



Home » Posts tagged 'Qualified Infectious Disease Product'
Tag Archives: Qualified Infectious Disease Product
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RIDINILAZOLE

RIDINILAZOLE
SMT19969
- Molecular FormulaC24H16N6
- Average mass388.424 Da
- ридинилазол [Russian] [INN]ريدينيلازول [Arabic] [INN]利地利唑 [Chinese] [INN]
- リジニラゾール;
10075
2,2′-Di(4-pyridinyl)-3H,3’H-5,5′-bibenzimidazole
308362-25-6[RN]6,6′-Bi-1H-benzimidazole, 2,2′-di-4-pyridinyl-
Summit Therapeutics (formerly Summit Corp ) is developing ridinilazole the lead compound from oral narrow-spectrum, GI-restricted antibiotics, which also include SMT-21829, for the treatment of Clostridium difficile infection and prevention of recurrent disease.
Ridinilazole (previously known as SMT19969) is an investigational small molecule antibiotic being evaluated for oral administration to treat Clostridioides difficile infection (CDI). In vitro, it is bactericidal against C. difficile and suppresses bacterial toxin production; the mechanism of action is thought to involve inhibition of cell division.[1] It has properties which are desirable for the treatment of CDI, namely that it is a narrow-spectrum antibiotic which exhibits activity against C. difficile while having little impact on other normal intestinal flora and that it is only minimally absorbed systemically after oral administration.[2] At the time ridinilazole was developed, there were only three antibiotics in use for treating CDI: vancomycin, fidaxomicin, and metronidazole.[1][2] The recurrence rate of CDI is high, which has spurred research into other treatment options with the aim to reduce the rate of recurrence.[3][4]
As of 2019, two phase II trials have been completed and two phase III trials comparing ridinilazole to vancomycin for CDI are expected to be completed in September 2021.[2][5][6] Ridinilazole was designated as a Qualified Infectious Disease Product (QIDP) and was granted Fast Track status by the U.S. FDA.[2] Fast Track status is reserved for drugs designed to treat diseases where there is currently a gap in the treatment, or a complete lack thereof.[7] The QIDP designation adds five more years of exclusivity for ridinazole upon approval.[8]


PATENT
WO-2021009514
Process for preparing ridinilazole useful for treating Clostridium difficile infection. Also claimed is the crystalline form of a compound.
The present invention relates to processes for the preparation of 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole (which may also be known as 5,5’-bis[2-(4-pyridinyl)-1/-/-benzimidazole], 2,2′-bis(4-pyridyl)-3/-/,3’/-/-5,5′-bibenzimidazole or 2-pyridin-4-yl-6-(2-pyridin-4-yl-3/-/-benzimidazol-5-yl)-1/-/-benzimidazole), referenced herein by the INN name ridinilazole, and pharmaceutically acceptable derivatives, salts, hydrates, solvates, complexes, bioisosteres, metabolites or prodrugs thereof. The invention also relates to various crystalline forms of ridinilazole, to processes for their preparation and to related pharmaceutical preparations and uses thereof (including their medical use and their use in the efficient large-scale synthesis of ridinilazole).
WO2010/063996 describes various benzimidazoles, including ridinilazole, and their use as antibacterials (including in the treatment of CDAD).
WO 2011/151621 describes various benzimidazoles and their use as antibacterials
(including in the treatment of CDAD).
W02007056330, W02003105846 and W02002060879 disclose various 2-amino benzimidazoles as antibacterial agents.
W02007148093 discloses various 2-amino benzothiazoles as antibacterial agents.
W02006076009, W02004041209 and Bowser et at. (Bioorg. Med. Chem. Lett., 2007, 17, 5652-5655) disclose various substituted benzimidazole compounds useful as anti-infectives that decrease resistance, virulence, or growth of microbes. The compounds are said not to exhibit intrinsic antimicrobial activity in vitro.
US 5,824,698 discloses various dibenzimidazoles as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp.and Enterococcus spp. However, this document does not disclose activity against anaerobic spore-forming bacteria and in particular does not disclose activity against any Clostridioides spp. (including C. difficile).
US 2007/0112048 A1 discloses various bi- and triarylimidazolidines and bi- and
triarylamidines as broad-spectrum antibiotics, disclosing activity against both Gram negative and Gram-positive bacteria, including Staphylococcus spp., Enterococcus spp. and Clostridioides spp. However, this document does not disclose compounds of formula (I) as described herein.
Chaudhuri et al. (2007) J.Org. Chem. 72, 1912-1923 describe various bis-2-(pyridyl)-1 H-benzimidazoles (including compounds of formula I as described herein) as DNA binding agents. This document is silent as to potential antibacterial activity.
Singh et al. (2000) Synthesis 10: 1380-1390 describe a condensation reaction for producing 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole using 4-pyridine
carboxaldehyde, FeCI3, 02, in DMF at 120°C.
Bhattacharya and Chaudhuri (2007) Chemistry – An Asian Journal 2: 648-655 describe a condensation reaction for producing 2,2′-di(pyridin-4-yl)-1/-/,T/-/-5,5′-bibenzo[d]imidazole using 4-pyridine carboxaldehyde and nitrobenzene at 120°C.
WO2019/068383 describes the synthesis of ridinilazole by metal-ion catalyzed coupling of 3,4,3’,4’-tetraaminobiphenyl with 4-pyridinecarboxaldehyde in the presence of oxygen, followed by the addition of a complexing agent.
PATENT
WO2010063996
claiming antibacterial compounds. Bicyclic heteroaromatic compounds, particularly bi-benzimidazole derivatives.
WO2007056330, WO2003105846 and WO2002060879 disclose various 2-amino benzimidazoles as antibacterial agents.
WO2007148093 discloses various 2-amino benzothiazoles as antibacterial agents.
WO2006076009, WO2004041209 and Bowser et al. (Bioorg. Med. Chem. Lett., 2007, 17, 5652-5655) disclose various substituted benzimidazole compounds useful as anti-infectives that decrease resistance, virulence, or growth of microbes. The compounds are said not to exhibit intrinsic antimicrobial activity in vitro.
US 5,824,698 discloses various dibenzimidazoles as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp.and Enterococcus spp. However, this document does not disclose activity against anaerobic spore-forming bacteria and in particular does not disclose activity against any Clostridium spp. (including C. difficile).
US 2007/0112048 A1 discloses various bi- and triarylimidazolidines and bi- and triarylamidines as broad-spectrum antibiotics, disclosing activity against both Gram-negative and Gram-positive bacteria, including Staphylococcus spp., Enterococcus spp.
and Clostridium spp. However, this document does not disclose compounds of general formula (I) as described herein.
Chaudhuri et al. (J.Org. Chem., 2007, 72, 1912-1923) describe various bis-2-(pyridyl)-1 H-benzimidazoles (including compounds of formula I as described herein) as DNA binding agents. This document is silent as to potential antibacterial activity.
PATENT
Product PATENT, WO2010063996 ,
protection in the EP until 2029 and expire in the US in December 2029.
PAPER
https://www.frontiersin.org/articles/10.3389/fmicb.2018.01206/full
PAPER
Synthesis (2000), (10), 1380-1390.
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-2000-7111
PAPERT
Chemistry – An Asian Journal (2007), 2(5), 648-655.
https://onlinelibrary.wiley.com/doi/abs/10.1002/asia.200700014
Studies of double‐stranded‐DNA binding have been performed with three isomeric bis(2‐(n‐pyridyl)‐1H‐benzimidazole)s (n=2, 3, 4). Like the well‐known Hoechst 33258, which is a bisbenzimidazole compound, these three isomers bind to the minor groove of duplex DNA. DNA binding by the three isomers was investigated in the presence of the divalent metal ions Mg2+, Co2+, Ni2+, Cu2+, and Zn2+. Ligand–DNA interactions were probed with fluorescence and circular dichroism spectroscopy. These studies revealed that the binding of the 2‐pyridyl derivative to DNA is dramatically reduced in the presence of Co2+, Ni2+, and Cu2+ ions and is abolished completely at a ligand/metal‐cation ratio of 1:1. Control experiments done with the isomeric 3‐ and 4‐pyridyl derivatives showed that their binding to DNA is unaffected by the aforementioned transition‐metal ions. The ability of 2‐(2‐pyridyl)benzimidazole to chelate metal ions and the conformational changes of the ligand associated with ion chelation probably led to such unusual binding results for the ortho isomer. The addition of ethylenediaminetetraacetic acid (EDTA) reversed the effects completely.
PAPER
Journal of Organic Chemistry (2007), 72(6), 1912-1923.
https://pubs.acs.org/doi/10.1021/jo0619433

Three symmetrical positional isomers of bis-2-(n-pyridyl)-1H-benzimidazoles (n = 2, 3, 4) were synthesized and DNA binding studies were performed with these isomeric derivatives. Like bisbenzimidazole compound Hoechst 33258, these molecules also demonstrate AT-specific DNA binding. The binding affinities of 3-pyridine (m-pyben) and 4-pyridine (p-pyben) derivatized bisbenzimidazoles to double-stranded DNA were significantly higher compared to 2–pyridine derivatized benzimidazole o-pyben. This has been established by combined experimental results of isothermal fluorescence titration, circular dichroism, and thermal denaturation of DNA. To rationalize the origin of their differential binding characteristics with double-stranded DNA, computational structural analyses of the uncomplexed ligands were performed using ab initio/Density Functional Theory. The molecular conformations of the symmetric head-to-head bisbenzimidazoles have been computed. The existence of intramolecular hydrogen bonding was established in o-pyben, which confers a conformational rigidity to the molecule about the bond connecting the pyridine and benzimidazole units. This might cause reduction in its binding affinity to double-stranded DNA compared to its para and meta counterparts. Additionally, the predicted stable conformations for p-, m-, and o-pyben at the B3LYP/6-31G* and RHF/6-31G* levels were further supported by experimental pKa determination. The results provide important information on the molecular recognition process of such symmetric head to head bisbenzimidazoles toward duplex DNA.
Patent
US 8975416
PATENT
WO 2019068383
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019068383
Clostridium difficile infection (CDI) is the leading cause of infectious healthcare-associated diarrhoea. CDI remains a challenge to treat clinically, because of a limited number of antibiotics available and unacceptably high recurrence rates. Because of this, there has been significant demand for creating innovative therapeutics, which has resulted in the development of several novel antibiotics.
Ridinilazole (SMT19969) is the INN name of 5,5’bis[2-(4-pyridinyl)-lH-benzimidazole], which is a promising non-absorbable small molecule antibiotic intended for oral use in the treatment of CDI. It has been shown to exhibit a prolonged post-antibiotic effect and treatment with ridinilazole has resulted in decreased toxin production. A phase 1 trial demonstrated that oral ridinilazole is well tolerated and specifically targets Clostridia whilst sparing other faecal bacteria.
Ridinilazole has the following chemical structure:
Bhattacharya & Chaudhuri (Chem. Asian J., 2007, No. 2, 648-655) report performing double-stranded DNA binding with three benzimidazole derivatives, including ridinilazole. The compounds have been prepared by dissolving the reactants in nitrobenzene, heating at 120°C for 8- 1 Oh and purifying the products by column chromatography over silica gel. The compounds were obtained in 65-70% yield. Singh et al., (Synthesis, 2000, No. 10, 1380-1390) describe a catalytic redox cycling approach based on Fe(III) and molecular oxygen as co-oxidant for providing access to benzimidazole and
imidazopyridine derivatives, such as ridinilazole. The reaction is performed at high temperatures of 120°C and the product is isolated in 91% yield by using silica flash chromatography.
Both processes are not optimal, for example in terms of yield, ease of handling and scalability. Thus, there is a need in the art for an efficient and scalable preparation of ridinilazole, which overcomes the problems of the prior art processes.
Example 1 : Preparation of crude ridinilazole free base
A solution of 3,4,3′,4′-tetraaminobiphenyl (3.28 g, 15.3 mmol) and isonicotinaldehyde (3.21 g, 30.0 mmol) in DMF (40 mL) was stirred at 23 °C for one hour. Then anhydrous ferric chloride (146 mg, 0.90 mmol), water (0.10 mL, 5.4 mmol) and additional DMF (2 mL) were added and fresh air was bubbled into the solution during vigorous stirring for 5 hours at room temperature. Next, water (80 mL) and EDTA (0.29 g) were added resulting in a brownish suspension, which was stirred overnight. The product was isolated by filtration, washed with water, and dried in a desiccator in vacuo as a brown powder (5.56 g; 95%). The addition of EDTA had held iron in solution and the crude ridinilazole contained significantly lower amounts of iron than comparative example 1.
Example 12: Formation of essentially pure ridinilazole free base
To a suspension von ridinilazole tritosylate (1 10 mg, 0.12 mmol) in water (35 mL) featuring a pH value of about 4.5 stirring at 70 °C sodium bicarbonate (580 mg, 6.9 mmol) were added and caused a change of color from orange to slightly tan. The mixture, now at a pH of about 8.5, was cooled down to room temperature and the solids were separated by filtration, washed with water (1 ML) and dried in vacuo providing 40 mg (85%) essentially pure ridinilazole as a brownish powder.
Spectroscopic analysis:
¾ NMR (DMSO-de, 300 MHz): δ 7.55 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.4 Hz, 2H), 7.88 (s, 2H), 8.13 (d, J = 5.8 Hz, 4H), 8.72 (d, J = 5.8 Hz, 4H) ppm.
13C NMR (DMSO-d6, 75 MHz): δ 1 13.4 (2C), 1 16.4 (2C), 120.4 (4C), 121.8 (2C), 135.7 (2C), 138.7 (2C), 140.7 (2C), 141.4 (2C), 150.3 (4C), 151.1 (2C) ppm.
IR (neat): v 3033 (w), 1604 (s), 1429 (m), 1309 (m), 1217 (m), 1 1 15 (w), 998 (m), 964 (m), 824 (m), 791 (s), 690 (s), 502 (s) cm .
UV-Vis (MeOH): 257, 341 nm.
The sharp peaks in the ¾ NMR indicated that iron had been efficiently removed.
Comparative example 1 : Preparation of ridinilazole
A solution of 3,4,3′,4′-tetraaminobiphenyl (0.69 g, 3.2 mmol) and isonicotinaldehyde (0.64 g, 6.0 mmol) in DMF (20 mL) was stirred at 80°C for one hour. Then ferric chloride hexahydrate (49 mg, 0.18 mmol), water (0.10 mL, 5.4 mmol) and additional DMF (2 mL) were added and fresh air was bubbled into the solution during vigorous stirring for 10 hours at 120 °C. After cooling to room temperature water (50 mL) and the mixture was stirred for one hour. A black crude product was isolated by filtration and comprised ridinilazole and iron.
References
- ^ Jump up to:a b Cho JC, Crotty MP, Pardo J (March 2019). “Clostridium difficile infection”. Annals of Gastroenterology. 32 (2): 134–140. doi:10.20524/aog.2018.0336. PMC 6394264. PMID 30837785.
- ^ Jump up to:a b c d Carlson TJ, Endres BT, Bassères E, Gonzales-Luna AJ, Garey KW (April 2019). “Ridinilazole for the treatment of Clostridioides difficile infection”. Expert Opinion on Investigational Drugs. 28 (4): 303–310. doi:10.1080/13543784.2019.1582640. PMID 30767587.
- ^ Bassères E, Endres BT, Dotson KM, Alam MJ, Garey KW (January 2017). “Novel antibiotics in development to treat Clostridium difficile infection”. Current Opinion in Gastroenterology. 33 (1): 1–7. doi:10.1097/MOG.0000000000000332. PMID 28134686.
These tables highlight the increased drug development directed towards CDI due to the rise in prevalence of infections and to attempt to reduce the number of recurrent infections.
- ^ Vickers RJ, Tillotson G, Goldstein EJ, Citron DM, Garey KW, Wilcox MH (August 2016). “Ridinilazole: a novel therapy for Clostridium difficile infection”. International Journal of Antimicrobial Agents. 48 (2): 137–43. doi:10.1016/j.ijantimicag.2016.04.026. PMID 27283730.
there exists a significant unmet and increasing medical need for new therapies to treat CDI, specifically those that can reduce the rate of disease recurrence.
- ^ Clinical trial number NCT03595553 for “Ri-CoDIFy 1: Comparison of Ridinilazole Versus Vancomycin Treatment for Clostridium Difficile Infection” at ClinicalTrials.gov
- ^ Clinical trial number NCT03595566 for “Ri-CoDIFy 2: To Compare Ridinilazole Versus Vancomycin Treatment for Clostridium Difficile Infection” at ClinicalTrials.gov
- ^ “Fast Track”. U.S. Food and Drug Administration. 2018-11-03.
- ^ “”HHS spurs new antibiotic development for biodefense and common infections””. Public Health Emergency. U.S. Department of Health and Human Services. Retrieved 2020-12-04.
| Clinical data | |
|---|---|
| Other names | SMT19969 |
| ATC code | None |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 308362-25-6 |
| PubChem CID | 16659285 |
| ChemSpider | 17592423 |
| UNII | 06DX01190R |
| KEGG | D11958 |
| Chemical and physical data | |
| Formula | C24H16N6 |
| Molar mass | 388.42 g/mol |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]c6cc(c5nc4ccc(c3ccc2nc(c1ccncc1)[nH]c2c3)cc4[nH]5)ccn6 |
/////////RIDINILAZOLE, SMT19969, SMT 19969, ридинилазол , ريدينيلازول , 利地利唑 , リジニラゾール , Qualified Infectious Disease Product, QIDP, Fast Track , PHASE 3, Clostridioides difficile infection ,
LYS 228

