
Home » Posts tagged 'SPECTRAL'
Tag Archives: SPECTRAL
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RIVAROXABAN 利伐沙班 ريفاروكسابان Ривароксабан SPECTRAL VISIT

RIVAROXABAN
 5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide
5-Chloro-N-{[(5S)-2-oxo-3-[4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide
Molecular formula: C19H18ClN3O5S, MW435.9
CAS 366789-02-8
BAY 59-7939, XARELTO

Patent Expiration Date:
Feb 8, 2021(US7157456),
Dec 11, 2020(US7585860 and US7592339)
Originator and Manufacturer:Bayer
Marketer in the US: Johnson & Johnson
Sales: $1.3 billion (2013)
Originator and Manufacturer:Bayer
Marketer in the US: Johnson & Johnson
Sales: $1.3 billion (2013)
Rivaroxaban (BAY 59-7939) is an oral anticoagulant invented and manufactured by Bayer;[3][4] in a number of countries it is marketed as Xarelto.[1] In the United States, it is marketed by Janssen Pharmaceutica.[5] It is the first available orally active direct factor Xa inhibitor. Rivaroxaban is well absorbed from the gut and maximum inhibition of factor Xa occurs four hours after a dose. The effects last approximately 8–12 hours, but factor Xa activity does not return to normal within 24 hours so once-daily dosing is possible.
In September 2008, Health Canada granted marketing authorization for rivaroxaban for the prevention of venous thromboembolism(VTE) in people who have undergone elective total hip replacement or total knee replacement surgery.[8]
In September 2008, the European Commission granted marketing authorization of rivaroxaban for the prevention of venous thromboembolism in adults undergoing elective hip and knee replacement surgery.[9]
On July 1, 2011, the U.S. Food and Drug Administration (FDA) approved rivaroxaban for prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE), in adults undergoing hip and knee replacement surgery.[5]
On November 4, 2011, the U.S. FDA approved rivaroxaban for stroke prophylaxis in patients with non-valvular atrial fibrillation.

The drug compound having the adopted name “Rivaroxaban” has chemical name, 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-l,3-oxazolidin-5- yljmethyl)-2-thiophenecarboxamide; and has the structural formula I,

Formula I
The commercial pharmaceutical product XARELTO® tablets, contains rivaroxaban as active ingredient. Rivaroxaban is a factor Xa inhibitor useful as oral anticoagulant. Rivaroxaban can be used for the prevention and treatment of various thromboembolic diseases, in particular of deep vein thrombosis (DVT), pulmonary embolism (PE), myocardial infract, angina pectoris and restenoses after angioplasty or aortocoronary bypass, cerebral stroke,
transitory ischemic attacks, and peripheral arterial occlusive diseases.
U.S. Patent No. 7, 157,456 describes Rivaroxaban and process for the preparation thereof. The process of US ‘456 for rivaroxaban involves reaction of 2-[(2S)-2-oxiranylmethyl]-lH-isoindole-l,3(2H)-dione with 4-(4-aminophenyl)-3-morpholinone to provide 2-((2R)-2-hydroxy-3- { [4-(3-oxo-4-morpholiny)phenyl]amino Jpropyl)- lH-isoindole- 1 ,3(2H)-dione, which on cyclization using Ν,Ν-carbonyl diimidazole to afford 2-({5S)-2-Oxo-3-[4-(3-oxo-4-morpholiny)phenyl]-l,3-oxazolidin-5-yl}methyl)-lH-isoindole-l,3(2H)-dione, which on reacted with methylamine followed by reaction with 5-chlorothiophene-2-carbonyl chloride to provide Rivaroxaban.
Various processes for the preparation of rivaroxaban, its intermediates, and related compounds are disclosed in U.S. Patent Nos. 7,585,860; 7,351,823, 7,816,355, and 8,101,609; patent application Nos. WO 2011/012321, WO 2012/156983, WO 2012/153155, WO 2013/053739, WO 2013/098833, WO 2013/156936, WO 2013/152168, WO 2013/120464, WO 2013/164833, US 2012/0283434 and US 2013/184457; and J. Med. Chem. 2005, 48, 5900-5908.

PAPER CONTAING SPECTRAL DATA
JOURNAL OF CHEMICAL RESEARCH v 35, issue 7, pg 400-4-1, 2011
An approach to the anticoagulant agent rivaroxaban via an isocyanate-oxirane cycloaddition promoted by MgI2.etherate
Chao Lia, Yingshuai Liua, Yongjun Zhangb and Xingxian Zhanga*
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
a College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310032, P. R. China
b Zhejiang Apeloa Medical Technology Co., Ltd, Dongyang 322118, P. R. China
A convergent and efficient synthesis of anticoagulant rivaroxaban was developed using the cycloaddition of commercially
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
available (R)-epichlorohydrin with 4-(morpholin-3-one)phenyl isocyanate catalysed by MgI2 etherate as the
key step, in 22% overall yield.
Keywords: (R)-epichlorohydrin, isocyanate, MgI2.etherate, rivaroxaban
* Correspondent. E-mail: mhmosslemin@yahoo.com
(Rivaroxaban) (1):1
rivaroxaban 1 (689 mg) in 88% yield, Rf = 0.30 (ethyl acetate), as a white solid,
m.p. 229.3–230.7 °C(lit.1, 230 °C).
[α]D20 = −37° (c = 0.5, DMSO) [lit.1, [α]D21 = –38°(c = 0.2985, DMSO)].
IR (KBr) (νmax /cm−1): 3343, 1724 (C=O), 1649(C=O), 1523, 1430, 808, 756
δH 3.60–3.62 (m, 2H), 3.71–3.73 (m,2H), 3.84–3.87 (dd, J = 6.5, 9.5 Hz, 1H), 3.96–3.98 (m, 2H), 4.20 (s,2H), 4.18–4.21 (m, 1H), 4.83–4.86 (m, 1H), 7.20 (d, J = 4.0 Hz, 1H),7.41 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 9.0 Hz, 2H), 7.69 (d, J = 4.0 Hz,1H), 8.99 (t, J = 5.5 Hz, 1H).
δC 42.19, 47.43, 49.00, 63.46, 67.71,71.30, 118.35, 125.92, 128.11, 128.43, 133.24, 136.48, 137.08,138.43, 154.08, 160.79, 165.95.
LIT REF 1=S. Roehrig, A. Straub, J. Pohlmann, T. Lampe, J. Pernerstorfer, K.Schlemmer, P. Reinemer and E. Perzborn, J. Med. Chem., 2005, 48, 5900.
STRUCTURE

SIMILARITY

Chemical structures of linezolid (top) and rivaroxaban (bottom). The shared structure is shown in blue.
Rivaroxaban bears a striking structural similarity to the antibiotic linezolid: both drugs share the same oxazolidinone-derived core structure. Accordingly, rivaroxaban was studied for any possible antimicrobial effects and for the possibility of mitochondrial toxicity, which is a known complication of long-term linezolid use. Studies found that neither rivaroxaban nor its metabolites have any antibiotic effect against Gram-positive bacteria. As for mitochondrial toxicity, in vitro studies found the risk to be low
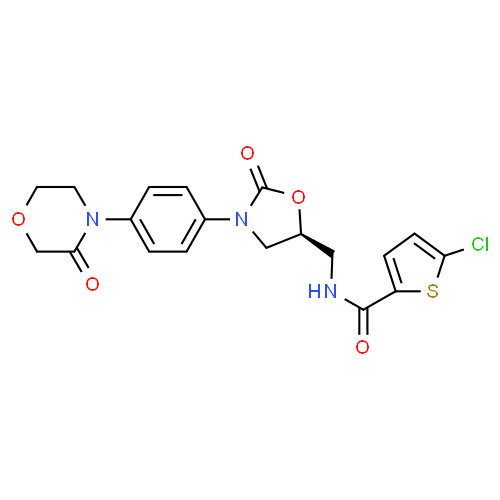
IH NMR PREDICT
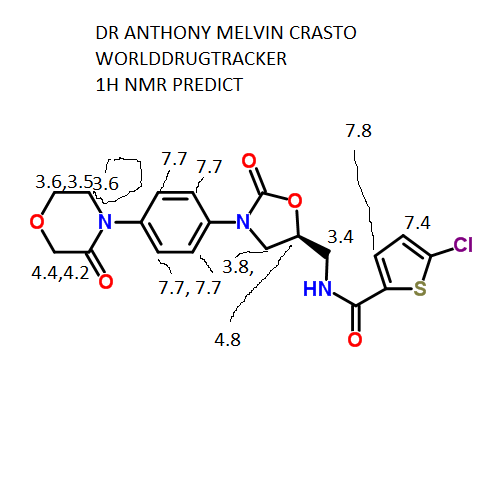
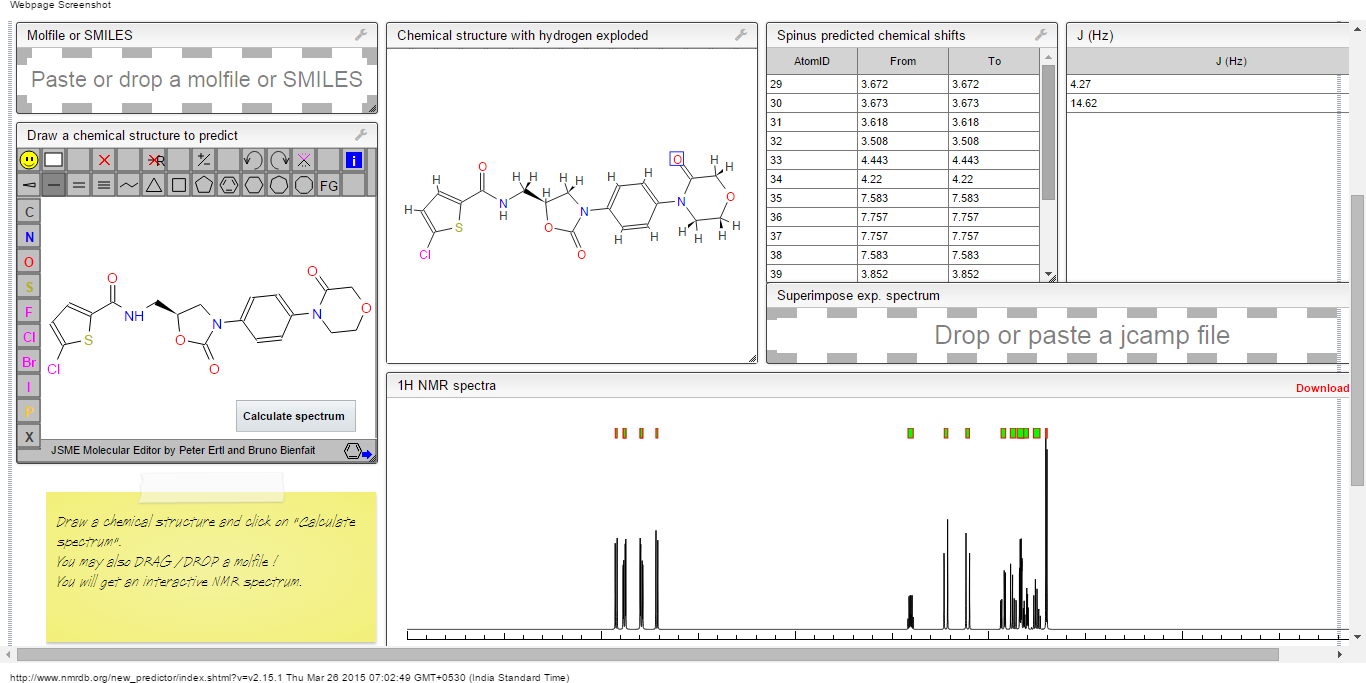

