as the “retention time” (“RT”). This time period varies daily based upon the condition of the instrumentation and many other factors. To mitigate the effect that such variations have upon accurate identification of an impurity, practitioners use “relative retention time” (“RRT”) to identify impurities. The RRT of an impurity is its retention time divided by the retention time of a reference marker.
It is known by those skilled in the art, the management of process impurities is greatly enhanced by understanding their chemical structures and synthetic pathways, and by identifying the parameters that influence the amount of impurities in the final product.
Therefore, there remains a need for improved process for the preparation of bendamustine hydrochloride, producing high yield and purity, and well-suited for use on an industrial scale. Despite the existence of various polymorphic forms of bendamustine hydrochloride, there exists a need for a simple process for the preparation of the stable form of bendamustine hydrochloride which is amenable to scale up and results in high yield and purity.
Bendamustine, (4-{5-[bis(2-chloroethyl)amino]-1-methyl-2-benzimidazolyl}butyric

Bendamustine
is an atypical structure with a benzimidazole ring, which structure includes an active nitrogen mustard. Bendamustine was initially synthesized in 1963 in the German Democratic Republic and was available from 1971 to 1992 in that location under the name Cytostasan®. Since that time, it has been marketed in Germany under the tradename Ribomustin®. It is currently available for use in the United States under the tradename Treanda® (Cephalon, Inc., Frazer, Pa.). It has been widely used to treat chronic lymphocytic leukemia, Hodgkin’s disease, non-Hodgkin’s lymphoma, multiple myeloma, and breast cancer.
Like other nitrogen mustards, bendamustine hydrolyzes in aqueous solution, with the major degradant being the primary alcohol HP1 (See U.S. application Ser. No. 11/330,868, the entirety of which is incorporated herein):
In light of its instability in aqueous solution, bendamustine is currently supplied as a lyophilized powder for injection. Just prior to its infusion, the medical practitioner reconstitutes the powder with Sterile Water for Injection. Reconstitution should yield a clear, colorless to pale yellow solution and the powder should completely dissolve in about 5 minutes. If particulate matter is observed, the reconstituted product should not be used and should be discarded. The reconstituted product is then transferred to a 0.9% Sodium Chloride Injection infusion bag within 30 minutes of reconstitution. This admixture should be a clear and colorless to slightly yellow solution. If the admixture comprises particulate matter or is discolored, it should be discarded and a fresh sample prepared.
The salt bendamustine hydrochloride is an alkylating agent, originally synthesized in 1963 at the Institute for Microbiology & Experimental Therapy in Jena, German Democratic Republic, with the intent to produce an agent with both alkylating and antimetabolite properties. Jenapharm (now Schering AG) formerly marketed it in Germany under the trade name Cytostasan from 1971 to 1992. Cytostasan was a lyophilised powder for solution for injection (vials) conatining 25 mg of Bendamustine HCI. It was widely used but never studied systematically in patients until the 1990s, then German investigators demonstrated its clinical activity in a number of malignancies. Since 1993, Ribosepharm was marketing bendamustine in Germany under the brand name Ribomustin RBO. Ribomustin is available as a lyophilized powder for injection, containing 100 mg of drug in each 50 ml_ vial, or 25 mg of drug in each 20 ml_ vial, also comprising mannitol, and indicated for the treatment of chronic lymphocytic leukemia. The lyophilized powder is reconstituted as close to the time of patient administration as possible with 40 ml_ (for a 100 mg product) or 10 mL (for a 25 mg product) of sterile water for injection. The reconstituted product then is further diluted to 500 mL with 0.9% sodium chloride for injection. The route of administration is by intravenous infusion over 30 to 60 minutes.
Another bendamustine product is sold in the United States by Cephalon, Inc. as TREANDA® for Injection, a lyophilized powder in a single-use vial indicated for the treatment of patients with chronic lymphocytic leukemia and indolent B-cell non-Hodgkin’s lymphoma. A 25 mg dose vial contains 25 mg of bendamustine hydrochloride and 42.5 mg of mannitol, and a 100 mg dose vial contains 100 mg of bendamustine hydrochloride and 170 mg of mannitol.
TREANDA is intended for intravenous infusion only after reconstitution with Sterile Water for Injection USP, and then further dilution with either 0.9% Sodium
Chloride Inj.ection, USP, or 2.5% Dextrose/0.45% Sodium Chloride Inj.ection, USP. The pH of the reconstituted solution is 2.5-3.5. TREANDA is supplied as a sterile non-pyrogenic white to off-white lyophilized powder, in a single-use vial.
Bendamustine hydrochloride is very unstable in an aqueous solution. The bis-2-chlorethylamino bond is hydrolyzed in weak acid, neutral, or alkaline solution. Monohydroxybendamustine [HP-1 ] is formed rapidly in the presence of water. Bendamustine ethyl ester [BM1 EE] is formed when bendamustine reacts with ethyl alcohol. BM1 EE can be formed during drug substance manufacturing, e.g., during recrystalization and/or purification processes. BM1 EE is a more potent cytotoxic drug than bendamustine.
FDA accepts new drug application for investigational compound Epanova for the treatment of severe hypertriglyceridaemia
LONDON, Sept. 18, 2013 – AstraZeneca today announced that the US Food and Drug Administration (FDA) has accepted for review a New Drug Application (NDA) for EpanovaTM, an investigational compound for the treatment for patients with severe hypertriglyceridaemia (triglyceride levels greater than or equal to 500mg/dL). The NDA submission for Epanova was filed by Omthera Pharmaceuticals, now a wholly-owned subsidiary of AstraZeneca, as a 505(b)(1) application in July 2013. The Prescription Drug User Fee Act (PDUFA) goal date for the FDA is 5 May 2014.http://www.pharmalive.com/fda-accepts-astrazeneca-nda-for-epanova
Forigerimod, (Rigerimod) also known as Lupuzor, CEP-3345 for treatment of systemic lupus erythematosus (SLE)


