

Roche is investing $775 million in cash and equity for access to Blueprint Medicines’ oncology drug candidate pralsetinib, which is under review by the US Food and Drug Administration.
Home » Posts tagged 'Pralsetinib'
Tag Archives: Pralsetinib
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
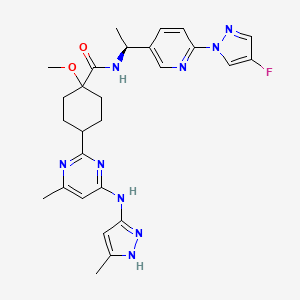
Pralsetinib
Pralsetinib
| Formula |
C27H32FN9O2
|
|---|---|
| CAS |
2097132-94-8
|
| Mol weight |
533.6005
|
Cyclohexanecarboxamide, N-[(1S)-1-[6-(4-fluoro-1H-pyrazol-1-yl)-3-pyridinyl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]-2-pyrimidinyl]-, cis–
2097132-94-8 [RN]
BLU-667
BS-15942
Other Names
- cis-N-[(1S)-1-[6-(4-Fluoro-1H-pyrazol-1-yl)-3-pyridinyl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]-2-pyrimidinyl]cyclohexanecarboxamide
- BLU 123244
- BLU 667
- Pralsetinib
- X 581238
- cis-N-{(1S)-1-[6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl]ethyl}-1-methoxy-4-{4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl}cyclohexane-1-carboxamide
N-[(1S)-1-[6-(4-fluoropyrazol-1-yl)pyridin-3-yl]ethyl]-1-methoxy-4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]pyrimidin-2-yl]cyclohexane-1-carboxamide
FDA APPROVED GAVRETO, 2020/9/4
Pralsetinib, sold under the brand name Gavreto, is a medication for the treatment of metastatic RET fusion-positive non-small cell lung cancer (NSCLC).[1] Pralsetinib is a tyrosine kinase inhibitor. It is taken by mouth.[1]
The most common adverse reactions include increased aspartate aminotransferase (AST), decreased hemoglobin, decreased lymphocytes, decreased neutrophils, increased alanine aminotransferase (ALT), increased creatinine, increased alkaline phosphatase, fatigue, constipation, musculoskeletal pain, decreased calcium, hypertension, decreased sodium, decreased phosphate, and decreased platelets.[1]
Pralsetinib was approved for medical use in the United States in September 2020.[1][2][3][4]
Medical uses
Pralsetinib is indicated for the treatment of adults with metastatic RET fusion-positive non-small cell lung cancer (NSCLC) as detected by an FDA approved test.[1][4]
History
Efficacy was investigated in a multicenter, open-label, multi-cohort clinical trial (ARROW, NCT03037385) with 220 participants aged 26-87 whose tumors had RET alterations.[1][4] Identification of RET gene alterations was prospectively determined in local laboratories using either next generation sequencing, fluorescence in situ hybridization, or other tests.[1] The main efficacy outcome measures were overall response rate (ORR) and response duration determined by a blinded independent review committee using RECIST 1.1.[1] The trial was conducted at sites in the United States, Europe and Asia.[4]
Efficacy for RET fusion-positive NSCLC was evaluated in 87 participants previously treated with platinum chemotherapy.[1] The ORR was 57% (95% CI: 46%, 68%); 80% of responding participants had responses lasting 6 months or longer.[1] Efficacy was also evaluated in 27 participants who never received systemic treatment.[1] The ORR for these participants was 70% (95% CI: 50%, 86%); 58% of responding participants had responses lasting 6 months or longer.[1]
The US Food and Drug Administration (FDA) granted the application for pralsetinib priority review, orphan drug, and breakthrough therapy designations[1]and granted approval of Gavreto to Blueprint Medicines.[1]
PATENT
US 20170121312

https://patents.google.com/patent/US20170121312A1/en
-
- Step 7: Synthesis of (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (Compound 129) and (1S,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (Compound 130)
- [0194]

