
如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'PHASE 3' (Page 18)
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

MACITENTAN
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide,
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl] -N’-propylsulfamide
CAS NO 441798-33-0
ACT-064992, Opsumit,UNII-Z9K9Y9WMVL
Mechanism of Action: Endothelin receptor antagonist (ERA)
Date of Approval: October 18, 2013(US)
Indication: Pulmonary Hypertension (PAH)
Company: Actelion Pharmaceuticals Ltd
PCT patent application: WO2002053557
FDA N204410, MACITENTANTABLET; ORAL10MG, OPSUMIT, ACTELION PHARMS LTD
Macitentan is achiral
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
Macitentan (Opsumit® )is a novel dual endothelin receptor antagonist that resulted from a tailored drug discovery process. Macitentan has a number of potentially key beneficial characteristics – i.e., increased in vivo preclinical efficacy vs. existing ERAs resulting from sustained receptor binding and tissue penetration properties. A clinical pharmacology program indicated a low propensity of macitentan for drug-drug interactions.
Macitentan (ACT-064992) is a tissue-targeting dual ET(A)/ET(B) endothelin (ET) receptor antagonist designed for tissue targeting. Macitentan inhibited ET-1-induced contractions in isolated endothelium-denuded rat aorta (ET(A) receptors) and sarafotoxin S6c-induced contractions in isolated rat trachea (ET(B) receptors). In diabetic rats, chronic administration of macitentan decreased blood pressure and proteinuria and prevented end-organ damage. Treatment with macitentan enhanced the cytotoxicity mediated by paclitaxel as measured by the degree of apoptosis in tumor cells and tumor-associated endothelial cells. A Phase III clinical trial of macitentan was successfully completed in 2012.


Macitentan is an investigational drug being studied for the treatment of pulmonary arterial hypertension. It acts as a dualendothelin receptor antagonist and is being developed by Actelion.[1] A Phase III clinical trial was successfully completed in 2012.[2]
on 22 October 2012 – Actelion (SIX: ATLN) announced that it has submitted a New Drug Application (NDA) to the US Food and Drug Administration (FDA) seeking approval for macitentan (Opsumit®) for the treatment of patients with pulmonary arterial hypertension
Actelion’s experimental lung drug macitentan prolonged overall survival by more than a third according to detailed study data, which the company hopes will convince investors it has a viable follow-up product to secure its commercial future.
Europe’s largest standalone biotech company wants the drug, which treats pulmonary arterial hypertension — a disease that causes high blood pressure in the arteries of the lungs — to replace blockbuster Tracleer.
Tracleer currently makes up 87 percent of sales but loses patent protection in 2015 and has also seen its market share eroded by Gilead’s Letairis.

Macitentan has an active metabolite, ACT-132577, which is an oxidative depropylation product. Both macitentan and ACT-132577 are mainly excreted in form of hydrolysis products via urine (about 2/3 of all metabolites) and faeces (1/3).[3]
Co-administration of ciclosporin has only a slight effect on the concentrations of macitentan and its active metabolite, whilerifampicin decreases the area under the curve (AUC) of the drug’s blood plasma concentration by 79%, and ketoconazoleapproximately doubles it. This corresponds to the finding that macitentan is mainly metabolised via the liver enzyme CYP3A4.[4]

SYNTHESIS
The synthesis begins with the reaction of chlorosulfonyl isocyanate (1) (dissolved in dichloromethane at 0 ° C) with one equivalent of tert-butanol. This produces a by BOC protected Aminosulfonylchlorid (2). With one equivalent of n-propylamine (dissolved in 3 eq. Of triethylamine, dichloromethane, at 0 ° C, RT 16 h) is produced by a hydrochloric acid elimination BOC-protected sulfamide (3). This is dissolved in 5 M HCl and dioxane (4-8 h), the BOC protecting group is cleaved. The sulfamide formed (4) is potassium tert-butoxide-(dissolved in MeOH, 3h) is converted to the potassium salt (5). Tert-butoxide potassium acts as a very strong base for deprotonation. This sulfamide potassium salt reacts with the nucleophilic substituents on the heteroaromatic Dichlorpyrimidinderivat (6) (dissolved in dimethyl sulfoxide, at room temperature, RT 42-72 h) under KCl-cleavage to a Monochlorpyrimidin intermediate (7). By treatment with ethylene glycol (dissolved in dimethyl ether, potassium-tert-butoxide,), the ethylene glycol side chain is generated (8). With 2-chloro-5-bromo-pyrimidine (dissolved in tetrahydrofuran, close, at 60-75 ° C) is formed under elimination of HCl in an S N 1 reaction Macitentan (9)…………Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .

Synthesis of Macitentan
…………………………………………….
SYNTHESIS


YOU CAN READ AT YAOPHA.COM, lovely site to see for drugs


如果您有关于yaopha.com的问题或建议请与我们联系,具体方式如下:
E-mail:contactus@yaopha.com
………………………….
SYNTHESIS
(WO2006/051502A2, JMC2012, 7849). Chlorosulfonyl isocyanate ( 1 ) reaction with tert-butyl alcohol 2 , which is then reacted with n-propylamine 3 . 3 de-boc protected through the acid after reaction with potassium t-butoxide 4 . Another compound 5 with NaH after acidic protons off with dimethyl carbonate ( 6 ) to obtain 7 . 7 and formamidine hydrochloride ( 8 ) to ring chlorinated later POCl3 9 . 9 and 4 SNAr reaction occurs 10 . 10under basic conditions with ethylene glycol SNAr reaction occurs again in alkaline conditions with11 SNAr reaction occurs MACITENTAN.

………………………
http://www.google.com/patents/WO2014155304A1?cl=en
LC-MS (Agilent MS detector G1956B with Agilent 1200 Binary Pump and DAD).
Parameters of the LC-MS method:
Injection volume: 2 |jL
Column: Kinetex C18, 2.6 μιη, 2.1 x 50 mm
Column flow rate: 1 mL/min
Eluents: Eluent A: water + 0.08% TFA
Eluent B: MeCN + 0.012% TFA
Gradient: 2.0 min 95% B
2.8 min 95% B
3.0 min 5% B
Temperature: 40°C Detector wavelength 210 nm

Preparation B: N-[5-(4-bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]- 4-pyrimidinyl] -N’-propylsulfamide (macitentan):
N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)propane- 1-sulfamide (200 g; 0.46 mol; see Example 2 or 3) and 5-bromo-2-chloropyrimidine (117 g; 0.60 mol; 1.3 eq) were dissolved in toluene (3 L) and DMF (400 mL). The reaction mixture was warmed up to 50°C and toluene (approx. 400 mL) was distilled our under reduced pressure. The mixture was cooled to 0 °C and tBuOK (156 g, 3 eq, 1.38 mol) was added portionwise. It was stirred at 20 °C for 1 h. Water (1 L) was added and the pH of the solution was adjusted to 3-5 using 33% aq. HC1. The mixture was heated to 50°C and the layers were separated. The org. phase was treated with charcoal at 50°C and filtered over Celite. The filter cake was rinsed with toluene. At 50°C, water (1 L) was added to the org. layer. The layers were separated. The org. layer was concentrated under reduced pressure to a total volume of 1 L and cooled to 0°C. The solid obtained was filtered off. It was rinsed with toluene and MeOH. The crude material was suspended in EA (1 L) and heated to 50°C. 300 mL of EA were distilled out and MeOH (400 mL) was added. The suspension was cooled down to 0°C. The solid was filtered off, rinsed with MeOH and dried under reduced pressure to afford the title compound as a white solid (225 g; 83% yield).