FORIGERIMOD
CHEMICAL NAMES
1. L-Tyrosine, L-arginyl-L-isoleucyl-L-histidyl-L-methionyl-L-valyl-L-tyrosyl-L-seryl-L-lysyl-L-arginyl-O-phosphono-L-serylglycyl-L-lysyl-L-prolyl-L-arginylglycyl-L-tyrosyl-L-alanyl-L-phenylalanyl-L-isoleucyl-L-α-glutamyl-
2. O3,140-phosphono(human U1 small nuclear ribonucleoprotein 70 kDa (snRNP70))-(131-151)-peptide
MOLECULAR FORMULA C117H181N34O32PS
MOLECULAR WEIGHT 2639
TRADEMARK Lupuzor
SPONSOR Cephalon, Inc.
CODE DESIGNATION IPP 201101
CAS REGISTRY NUMBER 497156-60-2
STRUCTURAL FORMULA

stucture, http://www.ama-assn.org/ama1/pub/upload/mm/365/forigerimod.pdf
-
Forigerimod nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/forigerimod.pdfSTATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD.
…………………………………………………………………………………………………………..
FORIGERIMOD ACETATE
CAS REGISTRY NUMBER 1160237-55-7 of acetate
http://www.ama-assn.org/resources/doc/usan/forigerimod-acetate.pdf
-
Forigerimod acetate nonproprietary drug name
http://www.ama-assn.org/resources/doc/usan/forigerimod-acetate.pdfSTATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN. FORIGERIMOD ACETATE
str is FORIGERIMOD ACETATE
FORIGERIMOD ACETATE
Forigerimod, also known as Lupuzor or CEP-33457, (SyB L-1001) is a CD4 T-cell modulator being investigated for the treatment of systemic lupus erythematosus (SLE). In the Phase II trials, Lupuzor was administered subcutaneously at a dose of 200 mcg once a month for 3 months. The Phase III study is anticipated to be complete in September 2012 and will measure the proportion of patients achieving a combined clinical response using the SLE responder index.