13 C NMR PREDICT


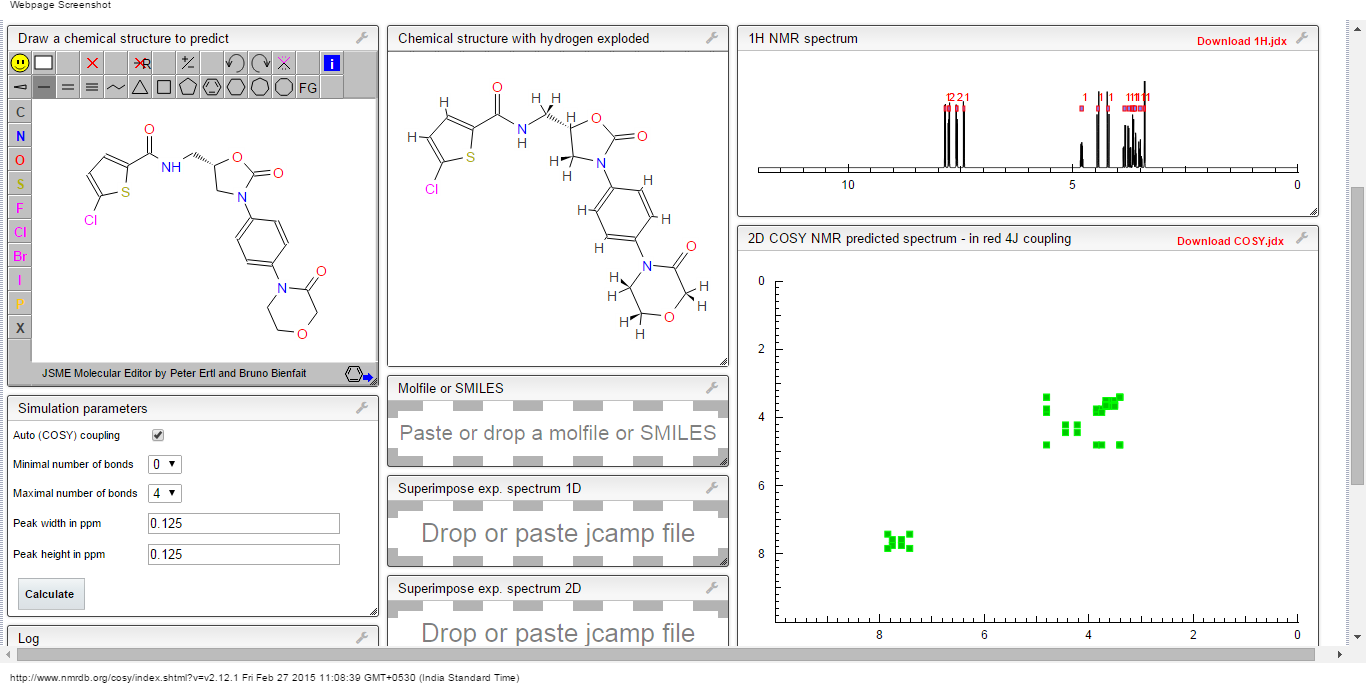
COSY NMR.
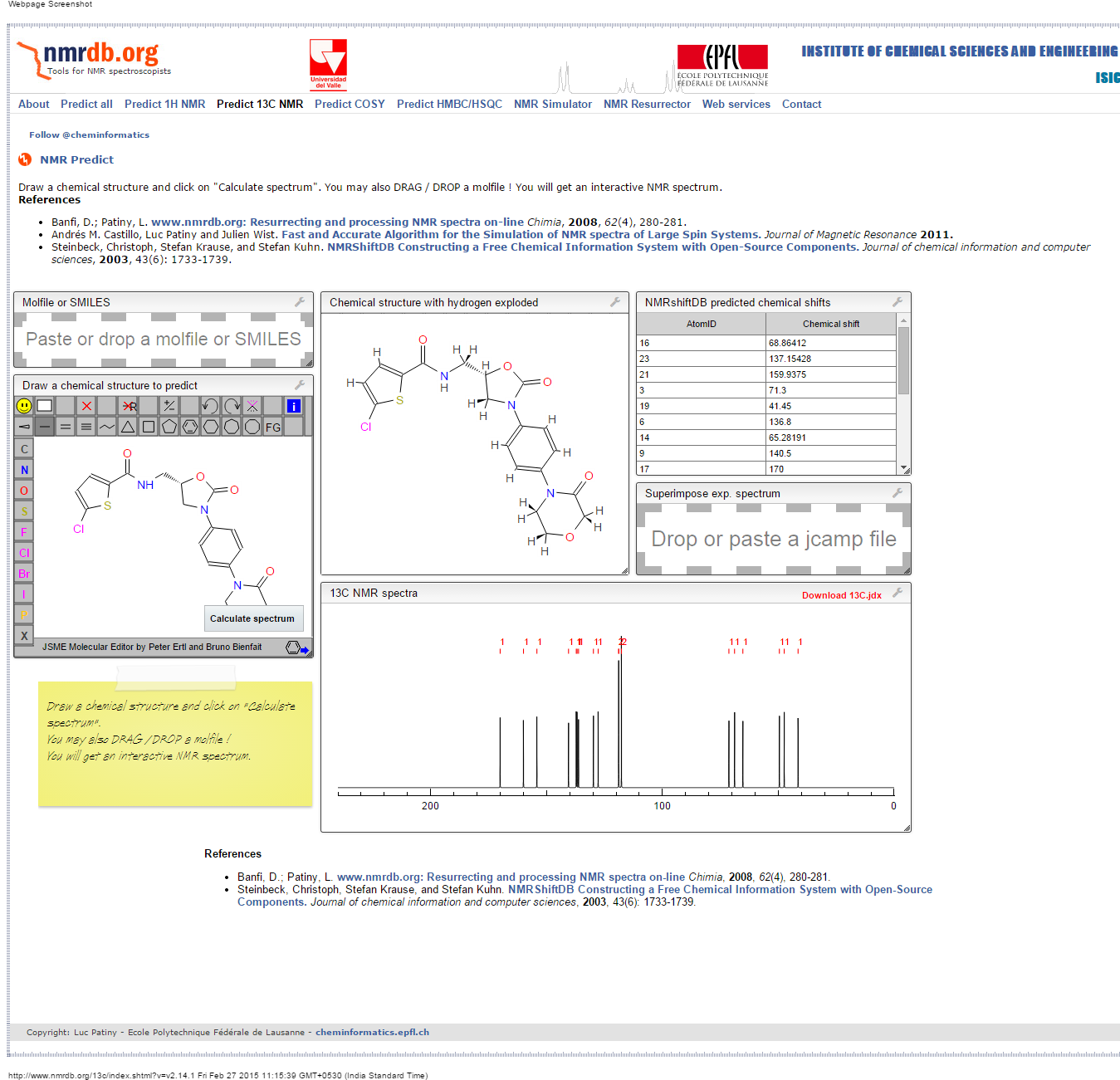
CLICK TO PREDICT..ALLOW SOME TIME TO LOAD ON NMRDB SITE…..CHECK JAVA AND FLASH SETTINGS
ABOVE PICTURES ARE THE ONES YOU WILL GET