……………………
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm3009103

Starting from the structure of bosentan (1), we embarked on a medicinal chemistry program aiming at the identification of novel potent dual endothelin receptor antagonists with high oral efficacy. This led to the discovery of a novel series of alkyl sulfamide substituted pyrimidines. Among these, compound 17 (macitentan, ACT-064992) emerged as particularly interesting as it is a potent inhibitor of ETA with significant affinity for the ETB receptor and shows excellent pharmacokinetic properties and high in vivo efficacy in hypertensive Dahl salt-sensitive rats. Compound 17 successfully completed a long-term phase III clinical trial for pulmonary arterial hypertension
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N′-propylsulfamide (17)
……………
WO 2015004265 click
Example 3 : N-(5-(4-bromophenyl)-6-(2-hydroxyethoxy)pyrimidin-4-yl)pr opane- 1- sulfamide (reaction in and work-up with MIBK):
EG (124 mL, 3.7 mol, 6.0 eq.) was added to a warm (40-50°C) suspension of the compound of Preparation A (150 g, 0.37 mol) in MIBK (600 mL). Solid KOtBu (114 g, 1.11 mol, 3.0 eq.) was added portionwise so that IT < 60°C. The mixture was stirred for
2- 3 h at 100-105°C. After completion of the reaction (LC-MS control), it was cooled to 50 °C. A 40%) aq. solution of citric acid monohydrate (300 mL) was added until pH 4 was reached. The layers were separated. The org. phase was washed with water (450 mL) and the layers were separated. Water (450 mL) was added and the mixture was warmed to 50°C. It was stirred at 50°C for 5 min. The layers were separated. The org. phase was concentrated under vacuum at 50°C until 200 mL of MIBK were removed. Hept (800 mL) was added dropwise at 70-75°C until turbidity was observed. The mixture was seeded with an analytically pure sample of N-(5-(4-bromophenyl)-6-(2 hydroxy ethoxy)pyrimidin-4-yl)propane-l-sulfamide and stirred at 60-65°C for 30 min. It was allowed to cool to 5°C within 5 h. It was filtered off, rinsed with a cold MIBK/Hept mixture (300 mL, 1 : 1) and dried under vacuum at 50°C to yield the title compound as a white solid (121 g; 76% yield).
The product had NMR data equivalent to those reported in Bolli et al, J. Med. Chem. (2012), 55, 7849-7861. [M+H]+ = 430 and 432. LC-MS: tR = 1.46 min; purity: 98.4% a/a. Residual ethylene glycol (GC-FID): 530 ppm.
…….
CN 104447572 click
(l) Martin H. Bolli et al. Reported the synthesis of Marcy cefotetan follows:

[0008] The method W 5- (4- desert phenyl) -4,6-dichloro-chewing clever as a starting material, N- propyl amine Lai ugly bell in DMS0 as a reaction solvent, an alcohol bell as t a base under substitution reaction conditions, the reaction temperature needs of 24-7 to give


The intermediate compound 15, compound 15 in hexylene glycol dimethyl off as the reaction solvent, a tertiary alcohol under conditions with a strong base clock as hexanediol substitution reaction, l〇 (TC Reaction of 18-2 to give compound 17, Compound 17 was then reacted with 5-chloro-chewing desert -2 clever substitution reaction at tetraammine Qiao Nan as a reaction solvent, ammoniated axis as the alkali conditions, the reaction to give the final product of Marcy cefotetan The route every step the higher the yield, the experimental use of N- propyl amine Lai ugly bell hygroscopic, unstable and a long time before the two-step reaction, the reaction at the second step requires l〇 (TC high temperature 18-2 technology is not suitable for industrial production.
[0009] International Patent W02002 / 053557 discloses some preparation methods and other Massey cefotetan column derivative method at each step of the preparation of the reaction times are longer, some reactions up to 4 days, and the resulting intermediate are purified by column chromatography method is not suitable for industrial production.



[00 pairs (3) N- [5- (4- desert) -6-mouth – [(5-desert -2- chew clever-yl) oxy] hexyl oxy] -4-chewing clever yl] -N ‘- Lai ugly propyl amine (Formula I) Synthesis
[0036] Weigh 20gN-5- (4- desert) -6- (2-2- light hexyl group -) 4- chew clever group -N ‘- Lai ugly propyl amine, 200ml dried DMS0 added to 1L H jar, add 20g of alcohol t-clock was added in portions, then add 17. 7g5- desert – dichloro chew clever, 30-4 (TC reduction reaction, the reaction and the reaction solution. a 10% sample skillfully acid to adjust PH value 3 to 4, the reaction mixture was added to 1000ml water, olive mix, suction. suction Massey cefotetan get wet crude product 42g, 450ml of methanol was added at room temperature and then beating 20min, filtration and dried 45C to give white solid was dried under vacuum to give 23.2 Marcy cefotetan yield;.. 85%
[0037] The compound (Formula I) relating to the physical and chemical properties, spectroscopic data are as follows:
[0038] branded point; 135-136 ° C; we NMR (300MHz, DMS0) 5 (egg m):… 9 8 (s, lH), 8 7 (s, 2H), 8 5 (s, l H,) 7. 5 (s, 2H), 7. 2 (s, IH), 7. 1 (s, 2H,) 4. 7 (s, 2H), 4. 6 (s, 2H,) 2. 8 (s, 2H,), 1. 5 (m, 2H,), 0. 81 (m, 3H), MS Qiaoqiao m / z 589 ([M + Tin +).
…………
see
WO 2002053557
http://www.google.com/patents/WO2002053557A1?cl=en
………..


Assignment of the signals mentioned in the text of the H-NMR spectrum of the drug Macitentan
Solvent: CDCl 3
δ 8.51 (s, 2H, CH) 11 , 8.49 (s, 1 H, CH) 10 , 7.58 to 7.63 (m, 2H, CH) 9 , 7.16 to 7.21 ( m, 2H, CH) 8 , 6.88 (s, 1H, NH) 7 , 5.61 (t, J = 6.2 Hz, 1H, NH) 6 , 4.72 to 4.76 (m, 2H , CH 2 ) 5 , 4.62 to 4.66 (m, 2H, CH 2 ) 4 , 2.99 (q, J = 6.8 Hz, 2H, CH 2 ) 3 , 1.61 (h, J = 7.3 Hz, 2H, CH2 ) 2 , 0.97 (t, J = 7.4 Hz, 3H, CH 3 ) 1 . [Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 .]

Solvent: CDCl 3
δ 11.6, 22.7, 46.1, 65.3, 65.9, 104.8, 112.4, 123.7, 128.0, 131.7, 133.0, 155.7, 156 , 4, 159.7, 163.5, 166.3. [ Journal of Medicinal Chemistry 55, 2012 S. 7849-7861, doi : 10.1021 / jm3009103 . ]
NMR PREDICT BY ME
1H NMR PREDICT

