
Home » Posts tagged 'R 16'
Tag Archives: R 16
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
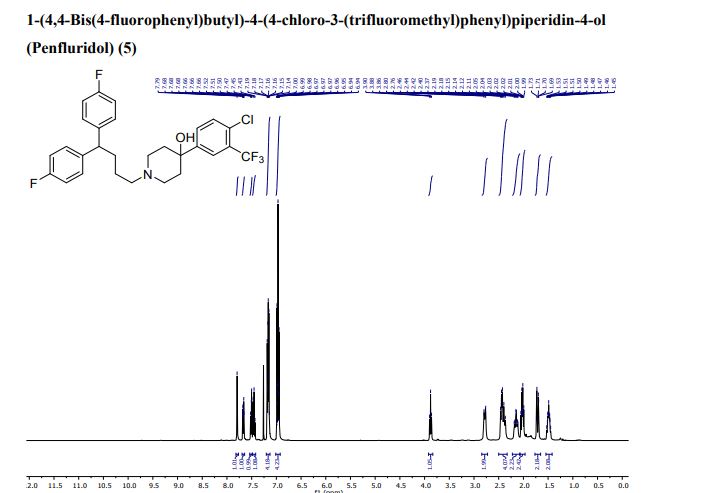
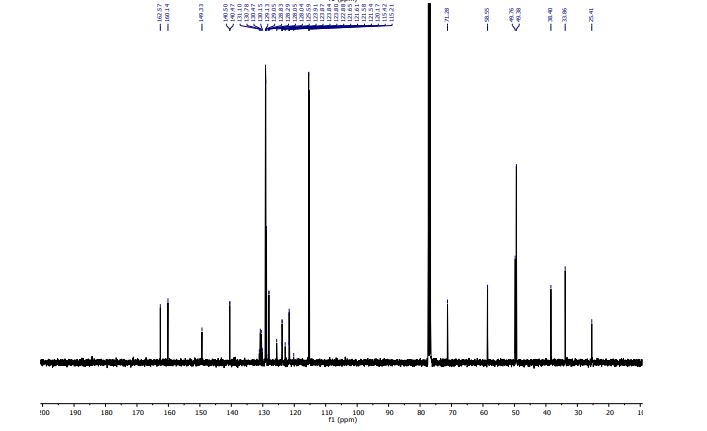
Penfluridol
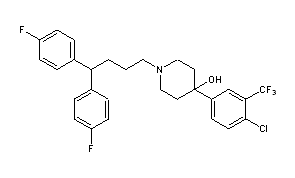
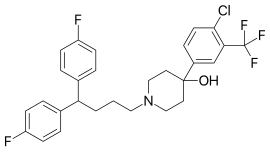
- Molecular FormulaC28H27ClF5NO
- Average mass523.965 Da
| Summary |
|---|
| Although there are shortcomings and gaps in the data, there appears to be enough overall consistency for different outcomes. The effectiveness and adverse effects profile of penfluridol are similar to other typical antipsychotics; both oral and depot. Furthermore, penfluridol is shown to be an adequate treatment option for people with schizophrenia, especially those who do not respond to oral medication on a daily basis and do not adapt well to depot drugs. One of the results favouring penfluridol was a lower drop out rate in medium term when compared to depot medications. It is also an option for people with long-term schizophrenia with residual psychotic symptoms who nevertheless need continuous use of antipsychotic medication. An additional benefit of penfluridol is that it is a low-cost intervention.[3] |
Penfluridol
-
- ATC:N05AG03
- Use:neuroleptic
- Chemical name:1-[4,4-bis(4-fluorophenyl)butyl]-4-[4-chloro-3-(trifluoromethyl)phenyl]-4-piperidinol
- Formula:C28H27ClF5NO
- MW:523.97 g/mol
- CAS-RN:26864-56-2
- EINECS:248-074-5
- LD50:87 mg/kg (M, p.o.);
160 mg/kg (R, p.o.)
Synthesis
PAPER
Late stage functionalization of secondary amines via a cobalt-catalyzed electrophilic amination of organozinc reagents
Org Lett 2019, 21(2): 494
https://pubs.acs.org/doi/10.1021/acs.orglett.8b03787
Scheme 6