New patent WO-2015104605
Process for preparing rivaroxaban – comprising the reaction of a thioester compound and its salts with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one.
Wockhardt Ltd
The synthesis of (II) via intermediate (I) is described (example 7, page 15)
4-{4-[(5S)-5-(Aminomethyl)-2-oxo-1,3-oxazolidin-3-yl]phenyl}morpholine-3-one (formula III) is (I) and rivaroxaban is (II) (claim 1, page 16).
The present invention relates to a process for the preparation of Rivaroxaban and its novel intermediates, or pharmaceutically acceptable salts thereof. The present invention provides novel intermediates, which may be useful for the preparation of Rivaroxaban or its pharmaceutically acceptable salts thereof. The process of preparation by using novel intermediate is very simple cost effective and may be employed at commercial scale. The product obtained by using novel intermediate yield the Rivaroxaban of purity 99% or more, when measured by HPLC. The present invention especially relates to a process for the preparation of Rivaroxaban from thioester of formula II, or a pharmaceutically acceptable salt thereof, wherein R is leaving group.
process includes the step of , reacting thioester of formula IIA or pharmaceutically acceptable salt thereof
Formula IIA

with 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one of formula III,
Formula III

Formula I
EXAMPLE 7: One pot process for Rivaroxaban
The triphenylphosphine (11.5g) and mercaptobenzothiazole disulphide (15.31g) were taken in methylene chloride and reaction mixture was stirred at 28°C -30°C for 1 hr. The 5-chlorothiophene-2-carboxylic acid (7.2g) and triethylamine (3.8 g) were added to the above reaction mixture. The reaction mixture is stirred at 0°C -25 °C for 1 hr. after 1 hr 4-{4-[(5S)-5-(aminomethyl)-2-oxo-l,3-oxazolidin-3-yl]phenyl}morpholine-3-one (lOg) and triethylamine (3.8g) were added. The resulting reaction mixture further stirred for 2 hrs. After completion of the reaction, water was added and stirred for 10 min. aqueous layer was separated and washed with methylene chloride. The organic layer was acidified to pH 6-7 with 2N hydrochloric acid and finally the organic layer was concentrated to get desired product. The product was purified and dried to yield Rivaroxaban.
Yield: 10.0 gm
Purity: 99.3 %
EXAMPLE 8: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent ethyl acetate and base potassium hydroxide were used to get the rivaroxaban.
EXAMPLE 9: One pot process for Rivaroxaban
Exemplified procedure in example 7 with the replacement of solvent acetonitile and base potassium carbonate were used, methylene chloride was added in the reaction mixture to extract the Rivaroxaban.
…………..
WO 01/47919 discloses ー species from 4_ (4_ aminophenyl) -3_ morpholinone (I) Preparation of rivaroxaban approach:

…………..
US 07/149522 discloses ー kind to 5_ chlorothiophenes _2_ carbonyl chloride (IV) is a method for preparing raw rivaroxaban in:

………….
http://www.google.com/patents/CN102786516A?cl=en

Preparation 6 rivaroxaban implementation

The 12.5 g (76.9 mmol) 5- chloro-thiophene-2-carboxylic acid was suspended in 35 g of toluene was heated to 80 で, at this temperature, a solution of 11.0 g (92.5 mmol) of thionyl chloride, reaction was continued for 30 min; then warmed to the boiling point of toluene was 120 ° C, and stirring was continued under reflux until cessation of gas; cooled to room temperature, the reaction mixture was concentrated under reduced pressure to remove excess thionyl chloride and toluene to give 5-chloro-thiophene-2-carbonyl chloride;
The 11.6 g (37.0 mmol) 4- {4 – [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3 -one hydrochloride was added 40ml of water, was added 4. 64 g (43 8 mmol.) Na2CO3 stirred and dissolved; then added 50 ml of toluene, was added dropwise at 10 ° C under the mixture, the mixture is 8. 0 g ( 44. 4 mmol) 5- chloro-thiophene-2-carbonyl chloride was dissolved in 15 ml of toluene, 20 min the addition was complete, then stirring was continued at room temperature, TLC monitoring progress of the reaction, 2 h after completion of the reaction; and the filter cake washed with water and washed with acetone to give a pale yellow solid 19. 6 g, used directly ko acid recrystallization, as a white solid 15. 2 g,
mp 227. 2 – 228. 1 ° C, [a] D21 = -38 2 ° (. c = 0. 30, DMS0), rivaroxaban yield of 94%, the total yield of 87.5% 0
1H-NMR (DMSO) 8: 3. 61 (. 2 H, t, / = 5 4 Hz), 3. 71 (2 H, t, / = 5 4 Hz.), 3.85 (IH, m ), 3.97 (2 H, t, J = 4. 5 Hz), 4. 19 (3 H, t, / = 7. 5 Hz), 4.84 (IH, m), 7. 19 (IH, d, / = 4. 2Hz), 7.40 (2 H, d, /=9.0 Hz), 7. 57 (2 H, t, /=9.0 Hz), 7. 69 (IH, d, J = 4. 19 Hz), 8. 96 (IH, t, / = 5. 7 Hz).
…………………
WO2013120465
EXAMPLE 28 (preparation of rivaroxaban)

10 g of the salt prepared according to Example 18 were suspended in 75 ml of N- methylpyrolidone, the suspension was heated at 50°C, then 14 ml of triethylamine was added and the mixture was heated at 60°C. This was followed by addition of 15.7 ml of a solution of 5-chlorothiophene-2-carboxylic acid chloride in toluene (2.46 M) and the reaction mixture was stirred and heated at 55°C for 15 minutes, then slowly cooled below 30°C, 75 ml were added and the turbid solution was filtered. The clear filtrate was stirred at 50°C, which was followed by addition of 15 ml of water and 75 ml of ethanol and stirring for 1 hour under slow cooling. The separated product was filtered off, washed with water (15 ml, 60°C), ethanol (2 x 25 ml) and dried in vacuo. 9.1 g (yield 81%) of rivaroxaban in the form of an off-white powder with the melt, point of 229.5-231°C was obtained, HPLC 99.95%, content of the ( )-isomer below 0.03%.
1H NMR (250 MHz, DMSO-D6), δ (ppm): 3.61 (t, 2H, CH2); 3.71 (m, 2H, CH2); 3.85 and 4.19 (m, 2×1 H, CH2); 3.97 (m, 2H, CH2); 4.19 (s, 2H, CH2); 4.84 (pent, 1H, CH); 7.18 (d, 1H); 7.40 (m, 2H); 7.56 (m, 2H); 7.68 (d, 1H); 8.95 (bt, 1H, NH).
13C NMR (250 MHz, DMSO-D6), δ (ppm): 42.2; 47.4; 49.0; 63.4; 67.7; 71.3; 1 18.3; 125.9; 128.1 ; 128.4; 133.2; 136.4; 137.0; 138.4; 154.0; 160.8; 165.9.
MS (m/z): 436.0729 (M+H)+. ation)

The optical isomer of rivaroxaban with the (R)- configuration was obtained by a process analogous to Example 28 starting from the salt prepared according to Example 19. The yield was 76%, HPLC 99.90%, content of the (5)-isomer below 0.03%. The NMR and MS spectra were in accordance with Example 28.
……………………..
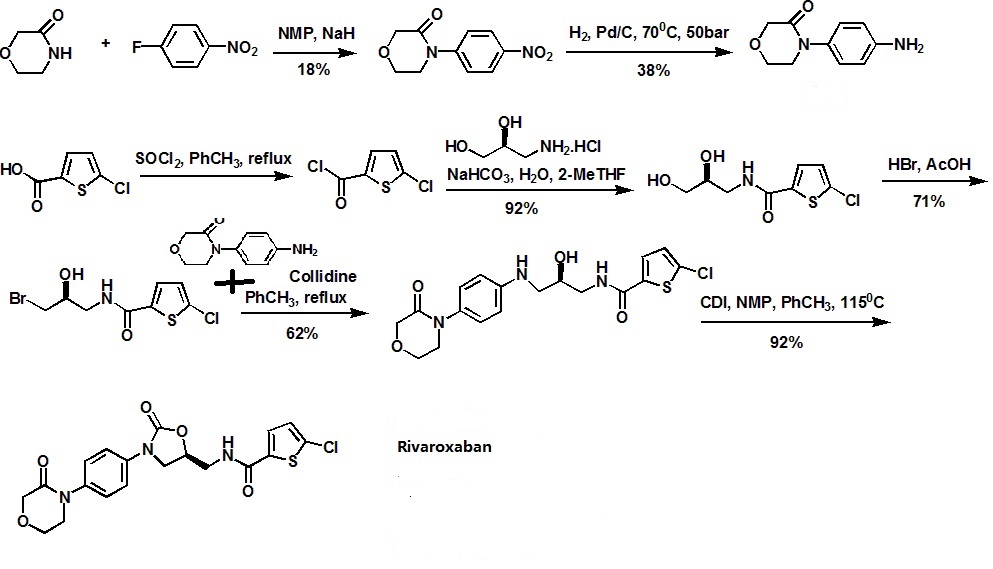
……………………
5- chloro-thiophene-2-chloride by condensation, bromide, with 4- (4-amino-phenyl) -3-morpholinone cyclization reaction rivaroxaban, the following reaction scheme 😦 References : W02005068456, US20070149522, DE10300111)

………………………
5- chloro-thiophene-2-chloride by condensation, oxidation, and 4- (4-amino-phenyl) -3-morpholinone cyclization reaction racemic rivaroxaban, since the epoxidation step is not give any stereoselectivity, the final chiral separation need to get rivaroxaban, the reaction scheme is as follows 😦 References: W0-0147919)

…………
4- (4- amino-phenyl) -3-morpholinone by condensation, cyclization, and potassium phthalimide after reaction with methyl chloroformate to give (S) -2 – hydroxy -3- (I, 3- dioxo – isoindoline-2-yl) propyl-4- (3-oxo –morpholino) phenyl carbamate, by condensation, methylamine and Ethanol action under profit rivaroxaban, the following reaction scheme (Ref: US20110034465):