LYS228
BOS-228
LYS-228
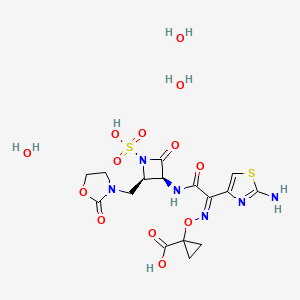
Molecular Formula, C16-H18-N6-O10-S2
Molecular Weight, 518.4783
(3S,4R)-3-((Z)-2-(2-Ammoniothiazol-4-yl)-2-((1-carboxycyclopropoxy)imino)acetamido)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)azetidine-1-sulfonate
RN: 1810051-96-7
UNII: 29H7N9XI1B
UNII-005B24W9YP
005B24W9YP
Lys-228 trihydrate
2091840-43-4
Yclopropanecarboxylic acid, 1-(((Z)-(1-(2-amino-4-thiazolyl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxo-3-oxazolidinyl)methyl)-1-sulfo-3-azetidinyl)amino)ethylidene)amino)oxy)-, hydrate (1:3)
1-[(Z)-[1-(2-amino-1,3-thiazol-4-yl)-2-oxo-2-[[(3S,4R)-2-oxo-4-[(2-oxo-1,3-oxazolidin-3-yl)methyl]-1-sulfoazetidin-3-yl]amino]ethylidene]amino]oxycyclopropane-1-carboxylic acid;trihydrate
BOS-228 (LYS-228) is a monobactam discovered at Novartis and currently in phase II clinical development at Boston Pharmaceuticals for the treatment of complicated urinary tract infection and complicated intraabdominal infections in adult patients.
The compound has been granted fast track and Qualified Infectious Disease Product (QIDP) designation from the FDA.
In October 2018, Novartis licensed to Boston Pharmaceuticals worldwide rights to the product.
Paper
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00330
Patent
US 20150266867
PATENT
WO 2017050218
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017050218&tab=FULLTEXT
Compound X: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0126]
Step 1: Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate. To a solution of (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetic acid (854 mg, 1.59 mmol) prepared according to published patent application US2011/0190254, Intermediate B (324 mg, 1.75 mmol) and HATU (785 mg, 2.07 mmol) in DMF (7.9 mL) , DIPEA was added (832 μL, 4.77 mmol) . After 1 h of stirring, it was poured into water and extracted with EtOAc. Brine was added to the aqueous layer, and it was further extracted with ethyl acetate (EtOAc) (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo. The crude residue was purified via silica gel chromatography (0-10%MeOH-DCM) to afford the title compound (1.09 g, 97%) as a beige foam. LCMS: R t = 0.97 min, m/z =705.3 (M+1) Method 2m_acidic.
[0127]
Instead of HATU, a variety of other coupling reagents can be used, such as any of the typical carbodiimides, or CDMT (2-chloro-4, 6-dimethoxy-1, 3, 5-triazine) and N-methylmorpholine to form the amide bond generated in Step 1.
[0128]
Step 2: (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid. Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate (1.00 g, 1.42 mmol) in DMF (7.0 mL) at 0 ℃ was treated with SO 3·DMF (448 mg, 2.84 mmol) . After 2 h of stirring at rt, the solution was poured into ice-cold brine and extracted with EtOAc (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo, affording the title compound (assumed quantitative) as a white solid. LCMS: Rt =0.90 min, m/z = 785.2 (M+1) Method 2m_acidic.
[0129]
Step 3: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0130]
[0131]
To a solution of (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid (1.10 g, 1.40 mmol) in DCM (1.5 mL) at 0℃, TFA (5.39 mL, 70.0 mmol) was added, and after 10 minutes, the ice bath was removed. Additional TFA (3.24 mL, 42.0 mmol) was added after 1 hr at rt and the solution was diluted with DCM and concentrated in vacuo after an additional 30 min. Optionally, anisole may be added to the TFA reaction to help reduce by-product formation, which may increase the yield of desired product in this step. The crude residue was purified by reverse phase prep HPLC (XSelect CSH, 30 x 100 mm, 5 μm, C18 column; ACN-water with 0.1%formic acid modifier, 60 mL/min) , affording the title compound (178 mg, 23%) as a white powder. LCMS: R t = 0.30 min, m/z = 518.9 (M+1) Method 2m_acidic; 1H NMR (400 MHz, DMSO-d 6) δ 9.27 (d, J = 9.0 Hz, 1H) 6.92 (s, 1H) 5.23 (dd, J = 9.1, 5.7 Hz, 1H) 4.12-4.23 (m, 3H) 3.72-3.62 (m, 2H assumed; obscured by water) 3.61-3.52 (m, 1H assumed; obscured by water) 3.26 (dd, J = 14.5, 5.9 Hz, 1H) 1.36 (s, 4H) . 1H NMR (400 MHz, D 2O) δ 7.23 (s, 1H) , 5.48 (d, J = 5.8 Hz, 1H) , 4.71-4.65 (m, 1H) , 4.44 (t, J = 8.2 Hz, 2H) , 3.89-3.73 (m, 3H) , 3.54 (dd, J = 14.9, 4.9 Hz, 1H) , 1.65-1.56 (m, 2H) , 1.56-1.46 (m, 2H) . The product of this process is amorphous. Compound X can be crystallized from acetone, ethanol, citrate buffer at pH 3 (50 mM) , or acetate buffer at pH 4.5 (50 mM) , in addition to solvents discussed below.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(4), 748-755.
https://www.sciencedirect.com/science/article/pii/S0960894X18300064
PATENT
WO 2019026004
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019026004&tab=PCTDESCRIPTION
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly disconcerting. Of the over 2 million (hospital-acquired) infections occurring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually.
Important causes of Gram-negative resistance include extended-spectrum 13- lactamases (ESBLs), serine carbapenemases (KPCs) and metallo-13-lactamases (for example NDM-1 ) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (AmpC) 13-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas. The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Klebsiella pneumonia harboring NDM-1 metallo-13- lactamase carries frequently additional serine-13-lactamases on the same plasmid that carries the NDM-1 .
Thus there is a need for new antibacterials, particularly antibacterial compounds that are effective against existing drug-resistant microbes, or are less susceptible to development of new bacterial resistance. Monobactam antibiotic, which is referred to herein as Compound X, is primarily effective against Gram-negative bacteria, including strains that show resistance to other monobactams.
The present invention relates to a process for the preparation of monobactam antibiotic Compound X and intermediates thereof.
More particularly, the present invention relates to a process for the preparation of Compound X
Compound X
also referred to as 1 -(((Z)-(1 -(2-aminothiazol-4-yl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)-1 -sulfoazetidin-3-yl)amino)ethylidene)amino)oxy)cyclopropanecarboxylic acid, or a salt thereof, or a solvate including hydrate thereof.
Patent application number PCT/US2015/02201 1 describes certain monobactam antibiotics. Compound X may be prepared using the method disclosed in PCT/US2015/02201 1 , in particular example 22, and in PCT/CN2016/099482.
A drawback from these processes is that they exhibit a large number of process steps and intermediate nitrogen protection/deprotection steps, reducing the overall yield and efficiency. Furthermore, these processes require several chromatographic purification steps to be carried out in course of the processes. We have found that the preparation of Compound X, as previously prepared on a manufacturing scale, possesses a number of disadvantages, in particular poor handling characteristics.
It would thus be beneficial to develop alternative or improved processes for the production of Compound X that do not suffer from some or all of these disadvantages.
Compound x Compound x
Scheme 1
Preparation of Compound X from Intermediates 22 and 2A
Scheme 3
Examples
The Following examples are merely illustrative of the present disclosure and they should not be considered as limiting the scope of the disclosure in any way, as these examples and other equivalents thereof will become apparent to those skilled in the art in the light of the present disclosure, and the accompanying claims.
Synthesis of Compound 8 (R = benzyl)
1 .50kg oxazolidin-2-one (7b) was charged into the reactor. 7.50kg THF was charged and the stirring started. The mixture was cooled to 10~20°C. 2.18kg potassium fert-butoxide was charged intol 2.00kg THF and stirred to dissolve.
The potassium fert-butoxide solution was added dropwise into the reactor while maintaining the temperature at 10-20 °C. The reaction was stirred for 1 ~2hrs at 10-20 °C after the addition. The solution of 2.36kg methyl-2-chloroacetate (7a) in 3.00kg of THF was added to the reactor while maintaining the temperature at 10-20 °C. The reaction mixture was stirred for 16-18 h at 20-25 °C. The IPC (in process control) showed completion of the reaction. The mixture was centrifuged and the wet cake was washed with 7.50kg THF. The filtrate was concentrated and the crude 7 was provided as reddish brown liquid, which was used for the next step without further purification,
1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.65 – 3.71 (m, 2 H) 3.74 (s, 3 H) 4.02 (s, 2 H) 4.34 – 4.45 (m, 2 H).
The dried reactor was exchanged with N2 three times. 3.71 kg LiHMDS solution in THF/Hep (1 M) and 1 .30kg THF were charged under nitrogen protection. The stirring was started and the solution was cooled to -70—60 °C. The solution of 0.71 kg benzyl acetate (6) in 5.20 kg THF was added dropwisely at -70— 60 °C, and the resulted mixture was stirred for 1 -1 .5 h after the addition. The solution of 0.65kg 7 in 3.90kg THF was added dropwise while maintaining the temperature at -70—60 °C, then stirred for 30-40 minutes. The reaction mixture was warmed to 20-25 °C and stirring was continued for 0.5-1 .0 h. IPC showed 6 was less than1 .0% (Otherwise, continue the reaction till IPC passes). The reaction mixture was poured into 13.65 kg aqueous citric acid below 10 °C. The mixture was stirred for 15-20 minutes after the addition. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (6.50kg * 2). The organic layer was combined, washed by 6.50 kg 28% NaCI solution and dried with 0.65
kg anhydrous MgSC . The mixture was filtered and the wet cake was washed with 1 .30kg EA. The filtrate was concentrated under vacuum to provide crude 8. The crude 8 was stirred in 2.60 kg MTBE at 20-25 °C for 1 -1 .5 h. The mixture was cooled to 0-10 °C and stirred for 1 .5-2.0 h and filtered. The filter cake was washed with 0.65kg pre-cooled MTBE and dried under vacuum (<-0.096Mpa) at 20-25 °C for 12~16hrs till a constant weight to give 513 g of 8 as a white solid, Yield: 45%, HPLC purity 96.4%,1 H NMR (400 MHz, CHLOROFORM-c δ ppm 3.48 – 3.55 (m, 1 H) 3.56 – 3.63 (m, 2 H) 3.66 – 3.74 (m, 1 H) 4.17 – 4.26 (m, 2 H) 4.31 – 4.44 (m, 2H) 5.12 – 5.24 (m, 2 H) 7.30 – 7.44 (m, 5 H).
Synthesis of Compound 9 (R = benzyl)
The dried reactor was charged with 3.75kg HOAc and 1 .50 kg 8. The stirring was started and the reaction mixture was cooled to 0-5 °C. 3.53kg aqueous NaN02 was added dropwise at 0-10 °C, and the reaction mixture was stirred for 15-30 minutes after the addition. IPC showed 8 was less than 0.2%. The reaction mixture was treated with 7.50kg EA and 7.50 kg water. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (7.50kg * 2). The organic layers were combined, washed with 7.50 kg 28% NaCI solution, and concentrated under vacuum to provide crude 9. The crude 9 was slurried with 5.25 kg water at 10-20 °C for 3~4hrs, and filtered. The wet cake was washed with 1 .50kg water. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .44 Kg of 9, yield: 86.9%, HPLC purity 92.9%,1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.60 – 3.76 (m, 2 H) 4.44 (t, J=8.07 Hz, 2 H) 4.60 (s, 2 H) 5.25 – 5.41 (m, 2 H) 7.30 – 7.43 (m, 5 H) 1 1 .62 (br s, 1 H).
Synthesis of Compound 9a (R = benzyl)
9
The dried reactor was charged with 0.58 kg Zn, 4.72kg (Βο Ο, 6.00 kg water, 1 .20 kg NH4CI and 6.00kg THF. The reaction mixture was stirred and heated to 50-55 °C. The solution of 0.60 kg 9 in 4.20kg THF
was added dropwisely while maintaining the temperature at 50-55 °C. The reaction mixture was stirred for 0.5-1 .Ohrs after the addition. IPC showed 9 was less than 0.1 %. The reaction mixture was treated withl .50 kg ethyl acetate and stirred for 15-20 minutes. Phase was separated and the water layer was extracted by1 .50 kg ethyl acetate. The organic layers were combined, washed with 6.00 kg 28% NaCI solution and concentrated under vacuum to provide crude 9a. The crude 9a was stirred with 3.60kg*2 n-heptane to remove excess (Βο Ο. The residue was purified by silica gel chromatography column eluted with ethyl acetate: Heptane= 1 :1 to provide crude 9a solution. The solution was concentrated under reduced pressure to obtain crude 9a. The crude 9a was slurried with 1 .80 kg MTBE for 2.0-3. Ohrs, filtered, and the wet cake was washed with MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 50-55 °C for 16-18 h till a constant weight to give 392 g of 9a as a white solid, Yield: 51 %, HPLC purity 98.1 %,1H NMR (400 MHz, DMSO-cfe) δ ppm 1.17 – 1 .57 (m, 9 H) 3.39 – 3.61 (m, 2 H) 4.20 – 4.45 (m, 3 H) 5.10 – 5.32 (m, 3 H) 5.75 (s, 1 H) 7.38 (br s, 5 H) 7.75 – 7.99 (m, 1 H).
Synthesis of compound (VII) (R = benzyl, X = CI)
9a VII
The dried reactor was charged with 13.0kg HCI in IPA and the stirring was started. 1 .33 kg 9a was charged in portions at 20-25 °C. The mixture was stirred at 20-25 °C for 3-4 h. IPC showed 9a was less than 0.1 %. The reaction solution was concentrated under vacuum 40-45 °C. The residue was treated with 21 .58kg MTBE at 20-25 °C for 3-4 h. The mixture was filtered and the wet cake was washed with 2.60kg MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .045 Kg of compound VII (R = benzyl, X = CI) as a yellow solid, Yield: 93.7%, HPLC purity 99.2%,1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.16 – 3.74 (m, 3 H) 4.10 – 4.35 (m, 4 H) 5.09 – 5.39 (m, 2 H) 7.27 – 7.60 (m, 5 H) 8.72 (br s, 2 H).
Synthesis of compound (Vile) (R = benzyl)
VII Vile
To an autoclave (3L) were added VII (R = benzyl, X = CI) (100 g, 304.2 mmol, 1 .0 equiv.), DCM (2650 g, 26.5 equiv., w/w) and (S-BINAP)RuCl2 (2.4 g, 3.04 mmol, 0.01 equiv.), successively. Air in the autoclave was replaced with N2 5 times. N2 in the autoclave was was replaced with H2 5 times. The solution was stirred with 250-260 r/min and H2 (2.1 ±0.1 MPa) at 40±5°C for 24 h. The reaction mixture was filtered, and the filter cake was washed with DCM (400 g, 4.0 equiv., w/w). The filter cake was slurried with IPA (785 g, 7.85 equiv., w/w) and H2O (40 g, 0.4 equiv., w/w) overnight (18-20 h). The mixture was filtered. The filter cake was washed with IPA (200 g, 2.0 equiv., w/w) and dried at 45±5°C overnight (18-20 h). Vile (R = benzyl) was obtained as off-white solid, 80.4 g, 79.9% yield, 95.5% purity, 97.6% de, >99.5% ee. 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.34-3.38 (m, 2 H) 3.50-3.52 (m, 1 H) 3.60-3.62 (m, 1 H) 4.18-4.24 (m, 4 H) 5.23 (s, 2H) 6.16 (s, 1 H) 7.32 (m, 5H) 8.74 (s, 1 H).
Alternative synthesis of compound 9a (R = benzyl)
5b
Mg(OtBu)2
To a flask was added 5a (1 .88 g, 12.93 mmol), THF (40 mL), and CDI (2.20 g, 13.58 mmol) at 25 °C. The mixture was stirred for 3 h. To the reaction mixture was added 5b (2.00 g, 6.47 mmol), and Mg(OfBu)2 (2.21 g, 12.93 mmol). The reaction mixture was stirred at 25 °C for 24 h. The reaction mixture was concentrated under vacuum to remove most of the THF solvent. To the concentrated solution was added MTBE (40 mL), followed by addition of an aqueous solution of HCI (1 M, 60mL) to adjust to pH = 2-3. Two phases were separated, and the water phase was extracted with MTBE (20 mL). The combined organic phase was washed with aqueous NaHCC (5%, 50 mL) and brine (20%, 40 mL). The organic phase was concentrated to a weight of -19 g, and a lot of white solid was obtained in the concentration process. The suspension was cooled to 0 °C, and filtered. The filter cake was washed with cold MTBE (5 mL) and dried under vacuum to obtain product 9a (1 .6g, 63% yield).
Synthesis of compound (Vile) (R = benzyl, PG = Cbz)
Vile Vile
To a flask (5 L) were added Vile (R = benzyl) (140 g, 423.2 mmol, LOequiv.), H20 (1273 g, 9.09 equiv., w/w) and toluene (2206 g, 15.76 equiv., w/w). The solution was stirred and cooled to 0-5 °C with ice bath. Then NaHCOa (78.4 g, 933 mmol, 2.22 equiv.) was added and CbzCI (89.6 g, 527 mmol, 1 .24 equiv.) was dropped into the stirring solution, respectively. The solution was stirred at 30±5 °C overnight (18-20 h). Heptane (3612 g, 25.8 equiv., w/w) was added dropwise to the stirring solution over 1 h at 20-30 °C. The mixture was filtered. The filter cake was washed with heptane (280 g, 2.00 equiv., w/w) and MTBE (377 g, 2.69 equiv., w/w), respectively. The filter cake was dried at 45±5°C overnight (18-20 h). Vile (R = benzyl, PG = Cbz) was obtained as an off-white solid, 169.4 g, 93% yield, 96.7% purity, 98% de, >99.5% ee, 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.23-3.24 (m, 1 H) 3.30 (m, 1 H) 3.51 -3.55 (m, 2 H) 3.99 (s, 1 H) 4.17-4.21 (m, 3 H) 5.02-5.03 (m, 2H) 5.12 (s, 2H) 5.46-5.48 (d, 1 H) 7.33-7.36 (m, 10H) 7.75-7.73 (d, 1 H).
Synthesis of compound (IV) (PG = Cbz)
Vile IV