- [0195]
The title compounds were prepared from methyl 1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxylate (192 mg, 0.53 mmol) using the same two-step procedure (hydrolysis and amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (190 mg), which was then dissolved in 6 mL methanol and purified by SFC (ChiralPak AD-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 2.5 mL injections, 70 mL/min). Peak 1 was concentrated to give (1R,4S)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexanecarboxamide (29 mg, 10%) as a white solid. Peak 2 was concentrated to give (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(4-methyl-6-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-2-yl)cyclohexane-carboxamide (130 mg, 46%) as a white solid.
- [0194]
Example 6. Synthesis of Compound 149Step 1: Synthesis of Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate
-
- [0196]

- [0197]
Methyl 4-iodo-1-methoxycyclohexanecarboxylate (3.37 g, 11.3 mmol) was dissolved in dimethylacetamide (38 mL) in a pressure vessel under a stream of N2. Rieke Zinc (17.7 mL of a 50 mg/mL suspension in THF, 13.6 mmol) was added quickly via syringe, and the vessel was capped and stirred at ambient temperature for 15 minutes. The vessel was opened under a stream of N2 and 2,4-dichloro-6-methylpyrimidine (1.84 g, 11.3 mmol) was added followed by PdCl2dppf (826 mg, 1.13 mmol). The vessel was capped and heated to 80° C. for one hour, then cooled to room temperature. The reaction mixture was diluted with EtOAc, filtered through celite, and the filtrate was washed with H2O (3×), brine, dried over sodium sulfate, filtered, and concentrated. The resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 50% EtOAc-hexanes) to give methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (74 mg, 2.2%) as a colorless oil. MS (ES+) C14H19ClN2O3 requires: 298, found: 299 [M+H]+.
- [0196]
Step 2: Synthesis of tert-Butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate
-
- [0198]

- [0199]
Methyl 4-(2-chloro-6-methylpyrimidin-4-yl)-1-methoxycyclohexane-1-carboxylate (70.5 mg, 0.236 mmol), tert-butyl 3-amino-5-methyl-1H-pyrazole-1-carboxylate (69.8 mg, 0.354 mmol), di-tert-butyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine (20.0 mg, 0.2 equiv.), Pd2(dba)3 (21.6 mg, 0.1 equiv.), and potassium acetate (70 mg, 0.71 mmol) were combined in a vial under nitrogen and 0.98 mL dioxane was added. The reaction mixture was heated to 115° C. for 2 h, then cooled to ambient temperature. The reaction mixture was diluted with EtOAc, filtered through celite, concentrated onto silica gel, and the resulting residue was purified by flash-column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-hexanes) to give tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (48 mg, 44%) as a yellow oil. MS (ES+) C23H33N5O5 requires: 459, found: 460 [M+H]+.
- [0198]
Step 3: Synthesis of 1-Methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid
-
- [0200]

- [0201]
Lithium hydroxide monohydrate (13 mg, 0.31 mmol) was added to a solution of tert-butyl 3-((4-(4-methoxy-4-(methoxycarbonyl)cyclohexyl)-6-methylpyrimidin-2-yl)amino)-5-methyl-1H-pyrazole-1-carboxylate (47.7 mg, 0.104 mmol) in THF/MeOH/H2O (17:1:1, 1.8 mL). The reaction mixture was heated to 60° C. and stirred for 16 h. The reaction mixture was then cooled to ambient temperature and concentrated to give crude 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, crude) which was used in the subsequent amide coupling without any further purification. MS (ES+) C17H23N5O3 requires: 345, found: 346 [M+H]+.
- [0200]
Step 4: Synthesis of (1s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (Compound 149)
-
- [0202]

- [0203]
The title compound was prepared from 1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxylic acid (57 mg, 0.104 mmol) using the same procedured (amide coupling) outlined in Synthetic Protocols 1 and 2, with PyBOP as the amide coupling reagent instead of HATU. The products were initially isolated as a mixture of diastereomers (36 mg), which was then dissolved in 6 mL methanol-DCM (1:1) and purified by SFC (ChiralPak IC-H 21×250 mm, 40% MeOH containing 0.25% DEA in CO2, 1.0 mL injections, 70 mL/min). Peak 1 was an undesired isomer, and Peak 2 was concentrated to give (1 s,4R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-1-methoxy-4-(6-methyl-2-((5-methyl-1H-pyrazol-3-yl)amino)pyrimidin-4-yl)cyclohexane-1-carboxamide (13.4 mg, 24%) as a white solid.
- [0202]
Synthesis of IntermediatesExample 7. Synthesis of Ketone and Boronate IntermediatesA. Methyl 1-methoxy-4-oxocyclohexane-1-carboxylate
-
- [0204]