……….
4- (4- amino-phenyl) -3-morpholinone (R) and – epichlorohydrin, in the DMF solvent phthalimide potassium salt was reacted with ammonia solution and then prepared to succeed amino compound, and 5-chloro-thiophene-2-chloride in pyridine catalyzed system benefit rivaroxaban, the following reaction scheme (Ref: W02009023233):

………….
4- (4- amino-phenyl) -3-morpholinone after condensation with (R) – epichlorohydrin, then the 5-chloro-thiophene-2-amide lithium chloride and tert-butyl the reaction of an alcohol potassium enrichment rivaroxaban, the following reaction scheme (Ref: US7816355):

……………….
3-chloro-1,2-propanediol by cyclization, the reaction with phthalimide, then with 4- (4-aminophenyl) -3-morpholinone reaction, CDI and hydrazine to give 4- {4- [(5S) -5- (aminomethyl) -2-oxo-1,3-oxazolidin-3-yl] phenyl} morpholin-3-one under the influence, in pyridine and under the action of tetrahydrofuran and 5-chloro-thiophene-2-chloride benefit rivaroxaban, the following reaction scheme (Reference: Gutcait, A. et al Tetrahedron:.. Asymmetry 1996, 7 (6), 1641-1648 Roehrig, .. S. et al J. Med Chem 2005,48 (19), 5900-5908)..:

…………..

http://www.google.com/patents/CN102702186A?cl=zh

Compound rivaroxaban Synthesis Example 7 formula (X), [0071] Example
[0072] Method One:

[0074] The compound of formula (VIII) of (180mg, 0. 618mmol), Ni chloride (5mL) and tris ko amine (187mg,
I. 85mmol) added to the reaction flask, stirred at room temperature for 10 minutes, cooled to 0 ° C, a solution of 5-chloro-2-thiophene chloride (224mg, 1.24mm0l), stirred at room temperature overnight; after the completion of the reaction, spin dry, rinse with anhydrous alcohol ko, filtered, washed ko anhydrous alcohol three times to obtain a white solid product rivaroxaban (215mg, embodiments of the total yield of 7,8 80%).
[0075] 1H-Mffi (DMSC) JOOMHz, δ d m):…. 3 61 (t, 2H, J = 5 6Hz), 3. 71 (t, 2H, J = 5 2Hz), 3 89 ( m, 1H), 3. 97 (t, 2H, J = 4. 4Hz), 4. 20 (m, 3H), 4. 85 (m, 1H), 7. 18 (d, 1H, J = 4. 0Hz), 7. 40 (d, 2H, J = 8. 8Hz), 7. 56 (d, 2H, J = 8. 8Hz), 7. 73 (d, 1H, J = 4. 0Hz).
The method of writing is:

[0078] The compound 5_ gas – oh -I- thiophene carboxylic acid (500mg, 3. 08mmol), MsCl (702mg, 6. 1 Bmmol) and sodium bicarbonate (. 517mg, 6 16mmol) was suspended in THF (20ml) in , heated to 60 ° C with stirring 45min, a large white suspension washed out; the reaction mixture was cooled to room temperature, the compound of formula VIII was added portionwise (800mg, 2 75mmol.), stirred for 5 hours, after completion of the reaction distilled THF, was added after the residue was cooled to room temperature, water (IOOml), at room temperature embrace Cheung 30min, filtered, and the filter cake washed with cold water, dried and added to a ko-ol (5ml) was heated at reflux for I hour. After cooling, stirred for 5 hours at room temperature After filtration to give the product of formula (X) of the compound rivaroxaban (719mg, 60%)
References
- “Xarelto: Summary of Product Characteristics”. Bayer Schering Pharma AG. 2008. Retrieved 2009-02-11.
- Abdulsattar, Y; Bhambri, R; Nogid, A (May 2009). “Rivaroxaban (xarelto) for the prevention of thromboembolic disease: an inside look at the oral direct factor xa inhibitor.”.P & T : a peer-reviewed journal for formulary management 34 (5): 238–44.PMID 19561868.
- Roehrig S, Straub A, Pohlmann J et al. (September 2005). “Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3- [4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene- 2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor”. Journal of Medicinal Chemistry 48 (19): 5900–8. doi:10.1021/jm050101d.PMID 16161994.
- Perzborn, Elisabeth; Roehrig, Susanne; Straub, Alexander; Kubitza, Dagmar; Misselwitz, Frank (17 December 2010). “The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor”. Nature Reviews Drug Discovery 10 (1): 61–75. doi:10.1038/nrd3185.
- “FDA Approves XARELTO® (rivaroxaban tablets) to Help Prevent Deep Vein Thrombosis in Patients Undergoing Knee or Hip Replacement Surgery” (Press release).Janssen Pharmaceutica. 2011-07-01. Retrieved 2011-07-01.
- Gómez-Outes, A; Terleira-Fernández, AI; Calvo-Rojas, G; Suárez-Gea, ML; Vargas-Castrillón, E (2013). “Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups.”. Thrombosis 2013: 640723. doi:10.1155/2013/640723. PMC 3885278.PMID 24455237.
- Brown DG, Wilkerson EC, Love WE (March 2015). “A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons”.Journal of the American Academy of Dermatology 72 (3): 524–34.doi:10.1016/j.jaad.2014.10.027. PMID 25486915.
- “Bayer’s Xarelto Approved in Canada” (Press release). Bayer. 2008-09-16. Retrieved2010-01-31.
- “Bayer’s Novel Anticoagulant Xarelto now also Approved in the EU” (Press release).Bayer. 2008-02-10. Retrieved 2010-01-31.
- “Medication Guide–Xarelto” (PDF). http://www.fda.gov/. U.S. Food and Drug Administration. Retrieved 1 September 2014.
- “Xarelto Side Effects”. http://www.webmd.com/. WebMD. Retrieved 1 September2014.
- “Xarelto Side Effects Center”. http://www.rxlist.com/. RxList. Retrieved 1 September2014.
- Eriksson BI, Borris LC, Dahl OE et al. (November 2006). “A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement”. Circulation 114 (22): 2374–81.doi:10.1161/CIRCULATIONAHA.106.642074. PMID 17116766.
- Eriksson BI, Borris LC, Friedman RJ et al. (June 2008). “Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty”. The New England Journal of Medicine 358(26): 2765–75. doi:10.1056/NEJMoa0800374. PMID 18579811.
- Kakkar AK, Brenner B, Dahl OE et al. (July 2008). “Extended duration rivaroxaban versus short-term enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: a double-blind, randomised controlled trial”. Lancet 372 (9632): 31–9.doi:10.1016/S0140-6736(08)60880-6. PMID 18582928.
- Lassen MR, Ageno W, Borris LC et al. (June 2008). “Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty”. The New England Journal of Medicine358 (26): 2776–86. doi:10.1056/NEJMoa076016. PMID 18579812.
- Turpie A, Bauer K, Davidson B et al. “Comparison of rivaroxaban – an oral, direct factor Xa inhibitor – and subcutaneous enoxaparin for thromboprophylaxis after total knee replacement (RECORD4: a phase 3 study) / European Federation of National Associations of Orthopaedics and Traumatology Annual Meeting; May 29 – June 1, 2008; Nice, France, Abstract F85”. Journal of Bone & Joint Surgery, British Volume 92–B (SUPP II): 329.
- Turpie AG, Lassen MR, Davidson BL et al. (May 2009). “Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty (RECORD4): a randomised trial”.Lancet 373 (9676): 1673–80. doi:10.1016/S0140-6736(09)60734-0. PMID 19411100.
- ClinicalTrials.gov. “Randomized, Double-Blind Study Comparing Once Daily Oral Rivaroxaban With Adjusted-Dose Oral Warfarin for the Prevention of Stroke in Subjects With Non-Valvular Atrial Fibrillation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “MAGELLAN – Multicenter, Randomized, Parallel Group Efficacy Superiority Study in Hospitalized Medically Ill Patients Comparing Rivaroxaban with Enoxaparin”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Once-Daily Oral Direct Factor Xa Inhibitor Rivaroxaban in the Long-Term Prevention of Recurrent Symptomatic Venous Thromboembolism in Patients With Symptomatic Deep-Vein Thrombosis or Pulmonary Embolism. The Einstein-Extension Study”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis (DVT) Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Pulmonary Embolism (PE) With Or Without Symptomatic Deep-Vein Thrombosis: Einstein-PE Evaluation”. Retrieved 2009-02-11.
- ClinicalTrials.gov. “A Randomized, Double-Blind, Placebo-Controlled, Event-Driven Multicenter Study to Evaluate the Efficacy and Safety of Rivaroxaban in Subjects With a Recent Acute Coronary Syndrome”. Retrieved 2009-02-11.
- “Venous Thromboembolic Event (VTE) Prophylaxis in Medically Ill Patients (MAGELLAN)”. ClinicalTrials.gov. 11 March 2011. Retrieved 15 April 2011.
- Hughes, Sue (5 April 2011). “MAGELLAN: Rivaroxaban prevents VTE in medical patients, but bleeding an issue”. theheart.org. Retrieved 15 April 2011.
- “About the MAGELLAN Study”. Bayer HealthCare. Retrieved 15 April 2011.
- Bauersachs, M.D., Rupert; The EINSTEIN Investigators (December 23, 2010). “Oral Rivaroxaban for Symptomatic Venous Thromboembolism”. The New England Journal of Medecine 363 (26): 2499–2510. doi:10.1056/NEJMoa1007903. PMID 21128814. Retrieved 4 April 2011.
- “Oral Direct Factor Xa Inhibitor Rivaroxaban In Patients With Acute Symptomatic Deep-Vein Thrombosis Without Symptomatic Pulmonary Embolism: Einstein-DVT Evaluation”. clinicaltrials.gov. Retrieved 15 April 2011.
- European Medicines Agency (2008). “CHP Assessment Report for Xarelto (EMEA/543519/2008)” (PDF). Retrieved 2009-06-11.
- Turpie AG (January 2008). “New oral anticoagulants in atrial fibrillation”. European Heart Journal 29 (2): 155–65. doi:10.1093/eurheartj/ehm575. PMID 18096568.
| WO2013120465A1 * | Feb 18, 2013 | Aug 22, 2013 | Zentiva, K.S. | A process for the preparation of rivaroxaban based on the use of (s)-epichlorohydrin |
| WO2001047919A1 | Dec 11, 2000 | Jul 5, 2001 | Bayer Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| WO2004060887A1 | Dec 24, 2003 | Jul 22, 2004 | Bayer Healthcare Ag | Method for producing 5-chloro-n-({5s)-2-oxo-3-[4-(3-oxo-4-morpholinyl)-phenyl]-1,3-oxazolidin-5-yl}-methyl)-2-thiophene carboxamide |
| WO2007116284A1 | Mar 26, 2007 | Oct 18, 2007 | Pfizer Prod Inc | Process for preparing linezolid |
| WO2009023233A1 | Aug 14, 2008 | Feb 19, 2009 | Concert Pharmaceuticals Inc | Substituted oxazolidinone derivatives |
| WO2010043110A1 | Oct 9, 2009 | Apr 22, 2010 | Changzhou Multiple Dimension Institute Of Industry Technology Co., Ltd. | A preparation method of high-purity l-carnitine |
| WO2010082627A1 | Jan 15, 2010 | Jul 22, 2010 | Daiso Co., Ltd. | Process for producing 2-hydroxymethylmorpholine salt |
| WO2010124835A1 | Apr 27, 2010 | Nov 4, 2010 | Belte Ag | Aluminium-silicon diecasting alloy for thin-walled structural components |
| WO2011080341A1 | Jan 3, 2011 | Jul 7, 2011 | Enantia, S.L. | Process for the preparation of rivaroxaban and intermediates thereof |
| WO2011098501A1 | Feb 10, 2011 | Aug 18, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2011102640A2 | Feb 16, 2011 | Aug 25, 2011 | Hanmi Holdings Co., Ltd. | Method for preparing sitagliptin and amine salt intermediates used therein |
| WO2012159992A1 * | May 18, 2012 | Nov 29, 2012 | Interquim, S.A. | Process for obtaining rivaroxaban and intermediate thereof |
| CN102786516A * | Aug 21, 2012 | Nov 21, 2012 | 湖南师范大学 | Method for synthesizing rivaroxaban |
| US7157456 | Dec 11, 2000 | Jan 2, 2007 | Bayer Healthcare Ag | Substituted oxazolidinones and their use in the field of blood coagulation |
| US7816355 * | Apr 28, 2009 | Oct 19, 2010 | Apotex Pharmachem Inc | Processes for the preparation of rivaroxaban and intermediates thereof |
| US20110034465 | Feb 10, 2011 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
| CN101560209A * | Apr 15, 2008 | Oct 21, 2009 | Shenyang, one hundred million Leo Pharmaceutical Co., Ltd. | Pyrimidine oxazolidinone compound and preparation method comprising |
| CN101619061A * | Aug 11, 2009 | Jan 6, 2010 | Shenyang Pharmaceutical University | Cyanopyridyl substituted oxazolidinone compounds |
| CN101821260A * | Aug 14, 2008 | Sep 1, 2010 | Consett Pharmaceuticals Ltd. | Substituted oxazolidinone derivatives |
| CN102250076A * | May 27, 2011 | Nov 23, 2011 | Hengdian Group homes Chemical Co., Ltd. | One kind of rivaroxaban Rivaroxaban intermediates and preparation methods |
| CN102250077A * | Jun 15, 2011 | Nov 23, 2011 | Zhejiang University | A method for intermediate and rivaroxaban Rivaroxaban for the synthesis of |
| CN102311400A * | Jun 29, 2010 | Jan 11, 2012 | Xiang really Biotechnology Co., Ltd. | Aminomethyl-3-aryl-2-oxazolidinone class method – Preparation of L-5- |
| CN102320988A * | Jun 3, 2011 | Jan 18, 2012 | Shanghai Institute of Organic Chemistry | 4- (4-aminophenyl) -3-morpholinone intermediate amide, the synthesis method and uses |
| EP2354128A1 * | Feb 10, 2010 | Aug 10, 2011 | Sandoz Ag | Method for the preparation of rivaroxaban |
| WO2010124385A1 * | Apr 28, 2010 | Nov 4, 2010 | Apotex Pharmachem Inc. | Processes for the preparation of rivaroxaban and intermediates thereof |
FROM THE NET
RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4 …
32 mins ago – RIVAROXABAN 5-Chloro-N-{[(5S) 2-oxo-3 [4-(3-oxo-4-morpholinophenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide (Rivaroxaban) (1):1 rivaroxaban 1 …
WO 2015104605.new patent on Rivaroxaban, Wockhardt …
1 hour ago – WO 2015104605.new patent on Rivaroxaban, Wockhardt Ltd Process for preparing rivaroxaban – comprising the reaction of a thioester compound and its salts …