English: DE patent 2040231
US patent 3575990
doi:10.1135/cccc19733879

SYN
References
-
- US 3 575 990 (Janssen; 20.4.1971; appl. 3.9.1969).
- DOS 2 040 231 (Janssen; appl. 13.8.1970; USA-prior. 3.9.1969).
-
alternative synthesis:
- FR-appl. 2 161 007 (Janssen; appl. 23.11.1972; J-prior. 25.11.1971).
PATENT
https://patents.google.com/patent/CN106187863A/en
Although Penfluridol listed for many years, but its chemical preparation technology abroad little studied in the earlier literature, there are several prepared as follows:
[0013] Process (a): 1971 Document Ger.0ffen [P], 2040231, (1971) Hermans.HKF first reported Penfluridol chemical synthesis, which process is as follows:
[0014]

[0015] The process of cyclopropyl methanol (ΙΠ) by 4,4, _-difluorophenyl-one ([pi) as a starting material, the reaction of cyclopropyl magnesium bromide-bis 4- (fluorophenyl), then the reaction with thionyl chloride to give 1,1_-bis (4-fluorophenyl) -4-chloro-butene (IV), obtained by catalytic hydrogenation 1,1_-bis (4-phenyl gas) burning chlorobutanol _4_ (V), and finally with 4-chloro-3-methylphenyl gas-4-piperidinol (X VH) in methyl isobutyl ketone was refluxed for three days the reaction to produce Penfluridol (the I), Document: Sindelar.K.et al, Collect Czech.Chem.Commun [J], 38 (12): 3879-3901, (1973).
[0016] In the above process, starting material and documentation of cyclopropyl magnesium bromide hardly prepared each reaction were not reported preparation yield, and therefore Document Sindelar · K · et al, Collect Czech · Chem · Commun [ J], 38 (12):. 3879-3901, (1973) that this technology is not very good.
[0017] Process (b): 1973, Sindelar.K successful research and the following other technology, which process is as follows:
[0018]

[0019] The process consists of 4,4_-bis (4-fluorophenyl) butoxy alkyl iodide as a starting material, 4,4_ ethylenedioxythiophene condensing piperidone removal of generated hydrogen iodide in N-pentanone – [4,4-bis (4-fluorophenyl) butoxy group] -4,4-dioxo-condensing vinyl piperidone, N-then obtained by acid hydrolysis [4,4-bis (4-fluorophenyl ) azetidinyl] -4-piperidone (W), the compound ![]() with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).
with 4-chloro-3-trifluoromethyl phenyl magnesium bromide reacted Penfluridol (I).
[0020] This process route may seem simple, but there are more desired to prepare intermediates, the process is more complex, with low yields reported in the literature.
[0021] Process (c): as follows:
[0022]

[0023] In this process, 4-chloro – (4-fluorophenyl) butyryl-one (Shan) starts, 4-fluorophenyl magnesium bromide reacts with 4-chloro – bis (4-fluorophenyl) butanol ( IX), and then boiling the reaction hydroiodic acid to give 4-iodo-in, red phosphorus catalyst – bis (4-fluorophenyl) butoxy left foot and finally burning ^^ – ^ – methyl ^ two gas – chlorophenyl Bu ‘piperidinol prepared products San ^ top five gas profitable ⑴.
[0024] This synthesis has the characteristics of high yield, but the intermediate (IX), (X) quality is not purified, many by-products, difficult to control the quality of products, and hydroiodic acid to be used, the source of raw material is difficult, therefore, not ideal technology.
[0025] Process (d), as follows:
[0026]