Positive final Lupuzor trial results. Marketwire. www.marketwire.com/press-release/Positive-Final-Lupuzor-Trial-Results-AIM-IMM-1176375.htm. Published November 19, 2009. Accessed June 18, 2011.
Rigerimod (IPP-201101, Lupuzor) is a polypeptide corresponding to the sequence 131-151 of the 70k snRNP protein with a serine phosphorylated in position 140.[1]
It gave encouraging results in a phase IIb trial for severe lupus.[1] Another phase IIb trial has started recruiting in the US.[2]
References
Lupuzor™ is a potential treatment for lupus, currently given the approval by the US FDA to start Phase III with a Special Protocol Assessment (SPA) and Fast Track designation. ImmuPharma holds all worldwide rights in this lead compound.
Background
Lupus (or Systemic Lupus Erythematosus) is a chronic, potentially life-threatening autoimmune disease. An estimated 1.4 million people are diagnosed in the 7 major world markets (the USA, Japan, Germany, France, Spain, the UK and Italy). Lupus is an inflammatory disease, which attacks multiple organs such as the skin, joints, kidneys, blood cells, heart and lungs. There is currently no cure.
The development of ImmuPharma’s Lupuzor™
ImmuPharma’s compound Lupuzor™ (previously known as IPP-201101 and also referred to as rigerimod or P140) has a novel mechanism of action aimed at modulating the body’s immune system so it does not attack healthy cells, without causing adverse side effects. It has the potential to halt the progression of the disease in a substantial proportion of patients.
Lupuzor™ has successfully completed Phase I, Phase IIa and Phase IIb studies and has now been given the approval by the US FDA to enter Phase III, the final testing phase.
The latest highlights of Lupuzor’s™ development as a treatment for lupus include:
- An ‘End of Phase 2’ meeting package with ImmuPharma’s Phase IIb data was submitted to the FDA and the FDA responded to all the questions
- The Investigational Medicinal Product Dossier (IMPD) submitted via the Voluntary Harmonized Procedure (VHP) in the EU was approved
- The Scientific Advice meeting with the European Medicines Evaluation Agency (EMEA) was held; the recommendations were very similar to those in the FDA’s ‘End Of Phase 2’ responses. Recommendations were incorporated into the Phase III pivotal programme
- The Japanese equivalent authorities (PMDA) have agreed to the initiation of clinical trials in Japan
- The FDA has granted Lupuzor™ the approval to start Phase III with a Special Protocol Assessment (SPA)
- The FDA has granted Lupuzor™ Fast Track designation
How Lupuzor™ works in the treatment of lupus
Lupuzor™ is a drug that specifically modulates the immune system of lupus patients by modifying the behaviour of some of the key cells involved in the pathogenesis of the disease. The clinical profile of lupus patients is generally assessed by standardised scales such as SLEDAI (SLE Disease Activity Index): the lower the score, the better the condition of the patient. During this Phase II study, the SLEDAI scores were assessed on multiple occasions even though the study was not designed or powered to demonstrate clinical benefit as the primary endpoint due to the short treatment period.
| forigerimod | IPP-201101 | oligopeptide | therapeutic | nucleolin |
| forigerimod acetate | CEP-33457, P-140, IPP-201101 | oligopeptide (salt) | therapeutic | nucleolin |
GLENMARK- ELOVERA , for dry skin disorders


Compositions:
Elovera extract 10% cream, Vitamin E 0.5%
Category–Locally Acting Skin Preparations
Description
* Aqueeze adequate amount of elovera moisturizing body wash onto wet hands or wet loran and work into a creamy lather. apply it all ovr the body, keep for some time and then rinse with water.
| Products Name : | Elovera Moisturizing Body Wash 150ml – (Glenmark) |
Elovera Cream, manufacture by Glenmark pharmaceuticals limited , is cream enriched with vitamin E and Aloe Vera. It’s a very special cream specially for treating scars and other minor pimple spots on the face.
reviews from net
My skin is very much oily hence I get these ugly Pimples very profoundly. On top of it i have the habit of bursting out the puss from these pimples. I always play it with my hands and as a result forms some very ugly scars on my face which are visible from distant away.Though I am bit dark with my completion ,even then It’s clearly visible and my mother scolds me like hell for bursting the pimples out.Honestly I just can’t stop my hands reaching out for them no matter how busy I am so Finally has to resort to some ointments to reduce the visibility of the scars.
I did try few popular products but were of no use basically. The spots didn’t get reduced but instead effected the completion of my face.Finally my mother came to my rescue. She had hear about this Elovera Cream from some one and bought home one for me.Initially i was a bit skeptic but finally I thought of trying it. For the first few days it had no effect what-so-ever , but slowly it started clearing the skin blemishes. My skin started showing it’s effects and the scars became less visible. Not only does it clear the scars but it helped me to fight the ugly pimples as well.
My face became much more glowing and healthy and i use the cream regularly even now.It’s really a magical product and should try it for clearing the blemishes and other skin problem.
EMA approves biosimilar Somatropin from Biopartners Gmbh
![OMNITROPE® (somatropin [rDNA origin] injection) Structural Formula Illustration](https://i0.wp.com/images.rxlist.com/images/rxlist/omnitrope1.gif)
SOMATROPIN
The European Medicine agency has approved a biosimilar somatropin from Biopartners GMBH. Somatropin biopartner would be the third biosimilar version of somatropin the European market. Other players selling somatropin inlcude Sandoz and Roche. Sandoz sells under the brand Omnitrope, while Roche which is the innovator of somatropin sells it under the brand name NutropinAq.
READ AT
DETAILS OF OMNITROPE
Omnitrope® (somatropin-[rDNA] origin) is a polypeptide hormone of recombinant DNA origin. It has 191 amino acid residues and a molecular weight of 22,125 daltons. The amino acid sequence of the product is identical to that of human growth hormone of pituitary origin (somatropin). Omnitrope® is synthesized in a strain of. Escherichia coli that has been modified by the addition of the gene for human growth hormone. Omnitrope® Cartridge is a clear, colorless, sterile solution for subcutaneous injection. Omnitrope® for Injection is a lyophilized powder that is reconstituted for subcutaneous injection.
Figure 1: Schematic amino acid sequence of human growth hormone including the disulfide bonds
|
Each Omnitrope® Cartridge or vial contains the following (see Table 4):
Table 4. Contents of Omnitrope® Cartridges and Vial
| Product | Cartridge 5 mg/1.5 mL | Cartridge 10 mg/1.5 mL | For Injection 5.8 mg/vial |
| Component | |||
| Somatropin | 5 mg | 10 mg | 5.8 mg |
| Disodium hydrogen phosphate heptahydrate | 1.3 mg | 1.70 mg | 2.09 mg |
| Sodium dihydrogen phosphate dihydrate | 1.6 mg | 1.35 mg | 0.56 mg |
| Poloxamer 188 | 3.0 mg | 3.0 mg | – |
| Mannitol | 52.5 mg | – | – |
| Glycine | – | 27.75 mg | 27.6 mg |
| Benzyl alcohol | 13.5 mg | – | – |
| Phenol | – | 4.50 mg | – |
| Water for Injection | to make 1.5 mL | to make 1.5 mL | – |
| Diluent (vials only) | Bacteriostatic Water for Injection | ||
| Water for injection | to make 1.14 mL | ||
| Benzyl alcohol | 17 mg | ||
Genzyme’s multiple sclerosis treatment approved by European Commission

