
Home » Posts tagged 'serine/threonine kinase inhibitor'
Tag Archives: serine/threonine kinase inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Famlasertib
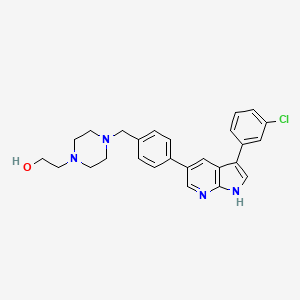
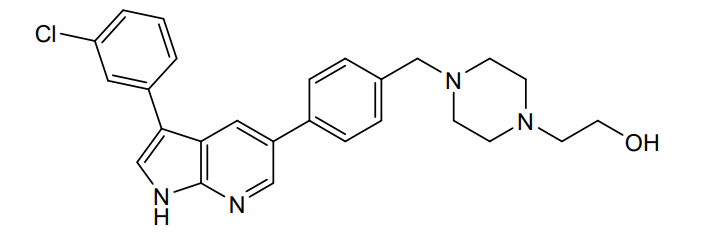
Famlasertib
CAS 2375591-69-6
MFC26H27ClN4O MW 447.0 g/mol
4-[[4-[3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl]phenyl]methyl]-1-piperazineethanol
2-[4-({4-[3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl]phenyl}methyl)piperazin-1-yl]ethan-1-ol
serine/threonine kinase inhibitor, amyotrophic lateral sclerosis, Prosetin, WJP32276AY
Prosetin is an orally administered blocker of MAP4K under investigation for the treatment of amyotrophic lateral sclerosis.
Famlasertib (also known as Prosetin or Prostetin/12k) is a highly potent, small-molecule inhibitor targeting the mitogen-activated protein kinase kinase kinase kinase (MAP4K) family. It is an experimental drug primarily under investigation for its neuroprotective capabilities in treating neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) and as an anti-invasive agent in certain cancers
- Target Pathways: MAP4K4 (HGK), MLK1, and MLK3
- Key Properties: Orally active, blood-brain barrier penetrant (CNS-penetrant)
Mechanism of Action
Famlasertib functions by blocking the activation of the MAP4K protein family, specifically demonstrating powerful inhibitory values (\(\text{IC}_{50}\)) against subfamilies like HGK (MAP4K4), MLK3, and MLK1. By inhibiting these kinases, the compound: [1]
- Reduces Endoplasmic Reticulum (ER) Stress: It helps mitigate the unfolded protein response that triggers programmed cell death in neurons affected by misfolded protein accumulation.
- Suppresses Inflammation: It blocks inflammatory pathways associated with neurodegeneration and cell damage.
- Restrains Cell Motility: In oncology contexts, it disrupts kinase signaling linked to actin cytoskeleton remodeling, preventing malignant cells from migrating.
Primary Areas of Research
1. Amyotrophic Lateral Sclerosis (ALS)
In motor neuron models of ALS, cellular stress frequently triggers neurodegeneration. Because famlasertib easily passes through the blood-brain barrier, it is capable of directly shielding motor neurons from ER-stress-mediated cell death, extending cell viability in laboratory models.
2. Oncology (Medulloblastoma)
Recent findings published on bioRxiv indicate that famlasertib acts as a “migrastatic” agent in medulloblastoma (a type of pediatric brain tumor). It suppresses the highly invasive behavior and single-cell motility of tumor cells without exhibiting developmental toxicity.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020163594&_cid=P21-MPGALG-31359-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US317630245&_cid=P21-MPGALG-31359-1
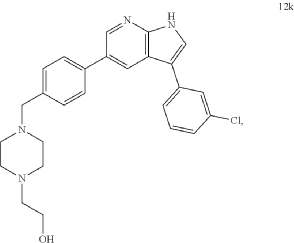
Preparation of 2-(4-(4-(3-(3-Chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (Compound 12k)
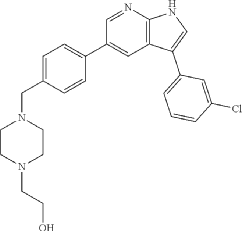
Following the general procedure described above, with 4-(3-(3-chlorophenyl)-1-tosyl-1H-pyrrolo[2,3-b]pyridin-5-yl)benzaldehyde (10c, 418 mg, 0.86 mmol) and 1-(2-hydroxyethyl)piperazine (224 mg, 211 μL, 1.72 mmol, 2.0 eq) as the starting materials, 2-(4-(4-(3-(3-chlorophenyl)-1H-pyrrolo[2,3-b]pyridin-5-yl)benzyl)piperazin-1-yl)ethan-1-ol (12k) was isolated as an off-white solid (139.9 mg, 36% yield over two steps). 1H NMR (400 MHz, Methanol-d 4) δ 8.57 (d, J=2.0 Hz, 1H), 8.54 (d, J=2.0 Hz, 1H), 7.80 (s, 1H), 7.76 (d, J=8.3 Hz, 2H), 7.65 (t, J=1.9 Hz, 1H), 7.64-7.57 (m, 3H), 7.42 (t, J=7.9 Hz, 1H), 7.29 (ddd, J=8.0, 2.1, 1.0 Hz, 1H), 4.27 (s, 2H), 3.93-3.86 (m, 2H), 3.62 (s, 4H), 3.41 (s, 4H), 3.35-3.31 (m, 2H) ppm. HRMS (APCI +, m/z): calcd. for C 26H 28N 40Cl [M+H +]: 447.1952, found: 447.1954.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US469942811&_cid=P21-MPGAUU-39605-1
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
- Design, synthesis of new 3H-imidazo[4,5-b]pyridine derivatives and evaluation of their inhibitory properties as mixed lineage kinase 3 inhibitorsPublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2024-03-15PMID: 38346577DOI: 10.1016/j.bmcl.2024.129652
- The emerging role of mixed lineage kinase 3 (MLK3) and its potential as a target for neurodegenerative diseases therapiesPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-09-05PMID: 37247505DOI: 10.1016/j.ejmech.2023.115511
- Development of MAP4 Kinase Inhibitors as Motor Neuron-Protecting AgentsPublication Name: Cell Chemical BiologyPublication Date: 2019-12-19PMCID: PMC7253076PMID: 31676236DOI: 10.1016/j.chembiol.2019.10.005
PAT
- Immunophilin binding agents and uses thereofPublication Number: US-2023063768-A1Priority Date: 2019-02-07
- Immunophilin binding agents and uses thereofPublication Number: WO-2020163594-A1Priority Date: 2019-02-07
- Immunophilin-dependent inhibitors and uses thereofPublication Number: US-2022193242-A1Priority Date: 2019-02-07
- Immunophilin-dependent inhibitors and uses thereofPublication Number: WO-2020163598-A1Priority Date: 2019-02-07
- Compounds, compositions and methods for inhibiting toxic endoplasmic reticulum stress – Patents.comPublication Number: JP-7487170-B2Priority Date: 2018-03-01Grant Date: 2024-05-20
- Compounds, compositions, and methods for suppressing toxic endoplasmic reticulum stress
- Publication Number: US-2021040091-A1
- Priority Date: 2018-03-01G
///////famlasertib, serine/threonine kinase inhibitor, amyotrophic lateral sclerosis, Prosetin, WJP32276AY
Engasertib
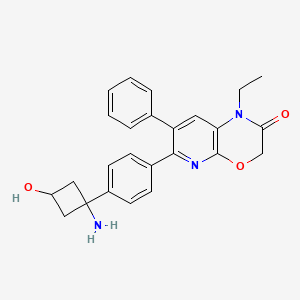
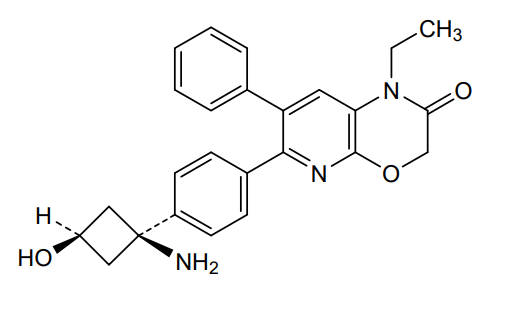
Engasertib
CAS 1313439-71-2
MF C25H25N3O3 MW415.5 g/mol
6-[4-(1-amino-3-hydroxycyclobutyl)phenyl]-1-ethyl-7-phenylpyrido[2,3-b][1,4]oxazin-2-one
6-{4-[(1S,3S)-1-amino-3-hydroxycyclobutyl]phenyl}-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
Engasertib is an oral, once-daily AKT inhibitor developed by Vaderis Therapeutics, primarily investigated as a targeted therapy for Hereditary Hemorrhagic Telangiectasia (HHT). Clinical trials show it safely reduces the frequency and duration of bleeding episodes without an FDA-approved equivalent currently available
Core Information
- Mechanism of Action: Engasertib is a highly selective inhibitor of AKT1 and AKT2. In HHT, mutations in the ALK1 pathway lead to abnormal blood vessel growth driven by an excess of the AKT protein. By inhibiting AKT, the drug promotes vascular stability and reduces vessel fragility.