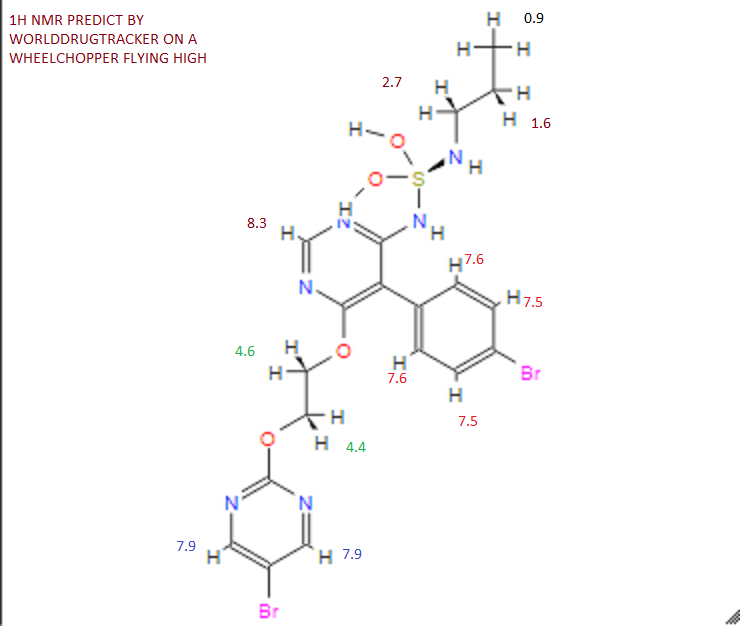
13C NMR PREDICT BY ME


COSY PREDICT BY ME, WORLDDRUGTRACKER ON A WHEELCHOPPER SCALING NEW HEIGHTS
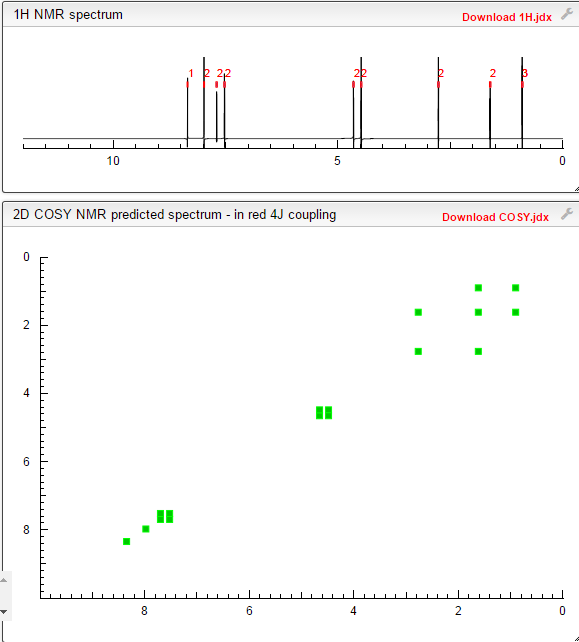
REFERENCES
Actelion Ltd
Actelion Ltd is a biopharmaceutical company with its corporate headquarters in Allschwil/Basel, Switzerland. Actelion’s first drug Tracleer®, an orally available dual endothelin receptor antagonist, has been approved as a therapy for pulmonary arterial hypertension. Actelion markets Tracleer through its own subsidiaries in key markets worldwide, including the United States (based in South San Francisco), the European Union, Japan, Canada, Australia and Switzerland. Actelion, founded in late 1997, is a leading player in innovative science related to the endothelium – the single layer of cells separating every blood vessel from the blood stream. Actelion’s over 2,400 employees focus on the discovery, development and marketing of innovative drugs for significant unmet medical needs. Actelion shares are traded on the SIX Swiss Exchange (ticker symbol: ATLN) as part of the Swiss blue-chip index SMI (Swiss Market Index SMI®).
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide
|
|
| Clinical data | |
| Trade names | Opsumit |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Metabolism | Hydrolysis, oxidation (CYP3A4) |
| Excretion | 2/3 urine, 1/3 faeces |
| Identifiers | |
| CAS Registry Number | 441798-33-0 |
| ATC code | C02KX04 |
| PubChem | CID: 16004692 |
| ChemSpider | 13134960 |
| ChEBI | CHEBI:76607 |
| Synonyms | ACT-064992 |
| Chemical data | |
| Formula | C19H20Br2N6O4S |
| Molecular mass | 588.273 g/mol |
| Patent | Submitted | Granted |
|---|---|---|
| Sulfamides and their use as endothelin receptor antagonists [US7094781] | 2004-04-22 | 2006-08-22 |
| Sulfamides and their use as endothelin receptor antagonists [US7285549] | 2006-08-10 | 2007-10-23 |
| Stable Pharmaceutical Compositions Comprising a Pyrimidine – Sulfamide [US2008233188] | 2008-09-25 | |
| Combination Comprising Paclitaxel for Treating Ovarian Cancer [US2010311774] | 2010-12-09 | |
| Stable pharmaceutical compositions comprising a pyrimidine-sulfamide [US2010004274] | 2010-01-07 | |
| SULFONYLUREA MODULATORS OF ENDOTHELIN RECEPTOR [US2011082151] | 2011-04-07 | |
| ENDOTHELIN RECEPTOR ANTAGONISTS FOR EARLY STAGE IDIOPATHIC PULMONARY FIBROSIS [US2010022568] | 2007-04-12 | 2010-01-28 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2011136818] | 2011-06-09 | |
| Therapeutic Compositions Comprising a Specific Endothelin Receptor Antagonist and a PDE5 Inhibitor [US2009318459] | 2009-12-24 |
Patent and Exclusivity
| Appl No | Prod No | Patent No | Patent Expiration |
Drug Substance Claim |
Drug Product Claim |
Patent Use Code |
|
|---|---|---|---|---|---|---|---|
| N204410 | 001 | US7094781 | Oct 12, 2022 | Y | Y | ||
| N204410 | 001 | US8268847 | Apr 18, 2029 | U – 1446 | |||
| N204410 | 001 | US8367685 | Oct 4, 2028 | Y | U – 1445 |
| Appl No | Prod No | Exclusivity Code | Exclusivity Expiration |
|---|---|---|---|
| N204410 | 001 | ODE | Oct 18, 2020 |
| N204410 | 001 | NCE | Oct 18, 2018 |
U1446 METHOD OF TREATING PULMONARY HYPERTENSION COMPRISING ADMINISTERING MACITENTAN IN COMBINATION WITH A COMPOUND HAVING PHOSPHODIESTERASE-5 INHIBITORY PROPERTIES
U1445 METHOD OF TREATING PULMONARY ARTERIAL HYPERTENSION BY ADMINISTERING A PHARMACEUTICAL COMPOSITION COMPRISING MACITENTAN AND A POLYSORBATE, WHERIN THE POLYSORBATE REPRESENTS 0.1 TO 1% OF THE WEIGHT OF SAID PHARMACEUTICAL COMPOSITION
OPSUMIT (macitentan) is an endothelin receptor antagonist. The chemical name of macitentan is N-[5-(4-Bromophenyl)-6-[2-[(5-bromo-2-pyrimidinyl)oxy]ethoxy]-4-pyrimidinyl]-N’-propylsulfamide. It has a molecular formula of C19H20Br2N6O4S and a molecular weight of 588.27. Macitentan is achiral and has the following structural formula:
 |
Macitentan is a crystalline powder that is insoluble in water. In the solid state macitentan is very stable, is not hygroscopic, and is not light sensitive.
OPSUMIT is available as a 10 mg film-coated tablet for once daily oral administration. The tablets include the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 80, povidone, and sodium starch glycolate Type A. The tablets are film-coated with a coating material containing polyvinyl alcohol, soya lecithin, talc, titanium dioxide, and xanthan gum.
//////

Amgen In Focus
Seeking Alpha
According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 …
http://seekingalpha.com/article/1510002-amgen-in-focus?source=google_news
Amgen has the second deepest pipeline of drugs of the three large cap biotechs. According to Amgen, they have 45 drugs in development from Phase 1 to Phase 3. Conversely, Gilead has 32 drugs in development and Pfizer has 64. Meanwhile, Gilead only has 8 drugs in Phase 3, Pfizer has 25, and Amgen has 14. 7 of those Phase 3 drugs are focused on cancer treatments for Amgen, more than either Pfizer or Gilead. Keep in mind that 12.4 million people learn they have cancer each year, while 7.6 million people lose that battle each year. The CDC predicts that the global number of cancer related deaths will increase by 80% by 2030. It doesn’t take a rocket scientist to know that cancer treating drugs presents the largest opportunity for any drug maker considering those statistics. Amgen has the inside track versus Gilead and Pfizer as far as quantity of drugs in late stage development.