Vile (R = benzyl) (220 g, 513.5 mmol, 1 .0 equiv.) was dissolved in THF (1464g, 6.65 equiv., w/w). The solution was filtered. The filter cake was washed with THF (488g, 2.22 equiv., w/w). The filtrate (Vile) was collected. To an autoclave (3L) were added the filtrate (Vile). The reactor was cooled down to -75 – -65 °C with dry-ice/EtOH bath, and bubbled with NH3 for not less than 4 h. Then the solution was stirred at 25±5 °C with NH3 (0.5-0.6 MPa) for 24 h. The autoclave was deflated to release NH3. The reaction solution was concentrated with a rotary evaporator to remove THF until the residue was around 440 g. The residue was slurried with EA (2200 g, 10 equiv., w/w) at 70±2 °C, then cooled to 25±5 °C and stirred for 16-18 h. The mixture was filtered. The filter cake was washed with EA (440 g). The filter cake was slurried with EA (1320 g, 6.00 equiv. w/w), and the temperature was raised to 70±2 °C, then cooled to 25±5 °C and stirred for 16-20 h. The mixture was filtered. The filter cake was washed with EA, and dried at 50±5 °C overnight (18-20 h). IV (PG = Cbz) was obtained as off-white solid, 141 g, 81 .5% yield, 99.1 % purity, >99.5% assay, 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.12 – 3.23 (m, 2 H) 3.31 (br s, 1 H) 3.56 (t, J=8.01 Hz, 2 H) 3.88 (quin, J=6.02 Hz, 1 H) 3.93 – 4.03 (m, 1 H) 4.20 (t, J=8.01 Hz, 2 H) 5.02 (s, 2 H) 5.27 (d, J=5.87 Hz, 1 H) 7.12 (s, 1 H) 7.22 – 7.45 (m, 5 H).
Synthesis of compound (III) (PG = Cbz, LG = S02CH3)
IV III
To a flask was added IV (PG = Cbz) (14.00 g, 41 .50 mmol, 1 .00 equiv), and dry 1 , 2-dimethoxyethane (300 mL) under N2. The mixture was stirred at -5°C ~ 0°C for 1 h to obtain a good suspension. MsCI (7.89 g, 68.89 mmol, 5.33 mL, 1 .66 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min, and Et3N (12.60 g, 124.50 mmol, 17.26 mL, 3.00 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min side to side. The reaction mixture was stirred for additional 5 min at -5°C ~ 0°C, and was quenched with water (6 mL). The reaction mixture was concentrated to remove DME. The solid was slurried in water (250 mL) and MTBE (125 mL) for 1 h. The solid was collected by filtration, and then slurried in water (250 mL) for 1 hr. The solid was collected by filtration, and washed with water (25 mL) to give white solid. The solid was slurried in EA (150 mL) and dried in vacuum at 60°C for 24 h to give III (PG = Cbz, LG = SO2CH3) (15.00 g, 36.1 1 mmol, 87.01 % yield), 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.17 (s, 3 H) 3.26 (br d, J=15.04 Hz, 1 H) 3.47 – 3.57 (m, 1 H) 3.64 (br d, J=6.36 Hz, 2 H) 4.22 (br dd, J=17.79, 8.50 Hz, 2 H) 4.50 (br s, 1 H) 4.95 – 5.17 (m, 3 H) 7.21 – 7.56 (m, 5H) 7.43 (s, 1 H) 7.63 – 7.89 (m, 2 H).
Synthesis of compound II (PG = Cbz, LG = SO2CH3, M+ = NBu4+)
O OMs o CISO3H, 2-picoline – ° O ?yO
HN Bu4NHS04< NHCbz
“Cbz
III II
To a flask was added 2-picoline (1 1 .50 g, 12.23 mL) and DMF (10 mL). The solution was cooled to 5 SC, followed by slow addition of chlorosulfonic acid (7.20 g, 4.14 mL). The temperature was increased to 20 SC. Ill (PG = Cbz, LG = SO2CH3) (5.13 g, 12.35 mmol) was added to the reaction mixture. The reaction mixture was heated to 42 SC for 18h. IPC (in process control) showed complete conversion of starting material. The reaction was cooled to 20 SC and dropwise added to a solution of tetrabutylammonium hydrogen sulfate (4.6 g, 13.6 mmol) in the mixed solvents of dichloromethane (100 mL) and water (100 mL) at 5SC. The phases were separated and the water phase was extracted with dichloromethane (2*50mL). The combined organic phase was washed with water (5*100mL). The organic phase was concentrated to dryness and purified by column chromatography (dichloromethane/methanol = 15/1 v/v) to afford II (PG = Cbz, LG = SO2CH3, M+ = NBii4+) (8.4 g, 92.30%), 1 H NMR (400 MHz, CHLOROFORM-c/) δ ppm 0.99 (t, J=7.34 Hz, 12 H) 1 .36 – 1 .50 (m, 8 H) 1 .54 – 1 .76 (m, 8 H) 3.15 (br d, J=8.31 Hz, 2 H) 3.21 – 3.35 (m, 8 H) 3.47 (br dd, J=14.73, 7.27 Hz, 1 H) 3.54 – 3.65 (m, 1 H) 3.67 – 3.81 (m, 2 H) 4.17 – 4.32 (m, 1 H) 4.39 – 4.62 (m, 1 H) 4.74 (br s, 1 H) 5.1 1 (s, 3 H) 5.32 – 5.50 (m, 1 H) 6.47 (br s, 1 H) 7.29 – 7.47 (m, 5 H) 8.69 – 8.94 (m, 1 H).
Synthesis of compound (IA)
A solution of II (PG = Cbz, LG = SO2CH3, M+ = NBu4+) (4.0 g) in dichloromethane (38 mL) was pumped to tube A at rate of 2.0844 mL/min, and a solution of KHCO3 (3.0 g) in water (100 mL) was pumped to tube B at a rate of 1 .4156 mL/min side to side. These two streams were mixed in a cross-mixer then flowed to a tube coil that was placed in an oil bath at 100 °C. The residence time of the mixed stream in the coil was 2 min. The reaction mixture flowed through a back-pressure regulator that was set at ~ 7 bars, and was collected to a beaker. After completion of the collection, two phases was separated. The organic phase was concentrated to dryness. The residue was slurried in ethyl acetate (5 mL). The solid was filtered and the filter cake was dried to give IA (2.6 g, 75%),
1H NMR (400 MHz, CHLOROFORM-c/) δ ppm 1.00 (t, J=7.27 Hz, 12 H) 1 .42 (sxt, J=7.31 Hz, 8 H) 1 .62 (quin, J=7.83 Hz, 8 H) 3.13 – 3.39 (m, 8 H) 3.54 – 3.69 (m, 2 H) 3.81 (dd, J=14.98, 2.51 Hz, 1 H) 3.96 – 4.13 (m, 1 H) 4.22 – 4.47 (m, 3 H) 4.99 – 5.23 (m, 3 H) 6.42 (br d, J=9.29 Hz, 1 H) 7.26 – 7.44 (m, 5 H).
Synthesis of compound 2A
Step 1
To a stirring solution of compound 16b (2 g, 10.14mmol, 1 .0 eq) in DMF (20 ml_) was added CS2CO3 (5.29g, 16.22 mmol, 1 .6 eq), then the resulting solution was stirred at room temperature for 10mins, then compound 16a (5.27g, 20.28mmol, 2eq) was added dropwise to the mixture for 2 minutes, then the resulting solution was stirred for another 2 hours. TLC showed the starting material was consumed completely. The mixture was added with water (60mL) and extracted with MTBE (20mL*3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The crude was slurried in heptane to give 1 .65 g 16 as a white solid (Yield: 57%), 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.48-7.28 (m, 10 H), 5.00-4.96 (t, J=6.0 Hz, 1 H), 3.81 (s, 3H), 3.44-3.42 (m, 2H), 2.40-2.37 (m, 2H).
Compound 16 (1 g, 2.66mmol, 1 eq) was dissolved in THF (20mL) under Nitrogen, and cooled to -40 °C. NaHMDS (1 .6mL, 2.0M THF solution, 1 .2 eq) was added dropwise. The reaction was stirred for 1 h at -40 °C. HPLC indicated the reaction was finished. The reaction was quenched with 10% Citric acid, extracted with MTBE (25 ml_ x 2). The combined organic layers were washed with brine (30 ml_), dried with Na2S04, filtered and concentrated to give 17 as a yellow solid, which was used for the next step without purification (assay yield: 65%); 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.27-7.13 (m, 10 H), 3.46 (s, 3H), 1 .21 -1 .17(dd, J=7.2, 10.4 Hz, 2H ); 1 .14-1 .1 1 (dd, J=7.2, 10.4 Hz, 2H).
Step 3
Compound 17 (100 mg) was dissolved in methanol (5 mL) and 2.0 M HCI IPAC solution (5 mL). The solution was heated at 45 °C for 3 days. HPLC indicated the reaction was finished. The reaction was cooled to room temperature and was diluted with 10 mL water. The reaction mixture was washed with MTBE (10 mL x 2), organic layer was discarded and the aqueous layer was concentrated to give compound 2A HCI (32 mg, 62% yield), 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.80-3.44 (br, 4H), 1 .56 (s, 2H), 1 .38 (s, 2H).
Step 4
To a solution of 2A HCI (0.70 g, 4.57 mmol) in methanol (5 mL) was added triethylamine (1 .26 mL, 9.14 mmol) at room temperature. The solution was stirred for 20 min, and the solvent was removed under vacuum. To the residue was added IPAC (10 mL) leading to precipitation. The solid was filtered, and the filtrate was concentrated to provide 2A (0.50g, 94% yield) containing ca. 6 wt% Et3N-HCI.
Synthesis of Compound X from compound of formula (I), (IA)
Compound x
To a flask was charged 21 (1 .00 g, 68.43 wt%, 2.50 mmol) and DMF (10 mL). The suspension was cooled to -20 °C, to which was added diphenylphosphinic chloride (0.52 mL, 2.75 mmol). The solution was stirred at -20 °C for 30 min, followed by addition of a mixed solution of (IA) (1 .52g, 3.00 mmol) and triethylamine (0.52 mL, 3.76 mmol) in DMF (2mL). The reaction mixture was stirred at 20 °C for 20 h, followed by addition of MTBE (20 mL). The reaction mixture was adjusted to pH = 2-3 using aqueous HCI solution (37%). To the mixture was added isopropanol (100 mL). The resulting mixture was stirred for 4 h to obtain a suspension. The suspension was filtered and the filter cake was dried under vacuum to afford crude 22 (1 .17 g). The crude 22 was slurried in a combined solvent of THF/H2O (= 12 mL / 3mL), and filtered to afford 22 (0.744 g, 75 wt% by Q-NMR, 53.3% yield). 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.47 – 3.55 (m, 2 H) 3.59 – 3.63 (m, 2 H) 4.13 – 4.21 (m, 3 H ) 5.05 (dd, J=8.8, 5.6 Hz, 1 H) 8.22 (s, 1 H) 9.73 (d, J=8.7 Hz, 1 H).
To a suspension of 22 (580 mg, 75 wt%, 1 .037 mmol) in DMAC (1 .5 mL) was added 2A (214.3 mg, 85 wt%, 1 .556 mmol). The reaction was stirred at 25 °C for 3 days, and in process control showed 22, Compound X = 4/96, and Z/E = 91 /9. the mixture was slowly added into 15ml acetone to precipitate yellowish solid. The reaction mixture was filtered to afford Compound X (0.7 g, 34 wt% by QNMR, 44% yield).
Synthesis of compound 3 (R2 = CH(Ph)2)
R2 = CH(Ph)2
2-(2-aminothiazol-4-yl)-2-oxoacetic acid (Y) (10.00 g, 47.93 mmol) and compound W (R2 = CH(Ph)2) (13.31 g, 46.98 mmol) were suspended in DMAC (40 mL), followed by addition of triethylamine (5.01 mL, 35.95 mmol). The reaction mixture was stirred at 20 °C for 5 h. HPLC showed completion of the reaction, and Z/E
= 97/3. To the reaction mixture was added water (120 mL) with stirring. The mixture was stirred for 20 min to obtain a suspension. The suspension was filtered and the filter cake was washed with water (50 mL).
The filter cake was slurried in a combined solvent of THF/ethyl acetate (50 mL / 50 mL) at 60 °C and cooled to 20 °C. The solid was filtered and dried at 50 °C for 3 h to get 3 (R2 = CH(Ph)2) (19.5 g, 88% yield). 1H
NMR (400 MHz, DMSO-cfe) δ ppm 1.37 -1 .42 (m, 2 H) 1 .44 – 1 .49 (m, 2 H) 6.87 (s, 1 H) 6.94 (s, 1 H) 7.22
– 7.30 (m, 6 H) 7.45 – 7.49 (m, 4 H).
Alternative Synthesis of Compound X from compound of formula (I), (IA)
Compound x
IA (40.14 g, 62.63 mmol) was dissolved in methanol (200 ml_), followed by addition of Pd/C (10%, 1 .1 g). The reaction mixture was maintained under hydrogen atmosphere (1 -2 bar) at 20 °C for 24 h. In process control showed completion of the reaction. The reaction mixture was filtered. The filtrate was concentrated to give an oil of IB (M+ = NBu4+) (58.20 g, 55 wt% by Q-NMR, 100% yield). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 0.93 (t, J=7.3 Hz, 12 H) 1 .23 – 1 .36 (m, 8 H) 1 .57 (m, 8 H) 2.99 – 3.28 (m, 8 H) 3.37 (dd, J=14.3, 7.5 Hz, 1 H) 3.65 – 3.70 (m, 3 H) 3.84 – 3.88 (m, 1 H) 4.08 (d, J=5.6 Hz, 1 H) 4.18 – 4.22 (m, 2 H).
3 (R2 = CH(Ph)2) (0.95 g, 2.17 mmol) was dissolved in THF (20 ml_). To the solution was added /V-methyl morpholine (0.77 g, 7.60 mmol) and 2-chloro-4,6-dimethoxy-1 ,3,5-triazine (0.57 g, 3.26 mmol). The reaction mixture was stirred at 20 °C for 1 h followed by addition of IB (M+ = NBu +) (2.70 g, 48.98 wt%, 2.61 mmol). The reaction was stirred at 20 °C for 5 h. In process control showed completion of the reaction. To the reaction mixture was added ethyl acetate (20 ml_). The organic phase was washed with brine (10 ml_). Solvent was removed. Acetone (40ml) was added to dissolve residue. TFA (1 .24 g, 10.86 mmol) dissolved in acetone (3 ml) was added slowly. The white solid was filtered and washed by acetone (10 ml) two times. Dried at 40 °C for 5h to get compound 4 (R2 = CH(Ph)2). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 1 .49 – 1 .55 (m, 4 H) 3.27 (dd, J=14.4, 6.2 Hz, 1 H) 3.49 – 3.65 (m, 2 H) 3.71 (dd, J=14.4, 6.2 Hz, 1 H) 4.04 – 4.10 (m, 1 H) 4.07 (dd, J=16.0, 8.6 Hz, 1 H) 4.17 (dd, J=1 1 .8, 6.0 Hz, 1 H) 5.28 (dd, J=9.0, 5.7 Hz, 1 H) 6.88 (s, 1 H) 7.03 (s, 1 H) 7.18 – 7.32 (m, 6 H) 7.43 (m, 4 H) 9.45 (d, J=9.0 Hz, 1 H).
Crude 4 (R2 = CH(Ph)2) (2.13 g) was dissolved in dichloromethane (20 ml_). The solution was cooled to 0 °C. To the solution was added anisole (0.68 ml_, 6.24 mmol) and trifluoroacetic acid (2.16 ml_, 28.08 mmol). The reaction was warmed to 20 °C, and stirred for 15 h. In process control showed completion of the
reaction. The aqueous phase was separated and added to acetone (40 mL) to obtain a suspension. The suspension was filtered to afford Compound X (0.98 g, 54.5% yield over two steps). 1 H NMR (400 MHz, DMSO-c/e) δ ppm 1.40 (m, 4 H) 3.26 (dd, J=14.4, 6.0 Hz, 1 H) 3.54 – 3.69 (m, 3 H) 4.14 – 4.21 (m, 3 H) 5.25 (dd, J= 8.9, 5.7 Hz, 1 H) 7.02 (s, 1 H) 9.38 (d, J=9.0 Hz, 1 H).
REF
Synthesis and optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – Identification of LYS228
57th Intersci Conf Antimicrob Agents Chemother (ICAAC) (June 1-5, New Orleans) 2017, Abst SATURDAY-297
//////////////LYS228, LYS 228, BOS-228, LYS-228, monobactam, Novartis, phase II, Boston Pharmaceuticals, complicated urinary tract infection, complicated intraabdominal infections, fast track, Qualified Infectious Disease Product, QIDP,
Nc1nc(cs1)\C(=N\OC2(CC2)C(=O)O)\C(=O)N[C@H]3[C@@H](CN4CCOC4=O)N(C3=O)S(=O)(=O)O
FDA approves new treatment for hospital-acquired and ventilator-associated bacterial pneumonia
The U.S. Food and Drug Administration today approved a new indication for the previously FDA-approved drug, Zerbaxa (ceftolozane and tazobactam) for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in patients 18 years and older. The FDA initially approved Zerbaxa in 2014 to treat complicated intra-abdominal infections and for complicated urinary tract infections.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections,” said FDA Principal Deputy Commissioner Amy Abernethy, M.D., Ph.D. “Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. New therapies to treat these infections are important to …
- June 03, 2019
The U.S. Food and Drug Administration today approved a new indication for the previously FDA-approved drug, Zerbaxa (ceftolozane and tazobactam) for the treatment of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia (HABP/VABP) in patients 18 years and older. The FDA initially approved Zerbaxa in 2014to treat complicated intra-abdominal infections and for complicated urinary tract infections.
“A key global challenge we face as a public health agency is addressing the threat of antimicrobial-resistant infections,” said FDA Principal Deputy Commissioner Amy Abernethy, M.D., Ph.D. “Hospital-acquired and ventilator-associated bacterial pneumonia are serious infections that can result in death in some patients. New therapies to treat these infections are important to meet patient needs because of increasing antimicrobial resistance. That’s why, among our other efforts to address antimicrobial resistance, we’re focused on facilitating the development of safe and effective new treatments to give patients more options to fight life-threatening infections.”
HABP/VABP occur in patients in hospitals or other health care facilities and can be caused by a variety of bacteria. According to data from the U.S. Centers for Disease Control and Prevention, HABP and VABP are currently the second most common type of hospital-acquired infection in the United States, and are a significant issue in patients in the intensive care unit (ICU).
The safety and efficacy of Zerbaxa for the treatment of HABP/VABP, administered via injection, was demonstrated in a multinational, double-blind study that compared Zerbaxa to another antibacterial drug in 726 adult patients hospitalized with HABP/VABP. The study showed that mortality and cure rates were similar between Zerbaxa and the comparator treatment.
The most common adverse reactions observed in the HABP/VABP trial among patients treated with Zerbaxa were elevated liver enzyme levels, renal impairment or failure, and diarrhea.
Zerbaxa should not be used in patients with known serious hypersensitivity to components of Zerbaxa, as well as hypersensitivity to piperacillin/tazobactam or other members of the beta lactam class of antibacterial drugs.
Zerbaxa received FDA’s Qualified Infectious Disease Product (QIDP) designation for the treatment of HABP/VABP. The QIDP designation is given to antibacterial and antifungal drug products intended to treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of QIDP designation, the Zerbaxa marketing application for the HABP/VABP indication was granted Priority Review under which the FDA’s goal is to take action on an application within an expedited time frame.
The FDA granted the approval of Zerbaxa for the treatment of HABP/VABP to Merck & Co., Inc.
//////////////ceftolozane, tazobactam, FDA 2019, Zerbaxa, HABP/VABP, Merck , Qualified Infectious Disease Product, (QIDP), Priority Review
Baloxavir marboxil, バロキサビルマルボキシル , балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 ,