- Target Indication: Hereditary Hemorrhagic Telangiectasia (HHT) — a rare, severe genetic disorder causing vascular abnormalities and frequent, heavy bleeding, particularly nosebleeds (epistaxis)
Clinical Efficacy & Safety
- Proof-of-Concept Trial: A 12-week, placebo-controlled study with 75 HHT patients evaluated daily doses of 30 mg and 40 mg.
- The 40 mg cohort demonstrated a 41% reduction in mean bleeding duration and a 28% reduction in bleeding frequency, compared to 24% and 18% in the placebo group.
- 61% of patients in the 40 mg group rated their clinical condition as “Much Better”.
- Extended Efficacy: In long-term open-label extensions, benefits were sustained and amplified over 12 months, resulting in a 66% reduction in mean bleeding duration and a 55% reduction in bleeding frequency.
- Side Effects: Generally well-tolerated. The most common side effects (reversible and manageable with supportive care) were mild-to-moderate rash and hyperglycemia
- OriginatorAlmac Discovery
- DeveloperVaderis Therapeutics
- ClassAntineoplastics; Small molecules; Vascular disorder therapies
- Mechanism of ActionProto-oncogene protein c-akt inhibitors
- Orphan Drug StatusYes – Hereditary haemorrhagic telangiectasia
- Phase IVascular disorders
- PreclinicalBreast cancer; Prostate cancer
- No development reportedHereditary haemorrhagic telangiectasia
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Belgium (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in France (PO, Capsule)
- 28 Dec 2025No recent reports of development identified for phase-I development in Hereditary haemorrhagic telangiectasia in Italy (PO, Capsule)
SYN
Example 139: 6-(4-((1s.3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
Step 1: tert-butyl ((1s.3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2.3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one (50 mg, 0.150 mmol), tert-butyl ((1s,3s)-3-hydroxy-1-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off-white solid (45 mg, 70% yleld). Ή-NMR (500 MHz, CDCl3) δ 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.
Step 2: 6-(4-((1s,3s)-1-amino-3-hydroxycyclobutyl)phenyl)-1-ethyl-7-phenyl-1H-pyrido[2,3-b][1,4]oxazin-2(3H)-one
tert-butyl ((1s,3s)-1-(4-(1-ethyl-2-oxo-7-phenyl-2,3-dihydro-1H-pyrido[2,3-b][1,4]oxazin-6-yl)phenyl)-3-hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drylng overnight to give the desired compound as an off-white solid (33 mg, 71% yleld).
1H-NMR (500 MHz, MeOD) δ 7.55 (1H, s), 7.39-7.42 (4H, m), 7.27-7.31 (3H, m), 7.20-7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.11 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.
SYN
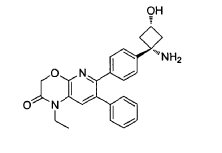
EXAMPLES
Example 1: Synthesis of 6-(4-(l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
6-(4-(l-amino-3-hydroxycyclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (referred to herein as “VAD044 free base”) was synthesized in accordance with the protocol as set out in W02011077098 – see in particular Examples 97, 113 and 139 (reproduced below):
Synthesis of 6-(4-((ls,3s)-l-amino-3-hvdroxycvclobutyl)phenyl)-l-ethyl-7-phenyl-lH-pyrido[2,3-bHl,41oxazin-2(3H)-one: from WO2Q11077098 Example 139:
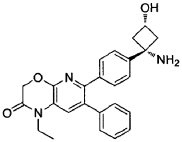
Step 1: tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3-hvdroxycvclobutyl)carbamate
In a 15 mL reaction tube was added 6-bromo-l-ethyl-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one* (50 mg, 0.150 mmol), tert-butyl((ls,3s)-3-hydroxy-l-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)cyclobutyl)carbamate** (49 mg, 0.125 mmol) and cesium carbonate (204 mg, 0.625 mmol) in a mixture of 1,4-dioxane (2.3 ml) and water (0.8 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 15 minutes, followed by the addition of [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (20 mg, 0.025 mmol) and degassing for a further 5 minutes. The reaction mixture was heated to 50°C under a nitrogen atmosphere for one hour then allowed to cool to room temperature, diluted with water (5 ml) and extracted into ethyl acetate (3 x 5 ml). The combined organic phases were dried over Na2SO4, filtered and concentrated to dryness under reduced pressure. The residue was purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 90:10 to 0:100) to give the desired product as an off- white solid (45 mg, 70% yield). 1H-NMR (500 MHz, CDCI3) 6 7.29-7.35 (5H, m), 7.28 (1H, s), 7.18-7.24 (4H, m), 4.96 (1H, br s), 4.88 (2H, s), 4.05 (1H, br s), 4.01 (2H, q), 2.98 (2H, br s), 2.75 (2H, br s), 1.20-1.51 (9H, br m), 1.32 (3H, t). LCMS (Method D) RT = 1.25 min, M+H+ = 516.20.

tert-butyl((ls,3s)-l-(4-(l-ethyl-2-oxo-7-phenyl-2,3-dihydro-lH-pyrido[2,3-b][l,4]oxazin-6-yl)phenyl)-3- hydroxycyclobutyl)carbamate (45 mg, 0.087 mmol) was dissolved in TFA (1 mL) and stirred for 30 seconds. The solution was immediately concentrated to dryness under reduced pressure. The residue was dissolved in diethyl ether (~3 mL) and concentrated to dryness under reduced pressure three times. The residue was then slurried in diethyl ether (3 mL) and after settling the supernatant solvent removed by pipette. This was repeated three times. The remaining solvent was removed by freeze drying overnight to give the desired compound as an off-white solid (33 mg, 71% yield). 1H-NMR (500 MHz, MeOD) 6 7.55 (1H, s), 7.39- 7.42 (4H, m), 7.27-7.31 (3H, m), 7.20- 7.24 (2H, m), 4.93 (2H, s), 4.01-4.11 (3H, m), 3.03-3.1 1 (2H, m), 2.42-2.50 (2H, m), 1.28 (3H, t). LCMS (Method D) RT = 0.74 min, M+H+ = 416.20.

To a suspension of sodium hydride (5.31 g, 133 mmol) in 1,4-dioxane (250 ml), ethyl glycolate (12.56 ml,
133 mmol) was added drop wise over a period of 30 minutes ensuring that the temperature was maintained below 30°C. The resulting thick suspension was stirred at room temperature for 15 minutes.
