
Home » Posts tagged 'Type 2 Diabetes Mellitus'
Tag Archives: Type 2 Diabetes Mellitus
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ebselen
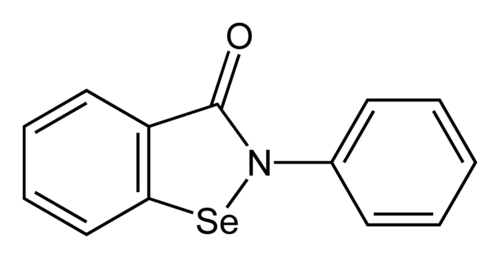
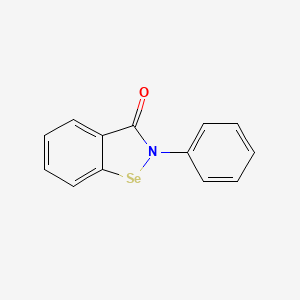
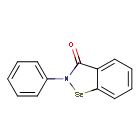
Ebselen
274.19 g/mol,
C13H9NOSe
2-phenyl-1,2-benzoselenazol-3-one
- CAS 60940-34-3
- 2-phenyl-1,2-benzoselenazol-3-one
- 2-Phenyl-1,2-benzisoselenazol-3(2H)-one
- Ebselene
- PZ 51, DR3305, and SPI-1005
- 40X2P7DPGH
Ebselen is a benzoselenazole that is 1,2-benzoselenazol-3-one carrying an additional phenyl substituent at position 2. Acts as a mimic of glutathione peroxidase. It has a role as a neuroprotective agent, an apoptosis inducer, an anti-inflammatory drug, an antioxidant, a hepatoprotective agent, a genotoxin, a radical scavenger, an enzyme mimic, an EC 1.3.1.8 [acyl-CoA dehydrogenase (NADP(+))] inhibitor, an EC 1.8.1.12 (trypanothione-disulfide reductase) inhibitor, an EC 1.13.11.33 (arachidonate 15-lipoxygenase) inhibitor, an EC 1.13.11.34 (arachidonate 5-lipoxygenase) inhibitor, an EC 2.5.1.7 (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) inhibitor, an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor, an EC 3.5.4.1 (cytosine deaminase) inhibitor, an EC 5.1.3.2 (UDP-glucose 4-epimerase) inhibitor, a ferroptosis inhibitor, an antifungal agent, an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor, an anticoronaviral agent, an antibacterial agent, an antineoplastic agent and an EC 3.1.3.25 (inositol–phosphate phosphatase) inhibitor.
Ebselen (also called PZ 51, DR3305, and SPI-1005), is a synthetic organoselenium molecule under preliminary investigation as a drug candidate.[1] It belongs to the class of compounds related to benzene and its derivatives.[1] It is being developed by the Seattle biotechnology company, Sound Pharmaceuticals, Inc.[1] It has also been reported to target tubulin, blocking its polymerization.[2]
Ebselen has been investigated for the treatment and basic science of Meniere’s Disease, Type 2 Diabetes Mellitus, and Type 1 Diabetes Mellitus.
Ebselen has been entered into clinical trials as a lead compound intended for the potential treatment of various diseases.[3] Its most advanced clinical trial is a Phase III study in people with Meniere’s disease, completed in July 2024.[4]
In vitro, ebselen is a mimic of glutathione peroxidase and reacts with peroxynitrite.[5] It is purported to have antioxidant and anti-inflammatory properties.[1][5]
Synthesis
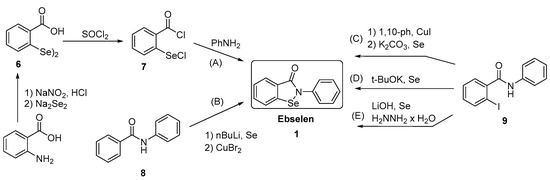
Generally, synthesis of the characteristic scaffold of ebselen, the benzoisoselenazolone ring system, can be achieved either through reaction of primary amines (RNH2) with 2-(chloroseleno)benzoyl chloride (Route I),[6] by ortho-lithiation of benzanilides followed by oxidative cyclization (Route II) mediated by cupric bromide (CuBr2),[7] or through the efficient Cu-catalyzed selenation / heterocyclization of o-halobenzamides, a methodology developed by Kumar et al.[8] (Route III).

SYN
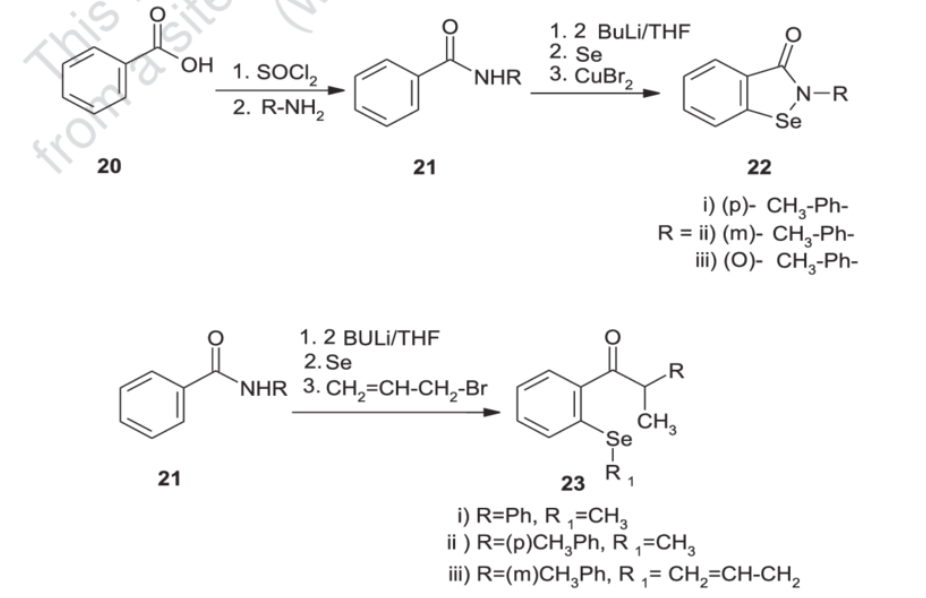
Synthesis of ebselen from benzoic acid by ortholithiation of benzanilide SOCl 2 =Thionyl chloride, R-NH 2 =Substituted aryl mine, BuLi/THF=n-butyllithium/ tetrahydrofuran, CuBr 2 =Cupper bromide, CH 2 =CH- CH 2 -Br = Allyl bromide.
SYN
New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential
Molecules, March 2017, 22(3):492
SYN
https://pubs.acs.org/doi/10.1021/ol102027j
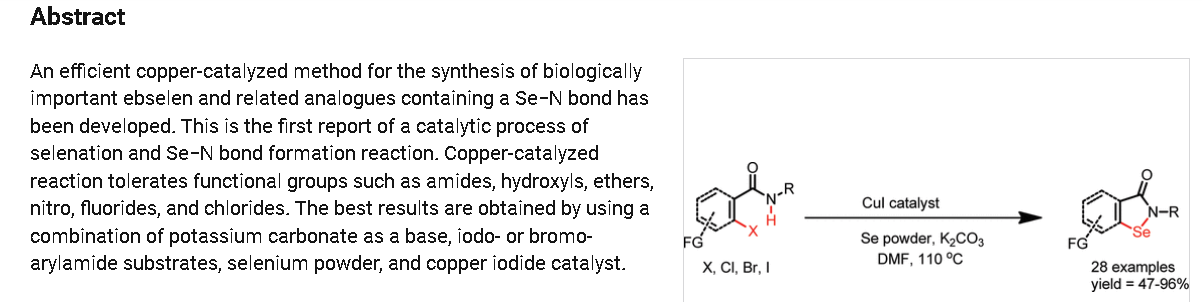

2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide (Typical
Procedure): Copper iodide (114 mg, 0.6 mmol) and 1,10-phenanthroline (108 mg, 0.6 mmol)
were added into DMF (3 mL) in a single neck flask. Resulted brownish solution was stirred for
15 min and then 2-iodo-N-phenylbenzamide1 (0.97 g, 3.0 mmol), selenium powder (0.29 g, 3.6
mmol), and potassium carbonate powder (0.65 g, 4.7 mmol) were added sequentially to same reaction flask. Brown colored reaction mixture was refluxed at 110oC using refluxing condenser
under nitrogen atmosphere. Progress of reaction was monitored by TLC. Reaction mixture was
refluxed for 8h. After this, reaction mixture poured over brine solution (60 mL) and stirred for 3
h. Product was precipitated as white solid which was collected by filtration over Buchner funnel,
product was washed with water (15 mL x 2), dried in air, dissolved in ethyl acetate, concentrated
over rotary evaporator, resulted brown solid which was purified by column chromatography
using hexane/ ethyl acetate (8:2) over silica gel. Yield 0.69 g (84%), mp 182-183 °C (180-181
°C).14,15 1H NMR (400 MHz, DMSO-d6) 8.09 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H),
7.71-7.62 (m, 3H), 7.51-7.43 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H). 1H NMR (400 MHz, CDCl3)
8.12 (d, 7.6 Hz, 1H), 7.68-7.62 (m, 4H), 7.52-7.41 (m, 3H), 7.29 (m, 1H). IR (plate): 3057, 2921,
1598, 1443, 1346, 1263, 1028 cm-1; ESMS m/z: 276 (M+H+).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide at 74 mmol
scale: Reaction was carried out at 74 mmol scale using 2-iodo-N-phenylbenzamide (24.00 g,
74.3 mmol), selenium powder (7.04 g, 89.1 mmol), CuI (2.83 g, 14.9 mmol), 1,10
phenanthroline (2.69 g, 14.9 mmol), and anhydrous potassium carbonate powder (15.40 g, 111.4
mmol) in DMF (50 mL) and procedure and workup followed are similar to 3.6 mmol scale
reaction. Yield 16.28 g (80%), Figure S1.
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Bromo-N-phenylbenzamide: Ebselen 1
was prepared from 2-bromo-N-phenylbenzamide2 (1.00 g, 3.6 mmol), selenium powder (0.34 g,
4.3 mmol), K2CO3 powder (0.74 g, 5.4 mmol), CuI (137 mg, 0.7 mmol), and 1,10-phenanthroline
(130 mg, 0.7 mmol) in DMF (3 mL). Reaction mixture was refluxed for 16 h at 110oC. Progress of reaction was monitored by TLC. After completion of reaction, mixture was poured into brine
solution (60 mL) and the resulted white precipitate was washed with water (20 mL x 2), and
dried in air. Purification by column chromatography on silica gel using CH2Cl2 provided white
crystalline solid (0.77 g, 78%).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Chloro-N-phenylbenzamide: Reaction
was carried out at 4 mmol scale using 2-chloro-N-phenylbenzamide3 (1.00 g, 4.3 mmol), CuI
(172 mg, 0.9 mmol), 1,10-phenanthroline (162 mg, 0.9 mmol), selenium powder (0.41 g, 5.2
mmol), K2CO3 (0.89 g, 6.4 mmol) in DMF (4 mL). Reaction mixture was refluxed for 24 h at
110oC. Workup procedure is similar as followed for bromo substrate. Yield 0.55 g (47%).
History
The first patent for 2-phenyl-1,2-benzoselenazol-3(2H)-one was filed in 1980 and granted in 1982.[9]
Research
Ebselen is in preliminary clinical development for the potential treatment of hearing loss and depression, among other medical indications.[3][10]



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Ebselen”. DrugBank. 29 January 2025. Retrieved 4 February 2025.
- Baksheeva VE, La Rocca R, Allegro D, Derviaux C, Pasquier E, Roche P, Morelli X, Devred F, Golovin AV, Tsvetkov PO (2025). “NanoDSF Screening for Anti-tubulin Agents Uncovers New Structure–Activity Insights”. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.5c01008.
- “Ebselen pipeline”. Sound Pharmaceuticals, Inc. 2025. Retrieved 4 February 2025.
- “SPI-1005 for the Treatment of Meniere’s Disease (STOPMD-3)”. ClinicalTrials.gov, US National Library of Medicine. 1 August 2024. Retrieved 4 February 2025.
- Schewe T (October 1995). “Molecular actions of ebselen – an antiinflammatory antioxidant”. General Pharmacology. 26 (6): 1153–69. doi:10.1016/0306-3623(95)00003-J. PMID 7590103.
- Kamigata N, Iizuka H, Izuoka A, Kobayashi M (July 1986). “Photochemical Reaction of 2-Aryl-1, 2-benzisoselenazol-3 (2 H)-ones”. Bulletin of the Chemical Society of Japan. 59 (7): 2179–83. doi:10.1246/bcsj.59.2179.
- Engman L, Hallberg A (1989-06-01). “Expedient synthesis of ebselen and related compounds”. The Journal of Organic Chemistry. 54 (12): 2964–2966. doi:10.1021/jo00273a035. ISSN 0022-3263.
- Balkrishna SJ, Bhakuni BS, Chopra D, Kumar S (December 2010). “Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles”. Organic Letters. 12 (23): 5394–7. doi:10.1021/ol102027j. PMID 21053969.
- DE3027073A1, Etschenberg, Eugen Dr; Renson, Marcel Prof Dipl-Chem Jupille & Winkelmann, Johannes Dr 5000 Köln, “2-phenyl-1,2-benzisoselenazol-3(2h)-on enthaltende pharmazeutische praeparate und ihre verwendung”, issued 1982-02-18
- “Ebselen search: list of clinical trials sponsored by Sound Pharmaceuticals”. ClinicalTrials.gov, US National Library of Medicine. 2025. Retrieved 4 February 2025.
External links
| Names | |
|---|---|
| Preferred IUPAC name2-Phenyl-1,2-benzoselenazol-3(2H)-one | |
| Identifiers | |
| CAS Number | 60940-34-3 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:77543 |
| ChEMBL | ChEMBL51085 |
| ChemSpider | 3082 |
| ECHA InfoCard | 100.132.190 |
| PubChem CID | 3194 |
| UNII | 40X2P7DPGH |
| CompTox Dashboard (EPA) | DTXSID7045150 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C13H9NOSe |
| Molar mass | 274.17666 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
////////Ebselen, Ebselene, PZ 51, DR 3305, SPI 1005, PHASE 3, 40X2P7DPGH, Meniere’s Disease, Type 2 Diabetes Mellitus, Type 1 Diabetes Mellitus
TOFOGLIFLOZIN 托格列净