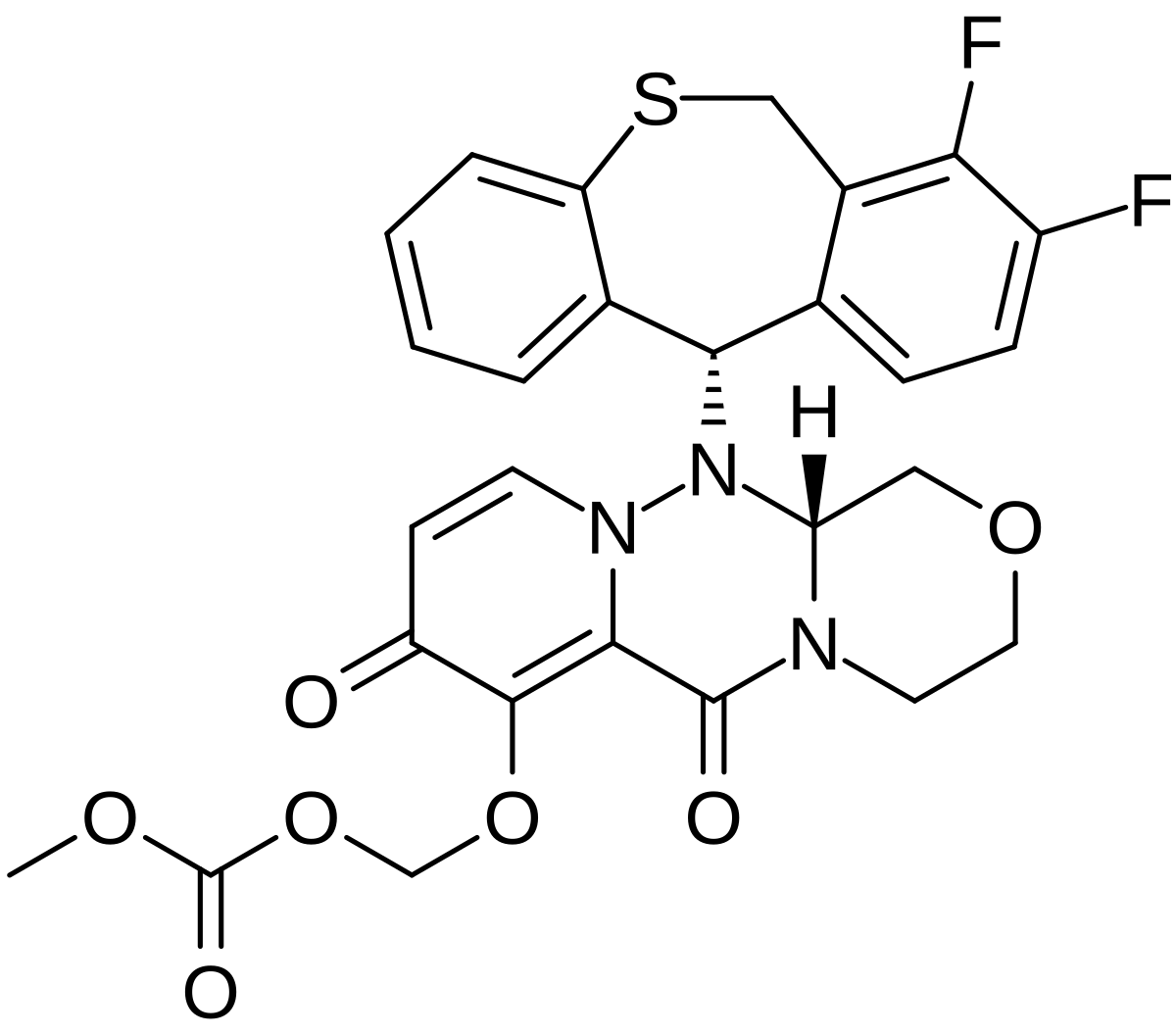
Baloxavir marboxil
バロキサビルマルボキシル
балоксавир марбоксил [Russian] [INN]
بالوكسافير ماربوكسيل [Arabic] [INN]
玛巴洛沙韦 [Chinese] [INN]
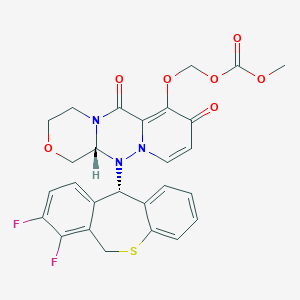
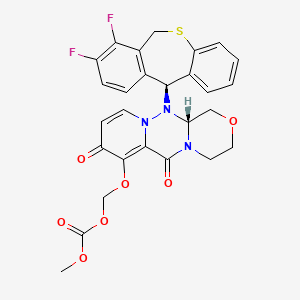
Carbonic acid, [[(12aR)-12-[(11S)-7,8-difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl]oxy]methyl methyl ester
({(12aR)-12-[(11S)-7,8-Difluoro-6,11-dihydrodibenzo[b,e]thiepin-11-yl]-6,8-dioxo-3,4,6,8,12,12a-hexahydro-1H-[1,4]oxazino[3,4-c]pyrido[2,1-f][1,2,4]triazin-7-yl}oxy)methyl methyl carbonate
- (((12aR)-12-((11S)-7,8-Difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-6,8-dioxo-3,4,6,8,12,12ahexahydro-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl carbonate
- Carbonic acid, (((12aR)-12-((11S)-7,8-difluoro-6,11-dihydrodibenzo(b,E)thiepin-11-yl)-3,4,6,8,12,12a-hexahydro-6,8-dioxo-1H-(1,4)oxazino(3,4-C)pyrido(2,1-F)(1,2,4)triazin-7-yl)oxy)methyl methyl ester
Antiviral
In Japan the product is indicated for treatment influenza types A and B in adults and children
RG-6152
- Originator Shionogi
- Developer Roche; Shionogi
- Class Antivirals; Dibenzothiepins; Esters; Pyridines; Small molecules; Triazines
- Mechanism of Action Endonuclease inhibitors
Highest Development Phases
- Marketed Influenza A virus infections; Influenza B virus infections
- Phase III Influenza virus infections
- Preclinical Influenza A virus H5N1 subtype
|
Xofluza (TN)
Antiviral
|
|
| Formula |
C27H23F2N3O7S
|
|---|---|
| Cas |
1985606-14-1
|
| Mol weight |
571.5492
|
| 2018/2/23 | PMDA | JAPAN | APPROVED | Baloxavir marboxil | Xofluza | Shionogi |