- [0205]
The title compound was prepared as described in WO 2014/130810 A1 page 86.
- [0204]
B. Ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
-
- [0206]

- [0206]
Step 1: Synthesis of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate
-
- [0207]
A solution of 1,4-dioxaspiro[4.5]decan-8-one (20.0 g, 128 mmol) in CHBr3 (3234 g, 1280 mmol) was cooled to 0° C. and potassium hydroxide (57.5 g, 1024 mmol) in EtOH (300 mL) was added dropwise over 2.5 hrs. After stirring the mixture for 23 h, the mixture was concentrated, and the residue was partitioned between EtOAc and H2O. The organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude product, which was purified by flash column chromatography on silica gel (gradient elution, PE:EA=15:1 to 10:1) to obtain the title compound (18.0 g).
- [0207]
Step 2: Synthesis of ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate
-
- [0208]
To a solution of ethyl 8-ethoxy-1,4-dioxaspiro[4.5]decane-8-carboxylate (10 g, 43 mmol) in 1,4-dioxane (250 mL) was added aqueous HCl (6 M, 92.5 mL), and the mixture was stirred for 23 h at ambient temperature. The mixture was then diluted with H2O and extracted with EtOAc.
- [0209]
The organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a crude residue, which was purified by flash column chromatography on silica gel (PE:EA=15:1) to obtain the product (8.0 g). 1H NMR (400 MHz, DMSO) δ 4.20-4.13 (m, 2H), 3.43 (q, J=6.9 Hz, 1H), 2.48-2.39 (m, 1H), 2.24-2.12 (m, 2H), 2.10-2.01 (m, 1H), 1.22 (t, J=7.1 Hz, 2H), 1.17 (t, J=7.0 Hz, 2H).
- [0208]
C. Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0210]

- [0210]
Step 1: Synthesis of ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate
-
- [0211]
A solution of methylmagnesium bromide (3M, 109.8 mL, 329.4 mmol) was added dropwise to a suspension of CuCN (14.75 g, 164.7 mmol) in diethyl ether (50 mL) at 0° C. The mixture was stirred for 30 min at 0° C. and then cooled to −78° C. The solution of ethyl 2-methyl-4-oxocyclohex-2-ene-1-carboxylate (10 g, 54.9 mmol) in diethyl ether (10 mL) was then added dropwise. The mixture was stirred between −40° C. to −20° C. for 2 h, then was warmed to ambient temperature for 16 h. The reaction mixture was carefully added to a saturated solution of ammonium chloride. The aqueous layer was extracted twice with diethyl ether, and the organic layers were combined. The combined organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (PE:EA=10:1) to give ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g).
- [0211]
Step 2: Synthesis of ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate
-
- [0212]
Ethyl 2,2-dimethyl-4-oxocyclohexane-1-carboxylate (1.16 g, 5.85 mmol) and DIPEA (3.03 g, 23.4 mmol) were dissolved in dry toluene (2 mL) and heated at 45° C. for 10 minutes. Trifluoromethanesulfonic anhydride (6.61 g, 23.4 mmol) in DCM (20 mL) was added dropwise over 10 min and the mixture was heated at 45° C. for 2 h. The mixture was allowed to cool to room temperature, concentrated, diluted with water (60 mL) and extracted with DCM (2×40 mL). The organic layer was washed with saturated sodium bicarbonate solution (20 mL) and brine (20 mL), dried over sodium sulfate, filtered, and concentrated. The crude product was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1-carboxylate (1 g).
- [0212]
Step 3: Synthesis of ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0213]
Ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (1 g, 3.03 mmol), 4,4,5,5-tetramethyl-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3,2-dioxaborolane (1.15 g, 4.54 mmol), Pd(dppf)Cl2 (73.5 mg, 0.09 mmol) and potassium acetate (891 mg, 9.08 mmol) were suspended in 1,4-dioxane (20 mL). The reaction mixture was flushed with nitrogen, then heated to 100° C. for 2 h. The mixture was cooled to room temperature, filtered, and concentrated, and the resulting brown oil was purified by flash column chromatography on silica gel (gradient elution, 0 to 100% ethyl acetate-petroleum ether) to afford ethyl 6,6-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate (618 mg).
- [0213]
D. Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate
-
- [0214]