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(S)-5-chloro-N-{[2-oxo-3-[4-(3-oxomorpholin-4-yl)
phenyl]oxazolidin-5-yl]methyl} thiophene-2-carboxamide |
|
| Clinical data | |
| Trade names | Xarelto |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Licence data | EMA:Link, US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
oral |
| Pharmacokinetic data | |
| Bioavailability | 80% to 100%; Cmax = 2 – 4 hours (10 mg oral)[1] |
| Metabolism | CYP3A4 , CYP2J2 and CYP-independent mechanisms[1] |
| Biological half-life | 5 – 9 hours in healthy subjects aged 20 to 45[1][2] |
| Excretion | 2/3 metabolized in liver and 1/3 eliminated unchanged[1] |
| Identifiers | |
| CAS Registry Number | 366789-02-8 |
| ATC code | B01AX06 |
| PubChem | CID: 6433119 |
| IUPHAR/BPS | 6388 |
| DrugBank | DB06228 |
| ChemSpider | 8051086 |
| UNII | 9NDF7JZ4M3 |
| ChEMBL | CHEMBL198362 |
| Synonyms | Xarelto, BAY 59-7939 |
| Chemical data | |
| Formula | C19H18ClN3O5S |
| Molecular mass | 435.882 g/mol |

Rivaroxaban, a FXa inhibitor, is the active ingredient in XARELTO Tablets with the chemical name 5-Chloro-N-({(5S)-2-oxo-3-[4-(3-oxo-4-morpholinyl)phenyl]-1,3-oxazolidin-5yl}methyl)-2-thiophenecarboxamide. The molecular formula of rivaroxaban is C19H18ClN3O5S and the molecular weight is 435.89. The structural formula is:
 |
Rivaroxaban is a pure (S)-enantiomer. It is an odorless, non-hygroscopic, white to yellowish powder. Rivaroxaban is only slightly soluble in organic solvents (e.g., acetone, polyethylene glycol 400) and is practically insoluble in water and aqueous media.
Each XARELTO tablet contains 10 mg, 15 mg, or 20 mg of rivaroxaban. The inactive ingredients of XARELTO are: croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. Additionally, the proprietary film coating mixture used for XARELTO 10 mg tablets is Opadry® Pink and for XARELTO 15 mg tablets is Opadry® Red, both containing ferric oxide red, hypromellose, polyethylene glycol 3350, and titanium dioxide, and for XARELTO 20 mg tablets is Opadry® II Dark Red, containing ferric oxide red, polyethylene glycol 3350, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.

SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
////////SEE ABAN SERIES AT…………http://organicsynthesisinternational.blogspot.in/p/aban-series.html
TOFACITINIB 的合成, トファシチニブ, Тофацитиниб, توفاسيتين يب SPECTRAL VISIT
Tofacitinib Citrate, 的合成
托法替布, トファシチニブクエン酸塩, Тофацитиниба Цитрат
3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt
CAS : 540737-29-9
ROTATION +
Tofacitinib; Tasocitinib;
477600-75-2 base ; CP-690550;
3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile;
3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt
CP 690550 Tofacitinib; CP-690550; CP-690550-10; Xeljanz; Jakvinus; Tofacitinib citrate
Trademarks: Xeljanz; Jakvinus
MF: C16H20N6O
CAS : 477600-75-2 BASE ; 540737-29-9(citrate) 3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
Molecular Weight: 312.369
SMILES: C[C@@H]1CCN(C[C@@H]1N(C)C2=NC=NC3=C2C=CN3)C(=O)CC#N
Activity: Treatment of Rheumatoid Arthritis; RA Treatment, JAK Inhibitor; Protein Kinase Inhibitor; JAK3 Inhibitor; Janus Kinase 3 Inhibitor; JAK-STAT Signaling Pathway; JAK1 Kinase Inhibitor; Selective Immunosuppressants
Status: Launched 2012
Originator: Pfizer 
Pfizer Inc’s oral JAK inhibitor tofacitinib was approved on November 6, 2012 by US FDA for the treatment of rheumatoid arthritis.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Tofacitinib (trade names Xeljanz and Jakvinus, formerly tasocitinib,[1] CP-690550[2]) is a drug of the janus kinase (JAK) inhibitor class, discovered and developed by Pfizer. It is currently approved for the treatment of rheumatoid arthritis (RA) in the United States,Russia, Japan and many other countries, is being studied for treatment of psoriasis, inflammatory bowel disease, and other immunological diseases, as well as for the prevention of organ transplant rejection. 