| バロキサビル マルボキシル Baloxavir Marboxil  C27H23F2N3O7S : 571.55 [1985606-14-1] |
![]()

https://chem.nlm.nih.gov/chemidplus/sid/1985606141
Baloxavir marboxil (trade name Xofluza, compound code S-033188/S-033447) is a medication being developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was in late-stage trials in Japan and the United States as of early 2018, with collaboration from Roche AG.[1].
It was approved for sale in Japan on February 23, 2018.[2]
Baloxavir marboxil is a medication developed by Shionogi Co., a Japanese pharmaceutical company, for treatment of influenza A and influenza B. The drug was approved for use in Japan in February 2018 and is in late phase trials in the United States as of early 2018. Roche, which makes Tamiflu, has acquired the license to sell Xofluza internationally, but it may not be until 2019 that it could be available in the United States [7]. Interestingly, a study has determined that administering Baloxavir marboxil with neuraminidase inhibitors leads to a synergistic effect in influenza treatment

It is an influenza therapeutic agent (cap-dependent endonuclease inhibitor), characterized by only taking one dose. Unlike neuraminidase inhibitors such as oseltamivir (Tamiflu) and zanamivir (Relenza) that inhibit the action of neuraminidase, which liberates viruses from the infected cells surface, baloxavir marboxil may prevent replication by inhibiting the cap-dependent endonuclease activity of the viral polymerase.[3]
In October 2015, the Japanese Ministry of Health, Labour and Welfare granted Sakigake status to Shionogi’s baloxavir marboxil for A type or B -type influenza virus infection . In October 2015, the drug was designated for Priority Review by the Ministry of Health, Labour and Welfare, presumably for the treatment of A type or B -type influenza virus infection .
This drug is a CAP endonuclease inhibitor [1]. The influenza endonuclease is an essential subdomain of the viral RNA polymerase enzyme. CAP endonuclease processes host pre-mRNAs to serve as primers for viral mRNA and therefore has been a common target for studies of anti-influenza drugs.
Viral gene transcription is primed by short-capped oligonucleotides that are cleaved from host cell pre mRNA by endonuclease activity. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription.The N-terminal domain of PA subunit (PAN) has been confirmed to accommodate the endonuclease activity residues, which is highly preserved among subtypes of influenza A virus and is able to fold functionally [4]. Translation of viral mRNAs by the host ribosome requires that they are capped at the 5′ end, and this is achieved in cells infected with influenza virus by a “cap-snatching” mechanism, whereby the endonuclease cleaves 5′ caps from host mRNA which then act as primers for transcription. The endonuclease domain binds the N-terminal half of PA (PAN) and contains a two-metal (Mn2+) active site that selectively cleaves the pre-mRNA substrate at the 3′ end of a guanine [3].
The administration of a CAP endonuclease inhibitor, such as Baloxavir marboxil, prevents the above process from occurring, exhibiting its action at the beginning of the pathway before CAP endonuclease may exert its action
It achieves this by inhibiting the process known as cap snatching[4], which is a mechanism exploited by viruses to hijack the host mRNA transcription system to allow synthesis of viral RNAs.
Shionogi, in collaboration with licensee Roche (worldwide except Japan and Taiwan), have developed and launched baloxavir marboxil
In March 2018, Shionogi launched baloxavir marboxil for the treatment of influenza types A and B in Japan . In September 2017, Shionogi was planning to file an NDA in the US; in February 2018, the submission remained in preparation
By September 2016, baloxavir marboxil had been awarded Qualified Infectious Disease Product (QIDP) designation in the US
In March 2017, a multicenter, randomized, double-blind, parallel-group, phase III study (NCT02954354; 1601T0831; CAPSTONE-1) was initiated in the US, Canada and Japan to compare a single dose of baloxavir marboxil versus placebo or oseltamivir bid for 5 days in influenza patients aged from 12 to 64 years of age (n = 1494). The primary endpoint was the time to alleviation of symptoms (TTAS).
PATENTS
JP 5971830
Kawai, Makoto; Tomita, Kenji; Akiyama, Toshiyuki; Okano, Azusa; Miyagawa, Masayoshi
PATENTS
WO 2017104691
Shishido, Takao; Noshi, Takeshi; Yamamoto, Atsuko; Kitano, Mitsutaka
In Japanese Patent Application No. 2015-090909 (Patent No. 5971830, issued on Aug. 17, 2016, Registered Publication), a compound having a CEN inhibitory action and represented by the formula:
[Chemical Formula 2]
is described. Anti-influenza agents of six mechanisms are enumerated as drugs that can be used together with the above compounds. However, no specific combinations are described, nor is it disclosed nor suggested about the combined effect.
Synthesis Example 2
[formula 39]
Compound III-1 (1.00g, 2.07mmol) to a suspension of DMA (5 ml) of chloromethyl methyl carbonate (0.483 g, 3.10 mmol) and potassium carbonate (0 .572 g, 4.14 mmol) and potassium iodide (0.343 g, 2.07 mmol) were added, the temperature was raised to 50 ° C. and the mixture was stirred for 6 hours. Further, DMA (1 ml) was added to the reaction solution, and the mixture was stirred for 6 hours. The reaction solution was cooled to room temperature, DMA (6 ml) was added, and the mixture was stirred at 50 ° C. for 5 minutes and then filtered. 1 mol / L hydrochloric acid water (10 ml) and water (4 ml) were added dropwise to the obtained filtrate under ice cooling, and the mixture was stirred for 1 hour. The precipitated solid was collected by filtration and dried under reduced pressure at 60 ° C. for 3 hours to obtain compound II-4 (1.10 g, 1.93 mmol, yield 93%).
1 H-NMR (DMSO-D 6) δ: 2.91-2.98 (1 H, m), 3.24-3.31 (1 H, m), 3.44 (1 H, t, J = 10.4 Hz) J = 10.8, 2.9 Hz), 4.06 (1 H, d, J = 14.3 Hz), 4.40 (1 H, dd, J = 11.5, 2.8 Hz), 3.73 (3 H, s), 4.00 , 5.67 (1 H, d, J = 6.5 Hz), 5.72 (1 H, d, J = 11.8 Hz), 4.45 (1H, dd, J = 9.9, 2.9 Hz), 5.42 J = 8.0, 1.1 Hz), 7.14 – 7.18 (1 H, m ), 7.23 (1 H, d, J = 7.8 Hz), 7.37 – 7.44 (2 H, m)
PATENTS
JP 6212678
PATENTS
JP 6249434
JP 5971830
SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF KEY INTERMEDIATE

SYNTHESIS OF FINAL PRODUCT

Japan’s New Drug: One Pill May Stop The Flu in Just One Day
Isao Teshirogi, president and chief executive officer of Shionogi & Co., speaks during an interview in Tokyo, Japan. Photographer: Kiyoshi Ota/Bloomberg
One day, you may be able to stop flu viruses in your body in just one day with just one pill. Based on an announcement yesterday, that day may be someday very soon in May in Japan.
On Friday, Japanese pharmaceutical company Shionogi announced that the flu medication that they have developed, Xofluza, otherwise known as baloxavir marboxil (which sounds a bit like a Klingon General), has been approved to be manufactured and sold in Japan. Beginning in October 2015, the medication underwent priority review by Japan’s Ministry of Health, Labor, and Welfare. Shionogi filed for approval in the autumn of 2017. Compared to Tamiflu, which requires two doses each day for five days, apparently only a single dose of Xofluza will be needed to treat the flu. Even though Xofluza has received approval, people will have to wait until the Japanese national insurance sets a price for the medication, which according to Preetika Rana writing for the Wall Street Journal, may not occur until May.
Xofluza works via a different mechanism from neuroaminidase inhibitors like Tamiflu (oseltamivir) and Relenza (zanamivir). Flu viruses are like squatters in your home that then use the furniture and equipment in your home to reproduce. Yes, I know, that makes for a lovely picture. A flu infection begins when flu viruses reach your lungs. Each flu virus will enter a cell in your lungs and then use your cell’s genetic material and protein production machinery to make many, many copies of itself. In order to do this, the flu virus uses “cap-snatching”, which has nothing to do with bottle caps or Snapchat. The virus employs an endonuclease enzyme to clip off and steal the caps or ends of your messenger RNA and then re-purposes these caps to reproduce its own genetic material. After the virus has made multiple copies of itself, the resulting viruses implement another enzyme called a neuroaminidase to separate themselves from parts of the host cell and subsequently spread throughout the rest of your body to cause havoc. While Tamiflu, Relenza, and other neuroaminidase inhibitors try to prevent the neuroaminidase enzyme from working, Xofluza acts at an earlier step, stopping the “cap-snatching” by blocking the endonuclease enzyme.