Alemtuzumab
Sanofi and its subsidiary Genzyme have been given marketing approval by the European Commission for Lemtrada (alemtuzumab), a treatment for multiple sclerosis. read all at
click on title below
Genzyme’s multiple sclerosis treatment approved by European Commission
Sanofi wins EU approval for second MS treatment Lemtrada
September 17,2013 | By Márcio Barra

Sanofi won today a marketing approval from the European commission for their second multiple sclerosis treatment, the injectable drug Lemtrada, following the approval of the pill Aubagio (teriflunomide) on August 30. This was the drug’s first regulatory approval worldwide.
View original post 283 more words
Generic versions of high cholesterol drug Lovaza can be developed, rules judge
LOVAZA, a lipid-regulating agent, is supplied as a liquid-filled gel capsule for oral administration. Each 1-gram capsule of LOVAZA (omega-3-acid ethyl esters) contains at least 900 mg of the ethyl esters of omega-3 fatty acids. These are predominantly a combination of ethyl esters of eicosapentaenoic acid (EPA – approximately 465 mg) and docosahexaenoic acid (DHA – approximately 375 mg).
The structural formula of EPA ethyl ester is:

The empirical formula of EPA ethyl ester is C22H34O2, and the molecular weight of EPA ethyl ester is 330.51.
The structural formula of DHA ethyl ester is:

The empirical formula of DHA ethyl ester is C24H36O2, and the molecular weight of DHA ethyl ester is 356.55.
LOVAZA capsules also contain the following inactive ingredients: 4 mg α-tocopherol (in a carrier of partially hydrogenated vegetable oils including soybean oil), and gelatin, glycerol, and purified water (components of the capsule shell).
Lovaza
A US appeals court ruled on this week that drug companies can develop generic versions of fish oil-derived, high-cholesterol drug Lovaza.
read all at