TOFOGLIFLOZIN
托格列净
CSG-452, R-7201, RG-7201
903565-83-3 (anhydrous)
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol hydrate (1:1)
PMDA Pharmaceuticals and Medical Devices Agency, Japan Approved mar24, 2014
THERAPEUTIC CLAIM Treatment of diabetes mellitus
CHEMICAL NAMES
1. Spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol, 6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-, hydrate (1:1), (1S,3’R,4’S,5’S,6’R)-
2. (1S,3’R,4’S,5’S,6’R)-6-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol monohydrate
3. (1S,3’R,4’S,5’S,6’R)-6-[(4-ethylphenyl)methyl]-3′,4′,5′,6′-tetrahydro-6′-(hydroxymethyl)-
spiro[isobenzofuran-1(3H),2′-[2H]pyran]-3′,4′,5′-triol monohydrate
(3S,3’R,4’S,5’S,6’R)-5-[(4-ethylphenyl)methyl]-6′-(hydroxymethyl)spiro[1H-2-benzofuran-3,2′-oxane]-3′,4′,5′-triol;hydrate
MW404.5, MF C22H26O6
INNOVATOR Chugai Pharmaceuticals
Sanofi, kowa
Deberza®………..KOWA/Apleway®……………SANOFI
CODE DESIGNATION CSG 452
Tofogliflozin (USAN, codenamed CSG452) is an experimental drug for the treatment of diabetes mellitus and is being developed byChugai Pharma in collaboration with Kowa and Sanofi.[1] It is an inhibitor of subtype 2 sodium-glucose transport protein (SGLT2), which is responsible for at least 90% of the glucose reabsorption in the kidney. As of September 2012, the drug is in Phase III clinical trials.[2][3]
Tofogliflozin is an SGLT-2 inhibitor first launched in 2014 in Japan by Sanofi and Kowa for the oral treatment of type II diabetes.
The product was discovered by Chugai and was licensed to Roche in 2007. In 2011, this license agreement was terminated. In 2012, the product was licensed to Kowa and Sanofi by Chugai Pharmaceutical in Japan for the treatment of diabetes type 2. In 2015, the license between Kowa and Chugai was expanded for developments and marketing of the agent in the U.S. and the E.U.
Chemistry
The active moiety or anhydrous form (ChemSpider ID: 28530778, CHEMBL2110731) has the chemical formula C22H26O6 and amolecular mass of 386.44 g/mol.
The United States Adopted Name tofogliflozin applies to the monohydrate, which is the form used as a drug.[4] The International Nonproprietary Name tofogliflozin applies to the anhydrous compound[5] and the drug form is referred to as tofogliflozin hydrate.

Several drugs are available for the treatment of type 2 diabetes mellitus (T2DM), but few patients achieve and maintain glycaemic control without weight gain and hypoglycaemias. Sodium glucose co-transporter 2 (SGLT-2) inhibitors are an emerging class of drugs with an original mechanism of action involving inhibition of renal glucose reabsorption. Two agents of this class, dapagliflozin and canagliflozin, have already been approved, although we need more data on cardiovascular outcomes along with bladder and breast cancer. Tofogliflozin is a further SGLT-2 inhibitor, which exhibits the highest selectivity for SGLT-2, the most potent antidiabetic action and a reduced risk of hypoglycaemia. Recently, a 52-week, multicentre, open-label, randomised controlled trial in Japanese T2DM patients has shown that tofogliflozin exhibits adequate safety and efficacy as monotherapy or as add-on treatment in patients suboptimally controlled with oral agents. Despite the very promising characteristics of this new drug, important questions remain to be answered, mainly additional data on safety outcomes and potential beneficial effects of tofogliflozin, for instance in prediabetes and diabetic nephropathy. Moreover, it would be welcome to examine the utility of its therapeutic use in combination with insulin and metformin.
Tofogliflozin has recently demonstrated safety and efficacy as monotherapy or add-on treatment . This is very important, granted our expectations of SGLT-2 inhibitors as useful alternative oral hypoglycaemic agents. Although important questions remain to be answered, the results of the new trial add to the importance of SGLT-2 inhibitors as a useful new class of oral hypoglycaemic agents.
CLIP
There are two scalable synthetic routes reported to prepare tofogliflozin.2 An efficient production synthesis of tofogliflozin hydrate from alcohol 2 was first described by Murakata et al. (Scheme 1, route 1).2a In 2016, Ohtake et al. reported an improved synthetic route, which achieved in just 7 linear steps (Scheme 1, route 2).2b They selected the optimal protecting groups for the purpose of chemoselective activation and crystalline purification, and obtained the pure tofogliflozin in a good overall yield. However, these methods suffer from several drawbacks. Firstly, some reagents, such as BH3 (Scheme 1, route 2) and 2-Methoxyproene (3, Scheme 1), are toxic or highly volatile. Meanwhile, the use of Palladium reagents may lead to an excess of residual heavy metal in the final product. Secondly, manufacturing costs in these methods are high due to the application of expensive raw materials and reagents. Last but not least, the key tactical stages that involve Br/Li exchange of aryl bromide followed by addition to gluconolactone 5 need the cryogenic conditions (< -60 oC), and this method is not suitable for industrial production. Herein, we report a newly developed synthetic method for tofogliflozin hydrate starting from readily available raw materials and affording good overall yield.
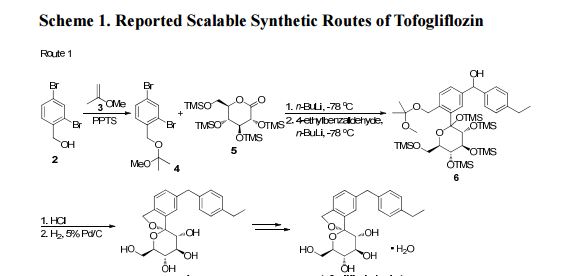
SCHEME 2 FOR

2. (a) Murakata, M.; Ikeda, T.; Kimura, N.; Kawase, A.; Nagase, M.; Yamamoto, K.; Takata, N.; Yoshizaki, S.; Takano, K. Crystal of spiroketal derivative, and process for production thereof. European Appl. EP 2308886 A1, April 13, 2011. (b) Ohtake, Y.; Emura, T.; Nishimoto, M.; Takano, K.; Yamamoto, K.; Tsuchiya, S.; Yeu, S.; Kito, Y.; Kimura, N.; Takeda, S.; Tsukazaki, M.; Murakata, M.; Sato, T. J. Org. Chem. 2016, 81, 2148.

CLIP
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
(1S,3′R,4′S,5′S,6′R)-6-[(4-Ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′- pyran]-3′,4′,5′-triol (1, tofogliflozin).
To a solution of 17b (89.9 g, 145 mmol) in DME (653 mL) and MeOH (73.0 mL), 2 N NaOH aq. solution (726 mL, 1.45 mol) was added dropwise for 1 h at waterbath temperature. After stirring at rt for 1 h, 2 N H2SO4 aq. solution (436 mL) was added slowly to the mixture. Water (700 mL) was added to the mixture, and the resultant mixture was extracted with AcOEt (500 mL × 2). The resultant organic layer was washed with brine (1.00 L) and then dried over anhydrous Na2SO4 (250 g). The mixture was concentrated in vacuo to obtain 1 (57.3 g, quant) as a colorless amorphous solid;
[α]D 26 +24.2° (c 1.02, MeOH);
1 H NMR (400 MHz, CD3OD) δ: 1.19 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42−3.47 (1H, m), 3.63−3.67 (1H, m), 3.75−3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.5 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07−7.14 (4H, m), 7.17−7.23 (3H, m);
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2;
MS (ESI) m/z: 387 [M + H]+ ; HRMS (ESI) calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
SGLT2 inhibitors inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with type 2 diabetes. Glucopyranosyl-substituted benzene derivative are described in the prior art as SGLT2 inhibitors, for example in
WO 01/27128, WO 03/099836, WO 2005/092877, WO 2006/034489,
WO 2006/064033, WO 2006/117359, WO 2006/117360,
WO 2007/025943, WO 2007/028814, WO 2007/031548,
WO 2007/093610, WO 2007/128749, WO 2008/049923, WO 2008/055870, WO 2008/055940.
PATENTS
Papers
Chinese Chemical Letters, 2013 , vol. 24, 2 pg. 131 – 133
Journal of Medicinal Chemistry, 2012 , vol. 55, 17 pg. 7828 – 7840
NMR
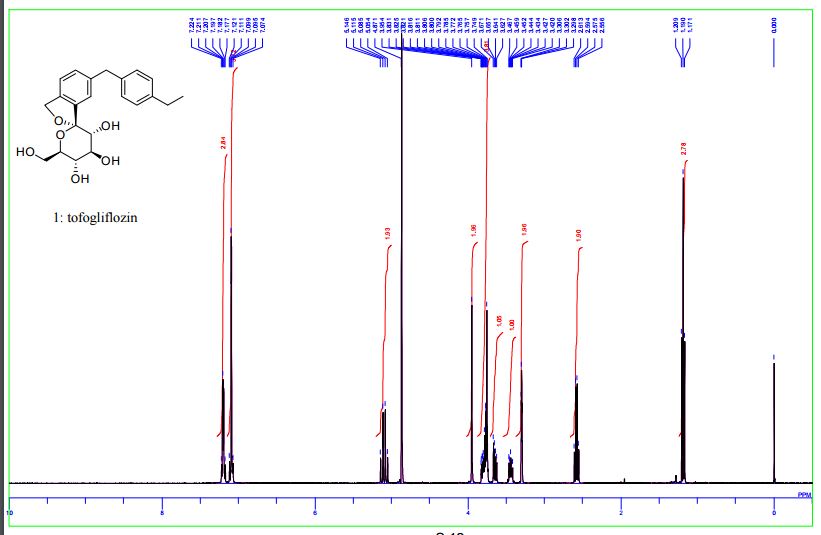
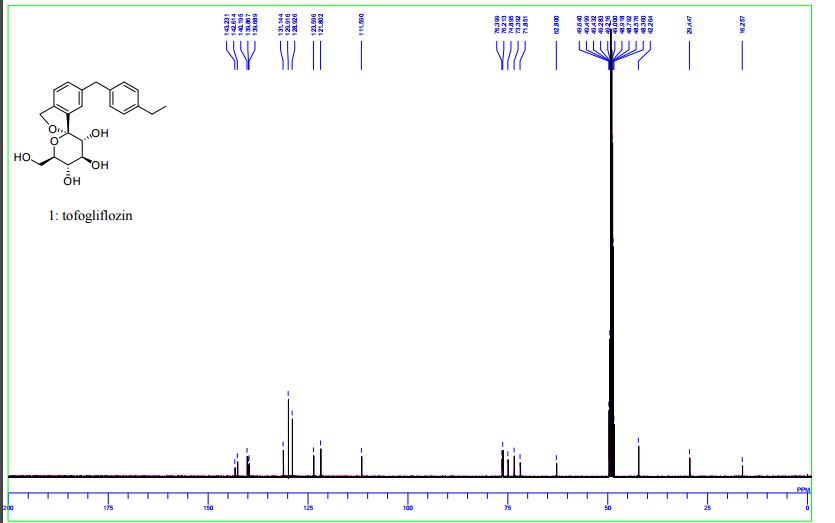

1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
Second set
http://pubs.acs.org/doi/full/10.1021/jm300884k
J. Med. Chem., 2012, 55 (17), pp 7828–7840
1H NMR (400 MHz, CD3OD) δ: 1.20 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42–3.47 (1H, m), 3.63–3.67 (1H, m), 3.75–3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.3 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07–7.14 (4H, m), 7.17–7.23 (3H, m).
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2.
MS (ESI): 387 [M + H]+. HRMS (ESI), m/z calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801.
THIRD SET
(1S,3′R,4′S,5′S,6′R)-6-[(4-Ethylphenyl)methyl]-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′- pyran]-3′,4′,5′-triol (1, tofogliflozin).
To a solution of 17b (89.9 g, 145 mmol) in DME (653 mL) and MeOH (73.0 mL), 2 N NaOH aq. solution (726 mL, 1.45 mol) was added dropwise for 1 h at waterbath temperature. After stirring at rt for 1 h, 2 N H2SO4 aq. solution (436 mL) was added slowly to the mixture. Water (700 mL) was added to the mixture, and the resultant mixture was extracted with AcOEt (500 mL × 2). The resultant organic layer was washed with brine (1.00 L) and then dried over anhydrous Na2SO4 (250 g). The mixture was concentrated in vacuo to obtain 1 (57.3 g, quant) as a colorless amorphous solid;
[α]D 26 +24.2° (c 1.02, MeOH);
1 H NMR (400 MHz, CD3OD) δ: 1.19 (3H, t, J = 7.6 Hz), 2.58 (2H, q, J = 7.6 Hz), 3.42−3.47 (1H, m), 3.63−3.67 (1H, m), 3.75−3.88 (4H, m), 3.95 (2H, s), 5.06 (1H, d, J = 12.5 Hz), 5.12 (1H, d, J = 12.5 Hz), 7.07−7.14 (4H, m), 7.17−7.23 (3H, m);
13C NMR (100 MHz, CD3OD) δ: 16.3, 29.4, 42.3, 62.8, 71.9, 73.4, 74.9, 76.2, 76.4, 111.6, 121.8, 123.6, 128.9, 129.9, 131.1, 139.7, 139.9, 140.2, 142.6, 143.2;
MS (ESI) m/z: 387 [M + H]+ ; HRMS (ESI) calcd for C22H27O6 [M + H]+ 387.1802, found 387.1801
DOI: 10.1021/acs.joc.5b02734 J. Org. Chem. 2016, 81, 2148−2153
Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn, K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828−7840
PATENT
Prepn
[Example 1] (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro- -6′-(hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran] -3 ‘, 4′, one of the preparation step [compound of formula (IX)] 5’-triol Preparation of methanol (2 – hydroxymethyl-phenyl – bromo-4)