- [0215]
Ethyl 6-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-ene-1-carboxylate was prepared using the same synthetic protocol as described above using ethyl 2-methyl-4-oxocyclohexane-1-carboxylate as the starting material.
- [0214]
E. Methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
-
- [0216]

- [0216]
Step 1: Synthesis of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate
-
- [0217]
A mixture of acrylaldehyde (120 g, 2.14 mol), methyl methacrylate (200 g, 2.00 mol) and hydroquinone (2.2 g, 20 mmol) were heated in a sealed steel vessel at 180° C. for one h. The mixture was then cooled to ambient temperature and concentrated. The residue was purified by silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=100:1 to 80:1) to give methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (70 g, 22% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 6.38 (d, J=6.4 Hz, 1H), 4.73-4.70 (m, 1H), 3.76 (s, 3H), 2.25-2.22 (m, 1H), 1.99-1.96 (m, 2H), 1.79-1.77 (m, 1H), 1.49 (s, 3H).
- [0217]
Step 2: Synthesis of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate
-
- [0218]
To a solution of methyl 2-methyl-3,4-dihydro-2H-pyran-2-carboxylate (20.0 g, 128 mmol) in anhydrous tetrahydrofuran (200 mL) was added borane (67 mL, 1 M in tetrahydrofuran) dropwise at −5° C. The reaction mixture was stirred at 0° C. for 3 hours. This reaction was monitored by TLC. The mixture was quenched by a solution of sodium acetate (10.5 g, 128 mmol) in water (15 mL). Then the mixture was treated with 30% hydrogen peroxide solution (23.6 g, 208.2 mmol) slowly at 0° C. and stirred at 30° C. for 3 h. The mixture was then partitioned between saturated sodium sulfite solution and tetrahydrofuran. The aqueous layer was further extracted with tetrahydrofuran (2×). The combined organic layers were washed with saturated brine, dried over sodium sulfate and concentrated in vacuo. The residue was purified by a silica gel column chromatography (gradient elution, petroleum ether:ethyl acetate=10:1 to 1:1) to give crude methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18 g, crude) as a pale yellow oil, which used directly for next step.
- [0218]
Step 3: Synthesis of methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate
-
- [0219]
To a solution of methyl 5-hydroxy-2-methyltetrahydro-2H-pyran-2-carboxylate (18.0 g, 103 mmol) in anhydrous dichloromethane (200 mL) was added PCC (45.0 g, 209 mmol) in portions. The reaction mixture was stirred at ambient temperature until TLC indicated the reaction was completed. Petroleum ether (500 mL) was then added and the mixture was filtered. The filter cake was washed with petroleum ether (100 mL), and the filtrate was concentrated under vacuum to give methyl 2-methyl-5-oxotetrahydro-2H-pyran-2-carboxylate (15 g, 84% yield) as a pale yellow oil. 1H-NMR (400 MHz, CDCl3): δ 4.25 (d, J=17.6 Hz, 1H), 4.07 (d, J=17.6 Hz, 1H), 3.81 (s, 3H), 2.52-2.44 (m, 3H), 2.11-2.04 (m, 1H), 1.53 (s, 3H).
- [0219]
Example 8. Synthesis of Iodide IntermediatesA. Methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
-
- [0220]