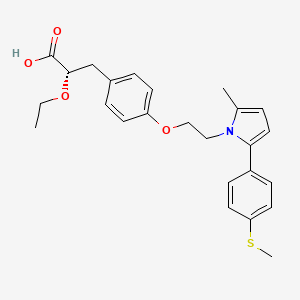
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
Saroglitazar, Lipaglyn
| Molecular Weight | 439.56706 g/mol |
|---|---|
| Molecular Formula | C25H29NO4S |


 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Cadila banks on diabetes drug
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet …http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

Mumbai, June 5: Cadila Healthcare will launch a homegrown drug against diabetes by the third quarter of this year.
The Drug Controller General of India has approved its drug — Lipaglyn — to treat “diabetic dyslipidemia”.
Diabetic dyslipidemia is a condition where a person is diabetic and has elevated levels of total cholesterol. Over 80 per cent of diabetic patients are dyslipidemic.
http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp
Zydus Cadila said it is looking for partnership to market its new chemical entity (NCE) Lipaglyn, to be used for treating a type of diabetes in developed and developing markets. “Lipaglyn is the first glitazar to be approved in the world and the first NCE discovered and developed indigenously by an Indian pharma company.
The new drug is expected to be launched in Q3 of this fiscal in the country,” Zydus Cadila Chairman and Manging Director Pankaj Patel told reporters.
The company has spent USD 250 million in developing Lipaglyn and aims to spend another USD 150-200 million to launch the drug in overseas markets in next 3-5 years period, Patel said, adding that the company is looking for marketing partnerships.
“We expect this to be a blockbuster drug, which means over USD 1 billion sales a year, when the drug is sold globally, he said. The market for this drug is estimated at Rs 100 crore in the local market over the next three years and having market potential size of over USD 30 billion in the world market, he said.
Zydus Cadila took about eight years to develop the molecule and conducted clinical trials on more than 1,000 patients in India, Patel said, adding that the company is yet to finalise the price, but believes that it will be reasonably priced in the local market.
The company said that the Indian drug regulator Drug Controller General of India (DCGI) has approved Lipaglyn to be used for treating ‘diabetic dyslipidemia’.
| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid | |
| Clinical data | |
| Trade names | Lipaglyn |
| Pregnancy cat. |
|
| Legal status |
|
| Routes | Oral |
| Identifiers | |
| CAS number | 495399-09-2 |
| ATC code | None |
| PubChem | CID 60151560 |
| ChemSpider | 32079086 |
| Chemical data | |
| Formula | C25H29NO4S |
| Mol. mass | 439.56 g/mol |
MORE DETAILS
Saroglitazar (INN, trade name Lipaglyn) is a drug for the treatment of type 2 diabetes mellitus and dyslipidemia. It is approved for use in India by the Drug Controller General of India.[1] Saroglitazar is indicated for the treatment of diabetic dyslipidemia andhypertriglyceridemia with type 2 diabetes mellitus not controlled by statin therapy. In clinical studies, saroglitazar has demonstrated reduction of triglycerides (TG), LDL cholesterol, VLDL cholesterol, non-HDL cholesterol and an increase in HDL cholesterol a characteristic hallmark of atherogenic diabetic dyslipidemia (ADD). It has also shown favorable glycemic control by reducing the fasting plasma glucose and HBA1c in diabetes patients.
Zydus-Cadila has developed and launched saroglitazar (ZYH-1; Lipaglyn; structure shown), a lipid metabolism modulator, a potent PPAR-alpha agonist with relatively weak PPAR-gamma activity, an insulin sensitizer (glucose-lowering agent), for the once-daily oral treatment of metabolic disorders, including diabetic dyslipidemia and hypertriglyceridemia
In June 2013, the Drug Controller General of India (DCGI) approved the drug for launch in India ; in September 2013, the drug was launched . The company is also developing the drug for the potential treatment of lipodystrophy. In May 2014, a phase III trial was initiated . In June 2012, the company was seeking to outlicense the drug for regional/global partnerships
By June 2012, an NDA filing had been made for dyslipidemia. In June 2013, the DCGI approved the drug for launch in India . By September 2013, the drug was launched for dyslipidemia and hypertriglyceridemia .
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]
_MoA.png)
The approval for saroglitazar was based on the results obtained from clinical studies, which were conducted for more than eight years.
The studies evaluated the efficacy, safety, pharmacokinetics and pharmacodynamics of the drug. Phase I clinical trials on saroglitazar were conducted in 2005. The highest dose of saroglitazar evaluated in a Phase I trial was 128 mg, several times the estimated therapeutic doses (1–4 mg). The pharmacokinetics of saroglitazar support a once daily dosage schedule. No serious adverse events were reported.[3] Phase II studies were completed in 2006.
The Phase III clinical trials were conducted between 2008 and 2011. The first Phase III clinical trials on saroglitazar compared saroglitazar 4 mg dose with pioglitazone 45 mg. The results of the study demonstrated that patients who were administered with saroglitazar 4 mg dose showed reduction in LDL cholesterol and triglycerides, and increase in HDL cholesterol. The study also showed that saroglitazar administered patients showed a reduction in fasting plasma glucose and glycosylated hemoglobin.
Saroglitazar 2 mg and 4 mg significantly reduced (P < 0.001) plasma triglycerides from baseline by 26.4% (absolute change ± SD: −78.2 ± 81.98 mg/dL) and 45% (absolute change ± SD −115.4 ± 68.11 mg/dL), respectively, as compared to pioglitazone -15.5% (absolute change ± SD: −33.3 ± 162.41 mg/dL) at week 24. Saroglitazar 4 mg treatment also demonstrated marked decrease in low-density lipoprotein (5%), very-low-density lipoprotein (45.5%), total cholesterol (7.7%), and apolipoprotein-B (10.9%).[4]
The second Phase III clinical trials on saroglitazar were conducted to evaluate the diabetic dyslipidemic patients insufficiently controlled with statin therapy. The second Phase III study results showed that patients treated with saroglitazar showed pronounced beneficial effect on both the lipid and glycaemic parameters.
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by -45.5±3.03% and -46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.[5]
Saroglitazar was found to be safe and well tolerated during the clinical program. In Phase III trials, There was no edema or weight gain reported in any of the study arms. During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no adverse events reported as far as cardiac safety is concerned.
After 12 weeks of treatment, there were a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, andγ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms.[6][7]
In Phase I clinical trials saroglitazar was used up to 128 mg and found well tolerated. No serious adverse events were reported. Adverse events were generally mild and moderate in nature and did not show any clinically relevant findings in clinical laboratory investigations, physical examinations, vital signs and electrocardiograms.[8]
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
http://www.sciencedirect.com/science/article/pii/S0040403914006200

PATENT
WO 2003009841
http://www.google.co.in/patents/WO2003009841A1?cl=en
PATENT
US 20030236254
http://www.google.com/patents/US20030236254
PATENT
US 20140099333
http://www.google.com/patents/US20140099333
PATENT
http://patentscope.wipo.int/search/en/WO2014174524
 (I)
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
PATENT
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.

Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.

wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1

In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)

wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2

Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).

Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3

Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
saroglitazar magnesium alongwith its intermediates may be prepared by the reaction scheme- 1, scheme-2 and scheme-3 as shown below, which is also the scope of the present invention.

Scheme-1

EXAMPLES
Example-l:
Preparation of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, sodium methoxide (165 g) and toluene (1000.0 ml) were added under nitrogen environment and cooled to 8°C to 12°C. Methyl acetoacetate (331.55 g) was added dropwise and stirred for 1 hour. 2-bromo-l-(4-methyl sulfonyl phenyl) ethanone (500.0 g) compound (El) in toluene (1500.0 ml) and sodium sulfate
(75.0 g) mixture was stirred for 10 min and filtered at 25° to 35°C. The filtrate as obtained was added dropwise into the previous reaction mixture and stirred at 30°C to 35°C for 30 min. The organic layer was collected and washed with 10% sodium bicarbonate solution. The separated organic layer was collected and washed with water. 2-[2-(4-Methyl sulfanyl-phenyl)-2-oxo-ethyl]-3-oxo-butynic acid methyl ester as obtained in toluene layer is diluted with methanol (2500 ml) and sodium hydroxide solution (89.75 g) in water (2500 ml) was added and heated to 50° to 55°C for 1 hour. The layers were separated and the toluene layer was collected and heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (57.3 g) and ethanol amine (143.9 g) were added and heated to 105° to 1 15°C for removing water azeotropically. The toluene layer was separated and triethyl amine (271.85 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (282.5 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum to obtain the residue. The residue was dissolved in toluene (1500 mL) and used for further process.
ExampIe-2:
Preparation of methanesulfonic acid 2-f2-methyl-5-(4-methylsulfanyl-pheny0-pyrrol- 1-viyethyl ester (Al)