To the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.1kg) in – bromoterephthalic was added at below 30 ℃ solution (7.5kg, 30.6mol) of the acid, and the mixture was stirred for 1 hour at 25 ℃. Then cooled to 19 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). In addition to methanol (15.0kg) in the mixture was kept for a while.
Again, to the mixing solution (1mol / L, 78.9kg, 88.4mol) of borane-tetrahydrofuran complex in tetrahydrofuran (6.34kg, 61.0mol) and, trimethoxyborane, two tetrahydrofuran (33.0kg) in – was added at below 30 ℃ solution (7.5kg, 30.6mol) of bromo terephthalic acid, and the reaction was carried out for 1 hour at 25 ℃. Then cooled to 18 ℃ The reaction mixture was stirred for 30 minutes and added a mixed solution of tetrahydrofuran and methanol (3.0kg) of (5.6kg). After addition of methanol (15.0kg) in the mixture is combined with the reaction mixture obtained in the previous reaction, and then the solvent was distilled off under reduced pressure. After addition of methanol (36kg) residue was obtained, and the solvent was evaporated under reduced pressure. Furthermore, (54 ℃ dissolved upon confirmation) which was dissolved by warming was added to methanol (36kg) to the residue. After cooling to room temperature the solution was stirred for 30 minutes added water (60kg). After addition of water (165kg) In addition to this mixture was cooled to 0 ℃, and the mixture was stirred for one hour. Centrifuge the obtained crystals were washed twice with water (45kg), and dried for 2 hours under reduced pressure to give (11.8kg, 54.4mol, 89% yield) of the title compound.
1 H-NMR (DMSO-d 6) δ: 4.49 (4H, t, J = 5.8Hz), 5.27 (1H, t, J = 5.8Hz), 5.38 (1H, t, J = 5.8Hz), 7.31 (1H, d, J = 7.5Hz), 7.47 (1H, d, J = 7.5Hz), 7.50 (1H, s).
Preparation of benzene (ethoxy methyl – methyl – – methoxy-1 1) – bromo-1 ,4 – 2:2 process bis

(- Bromo-4 – 2-hydroxyethyl methyl phenyl) in tetrahydrofuran (57kg) in the solution (8.0kg, 36.9mol) of methanol, I added (185.12g, 0.74mol) of pyridinium p-toluenesulfonate. After cooling to -15 ℃ below the mixture, 2 – was added at -15 ℃ or less (7.70kg, 106.8mol) methoxy propene, and the mixture was stirred 1 h at -15 ~ 0 ℃. Was added aqueous potassium carbonate (25 wt%, 40kg) and the reaction mixture was warmed to room temperature and separate the organic layer was added toluene (35kg). After washing with water (40kg) The organic layer was evaporated under reduced pressure. Was dissolved in toluene (28kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 1.42 (6H, s), 1.45 (6H, s), 3.24 (3H, s), 3.25 (3H, s), 4.45 ( 2H, s), 4.53 (2H, s), 7.28 (1H, dd, J = 1.5,8.0 Hz), 7.50 (1H, d, J = 8.0Hz), 7. 54 (1H, d, J = 1.5Hz).
MS (ESI +): 362 [M +2] +.
Preparation of on – (3R, 4S, 5R, 6R) -3,4,5 – tris (trimethylsilyloxy)-6 – trimethylsilyloxy methyl – tetrahydropyran-2: Step 3

Glucono -1,5 – – D-(+) in tetrahydrofuran (70kg) in the solution (35.8kg, 353.9mol) of N-methylmorpholine (7.88kg, 44.23mol) and lactone, chlorotrimethylsilane ( was added at 40 ℃ less 29.1kg, and 267.9mol), and the mixture was stirred for 2 hours at 30 ~ 40 ℃ resulting mixture. Was cooled to 0 ℃ the reaction mixture was added toluene (34kg) water (39kg), and the organic layer was separated. Twice sodium dihydrogen phosphate aqueous solution (5 wt%, 39.56kg) in, washed once with water (39kg) the organic layer the solvent was evaporated under reduced pressure. Was dissolved in toluene (34.6kg) and the residue obtained was obtained as a toluene solution of the title compound.
1 H-NMR (CDCl 3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74- 3.83 (3H, m), 3.90 (1H, t, J = 8.0Hz), 3.99 (1H, d, J = 8.0Hz), 4.17 (1H, dt, J = 2 .5,8.0 Hz).
Step 4: (1S, 3’R, 4’S, 5’S, 6’R) -3 ‘, 4’, 5 ‘, 6′-tetrahydro -6,6′ – bis (hydroxymethyl) – spiro [ (3H), 2’-[2H] pyran] -3 ‘, 4′, 5’-Preparation of triol isobenzofuran-1

(Methyl – – – methoxy 1-ethoxy-methyl) – bromo-1 ,4 – 2 prepared in step 2 bis cooled to below -10 ℃ toluene solution of benzene, hexane solution to (15 wt% n-butyl lithium , was added at below 0 ℃ 18.2kg, and 42.61mol), and the mixture was stirred 1.5 h at 5 ℃ resulting mixture. (10.5kg, 40.7mol), was added tetrahydrofuran (33.4kg) then magnesium bromide diethyl ether complex in the mixture, and the mixture was stirred for 1 hour at 25 ℃. Was added at below -10 ℃ toluene solution of the on – tris (trimethylsilyloxy) -6 – – 3,4,5 cooled to -15 ℃ below the mixture prepared in step 3 trimethylsilyloxy methyl – tetrahydropyran-2 was. After stirring 0.5 h at -15 ℃ or less, poured into 20% aqueous ammonium chloride solution to (80kg) of this solution, and the organic layer was separated. After washing with water (80kg) and the organic layer obtained, and the solvent was evaporated under reduced pressure. I was dissolved in methanol (43kg) residue was obtained. Was stirred for 1 hour at 20 ℃ was added (1.4kg, 7.4mol) and p-toluenesulfonic acid monohydrate in the mixture. Thereafter, it was stirred for another hour and cooled to 0 ℃, centrifuged crystals obtained was washed with methanol (25kg), and dried for 8 hours at reduced pressure under 40 ℃, (5.47kg, yield the title compound I got 50%) rate.
1 H-NMR (DMSO-d 6) δ :3.20-3 .25 (1H, m) ,3.41-3 .45 (1H, m) ,3.51-3 .62 (4H, m) , 4.39 (1H, t, J = 6.0Hz) ,4.52-4 .54 (3H, m), 4.86 (1H, d, J = 4.5Hz), 4.93 (1H, d, J = 5.5Hz), 4.99 (1H, d, J = 12.5Hz), 5.03 (1H, d, J = 12.5Hz), 5.23 (1H, t, J = 5 .8 Hz) ,7.24-7 .25 (2H, m), 7.29 (1H, dd, J = 1.5,8.0 Hz).
Step 5: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(methoxycarbonyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro-3’ , 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2’-[2H] pyran isobenzofuran] spiro

(1S, 3’R, 4’S, 5’S, 6’R) – tetrahydro -6,6 ‘- bis (hydroxymethyl) – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran ] -3 ‘, 4′, 5’-triol 4 (5.3kg, 17.8mol) and – dissolved in acetonitrile (35kg) (13.7kg, 112.1mol) a chloroformate, in the solution of dimethylaminopyridine I was added at 12 ℃ or less (10.01kg, 105.9mol) methyl. Heated to 20 ℃, After stirring for 1 h, was added ethyl acetate (40kg) and water (45kg), and the organic layer was separated and the mixture. Once (45.4kg) aqueous solution consisting of (9.01kg) sodium chloride and potassium hydrogen sulfate (1.35kg), sodium chloride aqueous solution (weight 10%, 44.5kg), sodium chloride aqueous solution (the organic layer was washed successively 20% by weight, in 45.0kg), and the solvent was evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (18kg) and the residue obtained was then evaporated under reduced pressure. Was dissolved in ethylene glycol dimethyl ether (13.2kg) again and the residue obtained was obtained as ethylene glycol dimethyl ether solution of the title compound. I was used as it was in the six step.
1 H-NMR (CDCl 3) δ: 3.54 (3H, s), 3.77 (6H, s), 3.811 (3H, s), 3.812 (3H, s), 4.23 ( 1H, dd, J = 2.8,11.9 Hz), 4.32 (1H, dd, J = 4.0,11.9 Hz) ,4.36-4 .40 (1H, m), 5.11 -5.24 (5H, m), 5.41 (1H, d, J = 9.8Hz), 5.51 (1H, t, J = 9.8Hz), 7.25 (1H, d, J = 7.5Hz), 7.42 (1H, d, J = 7.5Hz), 7.44 (1H, s).
MS (ESI +): 589 [M +1] +, 606 [M +18] +.
Step 6: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro-3 ‘4’, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – Preparation of [(3H), 2′-[2H] pyran isobenzofuran] spiro

[(Methoxycarbonyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro – (1S, 3’R, 4’S, 5’S, 6’R) -6 which had been prepared in Step 5 – 3 ‘, 4′, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – spiro [isobenzofuran -1 (3H), 2’-[2H] pyran] Ethylene glycol dimethyl ether in solution, 2 – (2.46kg, 17.8mol), 4 butanol (25kg), anhydrous potassium carbonate – – methyl-2 were sequentially added (3.73kg, 24.9mol) ethyl phenyl boronic acid, in the reaction vessel was replaced with argon atmosphere, was bubbled with argon mixture. To the mixture – after the addition (0.72kg, 0.88mol) and palladium (II) chloride dichloromethane adduct [1,1 ‘-bis (diphenylphosphino) ferrocene], it was replaced with argon again inside of the vessel, one at 80 ℃ I was stirring time. After cooling, I added sequentially (0.859kg, 5.3mol) of ethylene glycol dimethyl ether (9.85kg), ethyl acetate (19kg), N-acetyl-L-cysteine in the mixture. After stirring for 2.5 h the mixture was filtered and added Celite (5.22kg), and washed with ethyl acetate (78kg) and the filter residue. The combined washings and filtrate, and the solvent is evaporated off under reduced pressure, and in addition (0.58kg, 3.6mol) and ethanol (74kg), N-acetyl-L-cysteine residue was obtained, which is heated to 70 ℃ or I was dissolved residue is then. After addition of water (9.4kg) in the solution, cooled to 60 ℃, and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more The mixture was stirred for 1 hour or more at 5 ℃ less. Centrifuge the resulting solid was washed twice with a mixture of water (35kg) and ethanol (55kg). Was dissolved at 70 ℃ ethanol (77kg) again, wet powder was obtained (10.21kg), cooled to 60 ℃ added water (9.7kg), and the mixture was stirred for 1 h. After confirming solid precipitated, cooled to 0 ℃ from 60 ℃ over 2.5 hours or more, and the mixture was stirred for 1 hour or more at 5 ℃ less. (9.45kg, dry powder rate 8.47kg, 13.7mol which was centrifuged obtained crystals were washed with a mixture of water (32kg) and ethanol (51kg), was obtained as a moist powder the title compound, 77% overall yield from the previous step).
1 H-NMR (CDCl 3) δ: 1.20 (3H, t, J = 7.5Hz), 2.60 (2H, q, J = 7.5Hz), 3.50 (3H, s), 3 .76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J = 2.8,11 .9 Hz), 4.33 (1H, dd, J = 4.5,11.9 Hz) ,4.36-4 .40 (1H, m) ,5.11-5 .20 (3H, m), 5 .41 (1H, d, J = 10.0Hz), 5.51 (1H, t, J = 10.0Hz) ,7.07-7 .11 (4H, m), 7.14 (1H, d, J = 7.8Hz), 7.19 (1H, dd, J = 1.5,7.8 Hz), 7.31 (1H, d, J = 1.5Hz).
MS (ESI +): 619 [M +1] +, 636 [M +18] +.
Step 7: (1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6’-tetrahydro-6 , 4 ‘, 5′-Preparation of triol’ – -3 [(3H), 2′-[2H] pyran isobenzofuran] spiro – (hydroxymethyl) ‘

(1S, 3’R, 4’S, 5’S, 6’R) -6 – [(4 – ethyl-phenyl) methyl] -3 ‘, 4’, 5 ‘, 6′-tetrahydro-3’, 4 ‘, 5′-tris (methoxycarbonyl) oxy-6′-[(methoxycarbonyl) methyl] – wet powder spiro [(3H), 2’-[2H] pyran isobenzofuran -1] (8.92kg, In addition at 20 ℃ (4mol / L, 30.02kg, the 104.2mol) aqueous solution of sodium hydroxide, 1 hour the reaction mixture to a solution of (28kg) ethylene glycol dimethyl ether dry end conversion 8.00kg, of 12.9mol) the mixture was stirred. And the organic layer was separated by addition of water (8.0kg) in the mixture. The ethyl acetate aqueous sodium chloride solution (25 wt%, 40kg) and a (36kg) in the organic layer and the aqueous layer was removed after washing. The washed again aqueous sodium chloride solution (25 wt%, 40kg) in the organic layer was evaporated under reduced pressure. Were added and acetone (32.0kg) water (0.8kg) residue was obtained. After the solvent was evaporated under reduced pressure, dissolved in acetone (11.7kg) in water (15.8kg) and the residue obtained was cooled to below 5 ℃. Was added below 10 ℃ water (64kg) to the mixture, and the mixture was stirred for 1 hour at below 10 ℃. Centrifuge the resulting crystals were washed with a mixture of water (8.0kg) and (1.3kg) acetone. For 8 hours through-flow drying 13 ~ 16 ℃ temperature ventilation, under the conditions of 24-33% relative humidity the wet powder, the monohydrate crystal (3.94kg, 9.7mol, 75% yield) of the title compound I was obtained as: (4.502 wt% water content).
Method of measuring the amount of water:
Analysis: coulometric KF titration analyzer: trace moisture measurement device manufactured by Mitsubishi Chemical Corporation Model KF-100
Anolyte: Aqua micron AX (manufactured by Mitsubishi Chemical Corporation)
Catholyte: Aqua micron CXU (manufactured by Mitsubishi Chemical Corporation)
1 H-NMR (CD 3 OD) δ: 1.19 (3H, t, J = 7.5Hz), 2.59 (2H, q, J = 7.5Hz) ,3.42-3 .46 (1H , m), 3.65 (1H, dd, J = 5.5,12.0 Hz) ,3.74-3 .82 (4H, m), 3.96 (2H, s), 5.07 (1H , d, J = 12.8Hz), 5.13 (1H, d, J = 12.8Hz) ,7.08-7 .12 (4H, m) ,7.18-7 .23 (3H, m) .
MS (ESI +): 387 [M +1] +.
PATENT
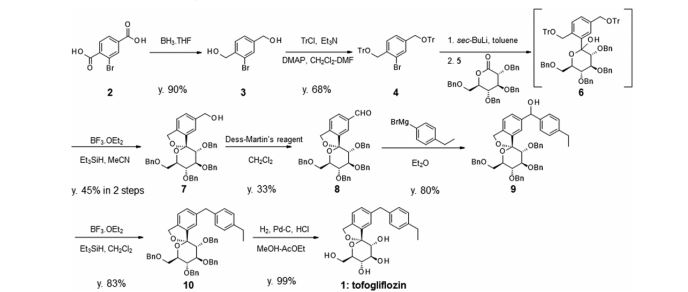
Example 1 Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose Step 1: Synthesis of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one

