
Home » Posts tagged 'RPX7009'
Tag Archives: RPX7009
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BIAPENEM

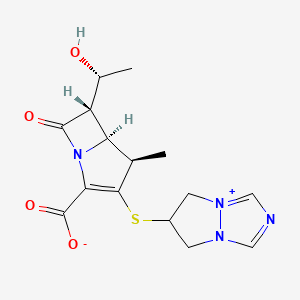
Biapenem
RPX7009
- Molecular FormulaC15H18N4O4S
- Average mass350.393 Da
Biapenern
CL 186-815LJ
C10,627LJ
C10627LJC 10627
omegacin
YR5U3L9ZH1
(4R,5S,6S)-3-((6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-yl)thio)-6-((R)-1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
120410-24-4[RN]
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
7074
(4R,5S,6S)-3-(6,7-Dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium-6-ylsulfanyl)-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylate
TL8000539UNII:YR5U3L9ZH1UNII-YR5U3L9ZH1биапенем
بيابينام比
阿培南
INDIA CDSCO APPROVED 25 SEPT 2021, BDR PHARMA, 
https://www.cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadCTApprovals/BDR.pdfhttps://medicaldialogues.in/news/industry/pharma/bdr-pharma-gets-dcgi-nod-for-generic-antibiotic-drug-biapenem-82384

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
Biapenem (INN) is a carbapenem antibiotic. It has in vitro activity against anaerobes.[1] 1-β-methyl-carbapenem antibiotic. Approved in Japan in 2001.
PATENT
EP 168707
EP 289801
JP 02088578
ZA 9100014
EP 533149
CN 1995040
IN 2006DE01555
CN 101121716
IN 2008CH00177
CN 101805359
CN 101851206
CN 101935321
CN 111875622
WO 2018074916
WO 2016059622
US 20150328323
WO 2015151081
WO 2015155753
WO 2015151078
US 20150284416
WO 2015151080
US 20150038726
WO 2014104488
IN 2013MU00181
WO 2014111957
CN 103570750
WO 2014097221
IN 2012CH01371
WO 2013150550
PAPERS
Journal of Organic Chemistry (1992), 57(15), 4243-9.
Heterocycles (1993), 36(8), 1729-34.
Journal of Antibiotics (1993), 46(12), 1866-82.
e-EROS Encyclopedia of Reagents for Organic Synthesis (2008), 1-3.
Bioorganic & medicinal chemistry letters (2009), 19(17), 5162-5.
IP.com Journal (2014), 14(12A), 1-3
IP.com Journal (2014), 14(10A), 1-2.
Bioorganic & medicinal chemistry (2013), 21(18), 5841-50.

NEW DRUG APPROVALS
one time
$10.00
PATENT
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.

Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80%
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
PATENT
https://patents.google.com/patent/WO2002057266A1/enEXAMPLE

PNB = p-nitrobenzyl

Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
References
- ^ Aldridge KE, Morice N, Schiro DD (April 1994). “In vitro activity of biapenem (L-627), a new carbapenem, against anaerobes”. Antimicrob. Agents Chemother. 38 (4): 889–93. doi:10.1128/aac.38.4.889. PMC 284564. PMID 8031067.
External links
- (in Japanese) Omegacin
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | IV |
| ATC code | J01DH05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 120410-24-4 |
| PubChem CID | 71339 |
| ChemSpider | 64442 |
| UNII | YR5U3L9ZH1 |
| ChEBI | CHEBI:3089 |
| ChEMBL | ChEMBL285347 |
| CompTox Dashboard (EPA) | DTXSID5046435 |
| Chemical and physical data | |
| Formula | C15H18N4O4S |
| Molar mass | 350.39 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
ClinicalTrials.gov
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| NCT04552444 | Clinical Efficacy of Combination Therapy Based on High-dose Biapenem in CRKP Infections | Recruiting | 2020-09-17 | |
| NCT01772836 | Safety Study of Intravenous Biapenem (RPX2003) and RPX7009 Given Alone and in Combination | Phase 1 | Completed | 2013-07-11 |
| NCT01702649 | Safety, Tolerability, Pharmacokinetics of Intravenous RPX2003 (Biapenem) in Healthy Adult Subjects | Phase 1 | Completed | 2012-12-03 |
NIPH Clinical Trials Search of Japan
| CTID | Title | Phase | Status | Date |
|---|---|---|---|---|
| UMIN000017219 | Feasibility and efficacy of the de-escalation therapy by Biapenem for postoperative bacterial pneumonia. | None | Recruiting | 2015-04-22 |
| UMIN000003964 | Clinical evaluation of Biapenem 0.3g, three times daily dosing in eldery patients with pneumonia (moderate and severe infection) | Not applicable | Complete: follow-up complete | 2010-07-29 |
/////////BIAPENEM, TL8000539, UNII:YR5U3L9ZH1, UNII-YR5U3L9ZH1, биапенем, بيابينام ,比阿培南 , Biapenern, CL 186-815, CL 186815, L 627, LJC 10627, Omegacin, Antibacterial, Antibiotics, Lactams, Carbapenems, ind 2021, india 2021, approvals 2021
CC1C2C(C(=O)N2C(=C1SC3CN4C=NC=[N+]4C3)C(=O)[O-])C(C)O
https://clinicaltrials.gov/search/intervention=Biapenem