In a separate II round- bottomed flask was added 5-bromo-2-chloro-3-nitropyridine (21 g, 88 mmol) in
1,4-dioxane (150 ml) to give a brown solution. The suspension of sodium hydride and ethyl glycolate was added drop wise over a period of 30 minutes at 0°C. The resulting reaction mixture was heated to 80°C overnight.
The reaction mixture was concentrated under reduced pressure and the crude residue was purified by
Biotage silica chromatography (gradient 0% to 10% ethyl acetate in n-hexanes) to give the title compound
(1 ,8g, 44%).1H NMR (500 MHz, CDCI3) 6 8.48 (1H, s), 8.42 (1H, s), 5.07 (2H, s), 4.28-4.24 (2H, q), 1.31-1.28
(3H, t).
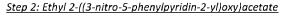
In a II round-bottomed flask was added ethyl 2-(5-bromo-3-nitropyridin-2-yloxy)acetate (18.33 g, 60.1 mmol), phenylboronic acid (10.99 g, 90 mmol), triphenylphosphine (4.73 g, 18.02 mmol) and cesium fluoride (45.6 g, 300 mmol) in 1,2-dimethoxyethane (300 ml) to give a yellow solution. The reaction mixture was degassed by bubbling nitrogen for 30 minutes. Pallad ium (II ) acetate (2.023 g, 9.01 mmol) was added and the mixture was heated to 75°C under a nitrogen atmosphere overnight. The reaction mixture was allowed to cool to room temperature and concentrated to dryness under reduced pressure to give a brown solid. This was re-dissolved in dichloromethane, filtered and concentrated to dryness under reduced pressure to give a brown solid The crude residue was purified via Biotage chromatography (gradient 5% to 60% ethyl acetate in n-hexanes) to give the title compound (6.9g, 38%). 1H NMR (500 MHz, CDCI3) 6 8.58 (1H, s), 8.56 (1H, s), 7.59-7.52 (2H, m), 7.48-7.46 (2H, m), 7.45-7.43 (1H, m), 5.13 (2H, s), 4.30-4.26 (2H, q), 1.33-1.30 (3H, t).
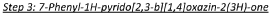
In a 500 ml round-bottomed flask was added ethyl 2-(3-nitro-5-phenylpyridin-2-yloxy)acetate (4.6 g, 15.22 mmol) in hydrochloric acid, 37% (40 ml) to give a yellow suspension. The mixture was cooled to 0-5°C followed by the portion wise addition of tin powder (9.94 g, 84 mmol). The addition proved to be exothermic. Caution should be taken while adding. The mixture was then stirred at room temperature for further 30 minutes until all foaming ceased. The reaction mixture was heated to 80°C under a nitrogen atmosphere for 3 hours. The reaction mixture cooled to room temperature and diluted with water (800ml). The white precipitate was isolated by filtration, washed with water (100 ml) and sucked dry to give a white solid. The solid was azeotroped with toluene (3 x 30 ml) to give a white solid as the title compound (2.6g, 77%). XH NMR (500 MHz, (CD3)2SO) 6 10.41 (1H, s), 8.10 (1H, s), 7.59 (2H, d), 7.49-7.42 (2H, t), 7.39-7-38 (1H, d), 4.83 (2H, s).
Step 4: 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one
In a 10ml microwave vial was 7-phenyl-l H-pyrido[2,3-b][l ,4]oxazin-2(3H)-one (50 mg, 0.221 mmol) and N-bromosuccinimide (78.6 mg, 0.441 mmol) in dimethylformamide (1 ml). The reaction mixture was heated to 80°C under microwave irradiation for 30 minutes. The reaction mixture was cooled to room
temperature and diluted with ethyl acetate (10ml). The organic solution was washed with water (2x10ml) and brine (2x10ml). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified via Biotage chromatography (gradient 0% to 5% methanol in dichloromethane) to give the title compound as a yellow solid (61 mg, 90%). 1H NMR (500 MHz, CD3OD) 6 7.48-7.32 (5H, m), 7.12 (1 H, s), 4.82 (2H, s).
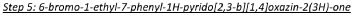
In a 15 mL reaction tube was added 6-bromo-7-phenyl-lH-pyrido[2,3-b][l,4]oxazin-2(3H)-one (300 mg, 0.983 mmol), iodoethane (0.095 mL, 1.180 mmol) and potassium carbonate (408 mg, 2.95 mmol) in anhydrous N,N-dimethylformamide (1 mL) to give a brown suspension. This was stirred at 50 °C under a nitrogen atmosphere for 60 minutes. The reaction mixture was diluted with saturated sodium bicarbonate solution (5 mL) and extracted into ethyl acetate (3 x 5 mL). The combined organic phases were washed with 50:50 water.brine (3 x 5 mL), dried over Na2SO4, filtered and concentrated to dryness under reduced pressure to give a brown solid. This was purified by Biotage chromatography (25g silica cartridge, cyclohexane:ethyl acetate, gradient elution from 90:10 to 20:80) to give the title compound as a beige solid (160 mg, 48.8 % yield). XH NMR (500 MHz, CDCI3) 6 7.58-7.37 (5H, m), 7.21 (1H, s), 4.86 (2H, s), 3.96 (2H, q), 1.27 (3H, t). LCMS (Method D) RT 1.293 min, M+l= 334.

n a 40 mL reaction tube was added tert-butyl(ls,3s)-l-(4-bromophenyl)-3- hydroxycyclobutylcarbamate*** (0.25 g, 0.731 mmol) in anhydrous tetrahydrofuran (14 ml) to give a colourless solution. This was degassed by bubbling nitrogen for 20 minutes, followed by the addition of [l,l’-bis(diphenylphosphino)ferrocene]dichloropalladium(ll) dichloromethane adduct (60 mg, 0.073 mmol). After bubbling nitrogen for a further 15 minutes, potassium acetate (143 mg, 1.461 mmol) and bis(pinacolato)diboron (223 mg, 0.877 mmol) were added. The reaction mixture was heated to reflux overnight then concentrated to dryness under reduced pressure and purified by Biotage chromatography (cyclohexane:ethyl acetate, gradient elution from 88:12 to 0:100) to give the desired product as a colourless oil that solidified upon standing (240 mg, 84% yield). 1H-NMR (500 MHz, CDCI3) 6 7.71 (2H, d), 7.44 (2H, d), 4.15 (1H, br s), 2.87-2.98 (2H, m), 2.27-2.44 (2H, m), 1.22-1.49 (21H, br m).