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 4-(methylthio)benzaldehyde (10 g), methyl vinyl ketone (3.63 g), triethylamine (9.95 g) and 3-methyl-5-(2-hydroxyethyl)-4-methyI thiazolium iodide (stetter
catalyst) (2.8 g) were heated to 70°C to 80°C and maintained overnight. The reaction mixture was cooled to room temperature and ethanol (100 mL) was added. The reaction mixture was stirred for 30 min and filtered. The product was washed with ethanol and dried to obtain 1 ,4-diketo compound (CI).
1 ,4-diketo compound (CI) obtained above and toluene (50 mL) were heated to 45° to 55°C and charcoalized. The reaction mixture was filtered and pivalic acid (5.7 g) and ethanol amine (14.4 g) were added and heated to 105° to 1 15°C and cooled to 25°C. Triethyl amine (27.2 g) was added at 25° to 35°C and the reaction mixture was cooled to 10° to 20°C. Methane sulphonyl chloride (28.3 g) was added dropwise, and stirred for 2 hours and heated to 35° to 45°C. The reaction mixture was filtered and washed with toluene. Toluene was distilled out completely under the vacuum, methanol (2500 ml) was added and heated to 55° to 65 °C and charcoalized for 30 min. The reaction mixture was filtered and washed with methanol. The reaction mixture was cooled to 25° to 35°C and stirred for 30 min. Reaction mass was further cooled to -5° to 5°C and filtered. The wet-cake was washed with methanol and dried to obtain compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.2).
Example-3:
Purification of methanesulfonic acid 2-r2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al)
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 70 g methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l -yl]-ethyl ester (Al) and 420 mL ethyl acetate were added at 25°C. The reaction mixture was stirred for 30 min to obtain clear solution. 3.5 g charcoal was added and stirred for 30 min. The reaction mixture was filtered and washed with ethyl acetate. The filtrate was concentrated and 315 mL methanol was added. The reaction mixture was stirred for 2 hours at 25°C and cooled to 0°C. The product precipitated was filtered and washed with methanol to obtain crystalline
compound (Al). The compound (Al) was characterized as crystalline solid by x-ray powder diffraction (FIG.3).
Example-4:
Preparation of saroglitazar magnesium (T)

In a 5 Liter three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) (100.0 g) and toluene (1300.0 ml) were charged and reaction mixture was heated to 45° to 55°C. Potassium carbonate (58.0 g) was added and stirred for 30 min. Toluene solution of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol- 1 -yl]-ethyl ester (Al) (150.24 g) obtained in example- 1, 18-Crown-6 (5.0 g) and THF (200.0 ml) were added and heated to 75°C to 85°C for 36 hour, The reaction mixture was cooled to 25° to 35°C and water (1000.0 ml) was added and stirred for 15 min. The separated aqueous layer was treated with toluene (200.0 ml) and stirred for 15 min. The organic, layers were combined and washed with caustic solution (600.0 ml). The separated organic layer was washed with water (600.0 ml) and characoalized with HP-120 (5.0 g) charcoal and stirred for 30 min and filtered. The filtrate was added sodium hydroxide 20.14 g solution in water (200.0 ml) and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with n-butyl acetate. The separated aqueous layer was added magnesium acetate tetrahydrate solution (90.0 g) in water (100.0 ml) and stirred for 1 hour. The aqueous layer was extracted with methylene dichloride (2000 ml). The separated organic layer was washed with sodium chloride solution and charcoalized. The charcoalized solution was filtered and filtrate was distilled to remove toluene completely. The residue was diluted with toluene (1000 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
The reaction of methanesulfonic acid 2-[2-methyl-5-(4-methylsulfanyl-phenyl)-pyrrol-l-yl]-ethyl ester (Al) and 2-ethoxy-3-(4-hydroxy-phenyl)-propionic acid ethyl ester (A) may also be performed in similar manner as above in absence of phase transfer catalyst 18-Crown-6.
ExampIe-5:
Preparation of saroglitazar (S)-(-)-phenyl ethylamine salt:

In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, residue-A obtained in example- 1 and ethanol (400 mL) were stirred for 15 min. Sodium hydroxide 20.14 g solution in water (200.0 ml) was added and the reaction mixture was stirred for 3 hours. The reaction mixture was diluted with water (1800.0 ml) and stirred for 15 min. The separated aqueous layer was washed with isopropyl acetate (400 mL). The separated aqueous layer was diluted with isopropyl acetate (500 mL) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with (S)-(-)-phenyl ethylamine (55.94 g) and stirred for 2 hours at 25°C and 30 min at 45°C. The reaction mixture was cooled to 0°C and stirred for 2 hours, filtered and washed with isopropyl acetate. The wet-cake was dried to obtain saroglitazar phenyl ethylamine salt.
ExampIe-6:
Preparation of saroglitazar magnesium from saroglitazar (SH-)-phenyl ethylamine salt:
In a 250 mL three necked round bottom flask equipped with nitrogen atmosphere facility, mechanical stirrer, thermometer and an addition funnel, saroglitazar phenyl ethylamine wet-cake obtained in example-7 and isopropyl acetate (800 mL) were added at 25°C. The reaction mixture was diluted with water (400.0 ml) and acidified with cone. HCI at adjust the pH 2-3. The separated aqueous layer was washed with isopropyl acetate. The combined organic layer was treated with sodium hydroxide solution (20.14 g) in water (200 mL) and stirred for 30 min. The separated aqueous layer was treated with magnesium acetate tetrahydrate (2.29 g) in water (5 mL) solution and stirred for 60 min. The reaction mixture was extracted with methylene dichloride (800 mL). The methylene dichloride was complete removed by distillation under vacuum below 40°C to obtain the residue. The residue was diluted with methylene dichloride (50 ml) and stirred for 30 min. The organic solution was added into n-heptane (1500 mL) and stirred for 3 hours. The product was filtered and washed with n-heptane and dried in vacuum tray dryer at 25°C to 30°C for 3 hours. The product was sieved through 0.5 mm sieve and milled through jet-milled. The product was further dried in vacuum tray drier at 40°C to 50°C for 6 hours followed by drying at 55°C to 65°C for 40 hours to obtain substantially amorphous saroglitazar magnesium (I). The compound is characterized by x-ray power diffraction (FIG.l).
by WORLD DRUG TRACKER
DR ANTHONY
do not miss out on updates
see my update at https://newdrugapprovals.org/2015/03/09/saroglitazar-magnesium-new-patent-wo-2015029066-cadila-healthcare-ltd/ 9 may 2015
////////////
CCO[C@@H](Cc1ccc(cc1)OCCn2c(ccc2c3ccc(cc3)SC)C)C(=O)O