updated
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Yield: 81.77%
HPLC Purity: 99.80%
PATENT
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
Vaborbactam, Ваборбактам , فابورباكتام , 法硼巴坦 ,


RN: 1360457-46-0
UNII: 1C75676F8V
- Originator Rempex Pharmaceuticals
- Developer The Medicines Company; US Department of Health and Human Services
- Class Antibacterials; Pyrrolidines; Small molecules; Thienamycins
- Mechanism of Action Beta lactamase inhibitors; Cell wall inhibitors
Highest Development Phases
- Registered Urinary tract infections
- Phase III Bacteraemia; Gram-negative infections; Pneumonia; Pyelonephritis
Most Recent Events
- 29 Aug 2017 Registered for Urinary tract infections (Treatment-experienced, Treatment-resistant) in USA (IV) – First global approval
- 29 Aug 2017 Updated efficacy and safety data from a phase III trial in Gram-negative infections released by The Medicines Company
- 09 Aug 2017 Planned Prescription Drug User Fee Act (PDUFA) date for Urinary tract infections (Treatment-experienced, Treatment-resistant) in USA (IV) is 2017-08-29

 Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.
Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.
Vaborbactam (INN)[1] is a non-β-lactam β-lactamase inhibitor discovered by Rempex Pharmaceuticals, a subsidiary of The Medicines Company. While not effective as an antibiotic by itself, it restores potency to existing antibiotics by inhibiting the beta-lactamase enzymes that would otherwise degrade them. When combined with an appropriate antibiotic it can be used for the treatment of gram-negative bacterial infections.[2]
According to a Medicines Company press release, as of June 2016 a combination of vaborbactam with the carbapenem antibiotic meropenem had met all pre-specified primary endpoints in a phase III clinical trial in patients with complicated urinary tract infections.[3] The company planned to submit an NDA to the FDAin early 2017.
Biochemistry
Carbapenemases are a family of β-lactamase enzymes distinguished by their broad spectrum of activity and their ability to degrade carbapenem antibiotics, which are frequently used in the treatment of multidrug-resistant gram-negative infections.[4] Carbapenemases can be broadly divided into two different categories based on the mechanism they use to hydrolyze the lactam ring in their substrates. Metallo-β-lactamases contain bound zinc ions in their active sites and are therefore inhibited by chelating agents like EDTA, while serine carbapenemases feature an active site serine that participates in the hydrolysis of the substrate.[4] Serine carbapenemase-catalyzed hydrolysis employs a three-step mechanism featuring acylation and deacylation steps analogous to the mechanism of protease-catalyzed peptide hydrolysis, proceeding through a tetrahedral transition state.[4][5]
Boronic acids are unusual in their ability to reversibly form covalent bonds with alcohols such as the active site serine in a serine carbapenemase. This property enables them to function as transition state analogs of serine carbapenemase-catalyzed lactam hydrolysis and thereby inhibit these enzymes. Based on data from Hecker et al., vaborbactam is a potent inhibitor of a variety of β-lactamases, exhibiting a 69-nanomolar {\displaystyle K_{i}}
Given their mechanism of action, the possibility of off-target effects brought about through inhibition of endogenous serine hydrolases is an obvious possible concern in the development of boronic acid β-lactamase inhibitors, and in fact boronic acids like bortezomib have previously been investigated or developed as inhibitors of various human proteases.[2] Vaborbactam, however, is a highly specific β-lactamase inhibitor, with an IC50 >> 1 mM against all human serine hydrolases against which it has been tested.[2] Consistent with its high in vitro specificity, vaborbactam exhibited a good safety profile in human phase I clinical trials, with similar adverse events observed in both placebo and treatment groups.[6] Hecker et al. argue this specificity results from the higher affinity of human proteases to linear molecules; thus it is expected that a boron heterocycle will have zero effect on them.
SYN
WO 2015171430