(*** synthesis described in WO2009148887 and WO2009148916)
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Inhibitors of akt activityPublication Number: EP-2516435-B1Priority Date: 2009-12-23Grant Date: 2014-08-06
- Inhibitors of akt activityPublication Number: WO-2011077098-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: EP-2516435-B8Priority Date: 2009-12-23Grant Date: 2014-10-15
- Inhibitors of akt activityPublication Number: EP-2516435-A1Priority Date: 2009-12-23
- Inhibitors of AKT activityPublication Number: US-9221838-B2Priority Date: 2009-12-23Grant Date: 2015-12-29
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: US-2024092801-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: WO-2022069552-A1Priority Date: 2020-09-30
- Allosteric akt inhibitors for use in the treatment of hereditary hemorrhagic telangiectasiaPublication Number: EP-4221713-A1Priority Date: 2020-09-30
- Inhibitors of akt activityPublication Number: US-2013116243-A1Priority Date: 2009-12-23
- Inhibitors of akt activityPublication Number: WO-2011077098-A9Priority Date: 2009-12-23
////////engasertib, anax labs, serine/threonine kinase inhibitor, ALM-301, VAD-044, ALM 301, VAD 044, Orphan Drug, K2US8HW4TQ
Alnodesertib
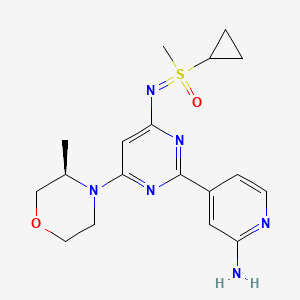
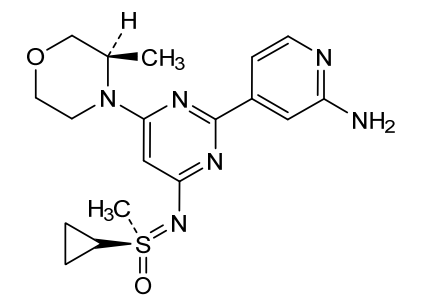
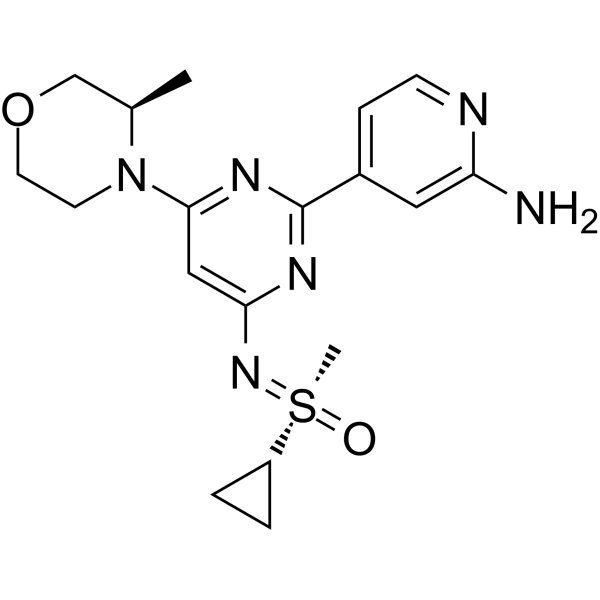
Alnodesertib
CAS 2267316-76-5
MF C18H24N6O2S MW388.49
4-[4-[(cyclopropyl-methyl-oxo-λ6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine
4-[4-[(cyclopropyl-methyl-oxo-lambda6-sulfanylidene)amino]-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-2-yl]pyridin-2-amine
(S)-({2-(2-aminopyridin-4-yl)-6-[(3R)-3-methylmorpholin-4-yl]pyrimidin-4-yl}imino)(cyclopropyl)(methyl)-λ6
-sulfanone
serine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085
Alnodesertib (formerly known as ART0380) is an investigational, orally administered drug designed to treat various types of cancer. It is a selective inhibitor of ATR (Ataxia-Telangiectasia and Rad3-related protein), a key kinase involved in DNA repair and cell cycle progression.
Mechanism of Action
Alnodesertib works by disrupting the DNA Damage Response (DDR):
- Targets ATR Kinase: It selectively inhibits ATR, which cancer cells rely on to fix DNA damage caused by rapid replication.
- Blocks Signaling: By blocking the phosphorylation of CHK1, it prevents the activation of DNA damage checkpoints.
- Induces Apoptosis: Inhibiting these repair pathways prevents cancer cells from surviving replication stress, ultimately leading to cell death (apoptosis).
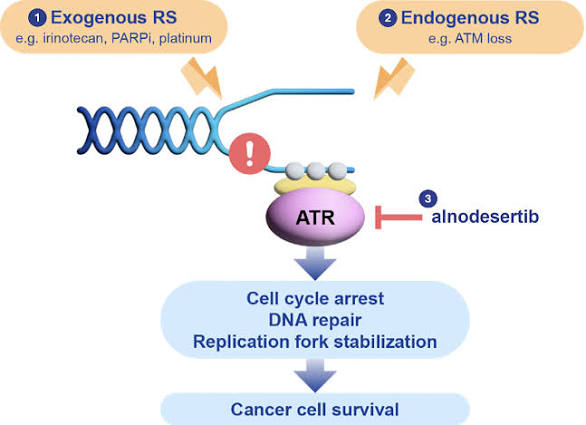

Clinical Status and Indications
As of early 2026, alnodesertib is undergoing several clinical trials:
- Metastatic Colorectal Cancer (mCRC): The FDA granted Fast Track designation in September 2025 for alnodesertib in combination with irinotecan for adult patients with ATM-negative mCRC in the third-line setting.
- Ovarian Cancer: In March 2026, Artios Pharma reported that a Phase 2a study reached its primary endpoint, showing that adding a low dose of alnodesertib to gemcitabine improved progression-free survival in patients with platinum-resistant high-grade serous ovarian carcinoma (HGSOC).
- Other Solid Tumours: It is being evaluated in the ongoing STELLA Phase 1/2a study for its potential across multiple solid tumour types characterized by high replication stress.
Key Facts
| Feature | Details |
|---|---|
| Developer | Artios Pharma Limited |
| Drug Class | ATR Kinase Inhibitor |
| Administration | Oral |
| Current Phase | Phase 2 clinical trials |
| FDA Status | Fast Track Designation (for ATM-negative mCRC) |
SYN
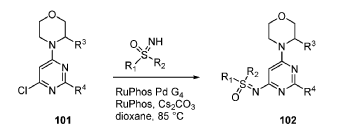
EXAMPLES 39a and 39b
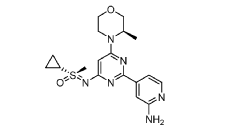
(R)-((2-(2-aminopyridin-4-yl)-6-((R)-3-methylmorpholino)pyrimidin-4- yl)imino)(cyclopropyl)(methyl)-λ6-sulfanone
and
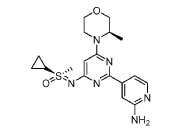
(S)-((2-(2-aminopyridin-4-yl)-6-((R)-3-methylmorpholino)pyrimidin-4- yl)imino)(cyclopropyl)(methyl)-λ6-sulfanone
[0648] Synthesis is similar to that described for Example 24. The mixture of diastereomers (26.8 mg, 0.069 mmol) was separated by Chiral SFC (Mobile phase: n-hexane (0.1% DEA):EtOH(0.1% DEA) = 70:30; Flow rate: 80 g / min; 20 min; Column temperature: 35 °C; Back pressure: 100 bar; Column: Gilson-281, AY 20 x 250mm, 10 μm) to afford the two diastereomers of unknown absolute stereochemistry at the sulfur atom, title compounds 39a (6.6mg, 25% yield, >99% ee) as a white solid and 39b (7.1mg, 27% yield, >99% ee) as a white solid.
[0649] 39a ((R)-cyclopropyl(methyl)-λ6-sulfanone or (S)-cyclopropyl(methyl)-λ6-sulfanone): 1H NMR (500 MHz, CD3OD) δ 8.03 – 7.91 (m, 1H), 7.53 (s, 1H), 7.49 (dd, J =5.5, 1.4 Hz, 1H), 5.97 (s, 1H), 4.48 (d, J = 4.6 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 4.02 (dt, J = 11.3, 3.6 Hz, 1H), 3.82 (d, J = 11.4 Hz, 1H), 3.75 (dt, J = 11.5, 3.0 Hz, 1H), 3.65 – 3.56 (m, 4H), 3.25 (tdJ, = 12.8, 3.8 Hz, 1H), 3.01 (td, J = 7.9, 4.0 Hz, 1H), 1.42 (dd, J = 10.2, 5.4 Hz, 1H), 1.31 (dt, J = 11.1, 6.2 Hz, 4H), 1.20 (dt,J = 11.3, 5.7 Hz, 2H); MS (ES+) C18H24N6O2S requires: 388, found: 389 [M+H]+; Rt = 11.35 min.