Lovaza is a brand name prescription drug. The capsule sold by GlaxoSmithKline but developed by Reliant Pharmaceuticals, contains esterified fish oils and is approved by the U.S. Food and Drug Administration to lower very high triglyceride levels. It is metabolized intoOmega-3 fatty acids. It is a dietary supplement that has been purified, chemically altered, branded, and been put through the approval process of the U.S. Food and Drug Administration (FDA); in these respects it is considered a pharmaceutical. Due to the esterification process during manufacturing there is no risk of contamination[citation needed] by methyl mercury, arsenic,[1] or other pollutants that are often seen in the world’s oceans. Each 1-gram capsule is 38% DHA, 47% EPA, and 17% other fish oils in the form of the ethyl ester.
Lovaza is named Omacor in Europe (and this name was once used in the US).[2]
Effectiveness
Lovaza is approved in the U.S. for treatment of patients with very high triglycerides (hypertriglyceridemia).[3]
In the European markets and other major markets outside the US Lovaza is known as Omacor, and is indicated for:
- Hypertriglyceridemia. Used as monotherapy, or in combination with a statin for patients with mixed dyslipidemia.
- Secondary prevention after myocardial infarction (heart attack)
in addition to other standard therapy (e.g. statins, antiplatelets medicinal products, beta-blockers, and ACE-I).
Lovaza has been demonstrated to reduce triglycerides in patients with high or very high triglycerides. [3]
Lovaza has also been demonstrated to reduce VLDL-cholesterol and non-HDL-cholesterol, and increase HDL-cholesterol. But, it can raise LDL-cholesterol up to 45%.[4] The LDL raising activity correlates with a reduction in ApoB levels, though. Lovaza, through the stimulation of Lipoprotein Lipase, seems to stimulate the production of less atherogenic LDL species. In some patients, it can elevatealanine transaminase levels, so liver enzymes should be checked, periodically.[4]
Effects on significant patient outcomes such as acute myocardial infarction, stroke, cardiovascular and all-cause mortality have been studied in patients who have suffered a myocardial infarction (this is in the US; however, data from GISSI-P showed a combined end-point of all-cause death, non-fatal MI, and non-fatal stroke was significantly reduced by 15%). Lovaza has not been shown to lower the rates of all cause mortality and cardiovascular mortality, or the combination of mortality and non-fatal cardiovascular events.[3]
GlaxoSmithKline‘s patent expired in September 2012. Generic versions may be made available at that time. Other DHA/EPA products containing similar amounts of Omega-3 fatty acids are currently sold over the counter in the United States as dietary supplements.
Competitors
In July 2012, Amarin Corporation received U.S. FDA marketing approval for Vascepa, also referred to as AMR-101.[5] Vascepa will undoubtedly become a major competitor for Lovaza.[6] In clinicial trials, Vascepa was shown to lower triglycerides; while Lovaza also lowers the triglyceride concentration, Vascepa also lowers LDL-C; Lovaza does not. Lovaza was approved to treat people with very high triglyceride levels (>500 mg/dl), Vascepa is also approved for this market; however the company has also demonstrated that the drug can impact levels in people with high triglyceride (> 200 mg/dl and < 500 mg/dl) levels and will file an sNDA for this indication late in 2012.[7]
In 2011, Ariix started selling an almost identical FDA-Certified Omega3 Ethyl Ester 1000 mg capsule ‘OmegaQ’ fish oil through direct marketing and online auto-ship at a discounted price, creating another major competitor for Lovaza and Amarin’s Vascepa. One capsule contains 295 mg EPA, and 235 mg DHA, but it is unique in that it is combined with 20 mg of the coenzyme CoQ-10, with reported ‘anti-aging’ effects on the cell’s telomeres, which are still under study.
Forms of Lovaza
Lovaza is available as 1-gram soft-gelatin capsules.[8]
Active Ingredient: Omega-3-acid ethyl esters
Inactive Ingredients: Gelatin, glycerol, purified water, alpha-tocopherol (in soybean oil)
References
- NIFES (Nasjonalt institutt for ernærings- og sjømatforskning – Norwegian National Institute for Nutrition and Seafood Research)
- University of Utah Pharmacy Services (August 15, 2007) “Omega-3-acid Ethyl Esters Brand Name Changed from Omacor to Lovaza”
- GSK Information for Medical Professionals
- Pharmacy & Therapeutics (May, 2008) “Omega-3-acid Ethyl Esters (Lovaza) For Severe Hypertriglyceridemia”
- “Amarin Prescription Fish-Oil Pill Approved – TheStreet”. Retrieved 26 July 2012.
- “http://www.reuters.com/article/2012/07/26/us-amarin-fda-vascepa-idUSBRE86P1SX20120726”. Reuters. 26 July 2012. Retrieved 27 July 2012.
- “Amarin’s AMR101 Phase 3 ANCHOR Trial Meets all Primary and Secondary Endpoints with Statistically Significant Reductions in Triglycerides at Both 4 Gram and 2 Gram Doses and Statistically Significant Decrease in LDL-C (NASDAQ:AMRN)”. Amarin. 18 April 2011. Retrieved 26 July 2012.
- http://www.rxwiki.com/lovaza
External links
The first generic version of the oral chemotherapy drug Xeloda (capecitabine) has been approved by the U.S. Food and Drug Administration to treat cancers of the colon/rectum or breast,

capecitabine
- R-340, Ro-09-1978, Xeloda
pentyl [1-(3,4-dihydroxy-5-methyltetrahydrofuran-2-yl)-5-fluoro-2-oxo-1H-pyrimidin-4-yl]carbamate

MONDAY Sept. 16, 2013 — The first generic version of the oral chemotherapy drug Xeloda (capecitabine) has been approved by the U.S. Food and Drug Administration to treat cancers of the colon/rectum or breast, the agency said Monday in a news release.