To a solution of D-(+)-glucono-1,5-lactone (7.88 kg) and N-methylmorpholine (35.8 kg) in tetrahydrofuran (70 kg) was added trimethylsilyl chloride (29.1 kg) at 40° C. or below, and then the mixture was stirred at a temperature from 30° C. to 40° C. for 2 hours. After the mixture was cooled to 0° C., toluene (34 kg) and water (39 kg) were added thereto. The organic layer was separated and washed with an aqueous solution of 5% sodium dihydrogen phosphate (39.56 kg×2) and water (39 kg×1). The solvent was evaporated under reduced pressure to give the titled compound as an oil. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 0.13 (9H, s), 0.17 (9H, s), 0.18 (9H, s), 0.20 (9H, s), 3.74-3.83 (3H, m), 3.90 (1H, t, J=8.0 Hz), 3.99 (1H, d, J=8.0 Hz), 4.17 (1H, dt, J=2.5, 8.0 Hz).
Step 2: Synthesis of 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene

Under a nitrogen atmosphere, to a solution of 2,4-dibromobenzyl alcohol (40 g, 0.15 mol) in tetrahydrofuran (300 ml) was added 2-methoxypropene (144 ml, 1.5 mol) at room temperature, and then the mixture was cooled to 0° C. At the same temperature, pyridinium p-toluenesulfonic acid (75 mg, 0.30 mmol) was added and the mixture was stirred for 1 hour. The reaction mixture was poured into a saturated aqueous solution of sodium hydrogen carbonate cooled to 0° C., and extracted with toluene. The organic layer was washed with a saturated aqueous solution of sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure to give the titled compound as an oil in quantitative yield. The product was used in the next step without further purification.
1H-NMR (CDCl3) δ: 1.44 (6H, s), 3.22 (3H, 4.48 (2H, s), 7.42 (1H, d, J=8.0 Hz), 7.44 (1H, dd, J=1.5, 8.0 Hz), 7.68 (1H, d, J=1.5 Hz).
Step 3: Synthesis of 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran

Under a nitrogen atmosphere, 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene (70 g, 207 mmol), which was obtained in the previous step, was dissolved in toluene (700 mL) and t-butylmethyl ether (70 ml), and n-butyllithium in hexane (1.65 M, 138 ml, 227 mmol) was added dropwise at 0° C. over 30 minutes. After the mixture was stirred for 1.5 hours at 0° C., the mixture was added dropwise to a solution of 3,4,5-tris(trimethylsilyloxy)-6-trimethylsilyloxymethyl-tetrahydropyran-2-one (Example 1, 108 g, 217 mol) in tetrahydrofuran (507 ml) at −78° C., and the reaction mixture was stirred for 2 hours at the same temperature. Triethylamine (5.8 ml, 41 mmol) and trimethylsilyl chloride (29.6 ml, 232 mmol) were added thereto, and the mixture was warmed to 0° C. and stirred for 1 hour to give a solution containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-bromo-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran.
The resulting solution was cooled to −78° C., and n-butyllithium in hexane (1.65 M, 263 ml, 434 mmol) was added dropwise thereto at the same temperature. After the mixture was stirred at −78° C. for 30 minutes, 4-ethylbenzaldehyde (62 ml, 455 mmol) was added dropwise at −78° C., and the mixture was stirred at the same temperature for 2 hours. A saturated aqueous solution of ammonium chloride was added to the reaction mixture, and the organic layer was separated, and washed with water. The solvent was evaporated under reduced pressure to give a product containing the titled compound as an oil (238 g). The product was used in the next step without further purification.
A portion of the oil was purified by HPLC (column: Inertsil ODS-3, 20 mm I.D.×250 mm; acetonitrile, 30 mL/min) to give four diastereomers of the titled compound (two mixtures each containing two diastereomers).
Mixture of Diastereomers 1 and 2:
1H-NMR (500 MHz, CDCl3) δ: −0.47 (4.8H, s), −0.40 (4.2H, s), −0.003-0.004 (5H, m), 0.07-0.08 (1314, m), 0.15-0.17 (18H, m), 1.200 and 1.202 (3H, each t, J=8.0 Hz), 1.393 and 1.399 (3H, each s), 1.44 (3H, s), 2.61 (2H, q, J=8.0 Hz), 3.221 and 3.223 (3H, each s), 3.43 (1H, t, J=8.5 Hz), 3.54 (1H, dd, J=8.5, 3.0 Hz), 3.61-3.66 (1H, m), 3.80-3.85 (3H, m), 4.56 and 4.58 (1H, each d, J=12.4 Hz), 4.92 and 4.93 (1H, each d, J=12.4 Hz), 5.80 and 5.82 (1H, each d, J=3.0 Hz), 7.14 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.50-7.57 (2H, m).
MS (ESI+): 875 [M+Na]+.
Mixture of Diastereomers 3 and 4:
1H-NMR (500 MHz, toluene-d8, 80° C.) δ: −0.25 (4H, s), −0.22 (5H, s), 0.13 (5H, s), 0.16 (4H, s), 0.211 and 0.214 (9H, each s), 0.25 (9H, s), 0.29 (9H, s), 1.21 (3H, t, J=7.5 Hz), 1.43 (3H, s), 1.45 (3H, s), 2.49 (2H, q, J=7.5 Hz), 3.192 and 3.194 (3H, each s), 3.91-4.04 (4H, m), 4.33-4.39 (2H, m), 4.93 (1H, d, J=14.5 Hz), 5.10-5.17 (1H, m), 5.64 and 5.66 (1H, each s), 7.03 (2H, d, J=8.0 Hz), 7.28-7.35 (3H, m), 7.59-7.64 (1H, m), 7.87-7.89 (1H, m).
MS (ESI+): 875 [M+Na]+.
Step 4: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

Under a nitrogen atmosphere, the oil containing 2,3,4,5-tetrakis(trimethylsilyloxy)-6-trimethylsilyloxymethyl-2-(5-(4-ethylphenyl)hydroxymethyl-2-(1-methoxy-1-methylethoxymethyl)phenyl)tetrahydropyran (238 g), which was obtained in the previous step, was dissolved in acetonitrile (693 ml). Water (37 ml) and 1N HCl aq (2.0 ml) were added and the mixture was stirred at room temperature for 5.5 hours. Water (693 ml) and n-heptane (693 ml) were added to the reaction mixture and the aqueous layer was separated. The aqueous layer was washed with n-heptane (693 ml×2), and water was evaporated under reduced pressure to give a product containing water and the titled compound (a diastereomer mixture) as an oil (187 g). The product was used in the next step without further purification.
1H-NMR (500 MHz, CD3OD) δ: 1.200 (3H, t, J=7.7 Hz), 1.201 (3H, t, J=7.7 Hz), 2.61 (2H, q, J=7.7 Hz), 3.44-3.48 (1H, m), 3.63-3.68 (111, m), 3.76-3.84 (4H, m), 5.09 (1H, d, J=12.8 Hz), 5.15 (1H, d, J=12.8 Hz), 5.79 (1H, s), 7.15 (2H, d, J=7.7 Hz), 7.24 and 7.25 (1H, each d, J=8.4 Hz), 7.28 (2H, d, J=7.7 Hz), 7.36 (1H, dd, J=8.4, 1.5 Hz), 7.40-7.42 (114, m).
MS (ESI+): 425 [M+Na]+.
Step 5: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (crude product)

To a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)hydroxymethyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (187 g), which was obtained in the previous step, in 1,2-dimethoxyethane (693 ml) was added 5% Pd/C (26 g, 6.2 mmol, water content ratio: 53%), and the mixture was stirred in the atmosphere of hydrogen gas at room temperature for 4 hours. After filtration, the filtrate was evaporated under reduced pressure to give an oil containing the titled compound (59 g). The purity of the resulting product was 85.7%, which was calculated based on the area ratio measured by HPLC. The product was used in the next step without further purification.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (3H, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Cadenza CD-C18 50 mm P/NCD032
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Gradient operation: Eluent B: 5% to 100% (6 min), 100% (2 min)
Flow rate: 1.0 mL/min
Temperature: 35.0° C.
Detection wavelength: 210 nm
Step 6: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose
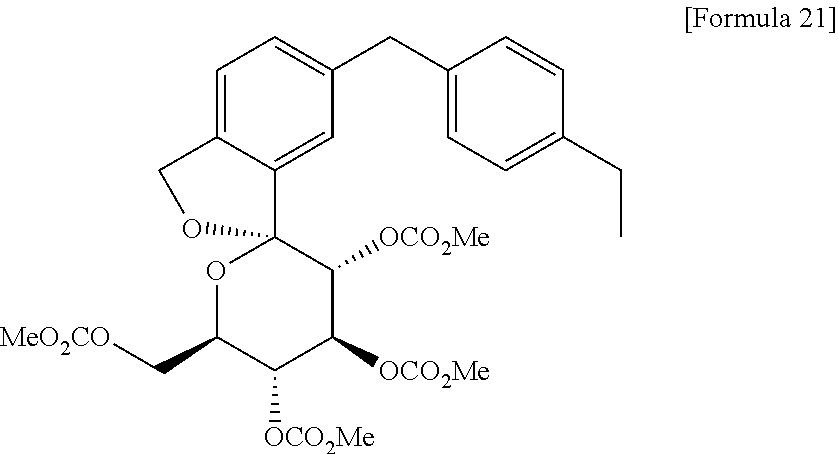
Under a nitrogen atmosphere, to a solution of the oil containing 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose (59 g) and 4-(dimethylamino)pyridine (175 g, 1436 mmol) in acetonitrile (1040 ml) was added dropwose methyl chloroformate (95 ml, 1231 mmol) at 0° C. The mixture was allowed to warm to room temperature while stirred for 3 hours. After addition of water, the mixture was extracted with isopropyl acetate. The organic layer was washed with an aqueous solution of 3% potassium hydrogensulfate and 20% sodium chloride (three times) and an aqueous solution of 20% sodium chloride, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. To the resulting residue was added ethanol (943 mL) and the mixture was heated to 75° C. to dissolve the residue. The mixture was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (472 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound (94 g). To the product (91 g) was added ethanol (1092 ml), and the product was dissolved by heating to 75° C. The solution was cooled to 60° C. and a seed crystal of the titled compound was added thereto. The mixture was cooled to room temperature and stirred for 1 hour. After precipitation of solid was observed, water (360 ml) was added thereto, and the mixture was stirred at room temperature for 2 hours. The resulting crystal was collected by filtration, washed with a mixture of water and ethanol (1:1), and dried under reduced pressure to give the titled compound [83 g, total yield from 2,4-dibromo-1-(1-methoxy-1-methylethoxymethyl)benzene used in Step 3: 68%].
1H-NMR (CDCl3) δ: 1.20 (3H, t, J=7.5 Hz), 2.60 (2H, q, J=7.5 Hz), 3.50 (3H, s), 3.76 (3H, s), 3.77 (3H, s), 3.81 (3H, s), 3.96 (2H, s), 4.23 (1H, dd, J=2.5, 11.8 Hz), 4.33 (1H, dd, J=4.5, 12.0 Hz), 4.36-4.40 (1H, m), 5.11-5.20 (3H, m), 5.41 (1H, d, J=10.0 Hz), 5.51 (1H, t, J=10.0 Hz), 7.07-7.11 (4H, m), 7.14 (1H, d, J=7.5 Hz), 7.19 (1H, dd, J=1.5, 7.8 Hz), 7.31 (1H, d, J=1.5 Hz).
MS (ESI+): 619 [M+1]+, 636 [M+18]+.
Another preparation was carried out in the same manner as Step 6, except that a seed crystal was not used, to give the titled compound as a crystal.
Step 7: Synthesis of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-β-D-glucopyranose