[0650] 39b ((R)-cyclopropyl(methyl)-λ6-sulfanone or (S)-cyclopropyl(methyl)-λ6-sulfanone): 1H NMR (500 MHz, CD3OD) δ 7.97 (d, J = 5.4 Hz, 1H), 7.53 (s, 1H), 7.49 (dt, J = 5.5, 1.3 Hz, 1H), 5.97 (s, 1H), 4.50 (s, 1H), 4.08 (d, J = 12.7 Hz, 1H), 4.02 (dd, J = 11.4, 3.7 Hz, 1H), 3.82 (d, J = 11.3 Hz, 1H), 3.75 (dt, J = 11.4, 3.0 Hz, 1H), 3.66 – 3.55 (m, 4H), 3.25 (tdJ, = 12.9, 3.9 Hz, 1H), 3.05 – 2.97 (m, 1H), 1.41 (dt, J = 10.6, 5.2 Hz, 1H), 1.31 (dd, J = 11.8, 5.8 Hz, 4H), 1.20 (dt, J = 11.1, 5.6 Hz, 2H); MS (ES+) C18H24N6O2S requires: 388, found: 389 [M+H]+; Rt = 15.22 min.
[0651] Alternatively, Example 39a can also be prepared from Int. CC, Isomer lb.
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- HETEROCYCLIC INHIBITORS OF KINASE ATRPublication Number: WO-2019014618-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of ATR kinasePublication Number: US-11434233-B2Priority Date: 2017-07-13Grant Date: 2022-09-06
- Heterocyclic inhibitors of ATR kinasePublication Number: US-10800769-B2Priority Date: 2017-07-13Grant Date: 2020-10-13
- Heterocyclic inhibitors of ATR kinasePublication Number: US-10392376-B2Priority Date: 2017-07-13Grant Date: 2019-08-27
- Heterocyclic inhibitors of atr kinasePublication Number: US-2019016713-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of atr kinasePublication Number: US-2020102296-A1Priority Date: 2017-07-13
- Heterocyclic inhibitors of atr kinasePublication Number: US-2021047311-A1Priority Date: 2017-07-13
///////alnodesertib, ANAX LAB, serine/threonine kinase inhibitor, antineoplastic, ART 0380, EX-A9085
Ocadusertib
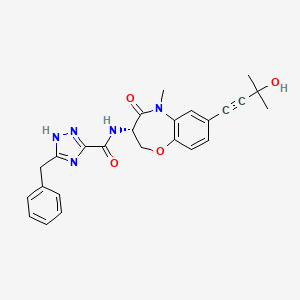
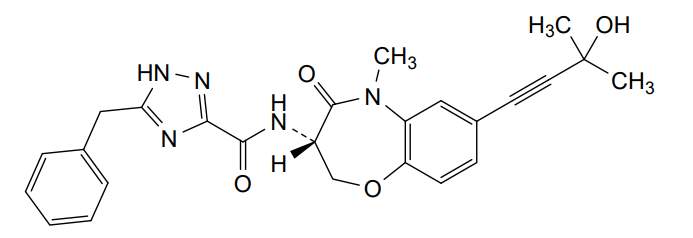
Ocadusertib
CAS 2382811-41-6
MF C25H25N5O4 MW 459.5 g/mol
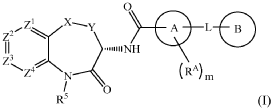
5-benzyl-N-[(3S)-7-(3-hydroxy-3-methylbut-1-ynyl)-5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
5-benzyl-N-[(3S)-7-(3-hydroxy-3-methylbut-1-yn-1-yl)-5-methyl-4-oxo-2,3,4,5-tetrahydro-1,5-benzoxazepin-3-yl]-1H-1,2,4-triazole-3-carboxamide
serine/threonine kinase inhibitor, LY3871801, R552, LY 3871801, R 552, S53J4A7ME4
- OriginatorRigel Pharmaceuticals
- DeveloperEli Lilly and Company; Rigel Pharmaceuticals
- ClassAnti-inflammatories; Antirheumatics; Small molecules
- Mechanism of ActionRIPK1 protein inhibitors
- Phase IIRheumatoid arthritis
- No development reportedUnspecified
- 28 Mar 2025No recent reports of development identified for phase-I development in Unspecified(In volunteers) in Singapore (PO, Suspension)
- 14 Nov 2024Pharmacodynamics data from preclinical trials in Rheumatoid arthritis presented at the ACR Convergence 2024 (ACR-2024)
- 14 Nov 2024Safety and pharmacokinetics data from a phase I trial in Rheumatoid arthritis presented at the ACR Convergence 2024 (ACR-2024)
Ocadusertib (LY3871801/R552) is an oral, potent, and selective small-molecule RIPK1 inhibitor developed by Rigel Pharmaceuticals and Eli Lilly for autoimmune and inflammatory diseases. It is currently in Phase 2 clinical trials for treating moderate-to-severe rheumatoid arthritis. 
Key Aspects of Ocadusertib:
- Mechanism of Action: It inhibits receptor-interacting serine/threonine-protein kinase 1 (RIPK1), which blocks necroptotic (cell death) responses and, consequently, reduces inflammation.
- Target Indications: Primarily focused on rheumatoid arthritis, it has also been investigated for psoriasis and general inflammatory joint conditions.
- Development Status: As of late 2025, it is in Phase 2 clinical trials (NCT05848258), with previous trials evaluating its safety, tolerability, and pharmacokinetics in healthy volunteers.
- Characteristics: It is designed to be a selective inhibitor, showing no significant inhibition in a broad panel of other kinases.
ACR Meeting Abstracts +3
Ocadusertib is a small molecule drug. The usage of the INN stem ‘-sertib’ in the name indicates that Ocadusertib is a serine/threonine kinase inhibitor. Ocadusertib is under investigation in clinical trial NCT05848258 (An Adaptive Phase 2a/2b Study of LY3871801 in Adult Participants With Rheumatoid Arthritis). Ocadusertib has a monoisotopic molecular weight of 459.19 Da.
- An Adaptive Phase 2a/2b Study of LY3871801 in Adult Participants With Rheumatoid ArthritisCTID: NCT05848258Phase: Phase 2Status: RecruitingDate: 2025-12-09
- A Study of LY3871801 in Healthy Asian and Non-Asian ParticipantsCTID: NCT05960851Phase: Phase 1Status: CompletedDate: 2024-01-10
- A Drug Interaction Study of LY3871801 in Healthy ParticipantsCTID: NCT05602675Phase: Phase 1Status: CompletedDate: 2023-04-18
- A Study of LY3871801 in Healthy ParticipantsCTID: NCT05222399Phase: Phase 1Status: CompletedDate: 2022-03-18
SYN
PAT
PAT
WO 2014/125444
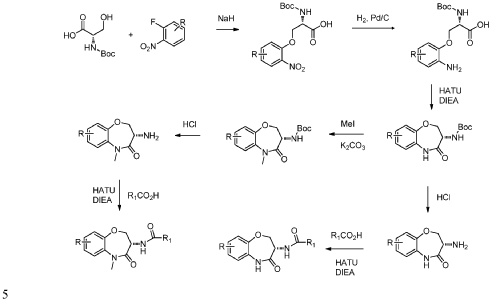
PAT
- [WO2021046407]
- https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021046407&_cid=P20-MM02H8-46041-1
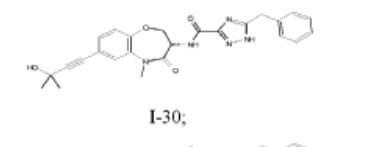
I-30: (S)-5-benzyl-N-(7-(3-hydroxy-3-methylbut-1-yn-1-yl)-5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]oxazepin-3-yl)-1H-1,2,4-triazole-3-carboxamide;
(S)-5-benzyl-N-(5-methyl-4-oxo-2,3,4,5-tetrahydrobenzo[b][1,4]oxazepin-3-yl)-4H-1,2,4-triazole-3-carboxamide (WO 2014/125444), having a structure as illustrated below, was used as a comparative compound and was examined using a similar protocol as described by WO 2014/125444. This comparison
compound exhibited 93% inhibition at a dose of 30 mg/kg according to WO 2014/125444; however, in the inventors hands, the compound inhibited only 70% at 30 mg/kg. In comparison, compound I-30 of the present disclosure achieved greater than 85% inhibition at a dose of just 5 mg/kg using the similar assay protocol described above.