An Improved and Efficient Process for the Preparation of Tofacitinib Citrate
Yogesh S. Patil, Nilesh L. Bonde, Ankush S. Kekan, Dhananjay G. Sathe*1, and Arijit Das
Publication Date (Web): November 17, 2014 (Article)
DOI: 10.1021/op500274j
MS m/z 313 (M+ + 1);
mp 201–202 °C;
|
1H NMR (CDCl3) δ 8.34 (s, 1H), δ 7.38 (d, 1H, J = 2.4 Hz), δ 6.93 (d, 1H, J = 2.4 Hz), δ 4.97 (m, 1H), δ 3.93–4.03 (m, 4H), δ 3.66 (m, 1H), δ 3.50 (m, 4H), δ 2.91 (d, 2H, J = 15.6 Hz), δ 2.80 (t, 2H, J = 12.8 Hz), δ 2.55 (m, 1H), δ 1.99 (m, 1H), δ 1.77 (m, 1H), δ 1.13–1.18 (m, 3H).
|



TEAMWORK
Part of the Pfizer group responsible for Xeljanz: Front row, from left: Sally Gut Ruggeri, Chakrapani Subramanyam, Eileen Elliott Mueller, and Frank Busch. Second row, from left: Matthew Brown, Mark Flanagan, and Robert Dugger. Back row, from left: Elizabeth Kudlacz and Douglas Ball.
Credit: Pfizer
Mark Flanagan, who was on the team at Pfizer that discovered Xeljanz, (tofacitinib citrate), an oral treatment for rheumatoid arthritis, remembers testing the drug in a rat model and seeing the drug decrease the level of inflammation in the rats’ footpads. “What we look for is physical measurements of the size of the joint. In the control animals, there was quite a bit of inflammation in the joints, whereas animals treated with different doses of the drug showed a dose-dependent decrease in the size of the joint. “Tofacitinib showed robust efficacy in the first such study run. I can remember the excitement that this data generated on the team,” he says.
Tofacitinib, chemically known as (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3- d]pyrimidin-4-ylamino)-B-oxo-l -piperidinepi panenitrile, is represented Formula I. Tofacitinib citrate, a janus kinase inhibitor, is approved as XELJANZ® tablets for treatment .of rheumatoid arthritis.

Various intermediates and processes for preparation of tofacitinib are disclosed in patents like US7301 023 and US8232394.

Formula I or isomers or a mixture of isomers thereof by following any method provided in the prior art, for example, by following Example 14 of U.S. Patent No. RE41,783 or by following Example 6 of U.S. Patent No. 7,301,023. Tofacitinib of Formula I or isomers of tofacitinib or a mixture of isomers thereof may be converted into a salt by following any method provided in the prior art, for example, by following Example 1 of U.S. Patent No. 6,965,027 or by following Example 1 or Example 8 of PCT Publication No. WO 2012/135338. The potential significance of JAK3 inhibition was first discovered in the laboratory of John O’Shea, an immunologist at the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH).[5] In 1994, Pfizer was approached by the NIH to form a public-private partnership in order to evaluate and bring to market experimental compounds based on this research.[5] Pfizer initially declined the partnership but agreed in 1996, after the elimination of an NIH policy dictating that the market price of a product resulting from such a partnership would need to be commensurate with the investment of public taxpayer revenue and the “health and safety needs of the public.”[5] The drug discovery, preclinical development, and clinical development of tofacitinib took place exclusively at Pfizer.[6] In November 2012, the U.S. Food and Drug Administration (FDA) approved tofacitinib for treatment of rheumatoid arthritis. Once on the market, rheumatologists complained that the $2,055 a month wholesale price was too expensive, though the price is 7% less than related treatments.[6] A 2014 study showed that tofacitinib treatment was able to convert white fat tissues into more metabolically active brown fat, suggesting it may have potential applications in the treatment of obesity.[7] It is an inhibitor of the enzyme janus kinase 1 (JAK1) and janus kinase 3 (JAK 3) , which means that it interferes with the JAK-STAT signaling pathway, which transmits extracellular information into the cell nucleus, influencing DNA transcription.[3] Recently it has been shown in a murine model of established arthritis that tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. This efficacy in this disease model correlated with the inhibition of both JAK1 and 3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to inhibition of JAK3.[4]
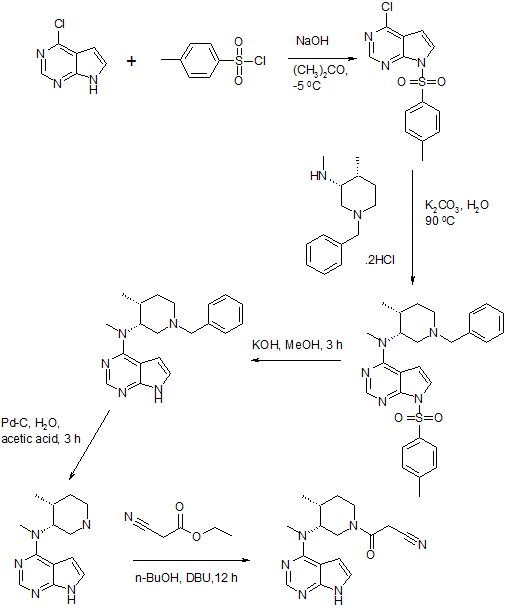
Preparation of 3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt (Tofacitinib citrate, Xeljanz, CP-690550-10)
To a round-bottomed flask fitted with a temperature probe, condenser, nitrogen source, and heating mantle, methyl-[(3R,4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (5.0 g, 20.4 mmol) was added followed by 1-butanol (15 mL), ethyl cyanoacetate (4.6 g, 40.8 mmol), and DBU (1.6 g, 10.2 mmol). The resulting amber solution was stirred at 40 °C for 20 h. Upon reaction completion, citric acid monohydrate (8.57 g, 40.8 mmol) was added followed by water (7.5 mL) and 1-butanol (39.5 mL). The mixture was heated to 81 °C and held at that temperature for 30 min. The mixture was then cooled slowly to 22 ºC and stirred for 2 h. The slurry was filtered and washed with 1-butanol (20 mL). The filter cake was dried in a vacuum oven at 80 °C to afford 9.6 g (93%) of tofacitinib citrate as an off-white solid.
1H NMR (500 MHz, d6-DMSO): δ 8.14 (s, 1H), 7.11 (d, J=3.6 Hz, 1H), 6.57 (d, J=3.6 Hz, 1H), 4.96 (q, J=6.0 Hz, 1H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1H), 2.45-2.41 (m, 1H), 1.81 (m, 1H), 1.69-1.65 (m, 1H), 1.04 (d, J=6.9 Hz, 3H).
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
PAPER
3-((3R,4R)-4-Methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (1) Monocitrate
J. Med. Chem., 2010, 53 (24), pp 8468–8484
DOI: 10.1021/jm1004286
1monocitrate as a white crystalline solid (mp = 201 dec).
LRMS: m/z 313.2 (MH+).
1H NMR (400 MHz) (D2O) δ HOD: 0.92 (2 H, d, J = 7.2 Hz), 0.96 (1 H, d, J = 7.6 Hz), 1.66 (1 H, m), 1.80 (1 H, m), 2.37 (1 H, m), 2.58 (2 H, 1/2 ABq, J = 15.4 Hz), 2.70 (2 H, 1/2 ABq, J = 15.4 Hz), 3.23 (2 H, s), 3.25 (1 H, s), 3.33 (1 H, m), 3.46 (1 H, m), 3.81 (4 H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Anal. Calcd for C22H28N6O8: C, 52.38; H, 5.59; N, 16.66. Found: C, 52.32; H, 5.83; N, 16.30. For additional characterization of the monocitrate salt of 1 see WO 03/048162.
NMR PREDICT


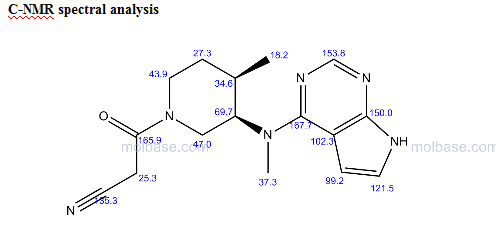

References:
Weiling Cai, James L. Colony,Heather Frost, James P. Hudspeth, Peter M. Kendall, Ashwin M. Krishnan,Teresa Makowski, Duane J. Mazur, James Phillips, David H. Brown Ripin, Sally Gut Ruggeri, Jay F. Stearns, and Timothy D. White; Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines; Organic Process Research & Development 2005, 9, 51−56
Ripin, D. H.B.; 3-amino-piperidine derivatives and methods of manufacture, US patent application publication, US 2004/0102627 A1
Ruggeri, Sally, Gut;Hawkins, Joel, Michael; Makowski, Teresa, Margaret; Rutherford, Jennifer, Lea; Urban,Frank,John;Pyrrolo[2,3-d]pyrimidine derivatives: their intermediates and synthesis, PCT pub. No. WO 2007/012953 A 2, US20120259115 A1, United States Patent US8232393. Patent Issue Date: July 31, 2012
Kristin E. Price, Claude Larrive´e-Aboussafy, Brett M. Lillie, Robert W. McLaughlin, Jason Mustakis, Kevin W. Hettenbach, Joel M. Hawkins, and Rajappa Vaidyanathan; Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates, Organic Letters, 2009, vol.11, No.9, 2003-2006
MORE NMR PREDICT