To a solution of 1,1-anhydro-1-C-[5-(4-ethylphenyl)methyl-2-(hydroxymethyl)phenyl]-2,3,4,6-tetra-O-methoxycarbonyl-β-D-glucopyranose (8.92 kg as wet powder, corresponding to 8.00 kg of dry powder) in 1,2-dimethoxyethane (28 kg) was added a solution of sodium hydroxide (4 mol/L, 30.02 kg) at 20° C., and the mixture was stirred for 1 hour. Water (8.0 kg) was added to the mixture and the layers were separated. To the organic layer were added an aqueous solution of 25% sodium chloride (40 kg) and ethyl acetate (36 kg). The organic layer was separated, washed with an aqueous solution of 25% sodium chloride (40 kg), and the solvent was evaporated under reduced pressure. The purity of the resulting residue was 98.7%, which was calculated based on the area ratio measured by HPLC. To the resulting residue were added acetone (32.0 kg) and water (0.8 kg), and the solvent was evaporated under reduced pressure. To the resulting residue were added acetone (11.7 kg) and water (15.8 kg), and the solution was cooled to 5° C. or below. Water (64 kg) was added to the solution at 10° C. or below, and the mixture was stirred at the same temperature for 1 hour. The resulting crystal was collected by centrifugation, and washed with a mixture of acetone (1.3 kg) and water (8.0 kg). The resulting wet powder was dried by ventilation drying under a condition at air temperature of 13 to 16° C. and relative humidity of 24% to 33% for 8 hours, to give a monohydrate crystal (water content: 4.502%) of the titled compound (3.94 kg). The purity of the resulting compound was 99.1%, which was calculated based on the area ratio measured by HPLC.
1H-NMR (CD3OD) δ: 1.19 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.42-3.46 (1H, m), 3.65 (1H, dd, J=5.5, 12.0 Hz), 3.74-3.82 (4H, m), 3.96 (2H, s), 5.07 (1H, d, J=12.8 Hz), 5.13 (1H, d, J=12.8 Hz), 7.08-7.12 (4H, m), 7.18-7.23 (311, m).
MS (ESI+): 387 [M+1]+.
Measurement Condition of HPLC:
Column: Capcell pack ODS UG-120 (4.6 mm I.D.×150 mm, 3 μm, manufactured by Shiseido Co., Ltd.)
Mobile phase: Eluent A: H2O, Eluent B: MeCN
Mobile phase sending: Concentration gradient was controlled by mixing Eluent A and Eluent B as indicated in the following table.
| TABLE 1 | ||||
| Time from | ||||
| injection (min) | Eluent A (%) | Eluent B (%) | ||
| 0 to 15 | 90→10 | 10→90 | ||
| 15 to 17.5 | 10 | 90 | ||
| 17.5 to 25 | 90 | 10 | ||
Flow rate: 1.0 mL/min
Temperature: 25.0° C.
Detection wavelength: 220 nm
Method for Measurement of Water Content:
Analysis method: coulometric titration method
KF analysis apparatus: Type KF-100 (trace moisture measuring apparatus manufactured by Mitsubishi Chemical Corporation)
Anode solution: Aquamicron AX (manufactured by Mitsubishi Chemical Corporation)
Cathode solution: Aquamicron CXU (manufactured by Mitsubishi Chemical Corporation)
PATENT
The compound of the present invention can be synthesized as shown in Scheme 1:

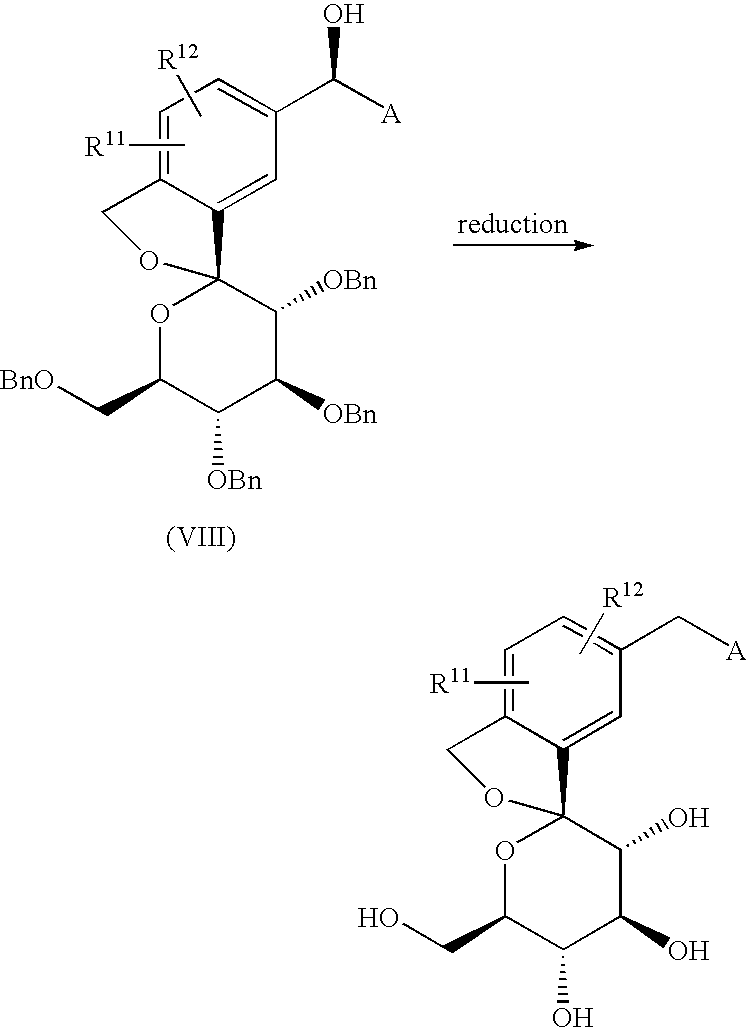
wherein R11 and R12 have the same meaning as defined above for substituents on Ar1, A is as defined above, and P represents a protecting group for a hydroxyl group.
CLIP
Tofogliflozin hydrate (Deberza)
Tofogliflozin hydrate, which is a sodium-glucose co-transporter 2 inhibitor, was approved in Japan for the treatment of type 2 diabetes
at the same time as luseogliflozin hydrate (XIX). The drug was discovered by Chugai Pharmaceutical and jointly developed
with Sanofi-Aventis and Kowa.263
Tofogliflozin hydrate reduces glucose levels by inhibiting the reuptake of glucose by selectively
inhibiting SGLT2, and plays a key role in the reuptake of glucose in the proximal tubule of the kidneys.264–266 The synthetic
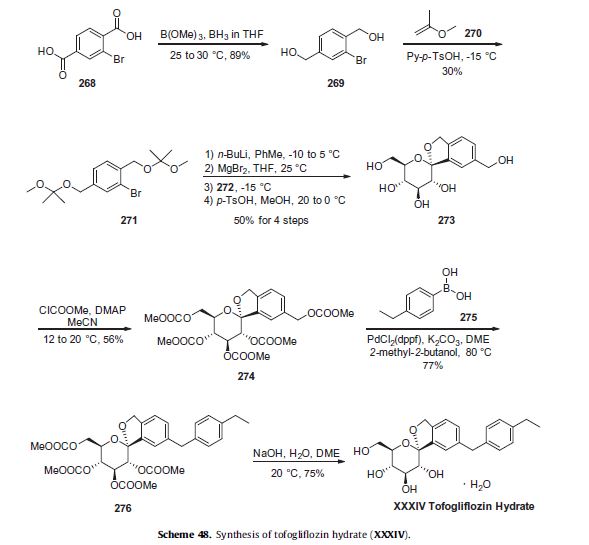
approach described in Scheme 48 represents the largest scale reported to date in a patent application.263,266–268
Reduction of commercially available 2-bromoterephtalic acid (268, Scheme 48) through the use of trimethoxyborane and borane-THF proceeded in 89% yield to afford diol 269.
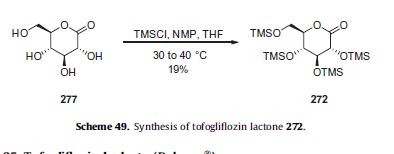
Subjection of this compound to 2-methoxypropene (270) under acidic conditions generated bis-acetonide 271. This bromide then underwent lithium–halogen exchange followed by exposure to magnesium bromide and treatment with lactone 272 (which was prepared by persilylation of commercially available (3R,4S,5S,6R)-3,4,5-trihydroxy-6-hydroxymethyl)tetrahydro-2Hpyran-2-one (277, Scheme 49).
This mixture was worked up with aqueous ammonium chloride and upon treatment with p-TsOH in methanol resulted in spiroacetal 273. Next, global protection of all alcohol functionalities within 273 was affected by reaction with methylchloroformate and DMAP in acetonitrile.
The benzyl carbonate within 274 was selectively exchanged via Suzuki coupling with 4-ethylphenylboronic acid (275) to afford methylene dibenzyl system 276. Subsequent treatment with aqueous sodium hydroxide in methanol followed by crystallization from 1:6 acetone and water furnished the desired product tofogliflozin hydrate (XXXIV) in 75% yield.
263 Takamitsu, K.; Tsutomu, S.; Masahiro, N. WO Patent 2006080421A1, 2006.
264. http://www.info.pmda.go.jp/shinyaku/P201400036/index.html.
265. Pafili, K.; Papanas, N. Expert Opin. Pharmacother. 2014, 15, 1197.
266. Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.;Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.;Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.;Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828.
267. Murakata, M.; Ikeda, T.; Kawase, A.; Nagase, M.; Kimura, N.; Takeda, S.;Yamamoto, K.; Takano, K.; Nishimoto, M.; Ohtake, Y.; Emura, T.; Kito, Y. WOPatent 2011074675A1, 2011.
268. Murakata, M.; Takuma, I.; Nobuaki, K.; Masahiro, N.; Kawase, A.; Nagase, M.;Yamamoto, K.; Takata, N.; Yoshizaki, S. WO Patent 2009154276A1, 2009.
Paper
A Scalable Synthesis of Tofogliflozin Hydrate

A newly process for the synthesis of tofogliflozin hydrate, a sodium-glucose cotransporter type 2 (SGLT2) inhibitor, was described. Three improvements were achieved, including the development of a regioselective Friedel–Crafts reaction, a high-yield reduction, and a mild metal–halogen exchange. These improvements ultimately resulted in the isolation of tofogliflozin hydrate as a white solid in >99% purity (HPLC area) and 23% overall yield after 12 steps without column chromatography.

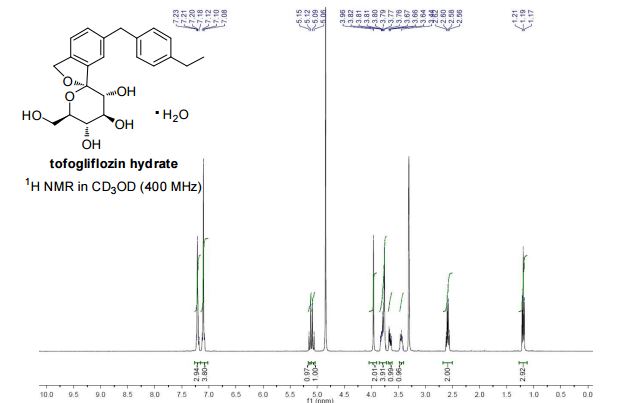
Tofogliflozin hydrate white solid with 99.56% purity by HPLC. Water content: 4.47%.
Mp: 71−80 oC. [α]20 D = +23.9 (c = 1.0, CH3OH).
1H NMR (400 MHz, CD3OD) δ 7.23-7.18 (m, 3H), 7.12-7.08(m, 4H), 5.13 (d, J = 12.4 Hz, 1H), 5.07 (d, J = 12.4 Hz, 1H), 3.96 (s, 2H), 3.83-3.73 (m, 4H), 3.65 (dd, J = 11.9, 5.5 Hz, 1H), 3.41-3.47 (m, 1H), 2.59 (q, J = 7.6 Hz, 2H), 1.19 (t, J = 7.6 Hz, 3H).
13C NMR (100 MHz, CD3OD) δ 143.2, 142.6, 140.2, 139.9, 139.7, 131.2, 129.9, 128.9, 123.6, 121.8, 111.6, 76.4, 76.2, 74.9, 73.4, 71.9, 62.8, 42.3, 29.5, 16.3.
HRMS (ESI) m/z: [M+H]+ Calcd for C22H27O6 387.1802; Found 387.1805.
IR (KBr, cm-1) ν: 3362, 2962, 2927, 1637, 1513, 1429, 1095, 1034, 808, 770. Spectroscopic data were identical with those reported.1b, 2
1. (a) Suzuki, M.; Honda, K.; Fukazawa, M.; Ozawa, K.; Hagita, H.; Kawai, T.; Takeda, M.; Yata, T.; Kawai, M.; Fukuzawa, T.; Kobayashi, T.; Sato, T.; Kawabe, Y.; Ikeda, S. J. Pharmacol. Exp. Ther. 2012, 341, 692.
(b) Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S.-H.; Ahn. K.-H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. J. Med. Chem. 2012, 55, 7828.
(c) Ikeda, S.; Takano, Y.; Cynshi, O.; Tanaka, R.; Christ, A. D.; Boerlin, V.; Beyer, U.; Beck, A.; Ciorciaro, C.; Meyer, M.; Kadowaki, T. Diabetes, Obesity and Metabolism 2015, 17, 984.
2. (a) Murakata, M.; Ikeda, T.; Kimura, N.; Kawase, A.; Nagase, M.; Yamamoto, K.; Takata, N.; Yoshizaki, S.; Takano, K. Crystal of spiroketal derivative, and process for production thereof. European Appl. EP 2308886 A1, April 13, 2011.
(b) Ohtake, Y.; Emura, T.; Nishimoto, M.; Takano, K.; Yamamoto, K.; Tsuchiya, S.; Yeu, S.; Kito, Y.; Kimura, N.; Takeda, S.; Tsukazaki, M.; Murakata, M.; Sato, T. J. Org. Chem. 2016, 81, 2148.
References
- Chugai Pharmaceutical: Development Pipeline
- Nagata, T.; Fukazawa, M.; Honda, K.; Yata, T.; Kawai, M.; Yamane, M.; Murao, N.; Yamaguchi, K.; Kato, M.; Mitsui, T.; Suzuki, Y.; Ikeda, S.; Kawabe, Y. (2012). “Selective SGLT2 inhibition by tofogliflozin reduces renal glucose reabsorption under hyperglycemic but not under hypo- or euglycemic conditions in rats”. AJP: Endocrinology and Metabolism 304 (4): E414–E423. doi:10.1152/ajpendo.00545.2012.PMID 23249697.
- Ohtake, Y.; Sato, T.; Kobayashi, T.; Nishimoto, M.; Taka, N.; Takano, K.; Yamamoto, K.; Ohmori, M.; Yamaguchi, M.; Takami, K.; Yeu, S. Y.; Ahn, K. H.; Matsuoka, H.; Morikawa, K.; Suzuki, M.; Hagita, H.; Ozawa, K.; Yamaguchi, K.; Kato, M.; Ikeda, S. (2012). “Discovery of Tofogliflozin, a NovelC-Arylglucoside with anO-Spiroketal Ring System, as a Highly Selective Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes”. Journal of Medicinal Chemistry 55 (17): 7828–7840. doi:10.1021/jm300884k.PMID 22889351.
- Statement on a nonproprietary name adopted by the USAN council: Tofogliflozin.
- http://www.who.int/entity/medicines/publications/druginformation/innlists/RL65.pdf
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(1S,3′R,4′S,5′S,6′R)-6-(4-Ethylbenzyl)-6′-(hydroxymethyl)-3′,4′,5′,6′-tetrahydro-3H-spiro[2-benzofuran-1,2′-pyran]-3′,4′,5′-triol hydrate (1:1)
|
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | 1201913-82-7 903565-83-3 (anhydrous) |
| ATC code | None |
| PubChem | CID 46908928 |
| ChemSpider | 28527871 |
| KEGG | D09978 |
| ChEMBL | CHEMBL2105711 |
| Synonyms | CSG452 |
| Chemical data | |
| Formula | C22H28O7 |
| Molar mass | 404.45 g/mol |
//////////TOFOGLIFLOZIN, 托格列净 , CSG-452, R-7201, RG-7201, 1201913-82-7 , 903565-83-3, oral hypoglycaemic agents, SGLT-2 inhibitors, type 2 diabetes mellitus, Deberza
CCc1ccc(cc1)Cc2ccc3c(c2)[C@]4([C@@H]([C@H]([C@@H]([C@H](O4)CO)O)O)O)OC3.O
The glucopyranosyl-substituted benzene derivatives are proposed as inducers of urinary sugar excretion and as medicaments in the treatment of diabetes.
The term “canagliflozin” as employed herein refers to canagliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2005/012326 and WO 2009/035969 for example. Preferred hydrates, solvates and crystalline forms are described in the patent applications WO 2008/069327 for example.
atigliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/007517 for example.
ipragliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2004/080990, WO 2005/012326 and WO 2007/114475 for example.
tofogliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