PAT
- RIP1 inhibitory compounds and methods of making and using the samePublication Number: CN-112368278-BPriority Date: 2018-05-03Grant Date: 2025-06-17
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021002237-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: AU-2019262144-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021002236-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021040053-A1Priority Date: 2018-05-03
- RIP1-inhibiting compounds and methods of making and using the samePublication Number: CN-112368278-APriority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2023113841-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11370765-B2Priority Date: 2018-05-03Grant Date: 2022-06-28
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11377428-B2Priority Date: 2018-05-03Grant Date: 2022-07-05
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021238153-A2Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: EP-4248975-A2Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-12116354-B2Priority Date: 2018-05-03Grant Date: 2024-10-15
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11332451-B2Priority Date: 2018-05-03Grant Date: 2022-05-17
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-11370764-B2Priority Date: 2018-05-03Grant Date: 2022-06-28
- RIP1 inhibitory compounds and methods of obtaining and using themPublication Number: MD-3788044-T2Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2019337907-A1Priority Date: 2018-05-03
- RIP1 inhibitory compounds and methods for making and using the samePublication Number: US-10815206-B2Priority Date: 2018-05-03Grant Date: 2020-10-27
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2021009537-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: US-2020407332-A1Priority Date: 2018-05-03
- Rip1 inhibitory compounds and methods for making and using the samePublication Number: EP-3788044-A1Priority Date: 2018-05-03

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
///////ocadusertib, serine/threonine kinase inhibitor, LY3871801, R552, LY 3871801, R 552, S53J4A7ME4
Nacresertib
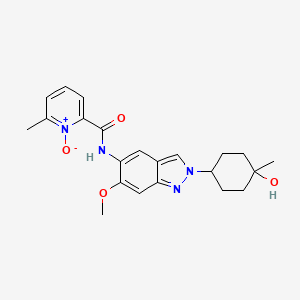
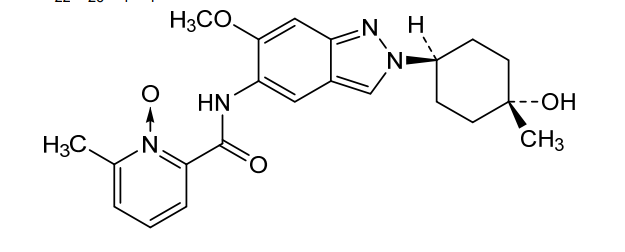
Nacresertib
CAS 2629977-59-7
MF C22H26N4O4, 410.5 g/mol
N-[2-(4-hydroxy-4-methylcyclohexyl)-6-methoxyindazol-5-yl]-6-methyl-1-oxidopyridin-1-ium-2-carboxamide
2-({2-[(1r,4r)-4-hydroxy-4-methylcyclohexyl]-6-methoxy-2H-indazol-5-yl}carbamoyl)-6-methylpyridine
1-oxide
serine/threonine kinase inhibitor, MB3QBD4BE7,
SYN
Example 1: Synthesis of Compound 001
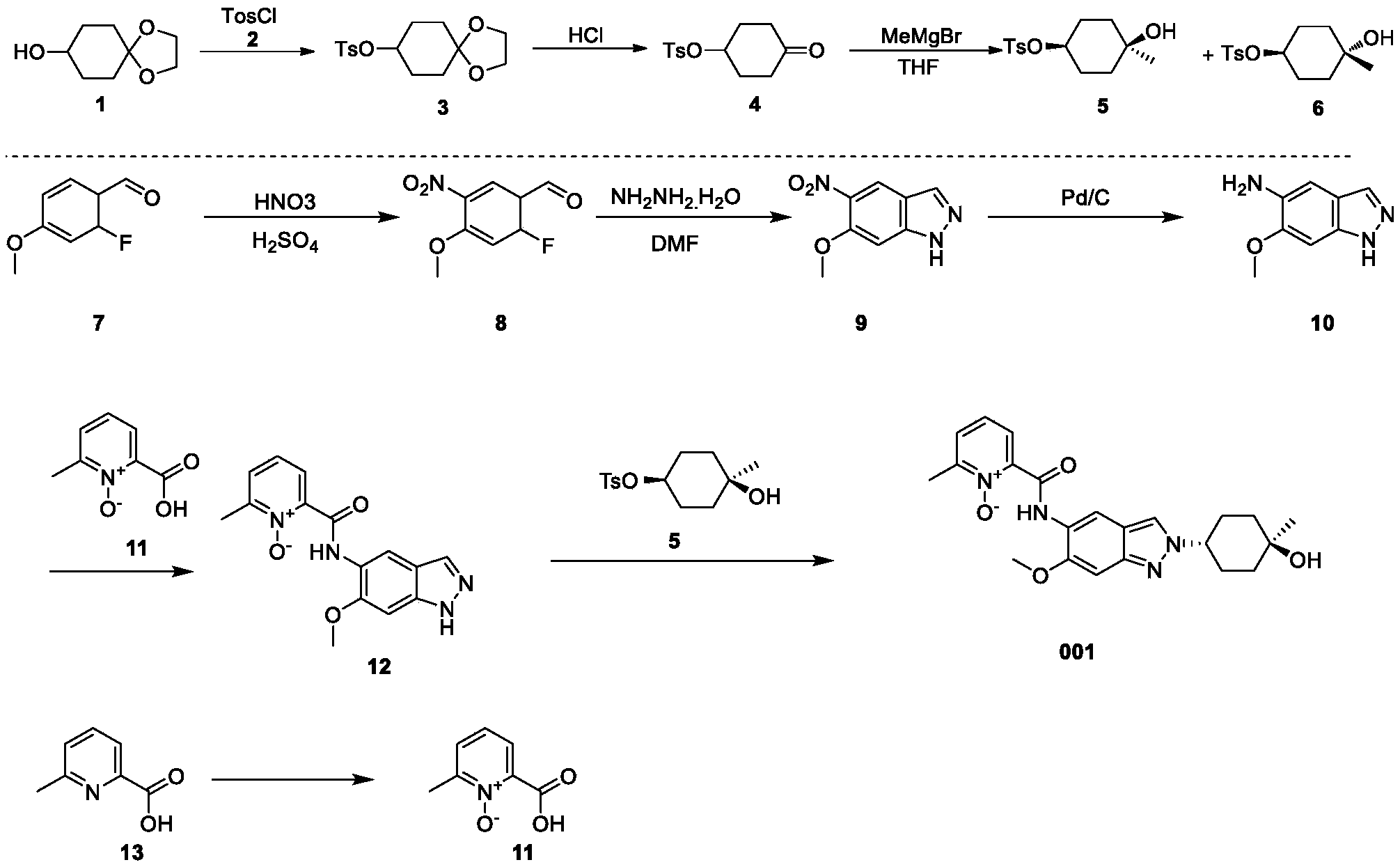
ynthesis of compound 001, namely 2-((2-(trans-4-hydroxy-cis-4-methylcyclohexyl)-6-methoxy-2H-indazol-5-yl)carbamoyl)-6-methylpyridine 1-oxide [0133]
[0134]Cesium carbonate (985 mg) was added to 5 mL of a DMF solution containing compound 12 (300 mg) and compound 5 (344 mg) at 25°C. The reaction mixture was stirred at 90°C for 16 hours. The reaction mixture was then added to 30 mL of water and extracted with ethyl acetate (10 mL * 3). The organic phase was concentrated under reduced pressure, and the residue was purified by preparative high-performance liquid chromatography (HPLC) using a column (CH3CN :
H2O ( 0.1 % NH4HCO3 ) = 15-45%, UV: 214 nm, flow rate: 15 mL/min) to obtain compound 001 (70 mg, yield 17%).[0135]
1 H NMR (400MHz, DMSO-d6): δ14.16(s,1H),8.78(s,1H),8.34(s,1H),8.32-8.30(m,1H),7.77(d,J=7.6Hz,1H),7.58(t,J=8.0Hz,1H),7.13(s,1 H),4.45(s,1H),4.43-4.40(m,1H),3.95(s,3H),2.53(s,3H),2.09-2.00(m,4H),1.68-1.58(m,4H),1.22(s,3H).LCMS: Rt=3.646min,[M+H] + =411.1.