[0027] The process begins by Stobber reaction with 4,4 – fluorophenyl ketone reaction product diethyl succinate and compound (XI), and then generates bis (4-fluorophenyl) methine acid or base hydrolysis after succinic acid (M), by catalytic hydrogenation to give 4,4_-bis (4-fluorophenyl) butanoic acid after, the reaction with thionyl chloride without isolating the compound (XIV) with the compound directly (XW), by reduction after obtain the final product – Penfluridol. The disadvantage of this process is that, in the above reaction step, Stobber the reaction yield is low; hydrogenation catalyst manufacturing operation more difficult and unsafe; reaction with thionyl chloride, large air pollution, and other refractory.
[0028] The various preparation techniques Penfluridol other drug earlier British Patent Brit. 1141664 and German patent Ger. Off en. 2040231 has been reported, but no other foreign patent reports. In neither country has patent coverage, and no magazine reported.
The reaction formula is as follows:
[0058]

[0059] Step (5), the preparation of compounds of formula (XW) as shown, may be employed a method reported in the literature, or prepared using a method specifically includes the following steps:
[0060]

0124] (6) Penfluridol drug (I) were prepared:
[0125]

[0126] In three 500ml reaction flask equipped with a mechanical stirrer, a condenser, a thermometer, a calcium chloride tube, was added 250ml of anhydrous diethyl ether, 2 · 4g (0 · 0631mol) tetrahydro lithium aluminum hydride, stirring was started, was added 20g (0 · 0372mol) amide (6), the addition was completed, 38 ° C for 6 hours.
[0127] completion of the reaction, water was added 4.2ml decomposition for 25 minutes, followed by addition of 5.4ml of 20% by weight concentration of sodium hydroxide solution decomposition for 20 minutes, 14.2ml decomposed with water for 15 minutes;
[0128] The decomposition was filtered, the filtrate (ethyl ether) and dried over anhydrous potassium carbonate. Filtered, the filter cake was washed with a little ether. The filtrate and the washings added to a distillation flask, recovery ether atmospheric distillation, vacuum drained, was added a mixed solvent l〇〇ml [chloroform: petroleum ether (60-90 ° C) = 1: 4, weight ratio, stirred and heated to reflux dissolution, filtered while hot, the filtrate was allowed to stand for crystallization at about 10 ° C, to be naturally deposited crystal after freezing -5 ° C overnight, filtered, the cake was washed with a mixed solvent, drain, ventilation pressure at 70 ° C dried to constant weight to give white crystalline product Penfluridol drug (I), mp 105-107 ° C, yield 81.5%.
[0129] Intermediate 4_ (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (XW) (referred piperidinol) Preparation:
[0130] (1) benzylamine (Beta) Preparation:
[0131]

[0132] equipped with a mechanical stirrer, a condenser, a thermometer 2000ml three reaction bottle, were added ammonium bicarbonate 240g (3.04mol), aqueous ammonia at a concentration of 20 wt %% of 15148 (17.812111〇1,
[0133] 1640ml), benzyl chloride 80g (0.632mol), reaction was stirred for 6 hours.Reaction to complete rested stratification. Aqueous layer was separated, and aqueous ammonia recovery bicarbonate atmospheric heating to 100 ° c, the water was distilled off under reduced pressure, with 50% sodium hydroxide PH12 above, extraction with benzene and dried solid sodium hydroxide. Recovery of benzene atmospheric distillation, vacuum distillation, collecting 33.4 g of the product obtained, yield 50.7%, content 99%,
[0134]

[0135] (3) N_ benzyl – bis ([beta] methoxycarbonyl-ethyl) amine (C) (referred to as diester thereof):
[0136]

[0137] The reaction flask equipped with a mechanical stirrer, a condenser, a thermometer three 250ml, 43g methyl acrylate (0.5111〇1) methanol 328 (401111), was added with stirring 21.48 benzylamine (0.2111〇1), The reaction was stirred for 7 hours. Completion of the reaction, recovery of excess methyl acrylate and methanol, water chestnut vacuum distillation until the internal temperature l〇〇-ll〇 ° C, to give the crude product as a yellow oil (C) 54g, yield 97%, content 94.3%.
[0138] (3) 1 – benzyl-4-piperidone (E) (referred to as the hydrolyzate) is prepared:
[0139]