The compound and methods of its synthesis are described in WO 2007/140191 and WO 2008/013280 for example.
remogliflozin and prodrugs of remogliflozin, in particular remogliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods of its synthesis are described in the patent applications EP 1213296 and EP 1354888 for example.
sergliflozin and prodrugs of sergliflozin, in particular sergliflozin etabonate, including hydrates and solvates thereof, and crystalline forms thereof. Methods for its manufacture are described in the patent applications EP 1344780 and EP 1489089 for example.
luseoghflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

ertugliflozin, including hydrates and solvates thereof, and crystalline forms thereof and has the following structure:

and is described for example in WO 2010/023594.
The compound of the formula

is described for example in WO 2008/042688 or WO 2009/014970.
Dapagliflozin

The compound is described for example in WO 03/099836. Crystalline forms are described for example in WO 2008/002824.
Remogliflozin and Remogliflozin Etabonate

The compound is described for example in EP 1354888 A1.
Sergliflozin and Sergliflozin Etabonate

The compounds are described in EP 1 329 456 A1 and a crystalline form ofSergliflozin etabonate is described in EP 1 489 089 A1.
1-Chloro-4-(β-D-glucopyranos-1-yl)-2-(4-ethyl-benzyl)-benzene

The compound is described in WO 2006/034489.
(1S)-1,5-anhydro-1-[5-(azulen-2-ylmethyl)-2-hydroxyphenyl]-D-glucitol

The compound (4-(Azulen-2-ylmethyl)-2-(β-D-glucopyranos-1-yl)-1-hydroxy-benzene) is described in WO 2004/013118 and WO 2006/006496. The crystalline choline salt thereof is described in WO 2007/007628.
(1S)-1,5-anhydro-1-[3-(1-benzothien-2-ylmethyl)-4-fluorophenyl]-D-glucitol

The compound is described in WO 2004/080990 and WO 2005/012326. A cocrystal with L-proline is described in WO 2007/114475.
Thiophen Derivatives of the Formula (7-1)

wherein R denotes methoxy or trifluoromethoxy. Such compounds and their method of production are described in WO 2004/007517, DE 102004063099 and WO 2006/072334.
1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene

The compound is described in WO 2005/012326. A crystalline hemihydrate is described in WO 2008/069327.
Spiroketal Derivatives of the Formula (9-1)

wherein R denotes methoxy, trifluoromethoxy, ethoxy, ethyl, isopropyl or tert. butyl. Such compounds are described in WO 2007/140191 and WO 2008/013280.
LY 2922470

LY 2922470

LY 2922470
Picture credit….Bethany Halford
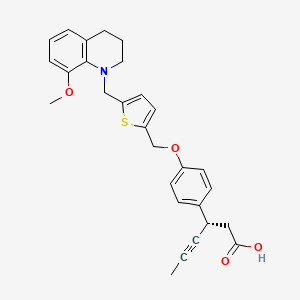
(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophen-2-yl]methoxy]phenyl]hex-4-ynoic acid
Benzenepropanoic acid, 4-[[5-[(3,4-dihydro-8-methoxy-1(2H)-quinolinyl)methyl]-2-thienyl]methoxy]-β-1-propyn-1-yl-, (βS)-
Glucose Lowering Agents, Signal Transduction Modulators
| CAS | 1423018-12-5 |
|---|---|
| Molecular Formula: | C28H29NO4S |
| Molecular Weight: | 475.59916 g/mol |
|---|
https://clinicaltrials.gov/ct2/show/NCT01867216
- Phase I Type 2 diabetes mellitus
Eli Lilly
Antihyperglycaemics
- 28 Jan 2014 Eli Lilly completes a phase I trial in Type-2 diabetes mellitus in USA (NCT01867216)
- 30 Jun 2013 Phase-I clinical trials in Type-2 diabetes mellitus in USA (PO)
- 14 Jun 2013 Eli Lilly plans a phase I trial for Type-2 diabetes mellitus in USA (NCT01867216)
PATENT
WO 2013025424
https://www.google.com/patents/US20130045990?cl=de
| Also published as | CA2843474A1, CA2843474C, CN103687856A, CN103687856B, EP2744806A1, US8431706, WO2013025424A1, Less « |
| Inventors | Chafiq Hamdouchi |
| Original Assignee | Eli Lilly And Company |



Preparation 18-Methoxyquinoline
Add potassium hydroxide (435 g, 7.76 mol) to a solution of 8-hydroxy quinoline (250 g, 1.724 mol) in THF (10 L) at ambient temperature and stir. Add methyl iodide (435 g, 2.58 mol) dropwise and stir overnight. Filter the reaction mixture and wash the solid with THF (2 L). Concentrate the solution to dryness; add water; extract with dichloromethane (2×3 L); combine the organic layers; and wash with brine. Collect the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a red oil, which solidifies on standing, to give the title compound (281 g, 102%), which can be used without further purification. ESI (m/z) 160(M+H).
Preparation 2
8-Methoxy-1,2,3,4-tetrahydroquinoline
Add sodium cyanoborohydride (505 g, 8.11 mol) in EtOH (1 L) to a solution of 8-methoxy quinoline (425 g, 2.673 mol) in EtOH (9 L), and stir. Cool the reaction mixture to an internal temperature of 0° C. and add HCl (35%, 1.12 L, 10.962 mol) dropwise over 60 min so that the internal temperature did not rise above 20° C. Allow the reaction mixture to warm to ambient temperature and then heat to reflux for 2.5 hours. Cool to ambient temperature and stir overnight. Add ammonium hydroxide (25%, 1 L); dilute with water (15 L); and extract the mixture with dichloromethane (3×10 L). Combine the organic layers and dry over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography, eluting with ethyl acetate: hexane (1:10) to give the title compound (357 g, 82%). ESI (m/z) 164(M+H).
Preparation 3
Methyl-5-methylthiophene-2-carboxylate
Add thionyl chloride (153 ml, 2.1 mol) dropwise over 20 min to a solution of 5-methylthiophene-2-carboxylic acid (100 g, 0.703 mol) in MeOH (1 L) at 0° C. and stir. After the addition is complete, heat the reaction mixture to reflux for 3.5 hours. Cool and concentrate in vacuo to give a thick oil. Dilute the oil with EtOAc (500 ml) and sequentially wash with water (300 ml) then brine (300 ml). Dry the organic layer over sodium sulfate. Remove the solids by filtration. Collect the filtrate and concentrate under reduced pressure to give the title compound (106 g, 97%), which is used without further purification. ESI (m/z) 156(M+H).
Preparation 4
Methyl 5-(bromomethyl)thiophene-2-carboxylate
Add freshly recrystallised NBS (323.8 g, 1.81 mol) to a solution of methyl-5-methylthiophene-2-carboxylate (258 g, 1.65 mol) in chloroform (2.6 L) at room temperature, and stir. Add benzoyl peroxide (3.99 g, 0.016 mol) and heat the reaction mixture to reflux for 7 hours. Cool the reaction mixture to ambient temperature and filter through diatomaceous earth. Wash the filter cake with chloroform (250 ml). Collect the organic layers and remove the solvent to give the title compound (388 g, 100%), which is used without further purification. ESI (m/z) 236(M+H).
Preparation 5
Methyl-5-[8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]thiophene-2-carboxylate
Add methyl-5-(bromoethyl)thiophene-2-carboxylate (432.5 g, 1.84 mol) in EtOH (500 ml) to a solution of 8-methoxy-1,2,3,4-tetrahydroquinoline (300 g 1.84 mol) in EtOH (1 L) and stir. Add DIPEA (641 ml, 3.67 mol) dropwise and stir at room temperature overnight. After completion of the reaction, remove the EtOH in vacuo, and add water (5 L). Extract the aqueous with EtOAc (3×3 L); combine the organic layers; and dry over sodium sulfate. Filter the solution and concentrate under reduced pressure to give a residue. Purify the residue by silica gel flash chromatography eluting with ethyl acetate: hexane (6:94) to give the title compound (325 g, 56%). ESI (m/z) 318(M+H).
Preparation 6
[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methanol
Add DIBAL-H (1 M in toluene 2.7 L, 2.66 mol) slowly via a cannula over a period of 1.5 h to a stirred solution of methyl-5-(8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-carboxylate (281 g, 0.886 mol) in THF (4 L) at −70° C. Monitor the reaction via thin layer chromatography (TLC) for completion. After completion of the reaction, allow the reaction mixture to warm to 20° C. and add a saturated solution of ammonium chloride. Add a solution of sodium potassium tartrate (1.3 Kg in 5 L of water), and stir overnight. Separate the organic layer; extract the aqueous phase with EtOAc (2×5 L); then combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration. Remove the solvent from the filtrate under reduced pressure to give the title compound as a white solid (252 g, 98%). ESI (m/z) 290(M+H).
Preparation 7
Ethyl(3S)-3-[4-[[5-[(8-methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoate
Add tributylphosphine (50% solution in EtOAc, 543 ml, 1.34 mol) to a solution of ADDP (282.5 g, 1.5 eq) in THF (3 L) and cool the mixture to an internal temperature of 0° C., then stir for 15 minutes. Add (S)-ethyl 3-(4-hydroxyphenyl)hex-4-ynoate (173.5 g, 0.747 mol) in THF (3 L) dropwise over 15 min; then add 5-((8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophene-2-yl)methanol (216 g, 0747 mol) in THF (5 L) dropwise. Allow the reaction mixture to warm to ambient temperature and stir overnight. Filter the reaction mixture through diatomaceous earth and wash the filter cake with ethyl acetate (2 L). Concentrate the organic filtrate to dryness. Add water (4 L); extract with ethyl acetate (3×5 L); combine the organic layers; and dry the combined organic layers over sodium sulfate. Remove the solids by filtration and concentrate under reduced pressure to give an oil. Purify the residue by silica gel flash chromatography by eluting with ethyl acetate: hexane (6:94) to give the title compound (167 g, 44%). ESI (m/z) 504(M+H).
Example 1
(3S)-3-[4-[[5-[(8-Methoxy-3,4-dihydro-2H-quinolin-1-yl)methyl]-2-thienyl]methoxy]phenyl]hex-4-ynoic acid

Add a solution of potassium hydroxide (49.76 g, 0.88 mol) in water (372 ml) to a solution of (S)-ethyl-3-(4-((5-8-methoxy-3,4-dihydroquinolin-1(2H)-yl)methyl)thiophen-2-yl)methoxy) phenyl)hex-4-ynoate (149 g, 0.296 mol) in EtOH (1.49 L) at room temperature and stir overnight. Concentrate the reaction mixture to dryness and add water (1.3 L). Extract the resulting solution with EtOAc (2×300 ml) and separate. Adjust the pH of the aqueous layer to pH=6 with 2 N HCl. Collect the resulting solids. Recrystallise the solids from hot MeOH (298 ml, 2 vol) to give the title compound (91 g, 65%). ESI (m/z) 476(M+H).
Abstract
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260