Tofacitinib 
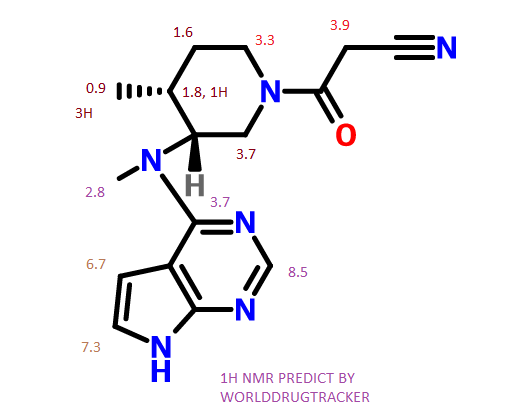
13C NMR PREDICT 

SEE…….https://newdrugapprovals.org/2015/07/24/tofacitinib-%E7%9A%84%E5%90%88%E6%88%90-spectral-visit/
COSY PREDICT 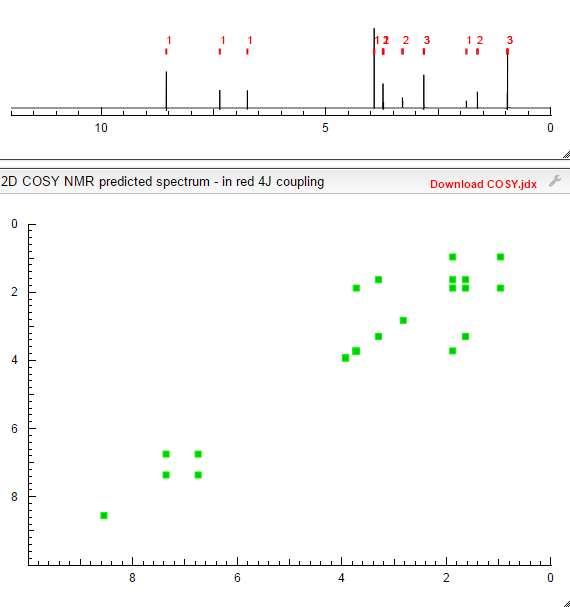 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
NMR PICTURE FROM THE NET

PAPER
Volume 54, Issue 37, 11 September 2013, Pages 5096–5098

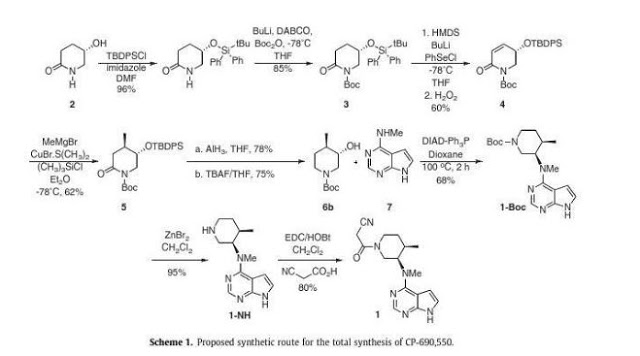
Asymmetric total synthesis of Tofacitinib
- a Laboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, University of Talca, P.O. Box 747, Talca, Chile
- b Laboratory of Natural Products, Department of Chemistry, University of Antofagasta, P.O. Box 170, Antofagasta, Chile
http://dx.doi.org/10.1016/j.tetlet.2013.07.042
Abstract
A novel stereoselective synthesis of Tofacitinib (CP-690,550), a Janus tyrosine kinase (JAK3) specific inhibitor, has been achieved starting from (5S)-5-hydroxypiperidin-2-one in 10 steps from 2 with a 9.5% overall yield. The potentiality of this synthetic route is the obtention of tert-butyl-(3S,4R)-3-hydroxy-4-methylpiperidine-1-carboxylate (6b) as a new chiral precursor involved in the synthesis of CP690,550, in a three-step reaction, without epimerizations, rather than the 5 or more steps used in described reactions to achieve this compound from analogues of 6b.
Graphical abstract

…………………. Tofacitinib synthesis: US2001053782A1

Tofacitinib synthesis: WO2002096909A1

Tofacitinib synthesis: Org Process Res Dev 2014, 18(12), 1714-1720 (also from a chinese publication, same procedure just slight changes in reagents/conditions)
References:
1. Blumenkopf, T. A.; et. al. Pyrrolo[2,3-d]pyrimidine compounds. US2001053782A1
2. Flanagan, M. E.; et. al. Optical resolution of (1-benzyl-4-methylpiperidin-3-yl) -methylamine and the use thereof for the preparation of pyrrolo 2,3-pyrimidine derivatives as protein kinases inhibitors. WO2002096909A1
3. Das, A.; et. al. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org Process Res Dev2014, 18(12), 1714-1720.
Synthesis of Tofacitinib Citrate

PATENT https://www.google.co.in/patents/WO2003048162A1?cl=en The crystalline form of the compound of this invention 3-{4-methyl-3-[methyl- (7H-pyrrolot2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile mono citrate salt is prepared as described below. Scheme 1


Scheme 2




Example 1 3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt Ethanol (13 liters), (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H-pyrrolo[2,3- d]pyrimidin-4-yl)-amine (1.3 kg), cyano-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (1.5 kg), and triethylamine (1.5 liters) were combined and stirred at ambient temperature. Upon reaction completion (determined by High Pressure Liquid Chromotography (HPLC) analysis, approximately 30 minutes), the solution was filtered, concentrated and azeotroped with 15 liters of methylene chloride. The reaction mixture was washed sequentially with 12 liters of 0.5 N sodium hydroxide solution, 12 liters of brine and 12 liters of water. The organic layer was concentrated and azeotroped with 3 liters of acetone (final pot temperature was 42°C). The resulting solution was cooled to 20°C to 25°C followed by addition of 10 liters of acetone. This solution was filtered and then aqueous citric acid (0.8 kg in 4 liters of water) added via in-line filter. The reaction mixture was allowed to granulate. The slurry was cooled before collecting the solids by filtration. The solids were dried to yield 1.9 kg (71 %) (3R, 4R)- 3-{4-Methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile mono citrate. This material was then combined with 15 liters of a 1:1 ratio of ethanol/water and the slurry was agitated overnight. The solids were filtered and dried to afford 1.7 kg (63% from (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine) of the title compound as a white crystalline solid. 1H NMR (400 MH2)(D20) δ HOD: 0.92 (2H, d, J = 7.2 Hz), 0.96 (1H, d, J = 7.6 Hz), 1.66 (1H, m), 1.80 (1H, m), 2.37 (1H, m), 2.58 (2H, 1/2 ABq, J = 15.4 Hz), 2.70 (2H, 3 ABq, J = 154 Hz), 3.23 (2H, s), 3.25 (1H, s), 3.33 (1H, m), 3.46 (1H, m), 3.81 (4H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Patent
http://www.google.co.in/patents/EP1913000A2?cl=en Example 10 Preparation of methyl-[(3R, 4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine:
KEY INTERMEDIATE
To a clean, dry, nitrogen-purged 2 L hydrogenation reactor were charged 20 wt% Pd(OH)2/C (24.0 g, 50% water wet), water (160 ml), isopropanol (640 ml), (1-benzyl-4-methyl-piperidin-3-yI)-methyi- (7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (160.0 g, 0.48 mol), and acetic acid (28.65 g, 0.48 mol). The reactor was purged with three times at 50 psi with nitrogen and three times at 50 psi with hydrogen. Once purging was complete, the reactor was heated to 45-55°C and pressurized to 50 psi with hydrogen through a continuous feed. The hydrogen uptake was monitored until no hydrogen was consumed for 1 hour. The reactor was cooled to 20-300C and purged three times at 50 psi with nitrogen. The reaction mixture was filtered through wet Celite and the filtrate was sent to a clean, dry, nitrogen-purged vessel. A solution of sodium hydroxide (39.33 g) in water (290 ml) was charged and the mixture was stirred for a minimum of 1 hour then heated to 75-900C. The isopropanol was removed by distillation. The reaction mixture was cooled to 20-30°C and 2-methyltetrahydrofuran (1.6 L) was added. The aqueous layer was drained off and the 2-methyltetrahydrofuran was displaced with toluene (1.6 L). The distillation was continued until the final volume was 800 ml. The slurry was cooled to 20-30°C and held for a minimum of 7 hours. The resulting solids were isolated by filtration and washed with toluene (480 ml). After drying under vacuum between 40-50DC for a minimum of 24 hours with a slight nitrogen bleed 102.3 g (87.3%) of the title compound were isolated. Mp 158.6-159.8°C. 1H NMR (400 MHz, CDCI3): δ 11.38 (bs, 1H), 8.30 (s, 1H), 7.05 (d, J=3.5 Hz, 1H), 6.54 (d, J=3.5 Hz, 1H), 4.89-4.87 (m, 1H), 3.39 (s, 3H), 3.27 (dd, J=12.0, 9.3 Hz, 1 H), 3.04 (dd, J=12.0, 3.9 Hz, 1H), 2.94 (td, J=12.6, 3.1 Hz, 1H0, 2.84 (dt, J=12.6, 4.3 Hz, 1H), 2.51-2.48 (m, 1H), 2.12 (bs, 2H), 1.89 (ddt, J=13.7, 10.6, 4 Hz, 1 H), 1.62 (dq, J=13.7, 4Hz, 1 H), 1.07 (d, J=7.3 Hz, 3H). 13C NMR (400 MHz, CDCI3): δ 157.9, 152.0, 151.0, 120.0, 103.0, 102.5, 56.3, 46.2, 42.4, 34.7, 33.4, 32.4, 14.3.  KEY INT
KEY INT
Example 11 Preparation of 3-{(3R, 4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3- oxo-propionitrile….TOFACITINIB BASE
To a clean, dry, nitrogen-purged 1.0 L reactor were charged methyl-(4-methyl-piperidin-3-yI)-(7H- pyrroIo[2,3-d]pyrimidin-4-yl)-amine (32.0 g, 0.130 mol), toluene (160 ml), ethyl cyanoacetate (88.53 g, 0.783 mol) and triethyl amine (26.4 g, 0.261 mol). The reaction was heated to 1000C and held for 24 hours. The reaction was washed with water (160 ml). The organic layer concentrated to a volume of 10 ml and water (20 ml) was added. The residual toluene was removed by distillation and the mixture was cooled to room temperature. Acetone (224 ml) was added followed by citric acid (27.57 g, 0.144 mol) in water (76 ml). The resulting slurry was stirred for 7 hours. The solids were isolate by filtration, washed with acetone (96 ml), and dried under vacuum to afford 42.85 g (65.3%) of the title compound. Example 13 Preparation of 3-{(3R, 4R)~4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile citrate salt:…………..TOFACITINIB CITRATE To a clean, dry, nitrogen-purged 500 ml reactor were charged methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine (25.0 g, 0.102 mol) and methylene chloride (250 ml). The mixture was stirred at room temperature for a minimum of 2.5 hours. To a clean, dry, nitrogen-purged 1 L reactor were charged cyanoacetic acid (18.2 g, 0.214 mol), methylene chloride (375 ml), and triethyl amine (30.1 ml, 0.214 mol). The mixture was cooled to -15.0— 5.00C over one hour and trimethylacetyl chloride (25.6 ml, 0.204 mol) was added at a rate to maintain the temperature below O0C. The reaction was held for a minimum of 2.5 hours, then the solution of the amine was added at a rate that maintained the temperature below O0C. After stirring for 1 hour, the mixture was warmed to room temperature and 1 M sodium hydroxide (125 ml) was added. The organic layer was washed with water (125 ml) The methylene chloride solution.was displaced with acetone until a volume of 500 ml and a temperature of 55-650C had been achieved. Water (75 ml) was charged to the mixture while maintaining the temperature at 55-65°C. A solution of citric acid (20.76 g, 0.107 mol) in water (25.0) was charged and the mixture was cooled to room temperature. The reactor was stirred for a minimum of 5 hours and then the resulting solids were isolated by filtration and washed with acetone (2×75 ml), which was sent to the filter. The salt was charged into a clean, dry, nitrogen-purged 1L reactor with 2B ethanol (190 ml) and water (190 ml). The slurry was heated to 75-850C for a minimum of 4 hours. The mixture was cooled to 20-300C and stirred for an additional 4 hours. The solids were isolated by filtration and washed with 2B ethanol (190 ml). After drying in a vacuum oven at 500C with a slight nitrogen bleed, 34.6 g (67.3%) of the title compound were isolated. 1H NMR (500 MHz, CZ6-DMSO): δ 8.14 (s, 1 H), 7.11 (d, J=3.6 Hz, 1 H), 6.57 (d, J=3.6 Hz, 1 H), 4.96 (q, J=6.0 Hz, 1 H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1 H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1 H), 2.45-2.41 (m, 1 H), 1.81 (m, 1 H), 1.69-1.65 (m, 1 H), 1.04 (d, J=6.9 Hz, 3H)
PAPER
Org. Lett., 2009, 11 (9), pp 2003–2006
DOI: 10.1021/ol900435t
http://pubs.acs.org/doi/full/10.1021/ol900435t 
PATENT
http://www.omicsonline.org/open-access/advances-in-the-inhibitors-of-janus-kinase-2161-0444.1000540.php?aid=29799  ……………..
……………..