[0140] In a reaction flask equipped with a 500ml three mechanical stirrer, thermometer, fractional distillation apparatus, was added 27% sodium methoxide 27g, crude diester was 33.4g (0.12mol), toluene 300ml, stirred and heated, the temperature reached 90 when ° C or more, additional 50ml toluene was reacted for 3 hours. Cooled to room temperature, and neutralized with acetic acid to PH6, standing layer. The toluene layer was separated and extracted with 150ml of 22% hydrochloric acid three times. Hydrochloric acid extracts were combined, heated with stirring for 4 hours. Recovered by distillation under reduced pressure and hydrochloric acid (about 120ml distilled dilute hydrochloric acid) was cooled to distillation l〇 ° C below, with 40% sodium hydroxide PH12 above. With 80ml ethyl acetate 3 times extracted with ethyl acetate extracts were combined, sub-net water, dried over anhydrous sodium sulfate. Sodium sulfate was removed by filtration, recovering ethyl acetate atmospheric distillation, vacuum drained hydrolyzed to give (E) and the crude product 19g, yield 84%.

[0141] (4) 1-ethoxycarbonyl-4-piperidone (F) (referred to as a carbonyl group-piperidone) Preparation:
[0142]

[0143] equipped with a mechanical stirrer, a condenser, 250ml three reaction flask thermometer, was added ethyl chloroformate 23.9g (0 · 22mo 1), benzene 100ml, stirring slowly added dropwise [The crude hydrolyzate (E ) 37 · 8g (0 · 2mo 1) + 20ml phenyl] solution dropwise, the reaction was heated with stirring for 5 hours.Water chestnut evaporated under reduced pressure and ethyl benzene chlorine, Li mechanical change stream distilled off under reduced pressure, low boiling point evaporated to give the product 268 was collected, yield 76%.
[0144] (5) 1 – ethoxycarbonyl-4- (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (G) (referred to as a carbonyl group-piperidinol) is:
[0145]

[0146] In three 500ml reaction flask equipped with a mechanical stirrer, a condenser, a thermometer, a dropping funnel and a calcium chloride drying tube over anhydrous anhydrous absolute, at room temperature was added magnesium metal shoulder 2.5g (0.103mol) 20ml of anhydrous ethyl ether and slowly stirring was started.
[0147] 2-chloro-5-bromo – trifluorotoluene (referred bromide) was dissolved under 27g (0.104mol) at room temperature in 130ml anhydrous diethyl ether and stirred to obtain a uniform liquid mass (W is);
[0148] When the liquid material taken ![]() 15ml was added to the above reaction, a solution of iodine 0.13g, 1,2- dibromoethane 0.2g, initiated Grignard reaction was heated until the iodine color disappeared, the reaction slowed down, slow slow dropping liquid material (W). The addition was completed, refluxing was continued for 1 hour. Completion of the reaction, cooled to room temperature, slowly added dropwise at room temperature carbonyl piperidone (F) water solution was cooled at normal [carbonyl-piperidone 13.6g (0.0795mol) + 40ml dry ether], dropwise, the reaction was heated with stirring 1.5 hour. L〇〇ml ammonium chloride solution concentration of 20% by weight was added, refluxed for 15 minutes and allowed to stand 30 minutes at room temperature stratification. Discharged aqueous layer (lower layer), the residual liquid was distilled (upper layer) at an external temperature of 55 ° C atmospheric distillation recovery ether, discharge hot, refrigerated overnight, the precipitated solid. Filtered, washed with a small amount of time, drained, and dried to give the product (G) 24.1g, yield 85.7%, mp 118-126Γ.
15ml was added to the above reaction, a solution of iodine 0.13g, 1,2- dibromoethane 0.2g, initiated Grignard reaction was heated until the iodine color disappeared, the reaction slowed down, slow slow dropping liquid material (W). The addition was completed, refluxing was continued for 1 hour. Completion of the reaction, cooled to room temperature, slowly added dropwise at room temperature carbonyl piperidone (F) water solution was cooled at normal [carbonyl-piperidone 13.6g (0.0795mol) + 40ml dry ether], dropwise, the reaction was heated with stirring 1.5 hour. L〇〇ml ammonium chloride solution concentration of 20% by weight was added, refluxed for 15 minutes and allowed to stand 30 minutes at room temperature stratification. Discharged aqueous layer (lower layer), the residual liquid was distilled (upper layer) at an external temperature of 55 ° C atmospheric distillation recovery ether, discharge hot, refrigerated overnight, the precipitated solid. Filtered, washed with a small amount of time, drained, and dried to give the product (G) 24.1g, yield 85.7%, mp 118-126Γ.
[0149] (6) 4- (3-trifluoromethyl-4-chlorophenyl) -4-piperidinol (X VH) (referred piperidinol) Preparation:
[0150]