PATENT

| Inventors | Gavin Hirst, Raja Reddy, Scott Hecker, Maxim Totrov, David C. Griffith, Olga Rodny, Michael N. Dudley, Serge Boyer, Less « |
| Applicant | Rempex Pharmaceuticals, Inc. |
Antibiotics have been effective tools in the treatment of infectious diseases during the last half-century. From the development of antibiotic therapy to the late 1980s there was almost complete control over bacterial infections in developed countries. However, in response to the pressure of antibiotic usage, multiple resistance mechanisms have become widespread and are threatening the clinical utility of antibacterial therapy. The increase in antibiotic resistant strains has been particularly common in major hospitals and care centers. The consequences of the increase in resistant strains include higher morbidity and mortality, longer patient hospitalization, and an increase in treatment costs
[0003] Various bacteria have evolved β-lactam deactivating enzymes, namely, β-lactamases, that counter the efficacy of the various β-lactams. β-lactamases can be grouped into 4 classes based on their amino acid sequences, namely, Ambler classes A, B, C, and D. Enzymes in classes A, C, and D include active-site serine β-lactamases, and class B enzymes, which are encountered less frequently, are Zn-dependent. These enzymes catalyze the chemical degradation of β-lactam antibiotics, rendering them inactive. Some β-lactamases can be transferred within and between various bacterial strains and species. The rapid spread of bacterial resistance and the evolution of multi- resistant strains severely limits β-lactam treatment options available.
[0004] The increase of class D β-lactamase-expressing bacterium strains such as Acinetobacter baumannii has become an emerging multidrug-resistant threat. A. baumannii strains express A, C, and D class β-lactamases. The class D β-lactamases such as the OXA families are particularly effective at destroying carbapenem type β-lactam antibiotics, e.g., imipenem, the active carbapenems component of Merck’s Primaxin® (Montefour, K.; et al. Crit. Care Nurse 2008, 28, 15; Perez, F. et al. Expert Rev. Anti Infect. Ther. 2008, 6, 269; Bou, G.; Martinez-Beltran, J. Antimicrob. Agents Chemother. 2000, 40, 428. 2006, 50, 2280; Bou, G. et al, J. Antimicrob. Agents Chemother. 2000, 44, 1556). This has imposed a pressing threat to the effective use of drugs in that category to treat and prevent bacterial infections. Indeed the number of catalogued serine-based β- lactamases has exploded from less than ten in the 1970s to over 300 variants. These issues fostered the development of five “generations” of cephalosporins. When initially released into clinical practice, extended- spectrum cephalosporins resisted hydrolysis by the prevalent class A β-lactamases, TEM-1 and SHV-1. However, the development of resistant strains by the evolution of single amino acid substitutions in TEM-1 and SHV-1 resulted in the emergence of the extended- spectrum β-lactamase (ESBL) phenotype.
[0005] New β-lactamases have recently evolved that hydrolyze the carbapenem class of antimicrobials, including imipenem, biapenem, doripenem, meropenem, and ertapenem, as well as other β-lactam antibiotics. These carbapenemases belong to molecular classes A, B, and D. Class A carbapenemases of the KPC-type predominantly in Klebsiella pneumoniae but now also reported in other Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii. The KPC carbapenemase was first described in 1996 in North Carolina, but since then has disseminated widely in the US. It has been particularly problematic in the New York City area, where several reports of spread within major hospitals and patient morbidity have been reported. These enzymes have also been recently reported in France, Greece, Sweden, United Kingdom, and an outbreak in Germany has recently been reported. Treatment of resistant strains with carbapenems can be associated with poor outcomes.
[0006] Another mechanism of β-lactamase mediated resistance to carbapenems involves combination of permeability or efflux mechanisms combined with hyper production of beta-lactamases. One example is the loss of a porin combined in hyperproduction of ampC beta-lactamase results in resistance to imipenem in Pseudomonas aeruginosa. Efflux pump over expression combined with hyperproduction of the ampC β-lactamase can also result in resistance to a carbapenem such as meropenem.
[0007] Because there are three major molecular classes of serine-based β- lactamases, and each of these classes contains significant numbers of β-lactamase variants, inhibition of one or a small number of β-lactamases is unlikely to be of therapeutic value. Legacy β-lactamase inhibitors are largely ineffective against at least Class A carbapenemases, against the chromosomal and plasmid-mediated Class C cephalosporinases and against many of the Class D oxacillinases. Therefore, there is a need for improved β-lactamase inhibitors.
The following compounds are prepared starting from enantiomerically pure (R)-tert-butyl 3-hydroxypent-4-enoate (J. Am. Chem. Soc. 2007, 129, 4175-4177) in accordance with the procedure described in the above Example 1.