- [0220]
Step 1: Synthesis of methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate
-
- [0221]
Methyl 1-methoxy-4-oxocyclohexanecarboxylate (4.00 g, 21.5 mmol) was dissolved in methanol (100 mL) and the solution was cooled to 0° C. Sodium borohydride (2.03 g, 53.7 mmol) was added in portions over 20 min. The reaction mixture was stirred for 30 min, then was quenched by addition of aqueous saturated NH4Cl solution. The quenched reaction mixture was evaporated to remove the MeOH, then the aqueous suspension was extracted with DCM (3×). The combined organic layers were dried over sodium sulfate, filtered, and concentrated to yield a residue that was purified by flash-column chromatography on silica gel (gradient elution, 5% to 100% ethyl acetate-hexanes) to afford methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 49.5%) as a colorless oil. MS (ES+) C9H16O4 requires: 188, found: 211 [M+Na]+.
- [0221]
Step 2: Synthesis of methyl 1-methoxy-4-iodocyclohexane-1-carboxylate
-
- [0222]
Methyl 1-methoxy-4-hydroxycyclohexane-1-carboxylate (2.00 g, 10.6 mmol) was dissolved in THF (20 mL) and imidazole (723 mg, 10.6 mmol) and triphenylphosphine (3.34 g, 12.8 mmol) were added. The mixture was cooled to 0° C., and then a solution of iodine (3.24 g, 12.8 mmol) in THF (10 mL) was added dropwise over 15 min. The reaction mixture was allowed to warm to ambient temperature and was then stirred for 2 days, after which it was poured over saturated sodium thiosulfate solution and extracted with EtOAc. The organic layer was dried over sodium sulfate, filtered, concentrated, and the residue was triturated with hexane (40 mL, stir for 20 min). The mixture was filtered, and the filtrate was evaporated to provide a residue that was purified by flash-column chromatography on silica gel (gradient elution, 0 to 30% ethyl acetate-hexanes) to give the title compound (2.37 g, 75%) as a pale yellow oil. MS (ES+) C9H15IO3 requires: 298, found: 299 [M+H]+.
- [0222]
B. Ethyl 1-ethoxy-4-iodocyclohexane-1-carboxylate
-
- [0223]

- [0224]
The title compound was prepared as described above using ethyl 1-ethoxy-4-oxocyclohexane-1-carboxylate as a starting material. C11H19IO3 requires: 326, found: 327 [M+H]−.
- [0223]
Example 9. Synthesis of Amine IntermediatesA. (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
-
- [0225]

