
Home » Posts tagged 'Wanbury'
Tag Archives: Wanbury
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd
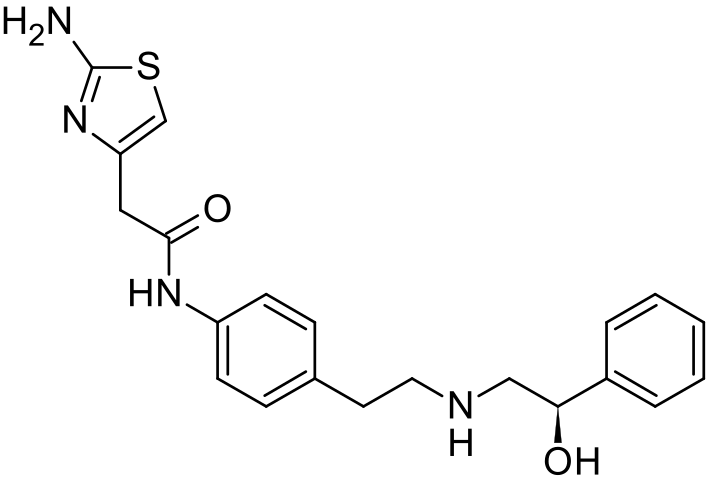
WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd

WANBURY LTD. [IN/IN]; BSEL tech park, B wing, 10th floor, sector 30A opp. Vashi Railway Station, Vashi Navi Mumbai 400703 Maharashtra (IN)
DR. NITIN SHARADCHANDRA PRADHAN; (IN).
DR. NILESH SUDHIR PATIL; (IN).
DR. RAJESH RAMCHANDRA WALAVALKAR; (IN).
MR. NILESH SUBHASH KULKARNI; (IN).
MR. SANTOSH NAMDEV RAWOOL; (IN).
MR. PURUSHOTTAM EKANATH AWATE; (IN)

LEFT , DR K CHANDRAN, DIRECTOR WANBURY
MR ASOK SHINKAR
The present invention relates to a novel process for preparation of Mirabegron of Formula (I) using intermediates of Formula (II), (IIIa), (Illb) and (IV).
The present invention relates to a process for preparation of Mirabegron of Formula
(I).

Formula (I)
The present invention further relates to the preparation of Mirabegron of Formula (I) by using compounds of Formula (II), (Ilia), (Illb) and (IV)

Formula (II)

Formula (IlIa) Formula (Illb)

Formula (IV)
Furthermore, the present invention relates to process for preparation of compound of Formula (II), (Ilia), (Illb) and (IV).
Background of the invention:
Mirabegron is chemically known as 2-amino-N-[4-[2-[[(2R)-2-hydroxy-2-phenylethyl]amino]ethyl]phenyl]-4-thiazoleactamide and is marketed under trade name Myrbetiq.
Mirabegron is a drug used for treatment of overactive bladder. It was first disclosed in US 6,346,532, wherein (R)-Styrene oxide is reacted with 4-nitrophenyl ethyl amine hydrochloride to obtain (R)-l- phenyl-2-[[2-(4-nitrophenyl)ethyl]amino]ethanol, the later is then protected with BOC anhydride and subjected to reduction in the presence of Pd/C to yield N-[2-(4-Aminophenyl)ethyl]-N-[(2R)-2-hydroxy-2-phenylethyljcarbamic acid tert-butyl ester. Thus formed compound was then coupled with (2-amino-l,3-thiazol-4yl) acetic acid to obtain BOC protected Mirabegron which is de-protected to give Mirabegron hydrochloride.
The synthetic route proposed in US 6,346,532 is presented in Scheme-I.
Scheme-I

The major draw-backs of the presented synthetic scheme are as follows:
1. Less atomic efficiency
2. Low yield and extensive impurities formations
3. Use of expensive and sensitive protecting agents
4. Column chromatographic techniques for purifications of intermediates.
One more synthetic route for the preparation of Mirabegron have been proposed US 6,346,532, however it is not exemplified.
US 7,342,117 disclose a process for preparation of Mirabegron. The process involves the step of condensation of 4-nitrophenyl ethylamine and (R)- mandelic acid in presence of tri ethylamine, hydroxybentriazole and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide in N,N-dimethylformamide to obtain compound of Formula (A). The second step involves conversion of compound of Formula (A) to compound of Formula (B) in presence of l,3-dimethyl-2-imidazolidone and borontetrahydro fluoride in tetrahydrofuran. In third step, compound of Formula (B) is subjected to reduction using 10% palladium-carbon in methanol to afford (R)-2-[[2′-(4-aminophenyl)-ethyl amino] -1-phenylethanol (Formula IV), which was further condensed with 2-aminothiazol-4-yl acetic acid in presence of l-(3-dimethylaminopropyl)-3 -ethyl carbodiimide and hydrochloric acid in water to obtain Mirabegron of Formula (I). The schematic representation is as Scheme-II