5
[0192] 2-((3R,6S)-2-hydroxy-3-(2-(thiophen-2-yl)acetamido)-l,2-oxaborinan-6-yl)acetic acid 5. 1H NMR (CD3OD) δ ppm 0.97-1.11 (q, IH), 1.47-1.69 (m, 2H), 1.69-1.80 (m, IH), 2.21-2.33 (td, IH), 2.33-2.41 (dd, IH), 2.58-2.67 (m, IH), 3.97 (s, 2H), 4.06-4.14 (m, IH), 6.97-7.04 (m, IH), 7.04-7.08 (m, IH), 7.34-7.38 (dd, IH); ESIMS found for Ci2Hi6BN05S m/z 28 -H20)+.
The following compounds are prepared starting from enantiomerically pure (R)-tert-butyl 3-hydroxypent-4-enoate (J. Am. Chem. Soc. 2007, 129, 4175-4177) in accordance with the procedure described in the above Example 1.

5
[0175] 2-((3R,6S)-2-hydroxy-3-(2-(thiophen-2-yl)acetamido)-l,2-oxaborinan-6- yl)acetic acid 5. 1H NMR (CD3OD) δ ppm 0.97-1.11 (q, 1H), 1.47-1.69 (m, 2H), 1.69-1.80 (m, 1H), 2.21-2.33 (td, 1H), 2.33-2.41 (dd, 1H), 2.58-2.67 (m, 1H), 3.97 (s, 2H), 4.06-4.14 (m, 1H), 6.97-7.04 (m, 1H), 7.04-7.08 (m, 1H), 7.34-7.38 (dd, 1H); ESIMS found for Ci2Hi6BN05S m/z 280 (100%) (M-H20)+.
EXAMPLES
Example 1 – Synthesis of Intermediate Compound 10
[0191] The compound of Formula 10 was synthesized as shown in Scheme 3, below:
Scheme 3