- [0225]
Step 1: Synthesis of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one
-
- [0226]
4-Fluoro-1H-pyrazole (4.73 g, 55 mmol) and potassium carbonate (17.27 g, 125 mmol) were combined and stirred in N,N-dimethylformamide (41.7 mL) for 10 minutes in an open sealed tube before addition of 2-bromo-5-acetylpyridine (10 g, 50 mmol). The reaction tube was sealed and stirred for 20 hours at 100° C. The reaction mixture was then cooled to room temperature and poured into water (˜700 mL). The mixture was sonicated and stirred for 20 minutes, after which a beige solid was isolated by filtration, washed with small amounts of water, and dried to yield 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.81 g, 96% yield). MS: M+1=206.0.
- [0226]
Step 2: Synthesis of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide
-
- [0227]
To a stirred room temperature solution of 1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-one (9.806 g, 47.8 mmol) in THF (96 mL) was added (R)-(+)-t-Butylsulfinamide (5.79 g, 47.8 mmol) followed by titanium (IV) ethoxide (21.8 g, 96 mmol). The solution was stirred at 75° C. on an oil bath for 15 hours. The reaction solution was cooled to room temperature and then to −78° C. (external temperature) before the next step. To the −78° C. solution was added dropwise over nearly 55 minutes L-Selectride (143 mL of 1N in THF, 143 mmol). During addition, some bubbling was observed. The reaction was then stirred after the addition was completed for 15 minutes at −78° C. before warming to room temperature. LC-MS of sample taken during removal from cold bath showed reaction was completed. The reaction was cooled to −50° C. and quenched slowly with methanol (˜10 mL), then poured into water (600 mL) and stirred. An off-white precipitate was removed by filtration, with ethyl acetate used for washes. The filtrate was diluted with ethyl acetate (800 mL), the layers were separated, and the organic layer was dried over sodium sulfate, filtered, and concentrated down. The crude was purified by silica gel chromatography to yield (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.5 g, 99% purity, 70.3% yield) as a light yellow solid. MS: M+1=311.1.
- [0227]
Step 3: Synthesis of (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine
- [0228]
A solution of (R)—N—((S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethyl)-2-methylpropane-2-sulfinamide (10.53 g, 33.9 mmol)) in methanol (79 mmol) and 4N HCl/dioxane (85 mL, 339 mmol) was stirred for 2.5 hours, at which point LC-MS showed reaction was complete. The reaction solution was poured into diethyl ether (300 mL) and a sticky solid was formed. The mixture was treated with ethyl acetate (200 mL) and sonicated. The solvents were decanted, and the sticky solid was treated with more ethyl acetate (˜200 mL), sonicated and stirred. The bulk of the sticky solid was converted to a suspension. A light yellow solid was isolated by filtration, washed with smaller amounts of ethyl acetate, and dried to yield (S)-1-(6-(4-fluoro-1H-pyrazol-1-yl)pyridin-3-yl)ethan-1-amine (7.419 g, 78% yield). LC-MS confirmed desired product in high purity. MS: M+1=207.1.
PATENT
CN 111440151
PATENT
CN 111362923
References
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves pralsetinib for lung cancer with RET gene fusions”. U.S. Food and Drug Administration (FDA). 4 September 2020. Retrieved 8 September 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Blueprint Medicines Announces FDA Approval of Gavreto (pralsetinib) for the Treatment of Adults with Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer” (Press release). Blueprint Medicines. 4 September 2020. Retrieved 8 September 2020 – via PR Newswire.
- ^ “Roche announces FDA approval of Gavreto (pralsetinib) for the treatment of adults with metastatic RET fusion-positive non-small cell lung cancer”. Roche (Press release). 7 September 2020. Retrieved 8 September 2020.
- ^ Jump up to:a b c d “Drug Trial Snapshot: Gavreto”. U.S. Food and Drug Administration. 4 September 2020. Retrieved 16 September 2020. This article incorporates text from this source, which is in the public domain.
External links
- “Pralsetinib”. Drug Information Portal. U.S. National Library of Medicine.
- “Pralsetinib”. NCI Drug Dictionary. National Cancer Institute.
- Clinical trial number NCT03037385 for “Phase 1/2 Study of the Highly-selective RET Inhibitor, Pralsetinib (BLU-667), in Patients With Thyroid Cancer, Non-Small Cell Lung Cancer, and Other Advanced Solid Tumors (ARROW)” at ClinicalTrials.gov
- “Understanding Metastatic RET Fusion-Positive Non-Small Cell Lung Cancer (NSCLC)” (PDF).
- “Understanding Metastatic RET-Driven Thyroid Cancers” (PDF).
| Clinical data | |
|---|---|
| Trade names | Gavreto |
| Other names | BLU-667 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Tyrosine kinase inhibitor |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H32FN9O2 |
| Molar mass | 533.612 g·mol−1 |
| 3D model (JSmol) | |
Roche buys into Blueprint’s RET inhibitor
The deal positions pralsetinib to compete against Lilly’s Retevmo
by Lisa M. Jarvis
JULY 18, 2020 | APPEARED IN VOLUME 98, ISSUE 28

Pralsetinib is a small-molecule inhibitor of RET alterations—rare genetic fusions or mutations that occur at low levels across lung, thyroid, and many other cancers.
The drug will go up against Eli Lilly and Company’s Retevmo, an RET inhibitor that received FDA approval in May for certain lung and thyroid cancers. Lilly acquired Retevmo in its $8 billion purchase of Loxo Oncology in 2019, a deal to obtain Loxo’s pipeline of small molecules for genetically defined tumors.
But SVB Leerink analyst Andrew Berens points out that Retevmo has side effects: it can cause an irregular heart rhythm called QT prolongation and hemorrhagic events. That leaves room for pralsetinib, which Roche will be better able to get in front of oncologists, Berens argues. In addition to a vast commercial network, Roche brings diagnostic tools to help identify cancer patients whose tumors feature RET alterations.
The FDA has a deadline of Nov. 23 to decide on approving the drug for lung cancer.
Roche’s move lowers the likelihood of a takeover of Blueprint, which had appeared on many investors’ short lists of acquisition targets. “We were surprised by the profuse language framing this deal as ensuring Blueprint’s independence,” Piper Sandler stock analyst Christopher J. Raymond told investors in a note.
//////////Pralsetinib, GAVRETO, 2020 APPROVALS, FDA 2020
CC1=CC(=NN1)NC2=NC(=NC(=C2)C)C3CCC(CC3)(C(=O)NC(C)C4=CN=C(C=C4)N5C=C(C=N5)F)OC