Another patent application CN103193730, discloses a novel process for preparation of Mirabegron wherein the amino group of 2-aminothiazole-5-acetic acid is protected with a protecting group and is condensed with 4-amino phenyl ethanol to obtain an intermediate (A); which on further oxidation yields intermediate (B). The intermediate B is subjected to reductive amination with (R)-2-amino-l -phenyl ethanol and deprotection, simultaneously to yield Mirabegron. The schematic representation is as Scheme-Ill.

Formula (I)
Scheme-Ill
Other references wherein process for preparation of Mirabegron are disclosed CN103387500 and CN103232352.
Most of the prior art reported for preparation of Mirabegron uses expensive and sensitive protecting agents thereby making process less feasible on industrial scale. Furthermore, the yield and purity of Mirabegron obtained by the processes known in art is not satisfactory. It is well known fact that pharmaceutical products like Mirabegron should have high purity due to the therapeutic advantages and also due to the stringent requirements of regulatory agencies. The purity requirements can be fulfilled either by avoiding the formation of by-products during the process or by purifying the end product of the process. The inventors of present invention have skillfully developed the process to provide Mirabegron with unachieved level of purity. Furthermore, the process of present invention is simple, industrially viable, and economic and avoids unfavorable reaction conditions.
![]()
According to present invention, the process for preparation of compound of Formula (IV), is depicted in Scheme IV