Presenter

Chafiq Hamdouchi
Senior Research Advisor at Eli Lilly and Company
https://www.linkedin.com/in/chafiq-hamdouchi-4988126
Summary
Dr. Hamdouchi earned his bachelor’s degree and doctorate in organic chemistry from Louis Pasteur University, Strasbourg-France.
Following two postdoctoral fellowships, sponsored by the National Science Foundation-USA and Ministerio de Educación y Ciencia-Spain, he joined Eli Lilly and Company in 1995.
Throughout his 20 years of career at Lilly, he has contributed to a sustainable drug discovery portfolio from preclinical hypothesis to clinical proof-of-concept that spans the oncology, neuroscience and endocrinology therapeutic areas. He has led multidisciplinary (chemistry, pharmacology, ADMET, PK, medical) scientific teams in USA, Europe and Asia to deliver a number of compounds that achieved first human dose.
He is a co-inventor of six innovative molecules being pursued in clinical development for the treatment of Diabetes, Cancer and Neurodegenerative Diseases.
He has an extensive patent and publication record and deep experience in conducting drug discovery and development in Asia through effective partnership and mentorship.
SEE AT…………ONE ORGANIC CHEMIST ONE DAY BLOG
LINK……http://oneorganichemistoneday.blogspot.in/2016/03/chafiq-hamdouchi-senior-research.html
| Patent ID | Date | Patent Title |
|---|---|---|
| US8431706 | 2013-04-30 | 1,2,3,4-tetrahydroqinoline derivative useful for the treatment of diabetes |
References
GPR40 agonists for the treatment of type 2 diabetes: From the laboratory to the patient
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 260
//////Phase 1, LY2922470, LY 2922470, Eli Lilly, Type 2 diabetes mellitus, 1423018-12-5, Chafiq Hamdouchi
CC#CC(CC(=O)O)C1=CC=C(C=C1)OCC2=CC=C(S2)CN3CCCC4=C3C(=CC=C4)OC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
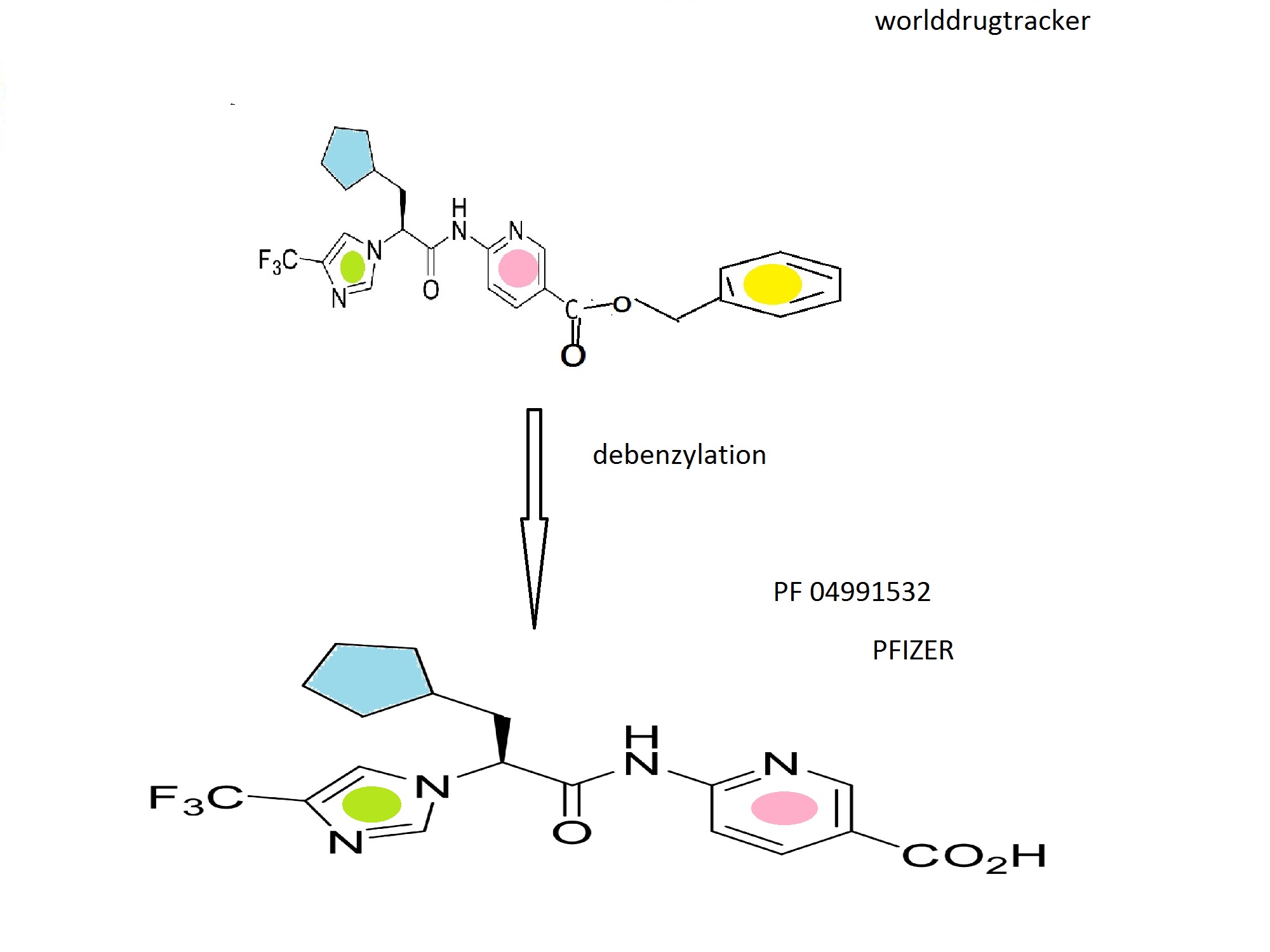
Pfizer’s PF 04991532 a Hepatoselective Glucokinase Activator Clinical Candidate for Treating Type 2 Diabetes Mellitus

PF 04991532
GKA PF-04991532
(S)-6-{3-cyclopentyl-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propanamido}nicotinic acid
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
MW 396.36, MF C18 H19 F3 N4 O3
CAS 1215197-37-7
3-Pyridinecarboxylic acid, 6-[[(2S)-3-cyclopentyl-1-oxo-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propyl]amino]-
http://www.biochemj.org/content/441/3/881
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public epidemic affecting over 300 million people worldwide. This disease is characterized by elevated fasting plasma glucose (FPG), insulin resistance, abnormally elevated hepatic glucose production (HGP), and reduced glucose-stimulated insulin secretion (GSIS). Moreover, long-term lack of glycemic control increases risk of complications from neuropathic, microvascular, and macrovascular diseases.
The standard of care for T2DM is metformin followed by sulfonylureas, dipeptidyl peptidase-4 (DPP-IV) inhibitors, and thiazolidinediones (TZD) as second line oral therapies. As disease progression continues, patients typically require injectable agents such as glucagon-like peptide-1 (GLP-1) analogues and, ultimately, insulin to help maintain glycemic control. Despite these current therapies, many patients still remain unable to safely achieve and maintain tight glycemic control, placing them at risk of diabetic complications and highlighting the need for novel therapeutic options.

Glucokinase (hexokinase IV) continues to be a compelling target for the treatment of type 2 diabetes given the wealth of supporting human genetics data and numerous reports of robust clinical glucose lowering in patients treated with small molecule allosteric activators. Recent work has demonstrated the ability of hepatoselective activators to deliver glucose lowering efficacy with minimal risk of hypoglycemia.
While orally administered agents require a considerable degree of passive permeability to promote suitable exposures, there is no such restriction on intravenously delivered drugs. Therefore, minimization of membrane diffusion in the context of an intravenously agent should ensure optimal hepatic targeting and therapeutic index.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type II diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM.
As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity.
Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type II diabetes and the metabolic syndrome” Nature 414; 821-827, (2001)): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents.
Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication Nos. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.

PATENT
US 20100063063
http://www.google.com/patents/US20100063063
SYNTHESIS CONSTRUCTION

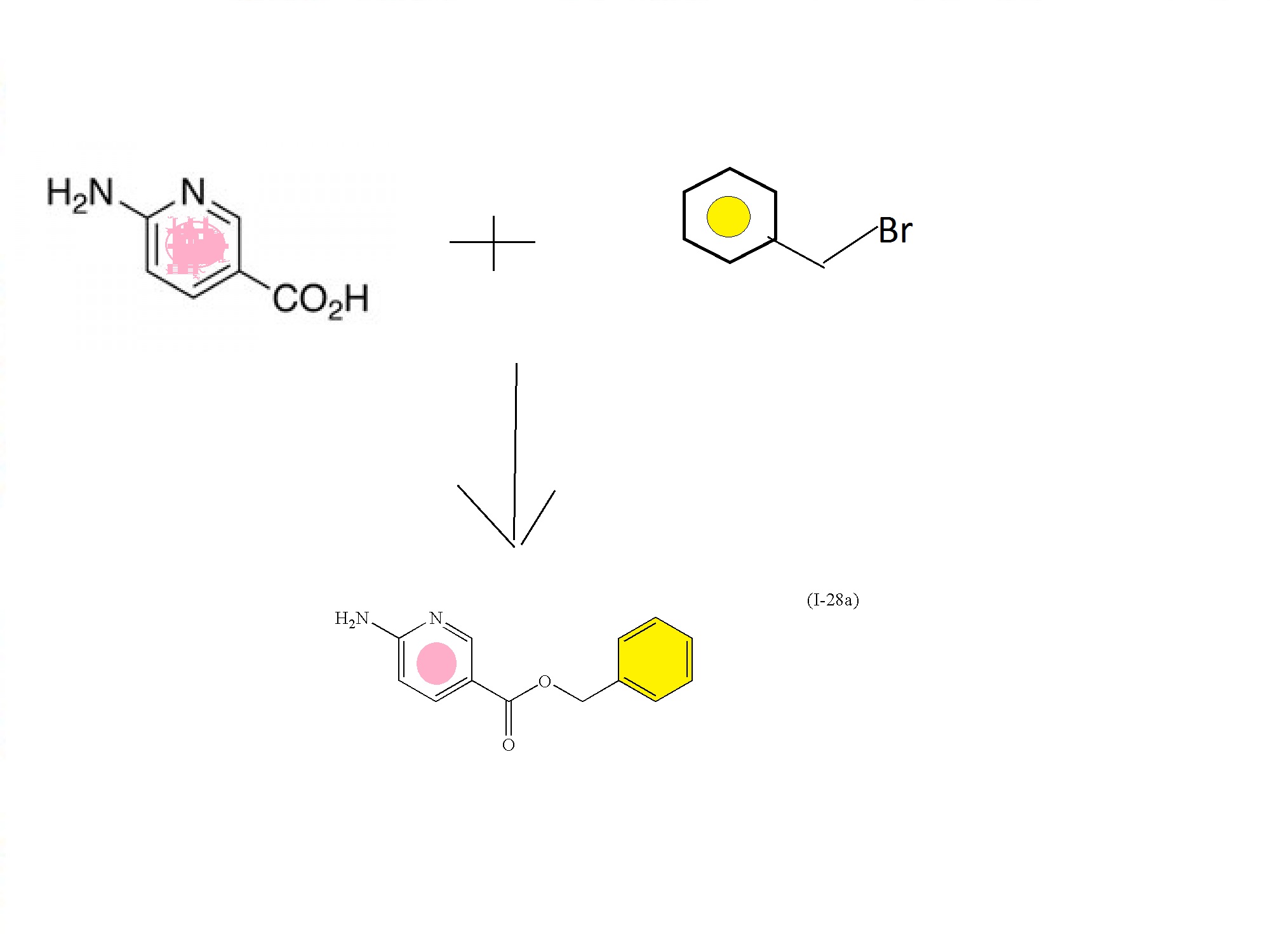
6-aminonicotinic acid

BENZYL BROMIDE

FIRST KEY INTERMEDIATE
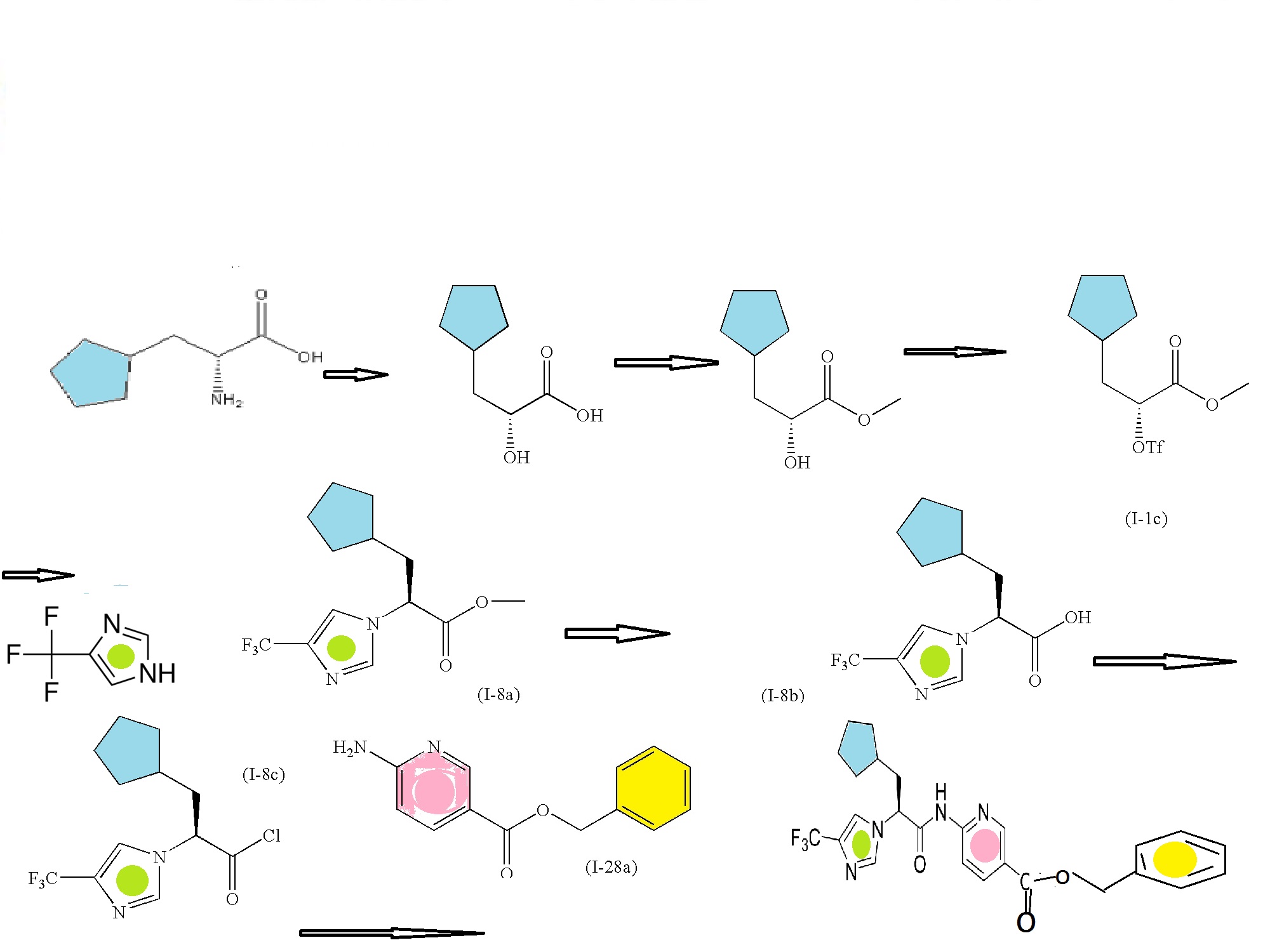
SECOND SERIES FOR NEXT INTERMEDIATE

(R)-2-amino-3-cyclopentylpropanoic acid

(R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)