95%

80% for 2 steps
(i?)-t-butyl 3-(trimethysilyloxy)-pent-4-enoate (7)
[0192] Chlorotrimethylsilane (4.6 mL, 36.3 mmol, 1.25 eq) was added to a solution of (R)-t-butyl 3-hydroxy-pent-4-enoate (1, 5 g, 29 mmol) and triethylamine (5.3 mL, 37.3 mmol, 1.3 eq) in dichloromethane (25 mL) keeping the temperature below 30 °C. After completion of the addition, the white heterogeneous mixture was stirred at rt for 20 minutes (TLC, GC, note 2) then quenched with MeOH (352 μί, 0.3 eq). After stirring at rt for 5 minutes, the white heterogeneous reaction mixture was diluted with heptane (25 mL). The salts were filtered off and rinsed with heptane (2 x 10 mL). The combined turbid filtrates were washed with a saturated solution of NaHC03 (2 x 25 mL) and concentrated to dryness. The residual oil was azeotroped with heptane (25 mL) to give a colorless oil that was used immediately.
QSVt-butyl 3-(trimethylsilyloxy)-5-(4,4,5,5-tetramethyl-[L3,21dioxaborolan-2-yl)-pentanoate (8)
[0193] A solution of bis-diphenylphosphino-ethane (46.3 mg, 0.2 mol%) and [Ir(COD)Cl]2 (39 mg, 0.1 mol%) in CH2C12 (5 mL) was added to a refluxing solution of crude TMS-protected pentenoate 7. Pinacol borane (9.3 mL,l .l eq) was added to the
refluxing solution. After stirring at reflux for 3 h, the reaction mixture was cooled to room temperature, concentrated to dryness and taken up in heptane (50 mL). The insolubles were filtered over Celite and rinse with heptane (10 mL).
Ethanolamine-boronic acid salt (10)
[0194] A mixture of fully protected boronate 8 (5.0 g, 13.4 mmol), 0.5 N HC1 (5 mL) and acetone (0.5 mL) was stirred vigorously at room temperature, providing intermediate 9. After complete consumption of the starting material, a solution of NaI04 (3.44 g, 1.2 eq) in water (15 mL) was added slowly keeping the temperature <30 °C. Upon the completion of the addition (30 min), the reaction mixture was allowed to cool to room temperature. After consumption of all pinacol, MTBE (5 mL) was added. After stirring at room temperature for 10 min, the white solids were filtered off and rinsed with MTBE (2 x 5 mL). The filtrate was partitioned and the aqueous layer was extracted with MTBE (10 mL). The combined organic extracts were washed sequentially with a 0.1 M NaHS03 solution (2 x 5 mL), a saturated NaHC03 solution (5 mL) and brine (5 mL). The organic layer was concentrated to dryness. The residue was taken up in MTBE (15 mL) and the residual salts filtered off. The filtrate was concentrated to dryness and the residue was taken up in MTBE (10 mL) and acetonitrile (1.7 mL). Ethanolamine (0.99 mL, 1.1 eq) was added. After stirring at room temperature for 1 hour, the heterogeneous mixture was stirred at 0 °C. After stirring at 0 °C for 2 hours, the solids were collected by filtration, rinsed with MTBE (2 x 5 mL), air dried then dried under high vacuum to give Compound 10 as a white granular powder.
Example 2 – Preparation of Beta-Lactamase Inhibitor (15)
[0195] The compound of Formula 15 was synthesized as shown in Scheme 4 below:
Scheme 4