The present invention further relates to a process for preparation of Mirabegron of Formula (I)
The schematic reaction scheme of Mirabegron according to present invention is depicted in Scheme-V.
 Wherein R is -OH or -CI
Wherein R is -OH or -CI
The detail of the invention provided in the following examples is given by the way of illustration only and should not be construed to limit the scope of the present invention.
EXAMPLES
Example 1: Preparation of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride; Formula (V); wherein R is -CI
20g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid was added to 250 ml of methylene dichloride and the mixture was cooled to -10°C followed by lot wise addition of 25g of phosphorous pentachloride. The mixture stirred while maintaining temperature of -10°C for 2-3 hours. After confirming completion of reaction, the product was filtered out, washed with methylene dichloride and dried to obtain 24g (Yield: 92%) of compound of Formula (V); wherein R is -CI
Example 2: Preparation of 4-nitrophenyl-[2-(formylamino)-l,3-thiazol-4-yl]acetate; Formula (IlIa)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.963g of potassium carbonate, the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (V) of example 1. After confirming completion of reaction, 5.87g (Yield: 99%) of compound of Formula (Ilia) was isolated. The obtained compound has been identified by;
HNMR(D20 Exchange)
8.614 (S,lH),7.359(d,2H),8.119(d,2H),6.561(S,lH),3.765(S,2H).
Example 3: Preparation of (2-amino-l,3-thiazol-4-yl)acetyl chloride; Formula (VI); wherein R is -CI
5g of (2-amino-l,3-thiazol-4-yl)acetic acid was added to 50 ml of methylene dichloride with few drops of dimethylformamide and 6g of oxalyl chloride at temperature ranging from 0-5°C. the mixture was maintained at 0-5°C for 4-5 hours and after completion of reaction, solid mass was filtered out, washed with methylene dichloride and dried to afford 5g (Yield: 89%) of compound of Formula (VI); wherein R is -CI
Example 4: Preparation of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate; Formula (Illb)
2g of p-nitrophenol was added to 40ml of methylene chloride and 4.96g of potassium carbonate, and the mixture was cooled to 10-15 °C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming completion of reaction, 6.18g (Yield: 99%) of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate of Formula (Illb) was isolated.
The obtained compound has been identified by
HNMR ( D2O Exchange)
7.359(d,2H),8.1 19(d,2H),6.425(S,lH).3.775(S,2H).
Example 5: In-situ preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol or its hydrochloride salt, of Formula (IV)
Step I – Preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
(R)-2-hydroxy-2-phenylacetic acid (75g), triethylamine (50g), hydroxybenzotriazole (HOBt) (33.3g) and l-(3-dimethylaminopropyl)-3-ethyl carbodiimide hydrochloride (EDC.HC1) (50g) were added to a mixture of 2-(4-nitrophenyl)ethylamine hydrochloride (100g) in Ν,Ν-dimethylformamide (375ml) at 25-30°C. The mixture was stirred for 30 minutes followed by addition of another lot of HOBt (33.3g) and EDC.HC1 (50g) in reaction mixture. The reaction mixture was maintained at 25-30°C for 15 hours under stirring. After completion of reaction, water (1850ml) was added to the reaction mixture and stirred. Subsequently, ethyl acetate (1500ml) was added to the reaction mixture at 25-30°C and stirred. The organic phase was separated from aqueous phase, and was washed sequentially with 1M HC1 solution, 20%aqueous potassium carbonate solution and water. The organic solvent was distilled out under reduced pressure to obtain residue comprising of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl] -2 -phenyl ethanamide of Formula (IX)
Step II – Preparation of (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
The residue from step I, methanol (740ml) and Raney Nickel (14.8g) were charged into an autoclave vessel, 10 kg/cm2 hydrogen gas pressure was applied to the reaction mixture at 25-30°C and the mixture was maintained under stiring 6 hours. Reaction mixture filtered through hyflo bed. Distilled off the solvent completely from the filtrate under reduced pressure to obtain residue comprising (2R)-2-hydroxy-N-[2-(4-aminophenyl)ethyl]-2-phenylethanamide of Formula (X)
Step III – Preparation of (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
The residue of step II was added in tetrahydrofuran (665ml) and the mixture was cooled to -5 to 0°C. To this cooled mixture was then successively added sodium borohydride (56.26g) and BF3-diethyl ether (466g), and the mixture was stirred for 15 minutes. The temperature of reaction mixture was gradually increased to 50-55°C and was maintained under stirring for 5 hours. After completion of reaction, the reaction mixture was cooled to 0-5°C and 50% sodium hydroxide solution was added till pH is basic. The temperature of reaction mixture is then raised to 25-30°C followed by addition of ethyl acetate (500ml). The organic layer was separated and subjected to distillation to afford a residue. To the residue was added isopropyl alcohol (665ml) and mixture was refluxed for 30 minutes. The mixture was then allowed to cool to 40-45°C, isopropyl alcohol hydrochloride (200ml) was added till pH acidic and mixture was stirred for 2 hours to afford precipitate. The precipitate was filtered out and washed with isopropyl alcohol. The wet cake thus obtained was added to 20% aqueous sodium hydroxide solution (till pH basic) followed by addition of dichloromethane (500ml). The organic layer was separated from aqueous layer and was subjected to distillation under reduced pressure to obtain residue. The residue was taken in toluene (500ml), heated to 55-60°C for 30 minutes and cooled to 10-15°C. The precipitate obtained was filtered, washed with toluene and to the wet cake afforded was added isopropyl alcohol (665ml). The mixture was refluxed for 30 minutes and then cooled to 50-55°C. At 50-55°C slowly isopropyl alcohol hydrochloride (200ml) till pH acidic was added and mixture was stirred for 2 hours to obtain precipitate. The precipitate was filtered out, washed with isopropyl alcohol and dried to get (lR)-2-[[2-(4-aminophenyl)ethyl]amino]-l-phenylethanol dihydrochloride salt, of Formula (IV)