(R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
Trifluoromethanesulfonic acid anhydride

(R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
CONDENSED WITH

4-Trifluoromethyl-1H-imidazole
TO GIVE PRODUCT SHOWN BELOW

(S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)

(S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)

CONVERTED TO ACID CHLORIDE, (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
AND CONDENSED WITH
WILL GIVE BENZYL DERIVATIVE AS BELOW

THEN DEBENZYLATION TO FINAL PRODUCT
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)
To a stirred solution of (R)-2-amino-3-cyclopentylpropanoic acid (5.0 grams; Chem-Impex International, Inc., Wood Dale, Ill.) and 1 M H2SO4 (45.1 mL) at 0° C., was added a solution of NaNO2 (3.12 g) in H2O (15.6 mL) drop wise over 10 minutes. The reaction mixture was stirred for 3 hours at 0° C., then for 2 hours at room temperature. The solution was then extracted (3 times) with diethyl ether. The combined organic extracts were dried over MgSO4, filtered, and the filtrate concentrated to afford 2.36 g of (I-1a). 1H NMR (400 MHz, CDCl3) δ 4.26-4.28 (1H), 1.99-2.07 (1H), 1.76-1.81 (4H), 1.60-1.62 (4H), 1.12-1.16 (2H); LCMS for C8H14O3 m/z 157.1 (M−H)−.
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
To a stirred solution of 2.36 g of (I-1a) in anhydrous methanol (15 mL) at room temperature was added SOCl2(1.64 mL). The resulting mixture was heated at reflux for 2 hours. It was then cooled and concentrated under reduced pressure. The residue was partitioned between ethyl acetate and aqueous saturated NaHCO3 solution. The biphasic mixture was separated and the aqueous portion was extracted with ethyl acetate. The combined extracts were dried over MgSO4, filtered, and the filtrate concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel, heptanes/ethyl acetate) to afford 1.5 g of (I-1b) as a clear oil.1H NMR (400 MHz, CDCl3) δ 4.15-4.20 (1H), 3.77 (3H), 2.62-2.63 (1H), 1.97-2.05 (1H), 1.49-1.86 (8H), 1.06-1.17 (2H); LCMS for C9H16O3 m/z 171.6 (M)+. Intermediate (I-1b) can alternatively be prepared by the method described below.
A 0.2M solution of Li2CuCl4 was prepared as follows: Anhydrous CUCl2 (26.9 g, 200 mol) and anhydrous LiCl (17.0 g, 400 mmol) were dissolved in THF (1000 mL). The mixture required gentle heating to completely dissolve the solids. After cooling the solution is ready for use.
A solution of Li2CuCl4 (0.2 M in THF, 125 mL, 25.0 mmol) was added slowly to a suspension of cyclopentylmagnesium bromide (2 M in diethyl ether, 135 mL, 270 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) and THF (500 mL) at −50° C. over 2-3 mins. The pale grey/brown suspension was then allowed to warm slowly to −10° C. over 30 mins, by which time the color had developed to a dark grey. The mixture was re-cooled to −78° C. and (R)-methyl oxirane-2-carboxylate (25.0 g, 245 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) was added neat via syringe over 90 seconds. The reaction was then stirred at −78° C. for 20 mins, before removing the ice-bath and allowing to warm to approximately −50° C. over 30 mins. Saturated NH4Cl (aq, 700 mL) was then added and the mixture stirred for 30 mins. The organic layer was collected and the aqueous layer extracted with diethyl ether (2×250 mL). The combined organics were washed with saturated NH4Cl (aq, 350 mL), dried over MgSO4, and evaporated. Distillation of the crude residue (68-70° C. at 0.8 mbar) yielded 65-70% of (I-1b) as a pale yellow oil. A small amount of less volatile material remained in the still pot. 1H NMR (400 MHz; CDCl3): δ 4.17(1H), 3.76 (3H), 2.67 (1H), 2.01 (1H), 1.48-1.88 (8H), 1.11 (2H).
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H).
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H)
Intermediate: (S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)
4-Trifluoromethyl-1H-imidazole (5.0 g, 37.0 mmol; Apollo Scientific Ltd., Bredbury, Cheshire, UK) was stirred in dry THF (180 mL) under nitrogen at room temperature. Lithium hexamethyldisilazide (1M in THF, 33.4 mL, 33.4 mmol) was added dropwise via addition funnel. The mixture was stirred at room temperature for 50 minutes and then chilled in an ice bath. A solution of (I-1c) (11.3 g, 37 mmol) in dry THF (45 mL), which had been chilled in an ice bath, was added in one portion. The reaction was allowed to warm to room temperature, stirred for 2 hours, quenched with saturated aqueous ammonium chloride solution (20 mL) and allowed to stir overnight. The aqueous layer was separated, and the organic layer was concentrated and then diluted with water and ethyl acetate. The organic layer was washed in series with dilute aqueous phosphoric acid, aqueous 10% potassium carbonate, and brine. The organic layer was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a brown oil. The crude material, containing the undesired regioisomer as a small impurity, was purified by chromatography on a 330 g pre-packed silica gel column, eluting with 10% ethyl acetate/heptane, linear gradient to 70% ethyl acetate/heptane. The product fractions were located by spotting on a silica TLC plate and visualizing with KMnO4 stain. TLC (1:1 ethyl acetate/heptane, developed in potassium permanganate) located the pure and mixed fractions. The clean product fractions were combined, evaporated, and dried under high vacuum to afford (I-8a) as a clear oil (6.61 g, 22.4 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 7.57 (1H), 7.38 (1H), 4.71-4.74 (1H), 3.76 (3H), 2.01-2.14 (2H), 1.45-1.79 (7H), 1.03-1.18 (2H); m/z 291.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
To a suspension of intermediate (I-8b) (0.25 g, 0.9 mmol) in dichloromethane (5 mL) was added oxalyl chloride (0.35 g, 2.7 mmol) and N,N-dimethylformamide (1 drop) at room temperature. The mixture was stirred for 2 hours at room temperature. The reaction mixture was concentrated in vacuo, and the residue was chased with dichloromethane two times and concentrated in vacuo to afford (I-8c) (0.27 g, 100%) as an oil, which was used in the next step directly.
Intermediate: (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido) nicotinoyl chloride (I-21a)
-
 Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.
Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.

(I-8b
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
(I-28a
Intermediate: benzyl 6-aminonicotinate (I-28a)
To a stirred suspension of 6-aminonicotinic acid (100 g, 0.72 mol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) in N,N-dimethylformamide (700 mL) with brisk mechanical stirring was added potassium carbonate (150 g, 1.08 mol) and the reaction was stirred for 10 min before the portionwise addition of benzyl bromide (95 mL, 0.80 mol). The reaction was stirred at room temperature overnight, then the solids were filtered off and washed thoroughly with ethyl acetate, and the solvent was removed under vacuum. The filter cake was dissolved in water and extracted with ethyl acetate. The residue after evaporation of N,N-dimethylformamide was combined with the ethyl acetate extracts (total volume 2 L of ethyl acetate) and the combined organic extracts washed with brine (5×500 mL), dried (MgSO4) and the solvent removed under reduced pressure. The crude product was refluxed with 1:1 diethyl ether:hexane for 30 min then the solids filtered off (warm), washed with diethyl ether:hexane (1:1), and dried. This solid was precipitated from hot toluene (hot filtration required to remove dibenzylated material) and dried to afford (I-28a) (107.2 g, 65%) as an off-white solid; 1H NMR (DMSO-d6): δ 8.50 (1H), 7.82 (1H), 7.34-7.29 (5H), 6.84 (2H), 6.43 (1H), 5.23 (2H); m/z 229.4 (M+H)+.
Example 47
(S)-benzyl 6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinate
Formula (1A-4) wherein R4 is
To Intermediate (I-8b) (16.28 g, 59.8 mmol) stirring in dry dichloromethane (400 mL) at room temperature under nitrogen was added 2 drops of DMF. Oxalyl chloride (11 mL, 130 mmol) was added dropwise. After the bubbling subsided the reaction was left stirring for 90 minutes and then concentrated under reduced pressure. Two successive portions of 1,2-dichloroethane were added and evaporated to remove all excess oxalyl chloride. The crude acid chloride was taken up in dichloromethane (150 mL) and stirred at room temperature. Intermediate (I-28a) (14.3 g, 62.5 mmol) and pyridine (10 mL, 130 mmol) were stirred in 400 mL dry dichloromethane. This was added to the acid chloride solution, using another 50 mL dry dichloromethane to complete the transfer. The mixture was left stirring at room temperature under nitrogen for 18 hours. The reaction was diluted with dichloromethane and water, and 1M aqueous phosphoric acid was added. The organic layer was separated and washed sequentially with dilute aqueous potassium carbonate, and brine. This was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a glass, which was taken up in hot ethyl acetate and stirred at room temperature. A precipitate appeared at about 30 minutes. The mixture was stirred for 16 hours and then filtered. The precipitate was washed with ethyl acetate and then diethyl ether and dried under high vacuum at 60° C. to afford the title compound as a white solid (17.8 g, 36.6 mmol, 61%). The mother liquor was evaporated and purified by silica gel chromatography on a 120 g pre-packed column, eluting with 40% ethyl acetate/heptane. The product fractions were combined, concentrated under reduced pressure, dried under high vacuum to a glass, and converted as previously described to additional product (3.5 g, 7.2 mmol, 12%, total yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (1H), 8.87-8.88 (1H), 8.29-8.32 (1H), 8.12-8.14 (1H), 7.93-7.94 (2H), 7.39-7.46 (2H), 7.30-7.37 (3H), 5.32 (2H), 5.21-5.25 (1H), 2.06-2.19 (2H), 1.26-1.63 (8H), 1.01-1.06 (1H); m/z 487.5 (M+H)+.
Example 48
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
Formula (1A-4) wherein R4 is

The compound of Example 47 (4.07 g, 8.35 mmol) was added to a 500 mL Parr bottle, followed by ethyl acetate (50 mL) and ethanol (100 mL). The mixture was warmed until all of the solid dissolved, and then cooled to room temperature. 10% Pd/C (450 mg) was added, and the mixture was shaken under 50 psi hydrogen for 90 minutes. The reaction was filtered through a microfiber filter. The filtrate was concentrated under reduced pressure and dried under high vacuum at 50° C. to afford product as a glassy solid (3.0 g, 7.75 mmol, 90.6%). The glassy solid was stirred overnight in diethyl ether. The white solid precipitate was filtered, washed with diethyl ether, suction dried, and dried under high vacuum at 50° C. to afford the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 13.10-13.25 (1H), 11.44 (1H), 8.83 (1H), 8.23-8.26 (1H), 8.09-8.12 (1H), 7.94-7.95 (2H), 5.22-5.26 (1H), 2.06-2.17 (2H), 1.29-1.64 (8H), 1.04-1.07 (1H);
m/z 397.3 (M+H)+.
THIS NMR IS FROM SUPPORTING INFO OF A JOURNAL
PAPER
Organic Process Research & Development (2012), 16(10), 1635-1645
http://pubs.acs.org/doi/abs/10.1021/op300194c

This work describes the process development and manufacture of early-stage clinical supplies of a hepatoselective glucokinase activator, a potential therapy for type 2 diabetes mellitus. Critical issues centered on challenges associated with the synthesis of intermediates and API bearing a particularly racemization-prone α-aryl carboxylate functionality. In particular, a T3P-mediated amidation process was optimized for the coupling of a racemization-prone acid substrate and a relatively non-nucleophilic amine. Furthermore, an unusually hydrolytically-labile amide in the API also complicated the synthesis and isolation of drug substance. The evolution of the process over multiple campaigns is presented, resulting in the preparation of over 110 kg of glucokinase activator.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (1)
Pressure Hydrogenation
PAPER
Journal of Medicinal Chemistry (2012), 55(3), 1318-1333
http://pubs.acs.org/doi/abs/10.1021/jm2014887

Glucokinase is a key regulator of glucose homeostasis, and small molecule allosteric activators of this enzyme represent a promising opportunity for the treatment of type 2 diabetes. Systemically acting glucokinase activators (liver and pancreas) have been reported to be efficacious but in many cases present hypoglycaemia risk due to activation of the enzyme at low glucose levels in the pancreas, leading to inappropriately excessive insulin secretion. It was therefore postulated that a liver selective activator may offer effective glycemic control with reduced hypoglycemia risk. Herein, we report structure–activity studies on a carboxylic acid containing series of glucokinase activators with preferential activity in hepatocytes versus pancreatic β-cells. These activators were designed to have low passive permeability thereby minimizing distribution into extrahepatic tissues; concurrently, they were also optimized as substrates for active liver uptake via members of the organic anion transporting polypeptide (OATP) family. These studies lead to the identification of 19 as a potent glucokinase activator with a greater than 50-fold liver-to-pancreas ratio of tissue distribution in rodent and non-rodent species. In preclinical diabetic animals, 19 was found to robustly lower fasting and postprandial glucose with no hypoglycemia, leading to its selection as a clinical development candidate for treating type 2 diabetes.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (19)

PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(24), 6588-6592
http://www.sciencedirect.com/science/article/pii/S0960894X13012638


Figure 1.
Structure of Hepatoselective GKA PF-04991532 (1).
References
Drug Metabolism & Disposition (2015), 43(2), 190-198
PLoS One (2014), 9(5), e97139/1-e97139/9,
Journal of Biological Chemistry (2012), 287(17), 13598-13610
Drug Discovery Today (2012), 17(9-10), 528-529
Biochemical Journal (2012), 441(3), 881-887.
///////////

Figure 1. Representative structures of glucokinase activators.

{kind=link}
{kind=link}
{kind=link}