 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Clinical trials
Rheumatoid arthritis
Phase II clinical trials tested the drug in rheumatoid arthritis patients that had not responded to DMARD therapy. In a tofacitinib monotherapy study, the ACR score improved by at least 20% (ACR-20) in 67% of patients versus 25% who received placebo; and a study that combined the drug with methotrexate achieved ACR-20 in 59% of patients versus 35% who received methotrexate alone. In a psoriasis study, the PASI score improved by at least 75% in between 25 and 67% of patients, depending on the dose, versus 2% in the placebo group.[8] The most important side effects in Phase II studies were increased blood cholesterol levels (12 to 25 mg/dl LDL and 8 to 10 mg/dl HDL at medium dosage levels) andneutropenia.[8] Phase III trials testing the drug in rheumatoid arthritis started in 2007 and are scheduled to run until January 2015.[9] In April 2011, four patients died after beginning clinical trials with tofacitinib. According to Pfizer, only one of the four deaths was related to tofacitinib.[10] By April 2011, three phase III trials for RA had reported positive results.[11] In November 2012, the U.S. FDA approved tofacitinib “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to, or who are intolerant of, methotrexate.”[12]
Psoriasis
As of April 2011 a phase III trial for psoriasis is under way.[11]
Alopecia
In June 2014, scientists at Yale successfully treated a male patient afflicted with alopecia universalis. The patient was able to grow a full head of hair, eyebrows, eyelashes, facial, armpit, genitalia and other hair. No side effects were reported in the study.[13]
Ulcerative colitis
The OCTAVE study of Tofacitinib in Ulcerative Colitis started in 2012. It is currently enrolling patients, though the NIH trials page states that they expect the trial to close in June 2015.[14]
Vitiligo
In a June 2015 study, a 53-year-old woman with vitiligo showed noticeable improvement after taking tofacitinib for five months.[15]
Development of Safe, Robust, Environmentally Responsible Processes for New Chemical Entities
– Dr. V. Rajappa, Director & Head-Process R&D, Bristol-Myers Squibb, India
A PRESENTATION
 The presentation will load below
The presentation will load below
![]()
Scroll with mouse to view 76 pages


- Herper, Matthew (2 March 2011). “Why Pfizer’s Biggest Experimental Drug Got A Name Change”. Forbes. Retrieved 3 March 2011.
- Kremer, J. M.; Bloom, B. J.; Breedveld, F. C.; Coombs, J. H.; Fletcher, M. P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R. N.; Wilkinson, B.; Zerbini, C. A. F.; Zwillich, S. H. (2009). “The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo”. Arthritis & Rheumatism 60 (7): 1895–1905. doi:10.1002/art.24567. PMID 19565475.
- “Tasocitinib”. Drugs in R&D 10 (4): 271–284. 2010. doi:10.2165/11588080-000000000-00000. PMC 3585773. PMID 21171673.
- Ghoreschi, K.; Jesson, M. I.; Li, X.; Lee, J. L.; Ghosh, S.; Alsup, J. W.; Warner, J. D.; Tanaka, M.; Steward-Tharp, S. M.; Gadina, M.; Thomas, C. J.; Minnerly, J. C.; Storer, C. E.; Labranche, T. P.; Radi, Z. A.; Dowty, M. E.; Head, R. D.; Meyer, D. M.; Kishore, N.; O’Shea, J. J. (2011). “Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550)”. J Immunol. 186 (7): 4234–4243. doi:10.4049/jimmunol.1003668. PMC 3108067. PMID 21383241.
- ^ Jump up to:a b c “Seeking Profit for Taxpayers in Potential of New Drug”, Jonathan Weisman, New York Times, March 18, 2013
- Ken Garber (9 January 2013). “Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market”. Nature Biotechnology 31 (1): 3–4. doi:10.1038/nbt0113-3. PMID 23302910.
- Jump up^ Moisan A, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology, 8 December 2014. DOI 10.1038/ncb3075
- “EULAR: JAK Inhibitor Effective in RA But Safety Worries Remain”. MedPage Today. June 2009. Retrieved 9 February 2011.
- Clinical trial number NCT00413699 for “Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis” at ClinicalTrials.gov
- Matthew Herper. “Pfizer’s Key Drug Walks A Tightrope”. Forbes.
- “Two Phase III Studies Confirm Benefits of Pfizer’s Tofacitinib Against Active RA”. 28 Apr 2011.
- “FDA approves Xeljanz for rheumatoid arthritis”. 6 Nov 2012.
- “Hairless man grows full head of hair in yale arthritis drug trial”. 19 Jun 2014.
- https://clinicaltrials.gov/ct2/show/NCT01465763?term=A3921094&rank=1
- “This Drug Brought Pigment Back for Woman with Vitiligo”. TIME. June 27, 2015. Retrieved June 29, 2015.
- Nordqvist, Christian (27 April 2013). “Pfizer’s Arthritis Drug Xeljanz (tofacitinib) Receives A Negative Opinion In Europe”. Medical News Today. Retrieved 2 August 2013.
- “”XALEJANZ PRESCRIBING INFORMATION @ Labeling.Pfizer.com””.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
|
|
| Clinical data | |
| Trade names | Xeljanz, Jakvinus |
| AHFS/Drugs.com | entry |
| Licence data | US FDA:link |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Bioavailability | 74% |
| Protein binding | 40% |
| Metabolism | Hepatic (via CYP3A4 andCYP2C19) |
| Biological half-life | 3 hours |
| Excretion | Urine |
| Identifiers | |
| CAS Registry Number | 477600-75-2 |
| ATC code | L04AA29 |
| PubChem | CID: 9926791 |
| IUPHAR/BPS | 5677 |
| DrugBank | DB08183 |
| ChemSpider | 8102425 |
| UNII | 87LA6FU830 |
| ChEBI | CHEBI:71200 |
| ChEMBL | CHEMBL221959 |
| Synonyms | CP-690550 |
| Chemical data | |
| Formula | C16H20N6O |
| Molecular mass | 312.369 g/mol |
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE

Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
 LIONEL MY SON
LIONEL MY SONHe was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy

सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////