Yield-70%
HPLC Purity: 98%
Example 6: Alternate method for preparation of (2R)-2-hydroxy-N-[2-(4-nitrophenyl)ethyl]-2-phenylethanamide of Formula (IX)
Step I – A mixture of (R)-2-hydroxy-2-phenylacetic acid (lOg), dichloromethane (50ml) and triethylamine (24ml) was cooled to 0-5°C and slowly para-toluene sulfonyl chloride (12.53g) was added to it. The temperature of reaction mixture was raised to 25-30°C and maintained for 12 hours. After completion of reaction, water (100ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distills out completely under reduced pressure to obtain [(R)-2-hydroxy -2-phenyl acetic tosyl ester].
Yield-56%
Step II – 2-(4-nitrophenyl)ethylamine hydrochloride (6g) was added to dichloromethane (50ml) and stirred for 30 minutes at 25-30°C. The mixture was
then cooled to 0-5 °C and triethylamine (13ml) was added. To say cooled mixture was then slowly added a mixture of (R)-2-hydroxy -2-phenyl acetic tosyl ester (lOg) and dichloromethane (50ml). The temperature of reaction mixture was then raised to reflux temperature and maintained for 5 hours. After completion of reaction, water (50ml) was added to the reaction mixture and the mixture was stirred for 15 minutes. The organic phase was separated and distill out completely under reduced pressure to obtain (R)-2-hydroxy-N-[2-(4-nitrophenyl) ethyl]-2-phenylacetamide
Yield-70%, Purity-96%
Example 7: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -OH
1.58g of [2-(formylamino)-l,3-thiazol-4-yl]acetic acid of Formula (V) was added solution of (1R )-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) in water (2g of Formula (IV) in 50ml water) followed by addition of 0.66g concentrated hydrochloric acid and 3.27g of l-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride. The mixture was stirred at 25-30°C for 0.5 hours. After completion of reaction, pH was adjusted to 8-9 using aqueous saturated solution of sodium carbonate. The solid precipitated out was filtered, washed with water and dried to obtain 2.1g of compound of Formula (II). (Yield: 72%) The obtained compound has been identified by HNMR
2.502(m,4H),2.599(m,2H),3.685(S,2H),4.9(S, NH protons),7.01(m, 10H, aromatic), 8.54(S,1H), 10.0(S, -OH proton),
HNMR(D20 Exchange) 2.502(m,4H),2.60(m,2H),4.57(m,lH),7.0(m, 10H, aromatic), 8.43(S,1H)
Example 8: Preparation of compound of Formula (II) from compound of Formula (V); wherein R is -CI
lOg of ( 1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), was added to 150ml of acetonitrile with 16.17g of potassium carbonate and the mixture was cooled to 10-15°C. 18.8g of Formula (V) of example 1 was added to above mixture at 10-15°C in lot wise. After completion of reaction, the reaction mixture was concentrated under vacuum and 90ml of water was added for isolation. The product was then filtered out, washed with water and dried to obtain 72g (Yield: 70%) of compound of Formula (II).
Example 9: Preparation of compound of Formula (II) from compound of Formula (IlIa)
5.87g of compound of Formula (IlIa) was added to 40 ml of methylene dichloride with 2.36 g of potassium carbonate and 3.67g of ( 1))-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol (Formula-IV ; prepared by methods known in prior art/ as given in example 5) . The mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 10: Insitu preparation of compound of Formula (II) without isolation of compound of Formula (IlIa)
2g of p-nitrophenol was added to 40 ml of methylene chloride with 4.963g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of [2-(formylamino)-l,3-thiazol-4-yl]acetyl chloride of Formula (V) of example 1. After confirming complete formation of compound of Formula (Ilia), 2.36g of potassium carbonate and 3.67g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-1 -phenyl ethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, the reaction mixture was concentrated followed by addition of 60 ml of water to isolate lg of compound of Formula (II).
Example 11: Preparation of Mirabegron from compound of Formula (II)
To 2g of compound of Formula (II) was added 30ml of 10% sodium hydroxide and the mixture was stirred at 55-60°C for 3 hours. After completion of reaction, the mixture was cooled to 25-30°C and the solid obtained was filtered, washed with water and dried to yield 1.3g of Mirabegron. (Yield: 70%)
Example 12: Preparation of Mirabegron from compound of Formula (Illb)
6.18g of 4-nitrophenyl-(2-amino-l,3-thiazol-4-yl)acetate was added to 40ml of methylene dichloride with 2.36g of potassium carbonate and 3.65g of (1R)-2-{ [2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5), and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 13: Insitu preparation of Mirabegron without isolation of compound of Formula (Illb)
To 40ml of methylene chloride was added 2g of p-nitrophenol and 4.96g of potassium carbonate, and the mixture was cooled to 10-15°C followed by lot wise addition of 3.95g of compound of Formula (VI) prepared in example 3. After confirming complete formation of compound of Formula (Illb), 2.36g of potassium carbonate and 3.65g of (1R)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) (prepared by methods known in prior art/ as given in example 5) was added insitu, and the mixture was stirred at 25-30°C for 1 hour. After completion of reaction, After completion of reaction, solid was filtered out, washed with methylene dichlrode and dried to yield lg of Mirabegron of Formula (I).
Example 14: Preparation of Mirabegron from compound of Formula (VI); wherein R is -CI
To 20ml of acetone was added 2g of (l/?)-2-{[2-(4-aminophenyl)ethyl]amino}-l-phenylethanol of Formula (IV) and 2.15g of potassium carbonate, and the mixture was cooled to 10-15°C followed by addition of (2-amino-l,3-thiazol-4-yl)acetyl chloride of Formula (VI). After completion of reaction, acetone was concentrated under vacuum and 90ml of water was added for for isolation. The product was then filtered out, washed with water and dried to obtain 2g (Yield: 70%) of Mirabegron.