CAS#: 1201902-80-8
Synonym: Ixazomib; MLN-9708.
IUPAC/Chemical name:
4-(carboxymethyl)-2-((R)-1-(2-(2,5-dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-1,3,2-dioxaborinane-4-carboxylic acid
UPDATES AT THE BOTTOM OF PAGE
CAMBRIDGE, Mass., May 23, 2013 – Takeda Pharmaceutical Company Limited (TSE:4502) today announced the initiation of an international phase 3 clinical trial evaluating once a week MLN9708 in combination with lenalidomide and dexamethasone in patients with newly diagnosed multiple myeloma who are not candidates for transplant. The multi-center study with MLN9708, an investigational, oral proteasome inhibitor, will be conducted in Europe and North America.———————-READ MORE AT
http://www.pharmalive.com/takeda-begins-phase-iii-trial-of-multiple-myeloma-drug
Description of Ixazomib: ixazomib is an orally bioavailable second generation proteasome inhibitor (PI) with potential antineoplastic activity. Ixazomib inhibits the activity of the proteasome, blocking the targeted proteolysis normally performed by the proteasome, which results in an accumulation of unwanted or misfolded proteins; disruption of various cell signaling pathways may follow, resulting in the induction of apoptosis. Compared to first generation PIs, second generation PIs may have an improved pharmacokinetic profile with increased potency and less toxicity. Proteasomes are large protease complexes that degrade unneeded or damaged proteins that have been ubiquinated
MLN9708 is an investigational proteasome inhibitor that, compared with bortezomib, has improved pharmacokinetics, pharmacodynamics, and antitumor activity in preclinical studies. MLN9708 rapidly hydrolyzes to MLN2238, the biologically active form. MLN9708 has a shorter proteasome dissociation half-life and improved pharmacokinetics, pharmacodynamics, and antitumor activity compared with bortezomib.MLN9708 has a larger blood volume distribution at steady state, and analysis of 20S proteasome inhibition and markers of the unfolded protein response confirmed that MLN9708 has greater pharmacodynamic effects in tissues than bortezomib. MLN9708 showed activity in both solid tumor and hematologic preclinical xenograft models, and we found a correlation between greater pharmacodynamic responses and improved antitumor activity. Moreover, antitumor activity was shown via multiple dosing routes, including oral gavage. Taken together, these data support the clinical development of MLN9708 for both hematologic and solid tumor indications. (source: Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16.).
| References |
1: Mullard A. Next-generation proteasome blockers promise safer cancer therapy. Nat Med. 2012 Jan 6;18(1):7. doi: 10.1038/nm0112-7a. PubMed PMID: 22227650.
2: Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol. 2012 Feb 1;30(4):445-52. Epub 2012 Jan 3. PubMed PMID: 22215754.
3: Appel A. Drugs: More shots on target. Nature. 2011 Dec 14;480(7377):S40-2. doi: 10.1038/480S40a. PubMed PMID: 22169800.
4: Lee EC, Fitzgerald M, Bannerman B, Donelan J, Bano K, Terkelsen J, Bradley DP, Subakan O, Silva MD, Liu R, Pickard M, Li Z, Tayber O, Li P, Hales P, Carsillo M, Neppalli VT, Berger AJ, Kupperman E, Manfredi M, Bolen JB, Van Ness B, Janz S. Antitumor activity of the investigational proteasome inhibitor MLN9708 in mouse models of B-cell and plasma cell malignancies. Clin Cancer Res. 2011 Dec 1;17(23):7313-23. Epub 2011 Sep 8. PubMed PMID: 21903769.
5: Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N, Richardson P, Anderson KC. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011 Aug 15;17(16):5311-21. doi: 10.1158/1078-0432.CCR-11-0476. Epub 2011 Jun 30. PubMed PMID: 21724551; PubMed Central PMCID: PMC3156932.
6: Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, Bolen J. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010 Mar 1;70(5):1970-80. Epub 2010 Feb 16. Erratum in: Cancer Res. 2010 May 1;70(9):3853. Hales, Paul [added]. PubMed PMID: 20160034.
7: Dick LR, Fleming PE. Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov Today. 2010 Mar;15(5-6):243-9. Epub 2010 Jan 29. Review. PubMed PMID: 20116451.8: Marblestone JG. Ubiquitin Drug Discovery & Diagnostics 2009 – First Annual Conference. IDrugs. 2009 Dec;12(12):750-3. PubMed PMID: 19943215.

http://www.cancernetwork.com/conference-reports/ash2012/content/article/10165/2119611
Nasopharyngeal cancer is a sub-type of head and neck cancer that arises from the epithelial cells that cover the surface and line the nasopharynx. The incidence of nasopharyngeal cancer has been reported at approximately 0.5 to 2 new cases per year per 100,000 in Europe and the USA. Rottey et ah, Curr. Opin. Oncol., 23(3): 254-258 (201 1). There are three subtypes of nasopharyngeal cancer recognized in the World Health Organization (WHO) classification: (i) Type 1 – squamous cell carcinoma, typically found in the older adult population; (ii) Type 2 non-keratinizing carcinoma; and (iii) Type 3 – undifferentiated carcinoma. Treatment for nasopharyngeal cancer often involves radiotherapy and/or chemotherapy. There remains a continuing need for new and improved treatments for patients with nasopharyngeal cancer. There remains a further need to identify nasopharyngeal patients most likely to benefit from treatment with a proteasome inhibitor.
Proteasome inhibition represents an important new strategy in cancer treatment. King et al. , Science 274: 1652-1659 ( 1996), describes an essential role for the ubiquitin-proteasome pathway in regulating cell cycle, neoplastic growth and metastasis. The authors teach that a number of key regulatory proteins, including cyclins, and the cyclin-dependent kinases p21 and p27K,P ! , are temporally degraded during the cell cycle by the ubiquitin-proteasome pathway. The ordered degradation of these proteins is required for the cell to progress through the cell cycle and to undergo mitosis.
The proteasome inhibitor VELCADE© (bortezomib; N-2-pyrazinecarbonyl-L -phenylalanine -L- leucineboronic acid) is the first proteasome inhibitor to achieve regulatory approval. Mitsiades et ai, Current Drug Targets, 7: 1341 (2006), reviews the clinical studies leading to the approval of bortezomib for the treatment of multiple myeloma patients who have received at least one prior therapy. Fisher et ai , J. Clin. Oncol, 30:4867, describes an international multi-center Phase II study confirming the activity of bortezomib in patients with relapsed or refractory mantle cell lymphoma. Ishii et al, Anti-Cancer Agents in Medicinal Chemistry, 7:359 (2007), and Roccaro et al., Curr. Pharm. Biotech., 7: 1341 (2006), discuss a number of molecular mechanisms that may contribute to the antitumor activities of bortezomib. The proteasome inhibitor MLN9708 [2,2′-{2-[(lR)- l -( {[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3- methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid] is currently undergoing clinical evaluation for hematological and solid cancers. MLN9708 is a citrate ester which rapidly hydrolyzes to the active form [(lR)-l -({[(2,5-dichlorobenzoyl)amino]acetyl}amino)-3-methylbutyl]boronic acid (MLN2238) on exposure to aqueous solution or plasma. MLN9708 has demonstrated anti-tumor activity in a range of hematological and solid tumor xenograft models (Kupperman et al. (2010) Cancer Res. 70: 1970- 1980),
Summary
The invention relates to the discovery that patients with nasopharyngeal cancer respond to treatment with MLN9708. In one aspect, the invention relates to the discovery of the increased expression of Nuclear Factor Kappa-B RelA 65,000 dalton subunit (NFKB p65) in biological samples comprising cells obtained from patients with nasopharyngeal cancer and responsive to MLN9708.
Accordingly, the invention features treating nasopharyngeal cancer patients withMLN9708 if a sample from the patient demonstrates an elevated expression of NFKB p65.
PATENT

or a pharmaceutically acceptable salt or a pharmaceutical composition or a boronic acid anhydride thereof.
[048| The compound of formula (II), [( l R)-l -( } [(2,5-dichlorobenzoyl)amino]acetyl} amino)-3- methylbutyljboronic acid (MLN2238) is disclosed in Olhava and Danca, U .S. Patent No. 7,442,830, herein incorporated by reference in its entirety. [049] In some other embodiments, Z and Z together form a moiety derived from a compound having at least two hydroxyl groups separated by at least two connecting atoms in a chain or ring, said chain or ring comprising carbon atoms and, optionally, a heteroatom or heteroatoms which can be N, S, or O, wherein the atom attached to boron in each case is an oxygen atom.
In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A) or (III-B):

(III-B), or a mixture thereof or a pharmaceutical composition thereof.
[054] In certain embodiments, wherein the alpha-hydroxy carboxylic acid or beta-hydroxy carboxylic acid is citric acid, the compound of formula (I) is characterized by formula (III-A):

or a pharmaceutical composition thereof.
[055] The compound of formula (III-A), 2,2′- {2-[( l i?)- l -( { [(2,5-dichlorobenzoyl)amino]acetyl } amino)- 3-methylbutyl]-5-oxo- l ,3,2-dioxaborolane-4,4-diyl} diacetic acid (MLN9708) is disclosed in Elliott et al. , WO 09/ 154737, herein incorporated by reference in its entirety
PATENT
http://www.google.com/patents/WO2009154737A1?cl=en
Example 1: Synthesis of 4-(/?,S)-(carboxymethyl)-2-( (R)-I -(2-(2,5- dichlorobenzamido)acetamido)-3-methylbutyl)-6-oxo-l,3,2-dioxaborinane-4- carboxylic acid (1-1)