This year, an estimated 142,820 people will be diagnosed with cancer of the colon/rectum, and 50,830 are predicted to die from the disease, the FDA said, citing the U.S. National Cancer Institute. An estimated 232,340 women will be diagnosed with cancer of the breast this year, and some 39,620 will die from it.
The most common side effects of the drug are diarrhea, vomiting; pain, redness, swelling or sores in the mouth; fever and infection, the FDA said.
The agency stressed that approved generics have the same high quality and strength as their brand-name counterparts.
License to produce the generic drug was given to Israel-based Teva Pharmaceuticals. The brand name drug is produced by the Swiss pharma firm Roche.
Capecitabine (INN) /keɪpˈsaɪtəbiːn/ (Xeloda, Roche) is an orally-administered chemotherapeutic agent used in the treatment of metastatic breast and colorectal cancers. Capecitabine is a prodrug, that is enzymatically converted to 5-fluorouracil in the tumor, where it inhibits DNA synthesis and slows growth of tumor tissue. The activation of capecitabine follows a pathway with three enzymatic steps and two intermediary metabolites, 5′-deoxy-5-fluorocytidine (5′-DFCR) and 5′-deoxy-5-fluorouridine (5′-DFUR), to form 5-fluorouracil

Indications
Capecitabine is FDA-approved for:
- Adjuvant in colorectal cancer Stage III Dukes’ C – used as first-line monotherapy.
- Metastatic colorectal cancer – used as first-line monotherapy, if appropriate.
- Metastatic breast cancer – used in combination with docetaxel, after failure of anthracycline-based treatment. Also as monotherapy, if the patient has failed paclitaxel-based treatment, and if anthracycline-based treatment has either failed or cannot be continued for other reasons (i.e., the patient has already received the maximum lifetime dose of an anthracycline).
In the UK, capecitabine is approved by the National Institute for Health and Clinical Excellence (NICE) for colon and colorectal cancer, and locally advanced or metastatic breast cancer.[1] On March 29, 2007, the European Commission approved Capecitabine, in combination with platinum-based therapy (with or without epirubicin), for the first-line treatment of advanced stomach cancer.
Capecitabine is a cancer chemotherapeutic agent that interferes with the growth of cancer cells and slows their distribution in the body. Capecitabine is used to treat breast cancer and colon or rectum cancer that has spread to other parts of the body.
Formulation
Capecitabine (as brand-name Xeloda) is available in light peach 150 mg tablets and peach 500 mg tablets.
- Lacy, Charles F; Armstrong, Lora L; Goldman, Morton P; Lance, Leonard L (2004). Lexi-Comp’s Drug Information Handbook (12th Edition). Lexi-Comp Inc. ISBN 1-59195-083-X
- Fischer, David S; Knobf, M Tish; Durivage, Henry J; Beaulieu, Nancy J (2003). The Cancer Chemotherapy Handbook (6th Edition). Mosby. ISBN 0-323-01890-4
- Thomson Centerwatch: Drugs Approved by the FDA (Xeloda) Retrieved 6/05
- Mercier C, Ciccolini J (2007). “Severe or lethal toxicities upon capecitabine intake: is DPYD genetic polymorphism the ideal culprit?”. Trends in pharmacological sciences 28 (12): 597–598. doi:10.1016/j.tips.2007.09.009. PMID 18001850.
- “Subtopics”. Nice.org.uk. Retrieved 2012-08-15.
- Fingerprints May Vanish With Cancer Drug – US News and World Report
- Cancer Drug Erases Man’s Fingerprints – CNN
- “Stritch School of Medicine”. Stritch.luc.edu. Retrieved 2012-08-15.
- Xeloda.com (patient information, tools, and resources)
- OralChemo Advisor (patient information)

Capecitabine is an orally-administered anticancer agent widely used in the treatment of metastatic breast and colorectal cancers. Capecitabine is a ribofuranose-based nucleoside, and has the sterochemical structure of a ribofuranose having an β-oriented 5-fluorocytosine moiety at C-I position.
US Patent Nos. 5,472,949 and 5,453,497 disclose a method for preparing capecitabine by glycosylating tri-O-acetyl-5-deoxy-β-D-ribofuranose of formula I using 5-fluorocytosine to obtain cytidine of formula II; and carbamoylating and hydrolyzing the resulting compound, as shown in Reaction Scheme 1 :
Reaction Scheme 1