In a clinical trial, Xofluza stopped an infected person from shedding flu virus sooner than Tamiflu. (Photo Illustration by Ute Grabowsky/Photothek via Getty Images)
By acting at an earlier step before the virus has managed to replicate, Xofluza could stop a flu virus infection sooner than neuroaminidase inhibitors. The results from Shionogi’s Phase III CAPSTONE-1 clinical trial compared Xofluza (then called Cap-dependent Endonuclease Inhibitor S-033188, which doesn’t quite roll off the tongue) with oseltamivir and placebo, with results being published in Open Forum Infectious Diseases. The study found that baloxavir marboxil (or Xofluza) stopped an infected person from shedding flu virus earlier (median 24 hours) than oseltamivir (median 72 hours). Those taking baloxavir marboxil also had lower measured amounts of viruses than those taking oseltamivir throughout the first 3 days of the infection. Baloxavir marboxil also seemed to shorten the duration of flu symptoms (median 53.7 hours compared to a median of 80.2 hours for those taking placebo). Since symptoms are largely your body’s reaction to the flu virus, you can begin shedding virus before you develop symptoms, and symptoms can persist even when you are no longer shedding the virus.
The key with any of these flu medications is early treatment, especially within the first 24 to 48 hours of infection, which may be before you notice any symptoms. Once the virus has replicated and is all over your body, your options are limited. The vaccine still remains the best way to prevent an infection.
In the words of Alphaville, this new drug could be big in Japan. While Xofluza won’t be available in time to help with the current flu season, this year’s particularly harsh flu season has highlighted the need for better ways to treat the flu. But will the United States see Xofluza anytime soon? Similar to Pokemon, Xofluza may need a year or two to reach the U.S. market. But one day, one pill and one day may be a reality in the U.S.
http://www.shionogi.co.jp/en/company/news/2018/pmrltj0000003nx1-att/e180223.pdf
XOFLUZA TM (Baloxavir Marboxil) Tablets 10mg/20mg Approved for the Treatment of Influenza Types A and B in Japan Osaka, Japan, February 23, 2018 – Shionogi & Co., Ltd. (Head Office: Osaka; President & CEO: Isao Teshirogi, Ph.D.; hereafter “Shionogi”) announced that XOFLUZATM (generic name: baloxavir marboxil) tablets 10mg/20mg was approved today by the Ministry of Health, Labour and Welfare for the treatment of Influenza Types A and B. As the cap-dependent endonuclease inhibitor XOFLUZATM suppresses the replication of influenza viruses by a mechanism different from existing anti-flu drugs, XOFLUZATM was designated for Sakigake procedure with priority review by the Ministry of Health, Labour, and Welfare of Japan in October 2015. Shionogi filed for approval to manufacture and sell XOFLUZATM in October 25, 2017. As the treatment with XOFLUZATM requires only a single oral dose regardless of age, it is very convenient, and is expected to improve adherence. XOFLUZATM is expected to be a new treatment option that can improve the quality of life in influenza patients. Shionogi will launch the product immediately after the National Health Insurance (NHI) price listing. Shionogi’s research and development targets infectious disease as one of its priority areas, and Shionogi have positioned “protecting people from the threat of infectious diseases” as one of its social mission targets. Shionogi strives constantly to bring forth innovative drugs for the treatment of infectious diseases, to protect the health of patients we serve.
References
- Jump up^ Rana, Preetika (10 February 2018). “Experimental Drug Promises to Kill the Flu Virus in a Day”. Wall Street Journal.
- Jump up^ “XOFLUZA (Baloxavir Marboxil) Tablets 10mg/20mg Approved For The Treatment Of Influenza Types A And B In Japan”. 23 February 2018 – via http://www.publicnow.com.
- Jump up^ Dias, Alexandre; Bouvier, Denis; Crépin, Thibaut; McCarthy, Andrew A.; Hart, Darren J.; Baudin, Florence; Cusack, Stephen; Ruigrok, Rob W. H. (2009). “The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit”. Nature. 458(7240): 914–918. doi:10.1038/nature07745. ISSN 0028-0836.
- Jump up^ “Cap snatching”.
 |
|
| Identifiers | |
|---|---|
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C27H23F2N3O7S |
| Molar mass | 571.55 g·mol−1 |
| 3D model (JSmol) | |

Shionogi & Company, Limited(塩野義製薬株式会社 Shionogi Seiyaku Kabushiki Kaisha) is a Japanesepharmaceutical company best known for developing Crestor. Medical supply and brand name also uses Shionogi (“シオノギ”).
Shionogi has business roots that date back to 1878, and was incorporated in 1919. Among the medicines produced are for hyperlipidaemia, antibiotics, and cancer medicines.
In Japan it is particularly known as a producer of antimicrobial and antibiotics. Because of antibiotic resistance and slow growth of the antibiotic market, it has teamed up with US based Schering-Plough to become a sole marketing agent for its products in Japan.
Shionogi had supported the initial formation of Ranbaxy Pharmaceuticals, a generic manufacturer based in India. In 2012 the company became a partial owner of ViiV Healthcare, a pharmaceutical company specialising in the development of therapies for HIV.[3]
The company is listed on the Tokyo Stock Exchange and Osaka Securities Exchange and is constituent of the Nikkei 225 stock index.[4]
Medicines
- Claritin, An anti-histamine marketed in alliance with Schering-Plough.
- Crestor, cholesterol drug
- Nitrazepam, a short-term treatment for insomnia.
- Differin, a topical retinoid for acne.
- Moxifloxacin, antibacterial antiseptic that treats a number of infections
- Cymbalta, an SNRI class anti-depressant, marketed in alliance with Eli Lilly
- Osphena, an estrogen receptor agonist
Media
- Shionogi has a close relationship with Fuji Television Network, Inc., because Shionogi is the sponsor of “Music Fair” (as of 2018, aired on 17 TV stations including TV Oita System Co.) started in 1964.
- Shionogi was a main sponsor of Team Lotus during the age 1991/1994.[5]
References
- “Shionogi Company Profile”. Retrieved March 18, 2014.
- “Shionogi Annual Report 2013” (PDF). Retrieved March 18, 2014.
- “Shionogi and ViiV Healthcare announce new agreement to commercialise and develop integrase inhibitor portfolio”. viivhealthcare.com. Retrieved 18 March 2014.
- “Components:Nikkei Stock Average”. Nikkei Inc. Retrieved March 11,2014.
- Perry, Alan. “Sponsor Company Profiles”. Retrieved 25 April 2012.
External links
- Official Website (in English)
/////////Baloxavir marboxil, バロキサビルマルボキシル, JAPAN 2018, Xofluza, S-033188, S-033447, RG-6152, Qualified Infectious Disease Product, Priority Review, SAKIGAKE, балоксавир марбоксил , بالوكسافير ماربوكسيل , 玛巴洛沙韦 , Shionogi, roche
COC(=O)OCOC1=C2C(=O)N3CCOCC3N(N2C=CC1=O)C4C5=C(CSC6=CC=CC=C46)C(=C(C=C5)F)F
FDA approves new antibacterial drug Vabomere (meropenem, vaborbactam)

Meropenem

Beta-lactamase inhibitor vaborbactam
08/29/2017
The U.S. Food and Drug Administration today approved Vabomere for adults with complicated urinary tract infections (cUTI), including a type of kidney infection, pyelonephritis, caused by specific bacteria. Vabomere is a drug containing meropenem, an antibacterial, and vaborbactam, which inhibits certain types of resistance mechanisms used by bacteria.
The U.S. Food and Drug Administration today approved Vabomere for adults with complicated urinary tract infections (cUTI), including a type of kidney infection, pyelonephritis, caused by specific bacteria. Vabomere is a drug containing meropenem, an antibacterial, and vaborbactam, which inhibits certain types of resistance mechanisms used by bacteria.
“The FDA is committed to making new safe and effective antibacterial drugs available,” said Edward Cox, M.D., director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research. “This approval provides an additional treatment option for patients with cUTI, a type of serious bacterial infection.”
The safety and efficacy of Vabomere were evaluated in a clinical trial with 545 adults with cUTI, including those with pyelonephritis. At the end of intravenous treatment with Vabomere, approximately 98 percent of patients treated with Vabomere compared with approximately 94 percent of patients treated with piperacillin/tazobactam, another antibacterial drug, had cure/improvement in symptoms and a negative urine culture test. Approximately seven days after completing treatment, approximately 77 percent of patients treated with Vabomere compared with approximately 73 percent of patients treated with piperacillin/tazobactam had resolved symptoms and a negative urine culture.
The most common adverse reactions in patients taking Vabomere were headache, infusion site reactions and diarrhea. Vabomere is associated with serious risks including allergic reactions and seizures. Vabomere should not be used in patients with a history of anaphylaxis, a type of severe allergic reaction to products in the class of drugs called beta-lactams.
To reduce the development of drug-resistant bacteria and maintain the effectiveness of antibacterial drugs, Vabomere should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria.
Vabomere was designated as a qualified infectious disease product (QIDP). This designation is given to antibacterial products that treat serious or life-threatening infections under the Generating Antibiotic Incentives Now (GAIN) title of the FDA Safety and Innovation Act. As part of its QIDP designation, Vabomere received a priority review.
The FDA granted approval of Vabomere to Rempex Pharmaceuticals.
//////////////FDA, antibacterial drug, Vabomere, meropenem, vaborbactam, fda 2017, Rempex Pharmaceuticals, qualified infectious disease product, QIDP, Generating Antibiotic Incentives Now, GAIN, priority review
Biafungin, CD 101, a Novel Echinocandin for Vulvovaginal candidiasis
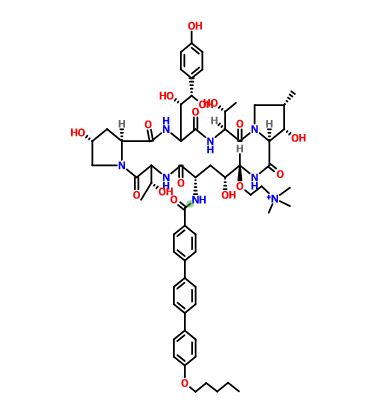
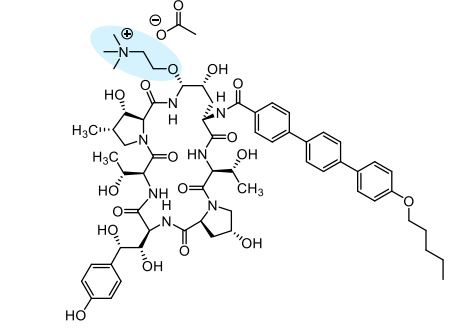
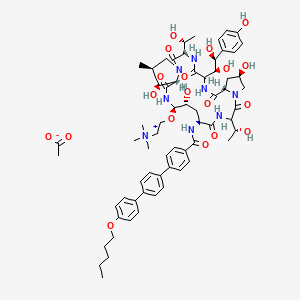
 as CH3COOH salt
as CH3COOH salt
CD 101
Several structural representations above
Biafungin™; CD 101 IV; CD 101 Topical; CD101; SP 3025, Biafungin acetate, Echinocandin B
UNII-G013B5478J FRE FORM,
CAS 1396640-59-7 FREE FORM
MF, C63-H85-N8-O17, MW, 1226.4035
Echinocandin B,
1-((4R,5R)-4-hydroxy-N2-((4”-(pentyloxy)(1,1′:4′,1”-terphenyl)-4-yl)carbonyl)-5-(2-(trimethylammonio)ethoxy)-L-ornithine)-4-((4S)-4-hydroxy-4-(4-hydroxyphenyl)-L-allothreonine)-
| Treat and prevent invasive fungal infections; Treat and prevent systemic Candida infections; Treat candidemia |
Biafungin acetate
CAS 1631754-41-0 ACETATE, Molecular Formula, C63-H85-N8-O17.C2-H3-O2, Molecular Weight, 1285.4472,
C63 H85 N8 O17 . C2 H3 O2
1-[(4R,5R)-4-hydroxy-N2-[[4”-(pentyloxy)[1,1′:4′,1”-terphenyl]-4-yl]carbonyl]-5-[2-(trimethylammonio)ethoxy]-L-ornithine]-4-[(4S)-4-hydroxy-4-(4-hydroxyphenyl)-L-allothreonine]-, acetate (1:1)
UNII: W1U1TMN677
CD101 – A novel echinocandin antifungal C. albicans (n=351) MIC90 = 0.06 µg/mL C. glabrata (n=200) MIC90 = 0.06 µg/mL Echinocandins have potent fungicidal activity against Candida species
- Originator Seachaid Pharmaceuticals
- Developer Cidara Therapeutics
- Class Antifungals; Echinocandins; Small molecules
- Mechanism of Action Glucan synthase inhibitors
- Orphan Drug Status Yes – Candidiasis
- On Fast track Candidiasis; Vulvovaginal candidiasis
- Phase II Candidiasis; Vulvovaginal candidiasis
-
Most Recent Events
- 01 Jun 2016 Phase-II clinical trials in Vulvovaginal candidiasis in USA (Topical) (9197627; NCT02733432)
- 31 May 2016 CD 101 receives Qualified Infectious Disease Product status for Vulvovaginal candidiasis in USA
- 31 May 2016 CD 101 receives Fast Track designation for Vulvovaginal candidiasis [Topical] in USA
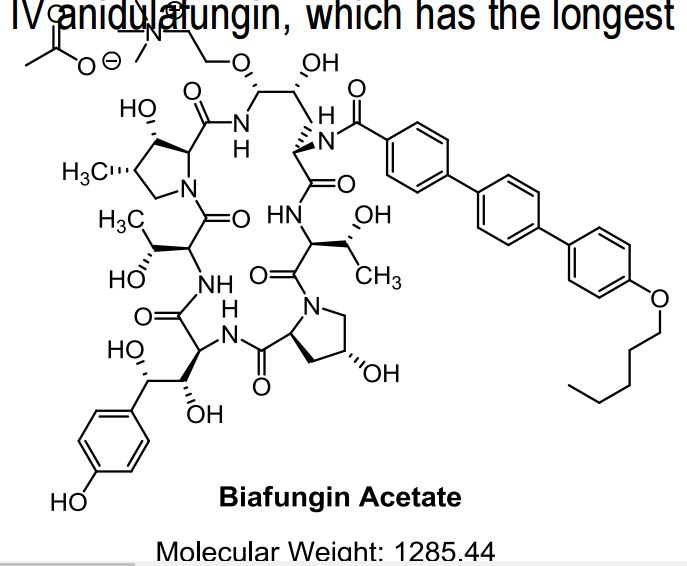
BIAFUNGIN, CD 101
Watch this space as I add more info…………….
U.S. – Fast Track (Treat candidemia);
U.S. – Fast Track (Treat and prevent invasive fungal infections);
U.S. – Orphan Drug (Treat and prevent invasive fungal infections);
U.S. – Orphan Drug (Treat candidemia);
U.S. – Qualified Infectious Disease Program (Treat candidemia);
U.S. – Qualified Infectious Disease Program (Treat and prevent invasive fungal infections)
Fungal infections have emerged as major causes of human disease, especially among the immunocompromised patients and those hospitalized with serious underlying disease. As a consequence, the frequency of use of systemic antifungal agents has increased significantly and there is a growing concern about a shortage of effective antifungal agents. Although resistance rates to the clinically available antifungal agents remains low, reports of breakthrough infections and the increasing prevalence of uncommon fungal species that display elevated MIC values for existing agents is worrisome. Biafungin (CD101, previously SP 3025) is a novel echinocandin that displays chemical stability and long-acting pharmacokinetics that is being developed for once-weekly or other intermittent administration (see posters #A-693 and A- 694 for further information). In this study, we test biafungin and comparator agents against a collection of common Candida and Aspergillus species, including isolates resistant to azoles and echinocandins.
The echinocandins are an important class of antifungal agents, but are administered once daily by intravenous (IV) infusion. An echinocandin that could be administered once weekly could facilitate earlier hospital discharges and could expand usage to indications where daily infusions are impractical. Biafungin is a highly stable echinocandin for once-weekly IV administration. The compound was found to have a spectrum of activity and potency comparable to other echinocandins. In chimpanzees single dose pharmacokinetics of IV and orally administered biafungin were compared to IV anidulafungin, which has the longest half-life (T1/2 ) of the approved echinocandins.
Background Vulvovaginal candidiasis (VVC) is a highly prevalent mucosal infection VVC is caused by Candida albicans (~85%) and non-albicans (~15%) 5-8% of women have recurrent VVC (RVVC) which is associated with a negative impact on work/social life Oral fluconazole prescribed despite relapse, potential DDIs and increased risk to pregnant women No FDA-approved therapy for RVVC and no novel agent in >20 years