Step l: 2,5-r(dichlorobenzoyI)aminolacetic acid
[0310] To a mixture of NaOH (12 g, 300 mmol) and glycine (18 g, 239 mmol) in water (120 mL) was added dropwise over 45 min a solution of 2,5-dichlorobenzoyl chloride (10 g, 48 mmol) in THF (15 mL) keeping the internal temperature below about 25 0C. After 1 h, the mixture was acidified with 2.0 M HCl (125 mL) keeping the internal temperature below about 5 0C. The resulting precipitate was collected by vacuum filtration. The crude product was recrystallized from water to give 2,5-[(dichlorobenzoyl)amino]acetic acid as a white, crystalline solid (6.1 g, 52%). mp 173.3 0C. 1H NMR (300 MHz, DMSOd6, δ): 12.72 (bs, IH), 8.89 (t, J = 6.0 Hz, IH), 7.54 (m, 2H), 7.48 (m, IH), 3.93 (d, J = 6.0 Hz). 13C NMR (75 MHz, DMSO-Ci6, δ): 41.6, 129.3, 129.6, 131.4, 132.2, 138.2, 171.4, 165.9. MS (ni/z): [M+H] calculated for C9H8Cl2NO3, 248.0; found, 248.0; [M+Na] calculated for C9H7Cl2NNaO3, 270.0; found 270.2.
2,5-[(dichlorobenzoyl)amino]acetic acid was also be prepared via the following procedure: To a mixture of glycine (21.5 g, 286 mmol) in water (437 mL), was added 2.0 M NaOH (130 mL) and the resulting solution was cooled to 0 0C. A solution of 2,5-dichlorobenzoyl chloride (50.0 g, 239 mmol) in THF (75 mL) was added dropwise at such a rate that the internal temperature was maintained at 0 ± 1 0C. During the addition, the pH was controlled at 11.0 ± 0.2 using a pH controller titrated with 2.0 M NaOH. After complete addition, the mixture was stirred at 0 ± 1 0C for an additional 2 h. The mixture was then acidified with 2.0 M HCl (176 mL) to a final pH of 2.5. The resulting precipitate was collected by filtration, washed with cold water (125 mL), and dried at 45 0C in a vacuum oven to afford 2,5-[(dichlorobenzoyl)amino]acetic acid as a white solid (57.6 g, 97.3%). Step 2: 2,5-dichloro-N-f2-(( (lR’)-3-niethyl-l-r(3aS,4S.6S.7aR)-3a,5,5-trimethylhexahvdro-
4,6-methano-l,3,2-benzodioxaborol-2-yllbutyl }amino)-2-oxoethvπbenzamide
To a solution of 2,5-[(dichlorobenzoyl)amino]acetic acid (6.10 g, 24.6 mmol) and TBTU (8.34 g, 26.0 mmol) in DMF (40 mL) with an internal temperature below about 5 0C was added (IR)- 3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5-trimethylhexahydro-4,6-methano-l,3,2-benodioxaborol-2- yl]butan-l-amine»TFA (9.35 g, 24.7 mmol). DIPEA (13 mL, 75 mmol) was then added dropwise over 2 h keeping the internal temperature below about 5 0C. After 40 min, the mixture was diluted with EtOAc (90 mL), washed with 5% NaCl (150 mL), twice with 10% NaCl (2 x 40 mL), once with 2% K2CO3 (1 x 40 mL), once with 1% H3PO4 (1 x 40 mL), and once with 10% NaCl (1 x 40 mL). The resulting organic layer was concentrated to a thick oil, diluted with heptane (40 mL) and evaporated to yield 2,5-dichloro-N-[2-({ (lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l ,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide as a white solid which was used in the next step without purification.
Step 3: N,N\N’Wboroxin-2A6-triyltrisir(lR)-3-methylbutane-l J-diyllimino(2-oxoethane- 2,l-diyl)^ ^tris(2,5-dichlorobenzamide)
To a solution of 2,5-dichloro-N-[2-({(lR)-3-methyl-l-[(3aS,4S,6S,7aR)-3a,5,5- trimethylhexahydro-4,6-methano-l,3,2-benzodioxaborol-2-yl]butyl }amino)-2-oxoethyl]benzamide (12.2 g, 24.6 mmol) in methanol/hexane (1 :1) (250 mL) were added IN HCl (30 mL, 30 mmol) and (2-methylpropyl)boronic acid (6.5 g, 64 mmol). The reaction mixture was allowed to stir overnight. The phases were separated and the methanol layer was washed twice with additional heptane (2 x 55 mL). The resulting organic layer was concentrated to about 10 mL and partitioned between 2.0M NaOH (30 mL) and DCM (25 mL). The DCM layer was washed once with additional 2.0M NaOH (5 mL). The basic aqueous layers were then combined, washed twice with DCM (2 x 25 mL) and acidified with IM HCl (60 mL). The resulting mixture was diluted with DCM (40 mL), the layers were separated, and the resulting aqueous layer was washed three times with DCM (3 x 10 mL). The combined DCM extracts were dried over MgSO4 (25 g) and evaporated to a thick oil. The product was precipitated with heptane (50 mL) and collected by filtration to yield N,N’,N”-{boroxin-2,4,6- -riyltris[[(lR)-3-methylbutane-l,l-diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) as a white solid (6.6 g, 74%). 1H NMR (300 MHz, DMSO-Cl6, δ): 8.93 (t, J – 6.0 Hz, IH), 8.68 (bs, IH), 7.63 (m, IH), 7.52 (m, 2H), 4.00 (d, J = 6.0 Hz, 2H), 2.62 (m, IH), 1.59 (m, IH), 1.33 (m, IH), 1.24 (m, IH), 0.81 (d, / = 5.9 Hz, 6H). 13C NMR (125 MHz, DMSO-Cl6, δ): 23.2, 25.8, 40.1, 40.7, 43.0, 129.0, 130.0, 131.0, 137.5, 165.0, 172.5. MS (m/z) in CH3CN: [M+H] calculated for C42H52B3Cl6N6O9, 1027.2; found, 1027.3; [M+Na] calculated for C42H51B3Cl6N6NaO9, 1049.2; found 1049.5.
Step 4: 4-(/?.S)-(carboxymethyl)-2-((/?)-l-(2-(2,5-dichlorobenzamido)acetamido)-3- methylbutyl)-6-oxo-l,3,2-dioxaborinane-4-carboxylic acid (1-1)
Form 1: To a solution of citric acid (2.75 g, 14.3 mmol) in EtOAc (85 mL) with an internal temperature of about 74 0C was added N,N’,N”-{boroxin-2,4,6-triyltris[[(lR)-3-methylbutane-l,l- diyl]imino(2-oxoethane-2,l-diyl)] }tris(2,5-dichlorobenzamide) (5.00 g, 4.87 mmol) as a solid. The solution was cooled uncontrolled until the internal temperature was about 25 0C and the mixture was stirred overnight. The resulting precipitate was collected by filtration to yield 2,2′-{2-[(lR)-l-({ [(2,5- dichlorobenzoyl)amino]acetyl }amino)-3-methylbutyl]-5-oxo-l,3,2-dioxaborolane-4,4-diyl}diacetic acid Form 1 as a crystalline solid (6.65 g, 88 %). 1H NMR (500 MHz, DMSOd6, δ 110 0C): 10.08 (s, IH), 8.69 (s, IH), 7.61 (s, IH), 7.52 (d, J = 1.3 Hz, 2H), 4.26 (d, J = 5.5 Hz, 2H), 2.70 (q, J = 14.5 Hz, 4H), 2.70 (bs, IH), 1.72 (sept, J – 6.5 Hz, IH), 1.42 (ddd, J = 5.2 Hz, J = 8.6 Hz, J = 13.9 Hz, IH), 1.28 (ddd, J = 5.3, J = 9.4 Hz, J = 14.3 Hz, IH), 0.91 (dd, J = 3.3 Hz, J = 6.6 Hz, 6H). MS (m/z) in CH3CN: [M+Na] calculated for C20H23BCl2N2NaO9, 539.1; found, 539.1.
Ixazomib citrate [USAN]
1,3,2-Dioxaborolane-4,4-diacetic acid, 2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo- [ACD/Index Name]
1,3,2-Dioxaborolane-4,4-diacetic acid,2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-
1239908-20-3 [RN]
2,2′-{2-[(1R)-1-{[N-(2,5-Dichlorbenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolan-4,4-diyl}diessigsäure [German] [ACD/IUPAC Name]
2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacetic acid [ACD/IUPAC Name]
2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diacetic acid
2-[4-(carboxymethyl)-2-[(1R)-1-[[2-[(2,5-dichlorobenzoyl)amino]acetyl]amino]-3-methyl-butyl]-5-oxo-1,3,2-dioxaborolan-4-yl]acetic acid
Acide 2,2′-{2-[(1R)-1-{[N-(2,5-dichlorobenzoyl)glycyl]amino}-3-méthylbutyl]-5-oxo-1,3,2-dioxaborolane-4,4-diyl}diacétique [French] [ACD/IUPAC Name]
MLN9708
Ixazomib (trade name Ninlaro) is a drug for the treatment of multiple myeloma, developed by Takeda Pharma. It acts as aproteasome inhibitor and has orphan drug status in the US. In November 2015, the U.S. Food and Drug Administration approved ixazomib for use in combination with lenalidomide and dexamethasone for the treatment of multiple myeloma after at least one prior therapy.[2]
Ixazomib is a peptide analogue that reversibly inhibits the protein proteasome subunit beta type-5 (PSMB5), which is part of the 20Sproteasome complex.[3]