Synthesis of pinanediol boronate (12)
[0196] Ethanolammonium boronate 11 (15 g, 61.7 mmol) and pinanediol (10.5 g, 61.7 mmol, 1 eq) were suspended in MTBE (75 mL). Water (75 mL) was added and the yellow biphasic heterogeneous mixture was stirred at room temperature. After stirring for 2 hours at room temperature, some pinanediol was still present and stirring was continued overnight. The layers were separated and the organic layer was washed with brine, concentrated under reduced pressure and azeotroped with MTBE (2 x 30 mL). The residual oil was taken up in dichloromethane (40 mL). In another flask, TBSC1 (1 1.6 g, 77.1 mmol, 1.25 eq) was added to a solution of imidazole (9.66 g, 141.9 mmol, 2.3 eq) in dichloromethane (25 mL). The white slurry was stirred at room temperature. After 5 minutes, the solution of pinanediol boronate was added to the white slurry and the flask was rinsed with dichloromethane (2 x 5 mL). The heterogeneous reaction mixture was heated at reflux temeprature. After stirring at reflux for 8 hours, the reaction mixture was cooled to 30 °C and TMSC1 (330 \JL) was added. After stirring 30 minutes at 30 °C, MeOH (15 mL) was added. After stirring at room temperature overnight, the reaction mixture was washed sequentially with 0.5 N HC1 (115 niL), 0.5 N HC1 (60 n L) and saturated NaHC03 (90 niL). The organic layer was concentrated under reduced pressure and azeotroped with heptane (150 n L) to give 12 as a yellow oil (27.09 g, 94.1%) which was used without purification.
Synthesis of chloroboronate (13)
[0197] A solution of n-butyllithium (2.5 M in hexane, 29.6 niL, 74.1 mmol, 1.3 eq) was added to THF (100 mL) at -80 °C. The resulting solution was cooled to -100 °C. A solution of dichloromethane (14.6 mL, 228 mmol, 4 eq) in THF (25 mL) was added via syringe pump on the sides of the flask keeping the temperature < -95 °C. During the second half of the addition a precipitate starts to appear which became thicker with the addition of the remaining dichloromethane solution. After stirring between -100 and -95 °C for 30 min, a solution of 12 (26.59 g, 57 mmol) in THF (25 mL) was added by syringe pump on the sides of the flask while maintaining the batch temperature < -95 °C to give a clear yellow solution. After stirring between -100 and -95 °C for 30 min, a solution of zinc chloride (1 M in ether, 120 mL, 120 mmol, 2.1 eq) was added keeping the temperature < -70 °C. The reaction mixture was then warmed to room temperature (at about -18 °C the reaction mixture became turbid/heterogeneous). After stirring at room temperature for 2 hours, the reaction mixture was cooled to 15 °C and quenched with 1 N HC1 (100 mL). The layers were separated and the organic layer was washed sequentially with 1 N HC1 (100 mL) and water (2 x 100 mL), concentrated to oil and azeotroped with heptane (3 x 150 mL) to provide 13 as a yellow oil (30.03 g, 102%) which was used without purification.
Synthesis of (14)
[0198] LiHMDS (1 M in THF, 63 mL, 62.7 mmol, 1.1 eq) was added to a solution of 13 (29.5 g, 57 mmol) in THF (60 mL) while maintaining the batch temperature at < -65 °C. After stirring at -78 °C for 2 hours, additional LiHMDS (5.7 mL, 0.1 eq) was added to consume the remaining starting material. After stirring at -78 °C for 30 minutes, the tan reaction mixture was warmed to room temperature. After stirring at room temperature for one hour, the solution of silylated amine was added via cannula to a solution of HOBT ester of 2-thienylacetic acid in acetonitrile at 0 °C (the solution of HOBT ester was prepared by adding EDCI (16.39 g, 85.5 mmol, 1.5 eq) to a suspension of recrystallized 2-thienylacetic acid (9.73 g, 68.4 mmol, 1.2 eq) and HOBT.H20 (11.35 g, 74.1 mmol, 1.3 eq) in acetonitrile (10 mL) at 0 °C. The clear solution was stirred at 0 °C for 30 minutes prior to the addition of the silylated amine). After stirring at 0 °C for one hour, the heterogeneous reaction mixture was placed in the fridge overnight. Saturated aqueous sodium bicarbonate (80 mL) and heptane (80 mL) were added, and after stirring 30 minutes at room temperature, the layers were separated. The organic layer was washed with saturated aqueous sodium bicarbonate (2 x 80 mL) and filtered through Celite. The filtrate was concentrated under reduced pressure and the tan oil was azeotroped with heptane (3 x 1 10 mL). The residue was taken up in heptane (60 mL) and seeds were added. After stirring at room temperature for one hour, the reaction mixture became heterogeneous. After stirring 4 hours at 0 °C, the solids were collected by filtration and washed with ice cold heptane (3 x 20 mL), air dried then dried under high vacuum to give 14 as an off white powder (10.95 g, 31%).
Synthesis of (15)
[0199] A mixture of 14 (10 g, 16.1 mmol), boric acid (1.3 g, 20.9 mmol, 1.3 eq), dioxane (20 mL), and 1 M sulfuric acid (10 mL) was heated at 75 °C. After stirring 7 hours at 75 °C, the cooled reaction mixture was diluted with water (10 mL) and MTBE (30 mL) and the residual mixture was cooled to 0 °C. The pH was adjusted to 5.0 with a solution of 2 N NaOH (14 mL). The layers were separated and the aqueous layer was extracted with MTBE (2 x 30 mL) then concentrated to dryness. The residue was taken up in water (10 mL) and the solution was filtered through a 0.45 μηι GMF syringe filter. The flask and filter were rinsed with water (7.5 mL). The pH of the filtrate was lowered to 4.0 with 2 M H2SO4 and seeds (5 mg) were added. After stirring at room temperature for 10 minutes, the pH was lowered to 1.9 over 1 hour with 2 M H2S04 (total volume 3.5 mL). After stirring at room temperature for 2 hours, the solids were collected by filtration. The filtrate was recirculated twice to rinse the flask and the cake was washed with water (2 x 12 mL), air dried then dried under high vacuum to give 15 as a white powder (3.63 g, 76%).