[0151] equipped with a mechanical stirrer, a condenser, 250ml three reaction flask thermometer, were added ethanol 40ml, 158 of sodium hydroxide (0.375111〇1), carbonyl piperidinol (6) 2 (^ (0.0569111〇1 ), heated to reflux, and the reaction stirred for 3.5 hours. the reaction was completed, 50ml of water was added, the reaction was refluxed for 10 minutes, the hot reaction solution was placed in 300g of crushed ice, stirred well, and the precipitated solid, -5 ° C frozen standing for 2 hours the above.
[0152] filtered, washed with water to pH 8-9, drained, and dried to give piperidinol (XVH) 15g, yield 94%, mp 137-144 ° C, ash content <5%.
[0153] Example 2
[0154] (a) 3- (4-fluorobenzoyl) propionic acid (2) (the acid) is prepared:
[0155]

[0156] The reaction flask equipped with a mechanical stirrer, a condenser, a thermometer three 500ml, was added 17.1g (0.171mol) of succinic anhydride, l〇5g (1 · 09mol) fluorobenzene, stirred and dissolved. Added in one portion 60g (0 · 306mol) in dry wrong trichloride, stirring, the reaction was stirred at 100 ° C for 2 hours, at a concentration of 10% by weight hydrochloric acid 165ml exploded 30 minutes;
[0157] Other embodiments with Example 1, the product, 111.? 105-107 ° (:, this step a yield of 81.5%, 46.7% overall yield.
References
- ^ van Praag HM, Schut T, Dols L, van Schilfgaarden R., Controlled trial of penfluridol in acute psychosis, Br Med J. 1971 December 18;4(5789):710-3
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, Van Nueten JM, Schaper WK., The pharmacology of penfluridol (R 16341) a new potent and orally long-acting neuroleptic drug, Eur J Pharmacol. 1970 July 15;11(2):139-54
- ^ Jump up to:a b Soares, B; Silva de Lima, M (2006). “Penfluridol for schizophrenia”. Cochrane Database of Systematic Reviews. 2: CD002923.pub2. doi:10.1002/14651858.CD002923.pub2.
Further reading
- Benkert O, Hippius H.: Psychiatrische Pharmakotherapie, Springer-Verlag, 1976, 2. Auflage. ISBN3-540-07916-5
- R Bhattacharyya, R Bhadra U Roy, S Bhattacharyya, J Pal S Sh Saha – Resurgence of Penfluridol:Merits and Demerits, Eastern Journal of Psychiatry, January-June 2015 vol 18, Issue 1 p 23 –29
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.043.689 |
| Chemical and physical data | |
| Formula | C28H27ClF5NO |
| Molar mass | 523.965 g·mol−1 |
| 3D model (JSmol) | |
/////////Penfluridol, Antipsychotic, Semap, Micefal, Longoperidol, MCN-JR-16,341, R 16,341, MCN-JR-16,341 / R 16,341,