SYN
2-((2-(trans-4-hydroxy-cis-4-methylcyclohexyl)-6-methoxy-2H-indazol-5-yl)carbamoyl)-6-methylpyridine 1-oxide as shown in the following formula:
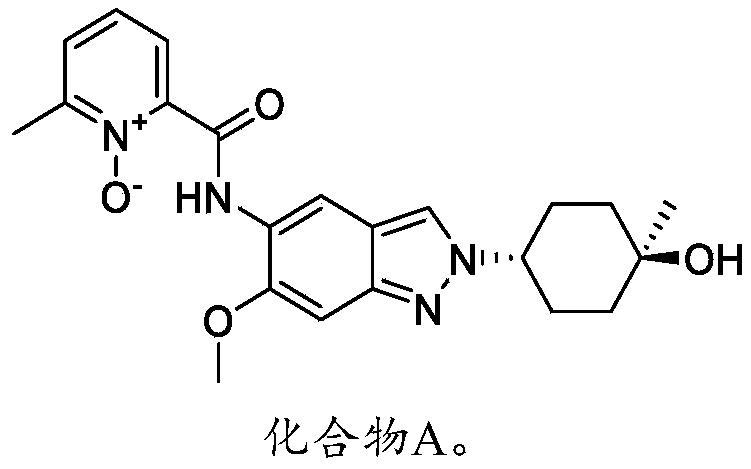
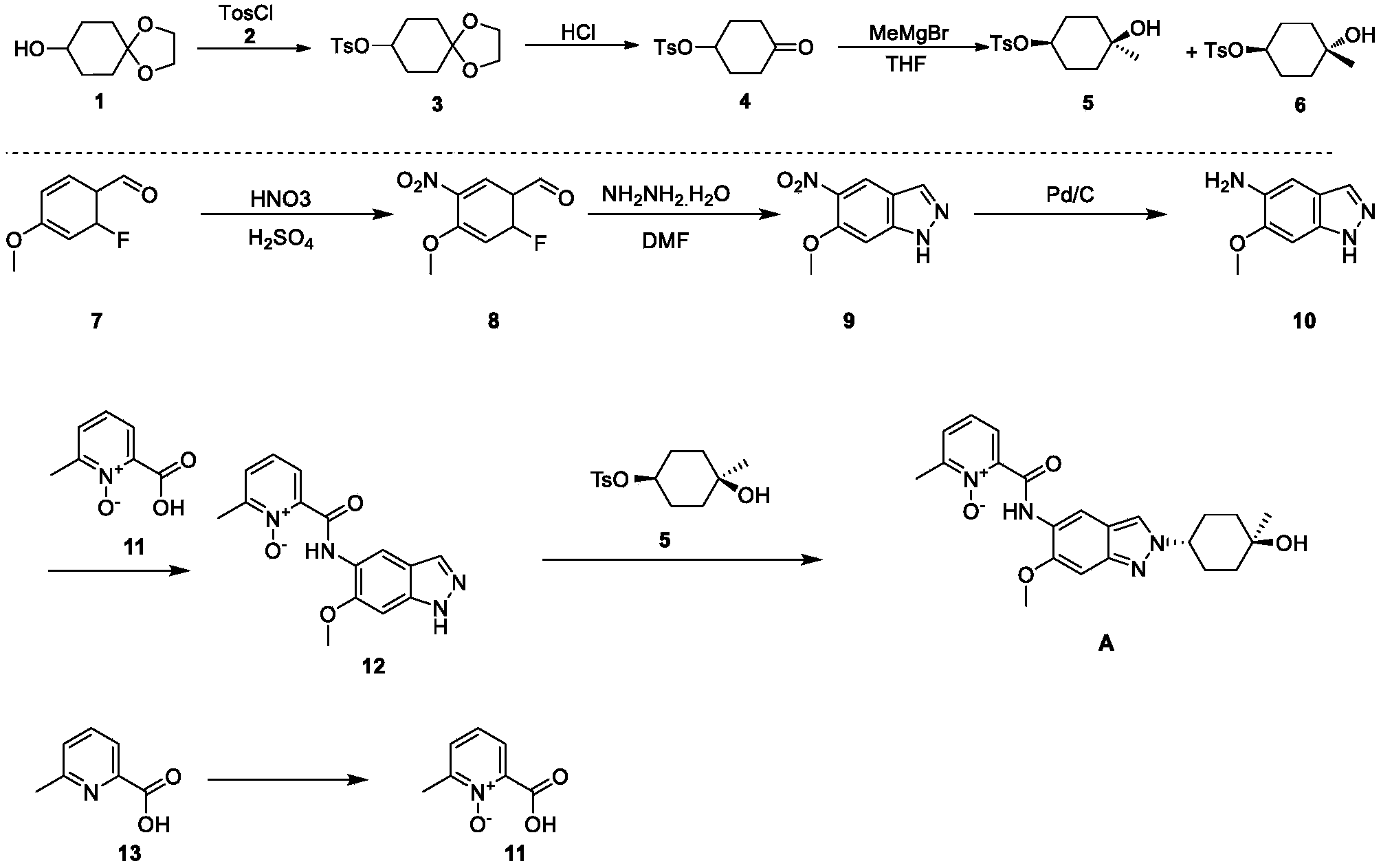
[0257](1) Synthesis of compound 3
[0258]DMAP (42.5 g), compound 2 (63.4 g), and triethylamine (63.9 g) were added sequentially to a 500 mL solution of compound 1 (50 g) in dichloromethane at 15°C, and the mixture was stirred at 25°C for 18 hours. Dichloromethane (200 mL) was added to the reaction mixture, followed by washing with water (300 mL × 2), then with 1 M dilute hydrochloric acid (300 mL × 3). The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain 98 g of a yellow solid, yield: 99% (i.e., compound 3).
[0259](2) Synthesis of compound 4
[0260]1M dilute hydrochloric acid (300 mL) was added to a tetrahydrofuran solution (50 g) of compound 3 at 15°C, and the mixture was stirred at 25°C for 20 hours. The mixture was cooled to 0°C. The pH was adjusted to 9 with 1M sodium hydroxide solution. Extraction was performed with ethyl acetate (200 mL × 3). The extract was washed with saturated sodium chloride solution (300 mL). The solution was dried over anhydrous sodium sulfate. The mixture was filtered. The solution was concentrated under reduced pressure, and the residue was slurried with petroleum ether (150 mL) to give 39 g of a white solid, 91% yield (i.e., compound 4).