1
The compound of formula I employed as an intermediate in Reaction
Scheme 1 is the isomer having a β-oriented acetyl group at the 1 -position, for the reason that 5-fluorocytosine is more reactive toward the β-isomer than the α-isomer in the glycosylation reaction due to the occurrence of a significant neighboring group participation effect which takes place when the protecting group of the 2-hydroxy group is acyl.
Accordingly, β-oriented tri-O-acetyl-5-deoxy-β-D-ribofuranose (formula
I) has been regarded in the conventional art to the essential intermediate for the preparation of capecitabine. However, such a reaction gives a mixture of β- and α-isomers from which cytidine (formula II) must be isolated by an uneconomical step.
Meanwhile, US Patent No. 4,340,729 teaches a method for obtaining capecitabine by the procedure shown in Reaction Scheme 2, which comprises hydrolyzing 1-methyl-acetonide of formula III to obtain a triol of formula IV; acetylating the compound of formula IV using anhydrous acetic anhydride in pyridine to obtain a β-/α-anomeric mixture of tri-O-acetyl-5-deoxy-D-ribofuranose of formula V; conducting vacuum distillation to purify the β-/α-anomeric mixture; and isolating the β-anomer of formula I therefrom:
Reaction Scheme 2
III IV
However, the above method is also hampered by the requirement to perform an uneconomical and complicated recrystallization steps for isolating the β-anomer from the mixture of β-/α-anomers of formula V, which leads to a low yield of only about 35% to 40% (Guangyi Wang et al., J. Med. Chem., 2000, vol. 43, 2566-2574; Pothukuchi Sairam et al., Carbohydrate Research, 2003, vol. 338, 303-306; Xiangshu Fei et al., Nuclear Medicine and Biology, 2004, vol. 31, 1033-1041; and Henry M. Kissman et al., J. Am. Chem. Soc, 1957, vol. 79, 5534-5540).
Further, US Patent No. 5,476,932 discloses a method for preparing capecitabine by subjecting 5′-deoxy-5-fluorocytidine of formula VI to a reaction with pentylchloroformate to obtain the compound of formula VII having the amino group and the 2-,3-hydroxy groups protected with C5Hi1CO2 groups; and removing the hydroxy-protecting groups from the resulting compound, as shown in Reaction Scheme 3 :
Reaction Scheme 3

Vl VII 1
However, this method suffers from a high manufacturing cost and also requires several complicated steps for preparing the 5′-deoxy-5-fluorocytidine of formula VI: protecting the 2-,3-hydroxy groups; conducting a reaction thereof with 5-fluorocytosine; and deprotecting the 2-,3-hydroxy groups.
Accordingly, the present inventors have endeavored to develop an efficient method for preparing capecitabine, and have unexpectedly found an efficient, novel method for preparing highly pure capecitabine using a trialkyl carbonate intermediate, which does not require the uneconomical β-anomer isolation steps.
synthesis

more info and description
Aspects of the present invention relate to capecitabine and processes for the preparation thereof.
The drug compound having the adopted name “capecitabine” has a chemical name 5′-deoxy-5-fluoro-N-[(pentyloxy) carbonyl] cytidine and has structural formula I.
H

OH OH I
This compound is a fluoropyrimidine carbamate with antineoplastic activity. The commercial product XELODA™ tablets from Roche Pharmaceuticals contains either 150 or 500 mg of capecitabine as the active ingredient.
U.S. Patent No. 4,966,891 describes capecitabine generically and a process for the preparation thereof. It also describes pharmaceutical compositions, and methods of treating of sarcoma and fibrosarcoma. This patent also discloses the use of ethyl acetate for recrystallization of capecitabine. The overall process is summarized in Scheme I.

Scheme I
U.S. Patent No. 5,453,497 discloses a process for producing capecitabine that comprises: coupling of th-O-acetyl-5-deoxy-β-D-hbofuranose with 5- fluorocytosine to obtain 2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine; acylating a 2′, 3′- di-O-acetyl-5′-deoxy-5-fluorocytidine with n-pentyl chloroformate to form 5′-deoxy- 2′,3′-di-O-alkylcarbonyl-5-fluoro-N-alkyloxycarbonyl cytidine, and deacylating the 2′ and 3′ positions of the carbohydrate moiety to form capecitabine. The overall process is summarized in Scheme II.