Cidara Therapeutics 6310 Nancy Ridge Drive, Suite 101 San Diego, CA 92121
The incidence of invasive fungal infections, especially those due to Aspergillus spp. and Candida spp., continues to increase. Despite advances in medical practice, the associated mortality from these infections continues to be substantial. The echinocandin antifungals provide clinicians with another treatment option for serious fungal infections. These agents possess a completely novel mechanism of action, are relatively well-tolerated, and have a low potential for serious drug–drug interactions. At the present time, the echinocandins are an option for the treatment of infections due Candida spp (such as esophageal candidiasis, invasive candidiasis, and candidemia). In addition, caspofungin is a viable option for the treatment of refractory aspergillosis. Although micafungin is not Food and Drug Administration-approved for this indication, recent data suggests that it may also be effective. Finally, caspofungin- or micafungin-containing combination therapy should be a consideration for the treatment of severe infections due to Aspergillus spp. Although the echinocandins share many common properties, data regarding their differences are emerging at a rapid pace. Anidulafungin exhibits a unique pharmacokinetic profile, and limited cases have shown a potential far activity in isolates with increased minimum inhibitory concentrations to caspofungin and micafungin. Caspofungin appears to have a slightly higher incidence of side effects and potential for drug–drug interactions. This, combined with some evidence of decreasing susceptibility among some strains ofCandida, may lessen its future utility. However, one must take these findings in the context of substantially more data and use with caspofungin compared with the other agents. Micafungin appears to be very similar to caspofungin, with very few obvious differences between the two agents.
Echinocandins are a new class of antifungal drugs[1] that inhibit the synthesis of glucan in the cell wall, via noncompetitive inhibition of the enzyme 1,3-β glucan synthase[2][3] and are thus called “penicillin of antifungals”[4] (a property shared with papulacandins) as penicillin has a similar mechanism against bacteria but not fungi. Beta glucans are carbohydrate polymers that are cross-linked with other fungal cell wall components (The bacterial equivalent is peptidoglycan). Caspofungin, micafungin, and anidulafungin are semisynthetic echinocandin derivatives with clinical use due to their solubility, antifungal spectrum, and pharmacokinetic properties.[5]


List of echinocandins:[17]
- Pneumocandins (cyclic hexapeptides linked to a long-chain fatty acid)
- Echinocandin B not clinically used, risk of hemolysis
- Cilofungin withdrawn from trials due to solvent toxicity
- Caspofungin (trade name Cancidas, by Merck)
- Micafungin (FK463) (trade name Mycamine, by Astellas Pharma.)
- Anidulafungin (VER-002, V-echinocandin, LY303366) (trade name Eraxis, by Pfizer)
History


Discovery of echinocandins stemmed from studies on papulacandins isolated from a strain of Papularia sphaerosperma (Pers.), which were liposaccharide – i.e., fatty acid derivatives of a disaccharide that also blocked the same target, 1,3-β glucan synthase – and had action only on Candida spp. (narrow spectrum). Screening of natural products of fungal fermentation in the 1970s led to the discovery of echinocandins, a new group of antifungals with broad-range activity against Candida spp. One of the first echinocandins of the pneumocandin type, discovered in 1974, echinocandin B, could not be used clinically due to risk of high degree of hemolysis. Screening semisynthetic analogs of the echinocandins gave rise to cilofungin, the first echinofungin analog to enter clinical trials, in 1980, which, it is presumed, was later withdrawn for a toxicity due to the solvent system needed for systemic administration. The semisynthetic pneumocandin analogs of echinocandins were later found to have the same kind of antifungal activity, but low toxicity. The first approved of these newer echinocandins was caspofungin, and later micafungin and anidulafungin were also approved. All these preparations so far have low oral bioavailability, so must be given intravenously only. Echinocandins have now become one of the first-line treatments for Candida before the species are identified, and even as antifungal prophylaxis in hematopoietic stem cell transplant patients.
CIDARA THERAPEUTICS DOSES FIRST PATIENT IN PHASE 2 TRIAL OF CD101 TOPICAL TO TREAT VULVOVAGINAL CANDIDIASIS
SAN DIEGO–(BUSINESS WIRE)–Jun. 9, 2016– Cidara Therapeutics, Inc. (Nasdaq:CDTX), a biotechnology company developing novel anti-infectives and immunotherapies to treat fungal and other infections, today announced that the first patient has been dosed in RADIANT, a Phase 2 clinical trial comparing the safety and tolerability of the novel echinocandin, CD101, to standard-of-care fluconazole for the treatment of acute vulvovaginal candidiasis (VVC). RADIANT will evaluate two topical formulations of CD101, which is Cidara’s lead antifungal drug candidate.
“There have been no novel VVC therapies introduced for more than two decades, so advancing CD101 topical into Phase 2 is a critical step for women with VVC and for Cidara,” said Jeffrey Stein, Ph.D., president and chief executive officer of Cidara. “Because of their excellent safety record and potency against Candida, echinocandin antifungals are recommended as first line therapy to fight systemic Candida infections. CD101 topical will be the first echinocandin tested clinically in VVC and we expect to demonstrate safe and improved eradication of Candida with rapid symptom relief for women seeking a better option over the existing azole class of antifungals.”
RADIANT is a Phase 2, multicenter, randomized, open-label, active-controlled, dose-ranging trial designed to evaluate the safety and tolerability of CD101 in women with moderate to severe episodes of VVC. The study will enroll up to 125 patients who will be randomized into three treatment cohorts. The first cohort will involve the treatment of 50 patients with CD101 Ointment while a second cohort of 50 patients will receive CD101 Gel. The third cohort will include 25 patients who will be treated with oral fluconazole.
The primary endpoints of RADIANT will be the safety and tolerability of a single dose of CD101 Ointment and multiple doses of CD101 Gel in patients with acute VVC. Secondary endpoints include therapeutic efficacy in acute VVC patients treated with CD101. Treatment evaluations and assessments will occur on trial days 7, 14 and 28.
The RADIANT trial will be conducted at clinical trial centers across the United States. More information about the trial is available at www.clinicaltrials.gov, identifier NCT02733432.
About VVC and RVVC
Seventy-five percent of women worldwide suffer from VVC in their lifetime, and four to five million women in the United Statesalone have the recurrent form of the infection, which is caused by Candida. Many women will experience recurrence after the completion of treatment with existing therapies. Most VVC occurs in women of childbearing potential (the infection is common in pregnant women), but it affects women of all ages. In a recent safety communication, the U.S. Food and Drug Administration(FDA) advised caution in the prescribing of oral fluconazole for yeast infections during pregnancy based on a published study concluding there is an increased risk of miscarriage. The Centers for Disease Control and Prevention (CDC) guidelines recommend using only topical antifungal products to treat pregnant women with vulvovaginal yeast infections. Vaginal infections are associated with a substantial negative impact on day-to-day functioning and adverse pregnancy outcomes including preterm delivery, low birth weight, and increased infant mortality in addition to predisposition to HIV/AIDS. According to the CDC, certain species of Candida are becoming increasingly resistant to existing antifungal medications. This emerging resistance intensifies the need for new antifungal agents.
About CD101 Topical
CD101 topical is the first topical agent in the echinocandin class of antifungals and exhibits a broad spectrum of fungicidal activity against Candida species. In May 2016, the FDA granted Qualified Infectious Disease Product (QIDP) and Fast Track Designation to CD101 topical for the treatment of VVC and the prevention of RVVC.
About Cidara Therapeutics
Cidara is a clinical-stage biotechnology company focused on the discovery, development and commercialization of novel anti-infectives for the treatment of diseases that are inadequately addressed by current standard-of-care therapies. Cidara’s initial product portfolio comprises two formulations of the company’s novel echinocandin, CD101. CD101 IV is being developed as a once-weekly, high-exposure therapy for the treatment and prevention of serious, invasive fungal infections. CD101 topical is being developed for the treatment of vulvovaginal candidiasis (VVC) and the prevention of recurrent VVC (RVVC), a prevalent mucosal infection. In addition, Cidara has developed a proprietary immunotherapy platform, Cloudbreak™, designed to create compounds that direct a patient’s immune cells to attack and eliminate pathogens that cause infectious disease. Cidara is headquartered inSan Diego, California. For more information, please visit www.cidara.com.
REF http://ir.cidara.com/phoenix.zhtml?c=253962&p=irol-newsArticle&ID=2176474
CLIP
Cidara Therapeutics raises $42 million to develop once-weekly anti-fungal therapy
Cidara Therapeutics (formerly K2 Therapeutics) grabbed $42 million in a private Series B funding round Wednesday to continue developing its once-weekly anti-fungal therapy. Just in June 2014, the company completed a $32 million Series A financing led by 5AM Ventures, Aisling Capital, Frazier Healthcare and InterWest Partners, which was the fourth largest A round in 2014 for innovative startups[1]. FierceBiotech named the company as one of 2014 Fierce 15 biotech startups.
Cidara has an impressive executive team. The company was co-founded by Kevin Forrest, former CEO of Achaogen (NASDAQ: AKAO), and Shaw Warren. Jeffrey Stein, former CEO of Trius Therapeutics (NASDAQ: TSRX) and Dirk Thye, former president of Cerexa, have joined Cidara as CEO and CMO, respectively. Trius successfully developed antibiotic tedizolid and was acquired in 2013 by Cubist Pharmaceuticals (NASDAQ: CBST) for $818 million.
Cidara’s lead candidate, biafungin (SP3025), was acquired from Seachaid Pharmaceuticals for $6 million. Biafungin’s half-life is much longer than that of similar drugs known as echinocandins (e.g., caspofungin, micafungin, anidulafungin), which may allow it to be developed as a once-weekly therapy, instead of once daily. The company is also developing a topical formulation of biafungin, namely topifungin. Cidara intends to file an IND and initiate a Phase I clinical trial in the second half of 2015.
Merck’s Cancidas (caspofungin), launched in 2001, was the first of approved enchinocandins. The drug generated annual sales of $596 million in 2008. The approved echinocandins must be administered daily by intravenous infusion. Biafungin with improved pharmacokinetic characteristics has the potential to bring in hundreds of millions of dollars per year.
[1] Nat Biotechnol. 2015, 33(1), 18.
CLIP
Biafungin is a potent and broad-spectrum antifungal agent with excellent activity against wild-type and troublesome azole- and echinocandin-resistant strains of Candida spp. The activity of biafungin is comparable to anidulafungin. • Biafungin was active against both wild-type and itraconazole-resistant strains of Aspergillus spp. from four different species. • In vitro susceptibility testing of biafungin against isolates of Candida and Aspergillus may be accomplished by either CLSI or EUCAST broth microdilution methods each providing comparable results. • The use of long-acting intravenous antifungal agents that could safely be given once a week to select patients is desirable and might decrease costs with long-term hospitalizations. Background: A novel echinocandin, biafungin, displaying long-acting pharmacokinetics and chemical stability is being developed for once-weekly administration. The activities of biafungin and comparator agents were tested against 173 fungal isolates of the most clinically common species. Methods: 106 CAN and 67 ASP were tested using CLSI and EUCAST reference broth microdilution methods against biafungin (50% inhibition) and comparators. Isolates included 27 echinocandin-resistant CAN (4 species) with identified fks hotspot (HS) mutations and 20 azole nonsusceptible ASP (4 species). Results: Against C. albicans, C. glabrata and C. tropicalis, the activity of biafungin (MIC50, 0.06, 0.12 and 0.03 μg/ml, respectively by CLSI method) was comparable to anidulafungin (AND; MIC50, 0.03, 0.12 and 0.03 μg/ml, respectively) and caspofungin (CSP; MIC50, 0.12, 0.25 and 0.12 μg/ml, respectively; Table). C. krusei strains were very susceptible to biafungin, showing MIC90 values of 0.06 μg/ml by both methods. Biafungin (MIC50/90, 1/2 μg/ml) was comparable to AND and less potent than CSP against C. parapsilosis using CLSI methodology. CLSI and EUCAST methods displayed similar results for most species, but biafungin (MIC50, 0.06 μg/ml) was eight-fold more active than CSP (MIC50, 0.5 μg/ml) against C. glabrata using the EUCAST method. Overall, biafungin was two- to four-fold more active against fks HS mutants than CSP and results were comparable to AND. Biafungin was active against A. fumigatus (MEC50/90, ≤0.008/0.015 μg/ml), A. terreus (MEC50/90, 0.015/0.015 μg/ml), A. niger (MEC50/90, ≤0.008/0.03 μg/ml) and A. flavus (MEC50/90, ≤0.008/≤0.008 μg/ml) using CLSI method. EUCAST results for ASP were also low for all echinocandins and comparable to CLSI results. Conclusions: Biafungin displayed comparable in vitro activity with other echinocandins against common wild-type CAN and ASP and resistant subsets that in combination with the long-acting profile warrants further development of this compound. 1. Arendrup MC, Cuenca-Estrella M, Lass-Florl C, Hope WW (2013). Breakpoints for antifungal agents: An update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp. Drug Resist Updat 16: 81-95. 2. Castanheira M, Woosley LN, Messer SA, Diekema DJ, Jones RN, Pfaller MA (2014). Frequency of fks mutations among Candida glabrata isolates from a 10-year global collection of bloodstream infection isolates. Antimicrob Agents Chemother 58: 577-580. 3. Clinical and Laboratory Standards Institute (2008). M27-A3. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: third edition. Wayne, PA: CLSI. 4. Clinical and Laboratory Standards Institute (2008). M38-A2. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Filamentous Fungi: Second Edition. Wayne, PA: CLSI. 5. Clinical and Laboratory Standards Institute (2012). M27-S4. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: 4th Informational Supplement. Wayne, PA: CLSI. 6. European Committee on Antimicrobial Susceptibility Testing (2014). Breakpoint tables for interpretation of MICs and zone diameters. Version 4.0, January 2014. Available at: http://www.eucast.org/clinical_breakpoints/. Accessed January 1, 2014. 7. Pfaller MA, Diekema DJ (2010). Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36: 1-53. 8. Pfaller MA, Diekema DJ, Andes D, Arendrup MC, Brown SD, Lockhart SR, Motyl M, Perlin DS (2011). Clinical breakpoints for the echinocandins and Candida revisited: Integration of molecular, clinical, and microbiological data to arrive at species-specific interpretive criteria. Drug Resist Updat 14: 164-176. ABSTRACT Activity of a Novel Echinocandin Biafungin (CD101) Tested against Most Common Candida and Aspergillus Species, Including Echinocandin- and Azole-resistant Strains M CASTANHEIRA, SA MESSER, PR RHOMBERG, RN JONES, MA PFALLER JMI Laboratories, North Liberty, Iowa, USA C
PATENT
https://www.google.com/patents/WO2015035102A2?cl=en
BIAFUNGIN ACETATE IS USED AS STARTING MATERIAL
Example 30b: Synthesis of Compound 31