[0261](3) Synthesis of compounds 5 & 6
[0262]At -40°C, a tetrahydrofuran solution (200 mL) of compound 4 (34.5 g) was added dropwise to a tetrahydrofuran solution (500 mL) of methyl magnesium bromide (85.8 mL). The mixture was stirred at -40°C for 4 hours. The reaction was quenched with a saturated ammonium chloride solution (100 mL). The mixture was extracted with ethyl acetate (500 mL × 3). The extract was washed with saturated brine (300 mL), dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 5:1) to give a colorless oily compound 5 (4.3 g, 10%), a colorless oily compound 6 (7.0 g, 17%), and a mixture of 12 g.
[0264]
1H NMR (400MHz,CDCl3 ) : δ7.79(d,J=8.0Hz,2H), 7.32(d,J=8.4Hz,2H), 4.52-4.41(m,1H), 2.44(s,3H), 1.95-1.80(m,2H), 1.77-1.61(m,4H), 1.46-1.35(m,2H), 1.19(s,3H).
[0266]
1H NMR (400MHz,CDCl3 ) : δ7.79(d,J=8.4Hz,2H), 7.33(d,J=8.0Hz,2H), 4.74-4.64(m,1H), 2.44(s,3H), 1.92-1.79(m,2H), 1.77-1.62(m,4H), 1.49-1.38(m,2H), 1.23(s,3H).
[0267](4) Synthesis of compound 8
[0268]A mixture of concentrated sulfuric acid (1.6 mL, 98%) and nitric acid (1.6 mL, 70%) was added dropwise to a solution of compound 7 (2.0 g) in concentrated sulfuric acid (12 mL, 98%) at -15°C. After the addition was complete, the mixture was stirred at -15°C for 2 hours. The reaction solution was then slowly poured into ice water and stirred for 5 minutes. The mixture was filtered, washed with water, and the solid was collected and dried under reduced pressure to give 2.5 g of a yellow solid, yield: 97% (i.e., compound 8).
[0269](5) Synthesis of compound 9
[0270]Hydrazine hydrate (2.4 mL, 98%) was added to a DMF (20 mL) solution of compound 8 (2.0 g) at room temperature. After the addition was complete, the mixture was heated to 120 °C and stirred for 16 hours. After cooling to room temperature, the mixture was slowly poured into ice water and stirred. The mixture was filtered, the solid was washed with water, and the solid was collected and concentrated under reduced pressure to obtain 1.3 g of yellow solid. Yield: 67% (i.e., compound 9).
[0271](6) Synthesis of compound 10
[0272]Compound 9 (12.4 g) and palladium on carbon (7 g, 10%) were added sequentially to 400 mL of ethyl acetate at 15°C. After the addition was complete, the mixture was stirred for 18 hours under hydrogen protection at 15°C. The palladium on carbon was filtered off from the reaction solution, and the filtrate was concentrated and evaporated to dryness to obtain 10.4 g of white solid product, with a yield of 99% (i.e., compound 10).
[0273](7) Synthesis of compound 12
[0274]EDCI.HCl (2.6 g) was added to a Py (15 mL) solution of compound 10 (1.5 g) and compound 11 (1.4 g) at 25°C. The reaction mixture was stirred at 25°C for 16 hours. The reaction mixture was concentrated and evaporated to dryness. The residue was slurried by passing MeOH/H₂O ( 20 mL/20 mL) to obtain 1.3 g of a yellow solid product, with a yield of 48% (i.e., compound 12).
[0275](8) Synthesis of compound A
[0276]
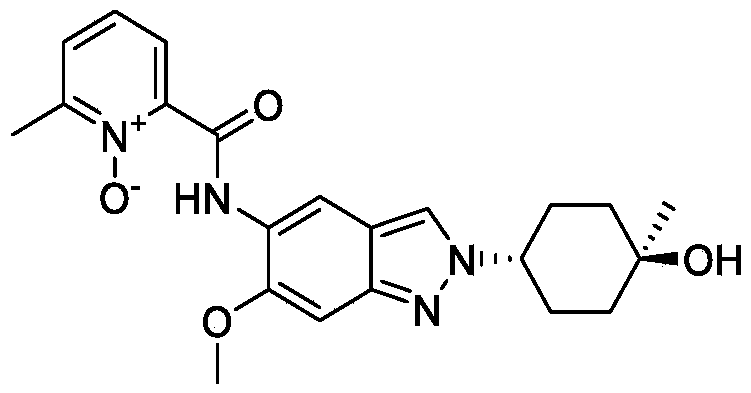
[0277]Cesium carbonate (985 mg) was added to 5 mL of a DMF solution containing compound 12 (300 mg) and compound 5 (344 mg) at 25°C. The reaction mixture was stirred at 90°C for 16 hours. The reaction mixture was then added to 30 mL of water and extracted with ethyl acetate (10 mL × 3). The organic phase was concentrated under reduced pressure, and the residue was purified by preparative high-performance liquid chromatography (HPLC) using a column (CH3CN :
H2O (
0.1 % NH4HCO3
) = 15-45%, UV: 214 nm, Flowrate: 15 mL/min) to obtain 70 mg of a yellow solid, with a yield of 17% (i.e., compound A).
[0278]
1H NMR(400MHz,DMSO-d6):δ14.16(s,1H),8.78(s,1H),8.34(s,1H),8.32-8.30(m,1H),7.77(d,J=7.6Hz,1H),7.58(t,J=8.0Hz,1H),7.13(s,1H),4.45(s,1H),4.43-4.40(m,1H),3.95(s,3H),2.53(s,3H),2.09-2.00(m,4H),1.68-1.58(m,4H),1.22(s,3H).LCMS:Rt=3.646min,[M+H] +=411.1.
PAT
- Irak inhibitor and preparation method therefor and use thereofPublication Number: EP-4015513-B1Priority Date: 2019-09-24Grant Date: 2023-11-01
- An IRAK inhibitor and its preparation method and usePublication Number: CN-114391013-BPriority Date: 2019-09-24Grant Date: 2024-01-26
- Irak inhibitor and preparation method therefor and use thereofPublication Number: US-2022298139-A1Priority Date: 2019-09-24
- A kind of IRAK inhibitor and its preparation method and usePublication Number: CN-114391013-APriority Date: 2019-09-24
- A kind of polymorphic form of compound and its preparation method and applicationPublication Number: CN-115109035-APriority Date: 2021-03-19
- Polymorphic forms of compound and preparation method therefor and application thereofPublication Number: EP-4310080-A1Priority Date: 2021-03-19
- Polymorphic forms of compound and preparation method therefor and application thereofPublication Number: US-2024182443-A1Priority Date: 2021-03-19
- Preparation method for fused pyrazole-type compoundPublication Number: US-2023250064-A1Priority Date: 2020-06-23
- An IRAK inhibitor and its preparation method and usePublication Number: CN-118146193-APriority Date: 2019-09-24
- IRAK4 inhibitor composition, and preparation method and application thereofPublication Number: CN-115252609-BPriority Date: 2022-08-01Grant Date: 2023-05-26
- Irak4 inhibitor composition, preparation method therefor and use thereofPublication Number: EP-4566607-A1Priority Date: 2022-08-01
- Composition of IRAK4 inhibitor, preparation method and application thereofPublication Number: CN-115252609-APriority Date: 2022-08-01
- Use of indazoles for treating psoriasisPublication Number: CN-114404415-APriority Date: 2022-02-25
- Use of indazole compound for treating psoriasisPublication Number: US-2025161283-A1Priority Date: 2022-02-25
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////nacresertib, serine/threonine kinase inhibitor, MB3QBD4BE7,

{kind=link}