/////WO-2016024284, WO 2016024284, New Patent, MIRABEGRON, Wanbury Ltd
WO 2015177807, New patent on AVANAFIL by Wanbury
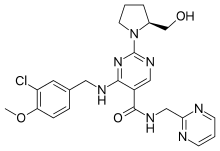

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd
![]()
The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group

It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)
m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)
CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
![]()
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
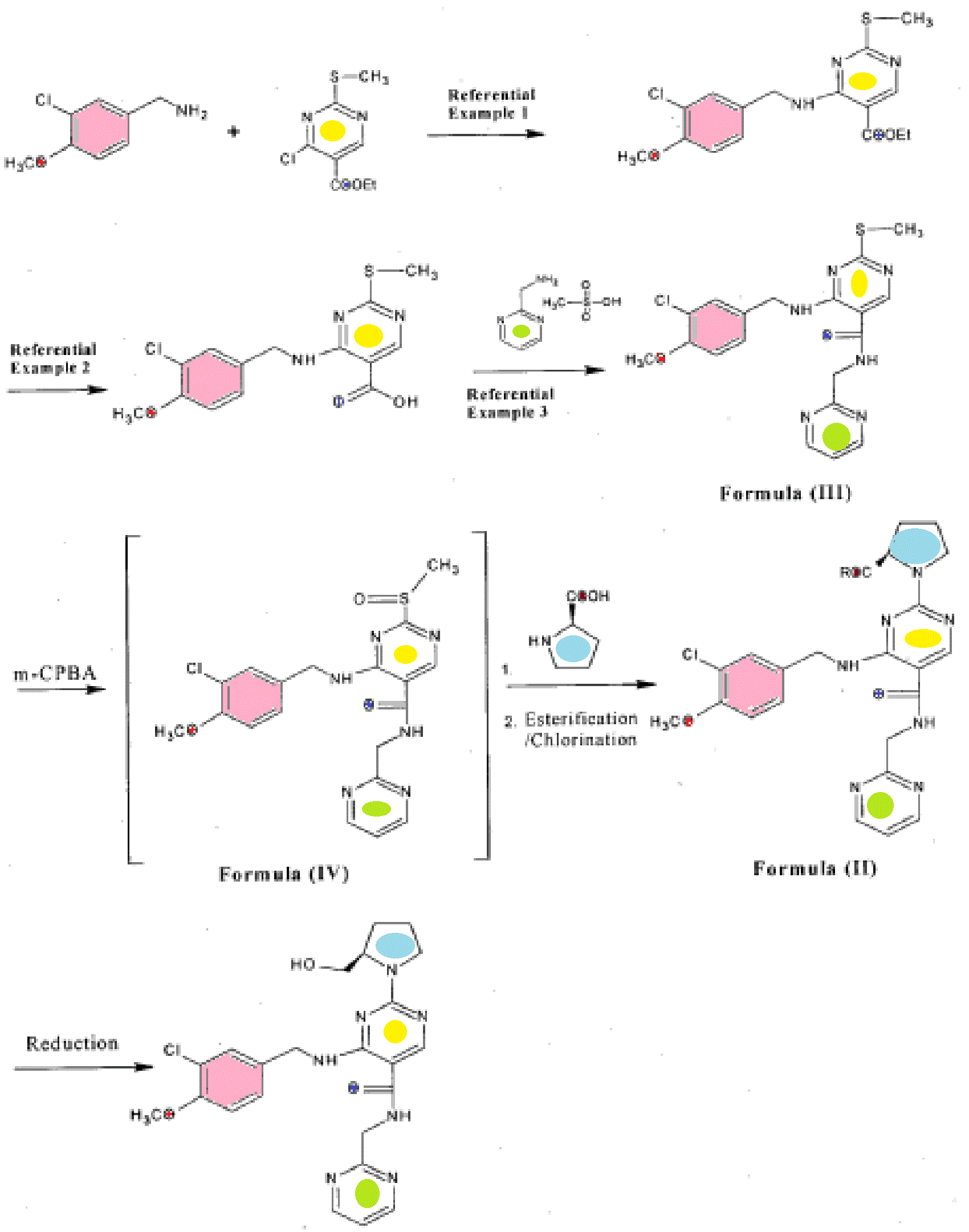
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.
![]()

| Mr. K. Chandran | ||
| Wholetime Director & Vice Chairman |
Tarapur plant

, Director, Wanbury, and Mr Asok Shinkar, Director-Corporate Finance, at a press conference held in Mumbai on Monday. Paul Noronha")
MR K. CHANDRAN (left), Director, Wanbury, and Mr Asok Shinkar, Director-Corporate Finance, at a press conference held in Mumbai on Monday.
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
//////////////