Capecitabine
Scheme Il
The preparation of capecitabine is also disclosed by N. Shimma et al., “The Design and Synthesis of a New Tumor-Selective Fluoropyrimidine Carbamate, Capecitabine,” Bioorganic & Medicinal Chemistry, Vol. 8, pp. 1697-1706 (2000). U.S. Patent No. 7,365,188 discloses a process for the production of capecitabine, comprising reacting 5-fluorocytosine with a first silylating agent in the presence of an acid catalyst under conditions sufficient to produce a first silylated compound; reacting the first silylated compound with 2,3-diprotected-5- deoxy-furanoside to produce a coupled product; reacting the coupled product with a second silylating agent to produce a second silylated product; acylating the second silylated product to produce an acylated product; and selectively removing the silyl moiety and hydroxyl protecting groups to produce capecitabine. The overall process is summarized in Scheme III. te

R: hydrocarbyl

Scheme III
Further, this patent discloses crystallization of capecitabine, using a solvent mixture of ethyl acetate and n-heptane. International Application Publication No. WO 2005/080351 A1 describes a process for the preparation of capecitabine that involves the refluxing N4– pentyloxycarbonyl-5-fluorocytosine with trimethylsiloxane, hexamethyl disilazanyl, or sodium iodide with trimethyl chlorosilane in anhydrous acetonitrile, dichloromethane, or toluene, and 5-deoxy-1 ,2,3-tri-O-acetyl-D-ribofuranose, followed by hydrolysis using ammonia/methanol to give capecitabine. The overall process is summarized in Scheme IV.

Scheme IV
International Application Publication No. WO 2007/009303 A1 discloses a method of synthesis for capecitabine, comprising reacting 5′-deoxy-5- fluorocytidine using double (trichloromethyl) carbonate in an inert organic solvent and organic alkali to introduce a protective lactone ring to the hydroxyl of the saccharide moiety; reacting the obtained compound with chloroformate in organic alkali; followed by selective hydrolysis of the sugar component hydrolytic group using an inorganic base to give capecitabine. The overall process is summarized in Scheme V.

Scheme V
Even though all the above documents collectively disclose various processes for the preparation of capecitabine, removal of process-related impurities in the final product has not been adequately addressed. Impurities in any active pharmaceutical ingredient (API) are undesirable, and, in extreme cases, might even be harmful to a patient. Furthermore, the existence of undesired as well as unknown impurities reduces the bioavailability of the API in pharmaceutical products and often decreases the stability and shelf life of a pharmaceutical dosage form.
nmr
1H NMR(CD3OD) δ 0.91(3H5 t), 1.36~1.40(4H, m), 1.41(3H, d), 1.68~1.73(2H, m), 3.72(1H, dd), 4.08(1H, dd), 4.13~4.21(3H, m), 5.7O(1H, s), 7.96(1H, d)

- The acetylation of 5′-deoxy-5-fluorocytidine (I) with acetic anhydride in dry pyridine gives 2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine (II), which is condensed with pentyl chloroformate (III) by means of pyridine in dichromethane yielding 2′,3′-di-O-acetyl-5′-deoxy-5-fluoro-N4-(pentyloxycarbonyl)cytidine (IV). Finally, this compound is deacetylated with NaOH in dichloromethane/water. The diacetylated cytidine (II) can also be obtained by condensation of 5-fluorocytosine (V) with 1,2,3-tri-O-acetyl-5-deoxy-beta-D-ribofuranose (VI) by means of trimethylchlorosilane in acetonitrile or HMDS and SnCl4 in dichloromethane..
-
- EP 602454, JP 94211891, US 5472949.
- Capecitabine. Drugs Fut 1996, 21, 4, 358,
- Bioorg Med Chem Lett2000,8,(7):1697,
- Capecitabine. Drugs Fut 1996, 21, 4, 358,
- EP 602454, JP 94211891, US 5472949.
Paclitaxel Against Cancer: A Short Review
| Priyadarshini K1* and 2Department of Biotechnology, Loyola Academy Degree & PG College, Secunderabad, IndiaKeerthi Aparajitha U2 | ||||||
| http://www.omicsonline.org/paclitaxel-against-cancer-a-short-review-2161-0444.1000130.php?aid=9996 | ||||||
| Corresponding Author : | Priyadarshini K Department of Biotechnology JSS College for Arts Commerce & Science Mysore, India E-mail: prits_bhargav88@yahoo.com |
|||||
| Received November 16, 2012; Accepted November 28, 2012; Published November 30, 2012 | ||||||
| Citation: Priyadarshini K, Keerthi Aparajitha U (2012) Paclitaxel Against Cancer: A Short Review. Med chem 2:139-141. doi:10.4172/2161-0444.1000130 | ||||||
| Copyright: © 2012 Priyadarshini K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. | ||||||
|
||||||