|
Patent ID
|
Patent Title
|
Submitted Date
|
Granted Date
|
|
CYCLIC BORONIC ACID ESTER DERIVATIVES AND THERAPEUTIC USES THEREOF
|
2013-07-29
|
2013-12-26
|
|
|
Cyclic boronic acid ester derivatives and therapeutic uses thereof
|
2011-08-08
|
2014-03-25
|
|
|
CYCLIC BORONIC ACID ESTER DERIVATIVES AND THERAPEUTIC USES THEREOF
|
2013-03-15
|
2013-12-12
|
|
|
METHODS OF TREATING BACTERIAL INFECTIONS
|
2013-02-11
|
2015-04-30
|
References
- Jump up^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 75” (PDF). World Health Organization. pp. 161–2.
- ^ Jump up to:a b c d Hecker, SJ; Reddy, KR; Totrov, M; Hirst, GC; Lomovskaya, O; Griffith, DC; King, P; Tsivkovski, R; Sun, D; Sabet, M; Tarazi, Z; Clifton, MC; Atkins, K; Raymond, A; Potts, KT; Abendroth, J; Boyer, SH; Loutit, JS; Morgan, EE; Durso, S; Dudley, MN (14 May 2015). “Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases”. Journal of Medicinal Chemistry. 58 (9): 3682–92. ISSN 0022-2623. doi:10.1021/acs.jmedchem.5b00127.
- Jump up^ “The Medicines Company Announces Positive Top-Line Results for Phase 3 TANGO 1 Clinical Trial of CARBAVANCE®“. Business Wire, Inc.
- ^ Jump up to:a b c Queenan, AM; Bush, K (13 July 2007). “Carbapenemases: the Versatile β-Lactamases”. Clinical Microbiology Reviews. 20 (3): 440–58. ISSN 0893-8512. PMC 1932750
 . PMID 17630334. doi:10.1128/CMR.00001-07.
. PMID 17630334. doi:10.1128/CMR.00001-07. - Jump up^ Lamotte-Brasseur, J; Knox, J; Kelly, JA; Charlier, P; Fonzé, E; Dideberg, O; Frère, JM (December 1994). “The Structures and Catalytic Mechanisms of Active-Site Serine β-Lactamases”. Biotechnology and Genetic Engineering Reviews. 12 (1): 189–230. ISSN 0264-8725. PMID 7727028. doi:10.1080/02648725.1994.10647912.
- Jump up^ Griffith, DC; Loutit, JS; Morgan, EE; Durso, S; Dudley, MN (October 2016). “Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of the β-Lactamase Inhibitor Vaborbactam (RPX7009) in Healthy Adult Subjects”. Antimicrobial Agents and Chemotherapy. 60 (10): 6326–32. ISSN 0066-4804. PMC 5038296 . PMID 27527080. doi:10.1128/AAC.00568-16.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
IV |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H16BNO5S |
| Molar mass | 297.13 g·mol−1 |
| 3D model (JSmol) | |
![]()
FDA approves new antibacterial drug Vabomere (meropenem, vaborbactam)

Meropenem

The U.S. Food and Drug Administration today approved Vabomere for adults with complicated urinary tract infections (cUTI), including a type of kidney infection, pyelonephritis, caused by specific bacteria. Vabomere is a drug containing meropenem, an antibacterial, and vaborbactam, which inhibits certain types of resistance mechanisms used by bacteria.
“The FDA is committed to making new safe and effective antibacterial drugs available,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides an additional treatment option for patients with cUTI, a type of serious bacterial infection.”
The safety and efficacy of Vabomere were evaluated in a clinical trial with 545 adults with cUTI, including those with pyelonephritis. At the end of intravenous treatment with Vabomere, approximately 98 percent of patients treated with Vabomere compared with approximately 94 percent of patients treated with piperacillin/tazobactam, another antibacterial drug, had cure/improvement in symptoms and a negative urine culture test. Approximately seven days after completing treatment, approximately 77 percent of patients treated with Vabomere compared with approximately 73 percent of patients treated with piperacillin/tazobactam had resolved symptoms and a negative urine culture.
The most common adverse reactions in patients taking Vabomere were headache, infusion site reactions and diarrhea. Vabomere is associated with serious risks including allergic reactions and seizures. Vabomere should not be used in patients with a history of anaphylaxis, a type of severe allergic reaction to products in the class of drugs called beta-lactams.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of antibacterial drugs, Vabomere should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria.
Vabomere was designated as a qualified infectious disease product (QIDP). This designation is given to antibacterial products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of its QIDP designation, Vabomere received a priority review.
The FDA granted approval of Vabomere to Rempex Pharmaceuticals.
 Moxalactam synthesis
Moxalactam synthesis
Latamoxef (or moxalactam)
http://www.wikiwand.com/en/Latamoxef
////////////////RPX7009, RPX 7009, VABORBACTAM, Vaborbactam, Ваборбактам , فابورباكتام , 法硼巴坦 , FDA 2017