Step a. Nitration of Biafungin Acetate

To a stirring solution of biafungin (1 00 mg, 0.078 mmol) in glacial acetic acid(1 .5 ml_) was added sodium nitrite (1 1 mg, 0.159 mmol) and the reaction was stirred at ambient temperature for 20 hours. The mixture was applied directly to reversed phase H PLC (Isco CombiFlash Rf; 50g RediSep C1 8 column, 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 85 mg of the desired product as a light yellow solid, formate salt. 1 H-NMR (300 M Hz, Methanol-d4) δ 8.58 (d, 1 H, J = 1 1 .7 Hz), 8.47 (t, 2H, J = 8.7Hz), 8.05 (d, 1 H, J = 2.1 Hz), 7.99 (d, 2H, J = 9.3 Hz), 7.82 (d, 2H, J = 8.7 Hz), 7.79-7.60 (m, 12H), 7.1 7 (d, 1 H, J = 8.7 Hz), 7.03 (d, 2H, J = 9 Hz), 5.48 (d, 1 H, J = 6 Hz), 5.08 (dd, 1 H, J = 1 .2, 5.7 Hz), 4.95-4.73 (m, 5H), 4.68-4.56 (m, 2H), 4.53 (d, 1 H, J = 5.7 Hz), 4.48-4.39 (m, 2H), 4.31 -3.79 (m, 6H), 4.04 (t, 2H, J = 5.7 Hz), 3.72-3.44 (m,3H), 3.1 8 (s, 9H), 2.60-1 .99 (m, 5H), 1 .83 (m, 2H, J = 8.7 Hz), 1 .56-1 .35 (m, 5H), 1 .28 (d, 6H, J = 4.2 Hz), 1 .09 (d, 3H, J = 1 0.2 Hz), 0.99 (t, 3H, J = 8.7 Hz) ; LC/MS, [M/2+H]+: 635.79, 635.80 calculated.
Step b. Reduction of Nitro-Biafungin To Amino-Biafungin

To a stirring solution of Nitro-Biafungin (1 00 mg, 0.075 mmol) in glacial acetic acid(1 .5 ml_) was added zinc powder (50 mg, 0.77 mmol) and the reaction was stirred at ambient temperature for 1 hour. The mixture was filtered and applied directly to reversed phase HPLC (Isco CombiFlash Rf, 50g Redisep C18 column; 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 55 mg of the desired product as a white solid, formate salt. 1 H-NMR (300 MHz, Methanol-d4) 5 8.47 (bs, 1 H), 7.99 (d, 2H, J = 1 0.8Hz), 7.82 (d, 2H, J = 7.5 Hz), 7.80-7.67 (m, 6H), 7.62 (d, 2H, J = 8.7 Hz), 7.03 (d, 2H, J = 7.5 Hz), 6.77 (d, 1 H, J = 1 .9 Hz), 6.68 (d, 1 H, J = 8.2 Hz), 6.55 (dd, 2H, J = 8.2, 1 .9 Hz), 5.43 (d, 1 H, J = 2.5 Hz), 5.05 (d, 1 H, J = 3 Hz), 4.83-4.73 (m, 2H), 4.64- 4.56 (m, 2H), 4.43-4.34 (m, 2H), 4.31 -4.15 (m, 4H), 4.03-4.08 (m, 1 H), 4.1 1 -3.89 (m, 8H), 3.83 (d, 1 H, J = 1 0.8 Hz), 3.68-3.47 (m, 3H), 3.1 7 (s, 9H), 2.57-2.42 (m, 2H), 2.35-2.27 (m, 1 H), 2.14-1 .98 (m, 2H), 1 .83 (m, 2H, J = 6 Hz), 1 .56-1 .38 (m, 4H), 1 .28 (dd, 6H, J = 6.5, 2 Hz), 1 .09 (d, 3H, J = 7 Hz), 0.986 (t, 3H, J = 7 Hz); High Res LC/MS: [M+H]+ 1241 .61 63; 1241 .6136 calculated.
Step c. Reaction of Amino-Biafungin with lnt-2 to Produce Compound 31

To a stirring solution of Amino-Biafungin (50 mg, 0.04 mmol) in DM F (1 ml_) was added formyl-Met-Leu-Phe- -Ala-OSu (lnt-2) (36 mg, 0.06 mmol) and DI PEA (7 uL, 0.04 mmol). The reaction was stirred at ambient temperature for 1 8 hours. The mixture was applied directly to reversed phase HPLC (Isco CombiFlash Rf; 50g Redisep C1 8 column; 5 to 95% acetonitrile in Dl water containing 0.1 % formic acid: 15 minute gradient). The pure fractions were pooled and lyophilized to yield 26 mg of a white solid as a formate salt. 1 H-NMR (300 M Hz, Methanol-d4) 5 8.55 (bs, 1 H), 8.44 (t, 1 H, J = 10 Hz), 8.1 8 (d, 1 H, J = 6 Hz), 8.1 1 (s, 1 H), 7.99 (d, 2H, J = 1 0 Hz), 7.84-7.70 (m, 6H), 7.63 (d, 2H, J = 7.8 Hz), 7.32-7.1 9 (m, 6H), 7.03 (d, 4H, J = 9 Hz), 6.87 (d, 1 H, J = 8.1 Hz), 5.44 (d, 1 H, J = 1 0.5 Hz), 5.05 (d, 1 H, J = 4.5 Hz), 4.83-4.74 (m, 2H), 4.66-4.50 (m, 6H), 4.45-4.29 (m, 10H), 4.1 9-3.82 (m, 1 0H), 3.67-3.57 (m, 6H), 3.1 7 (s, 9H), 2.64-2.46 (m, 6 H), 2.14-1 .92 (m, 6H), 1 .84 (m, 4H, J = 6 Hz), 1 .62-1 .40 (m, 8H), 1 .32-1 .22 (m, 6H), 1 .09 (d, 3H, J = 9 Hz), 0.99 (t, 3H, J = 7.5 Hz), 0.88 (m, 6H, J = 6.8 Hz) ; High Res LC/MS, [M/2+H]+ 865.4143, 865.4147 calculated.
REFERENCES
- Denning, DW (June 2002). “Echinocandins: a new class of antifungal.”. The Journal of antimicrobial chemotherapy 49 (6): 889–91. doi:10.1093/jac/dkf045. PMID 12039879.
- Morris MI, Villmann M (September 2006). “Echinocandins in the management of invasive fungal infections, part 1”. Am J Health Syst Pharm 63 (18): 1693–703.doi:10.2146/ajhp050464.p1. PMID 16960253.
- Morris MI, Villmann M (October 2006). “Echinocandins in the management of invasive fungal infections, Part 2”. Am J Health Syst Pharm 63 (19): 1813–20.doi:10.2146/ajhp050464.p2. PMID 16990627.
- ^ Jump up to:a b “Pharmacotherapy Update – New Antifungal Agents: Additions to the Existing Armamentarium (Part 1)”.
- Debono, M; Gordee, RS (1994). “Antibiotics that inhibit fungal cell wall development”.Annu Rev Microbiol 48: 471–497. doi:10.1146/annurev.mi.48.100194.002351.
17 Eschenauer, G; Depestel, DD; Carver, PL (March 2007). “Comparison of echinocandin antifungals.”. Therapeutics and clinical risk management 3 (1): 71–97. PMC 1936290.PMID 18360617.
///////////Biafungin™, CD 101 IV, CD 101 Topical, CD101, SP 3025, PHASE 2, CIDARA, Orphan Drug, Fast Track Designation, Seachaid Pharmaceuticals, Qualified Infectious Disease Product, QIDP, UNII-G013B5478J, 1396640-59-7, 1631754-41-0, Vulvovaginal candidiasis, Echinocandin B, FUNGIN
FREE FORM
CCCCCOc1ccc(cc1)c2ccc(cc2)c3ccc(cc3)C(=O)N[C@H]4C[C@@H](O)[C@H](NC(=O)[C@@H]5[C@@H](O)[C@@H](C)CN5C(=O)[C@@H](NC(=O)C(NC(=O)[C@@H]6C[C@@H](O)CN6C(=O)C(NC4=O)[C@@H](C)O)[C@H](O)[C@@H](O)c7ccc(O)cc7)[C@@H](C)O)OCC[N+](C)(C)C
AND OF ACETATE
CCCCCOc1ccc(cc1)c2ccc(cc2)c3ccc(cc3)C(=O)N[C@H]4C[C@@H](O)[C@H](NC(=O)[C@@H]5[C@@H](O)[C@@H](C)CN5C(=O)[C@@H](NC(=O)C(NC(=O)[C@@H]6C[C@@H](O)CN6C(=O)[C@@H](NC4=O)[C@@H](C)O)[C@H](O)[C@@H](O)c7ccc(O)cc7)[C@@H](C)O)OCC[N+](C)(C)C.CC(=O)[O-]
Three antifungal drugs approved by the United States Food and Drug Administration, caspofungin, anidulafungin, and micafungin, are known to inhibit β-1 ,3-glucan synthase which have the structures shown below.

caspofungin

Anidulafungin

Other exemplary p-1 ,3-glucan synthase inhibitors include,

echinocandin B

cilofungin

pneumocandin A0

pneumocandin B0

L-705589

L-733560

A-174591 

 or a salt thereof,
or a salt thereof,
Biafungin

or a salt thereof,
Amino-biafungin

or a salt thereof,

Amino-AF-053 


ASP9726
Yet other exemplary p-1 ,3-glucan synthase inhibitors include, without limitation:

Papulacandin B


Ergokonin
//////////////

{kind=link}