Ixazomib citrate—a prodrug for ixazomib
NINLARO Provides a New Option for Patients Living with Multiple Myeloma Who Have Received at Least One Prior Therapy
Cambridge, Mass. and Osaka, Japan, November 20, 2015 – Takeda Pharmaceutical Company Limited (TSE: 4502) today announced that the U.S. Food and Drug Administration (FDA) has approved NINLARO®(ixazomib) capsules, the first and only oral proteasome inhibitor, indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is a once-weekly pill. More information is available at www.NINLARO.com.
Takeda submitted a New Drug Application for NINLARO to the FDA in July 2015, and in September NINLARO was granted Priority Review status with a PDUFA date of March 10, 2016, reflecting the profound and continuing unmet need for new treatments for multiple myeloma, a devastating, relapsing and incurable rare cancer.
“With the approval of NINLARO, we can now offer patients a once-weekly oral proteasome inhibitor as part of a highly active triplet therapy,” said Paul Richardson, M.D., Clinical Program Leader and Director of Clinical Research, Jerome Lipper Multiple Myeloma Center Institute Physician at Dana-Farber Cancer Institute, and investigator for TOURMALINE-MM1, the pivotal Phase 3 trial on which today’s approval is based. “We, as investigators of the TOURMALINE-MM1 trial, felt it was vital to conduct a comprehensive ‘real world’ evaluation of this combination that included some of the most common patient types in the relapsed/refractory multiple myeloma setting, such as older patients, patients with moderate renal impairment, light chain disease, and high risk cytogenetics. Further, we treated patients until disease progression to determine the sustainability of NINLARO in treating their relapsed/refractory disease. The TOURMALINE-MM1 data demonstrate convincingly that oral NINLARO-based triplet treatment is effective at extending progression-free survival, over and above the clinical benefit seen with lenalidomide and dexamethasone, with a tolerable safety profile.”
“We introduced the first proteasome inhibitor for multiple myeloma, VELCADE, into clinical research approximately 20 years ago. Since that time, we’ve significantly advanced scientific understanding of this rare cancer, culminating in the introduction of NINLARO,” said Andy Plump, M.D., Ph.D, Takeda Chief Medical and Scientific Officer. “NINLARO is an entirely new molecule that offers the efficacy of this proteasome inhibitor in a convenient once-weekly pill with a tolerable safety profile. Takeda is delighted to bring this significant innovation to multiple myeloma patients today, and we continue to examine the potential of NINLARO through a robust clinical development program.”
Dr. Brian Durie, Chairman of the International Myeloma Foundation, said, “The IMF is pleased by the approval of ixazomib. This opens the door for a fully oral proteasome inhibitor-based triplet combination therapy. Having worked in multiple myeloma for decades, I’ve seen notable progress, yet significant unmet needs remain. With today’s approval, we now have another attractive option for many patients living with multiple myeloma.”
The FDA approval of NINLARO is based on results from the TOURMALINE-MM1 Phase 3 clinical trial, the first double-blind, placebo-controlled trial with a proteasome inhibitor. TOURMALINE-MM1 is the first of five ongoing Phase 3 clinical trials with study results available. The TOURMALINE program has enrolled approximately 3,000 patients to date in 40 countries. Data from the NINLARO Phase 3 TOURMALINE-MM1 pivotal trial will be presented at the upcoming 57th Annual Meeting of the American Society of Hematology on December 7, 2015.
“The approval of ixazomib offers a much-needed additional option in the multiple myeloma treatment landscape. It is developments such as these that help us to better understand the disease and provide continued hope for patients,” said Kathy Giusti, Founder and Executive Chairman of the Multiple Myeloma Research Foundation (MMRF). “A cancer diagnosis today is different from what it was just a few years ago and it’s exciting to see continued progress. As a patient, I understand the urgent need for advancing research through partnerships that bring new treatment options, as we’ve done with Takeda.”
“NINLARO is a first-of-its-kind innovation that is supported by a global development program, unprecedented for us at Takeda Oncology, and we would like to express our immense appreciation for all patients involved for their incredible strength and invaluable participation. The introduction of NINLARO marks an important step forward, as its efficacy and safety profile – coupled with its completely oral administration – potentially can reduce some logistical burdens, and help enable patients to reap the full benefits of this sustainable therapy,” explained Christophe Bianchi, M.D., President, Takeda Oncology. “As part of our unwavering 20-year commitment, Takeda will continue to pursue advances for these patients, and we look forward to introducing and expanding access to NINLARO in other markets around the world.”
About the TOURMALINE-MM1 Trial
TOURMALINE-MM1 is an international, randomized, double-blind, placebo-controlled clinical trial of 722 patients, designed to evaluate NINLARO plus lenalidomide and dexamethasone compared to placebo plus lenalidomide and dexamethasone in adult patients with relapsed and/or refractory multiple myeloma. Results showed NINLARO is effective in extending Progression Free Survival (PFS) and has a manageable safety profile. The trial achieved its primary endpoint and demonstrated a clinically meaningful and statistically significant prolongation in PFS at this analysis, which showed that patients treated in the NINLARO arm lived without their disease worsening for a significantly longer time compared to patients in the control arm. Patients continue to be treated to progression in this trial and will be evaluated for long term outcomes.
In the TOURMALINE-MM1 trial, the most common adverse reactions (≥20%) in patients receiving NINLARO included diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting and back pain. Serious adverse reactions reported in ≥2% patients included thrombocytopenia (2%) and diarrhea (2%).
Efficacy and safety data were reviewed by an Independent Data Monitoring Committee (IDMC), who recommended the study be continued in blinded fashion to allow further maturation of long term outcomes, including overall survival (OS) and long-term safety.
About NINLARO (ixazomib) capsules
NINLARO (ixazomib) is the first and only oral proteasome inhibitor indicated in combination with lenalidomide and dexamethasone for the treatment of patients with multiple myeloma who have received at least one prior therapy. NINLARO is administered orally, once-weekly on days 1, 8, and 15 of a 28-day treatment cycle. NINLARO is currently under review by the European Medicines Agency (EMA) and was granted an accelerated assessment by the Committee for Medicinal Products for Human Use (CHMP). NINLARO also received Breakthrough Therapy status by the U.S. FDA for relapsed or refractory systemic light-chain (AL) amyloidosis, a related ultra orphan disease, in 2014.
The TOURMALINE clinical development program further reinforces Takeda’s ongoing commitment to developing innovative therapies for people living with multiple myeloma worldwide and the healthcare professionals who treat them. Five global Phase 3 trials are ongoing:
In addition to the TOURMALINE program, a large number of investigator initiated studies are evaluating ixazomib for patients globally.
For additional information on the ongoing Phase 3 studies please visit www.clinicaltrials.gov. To learn more about NINLARO, please visit www.NINLARO.com or call 1-844-N1POINT (1-844-617-6468).
![]()
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
N2-(2,5-Dichlorobenzoyl)-N-[(1R)-1-(dihydroxyboryl)-3-methylbutyl]glycinamide
|
|
| Clinical data | |
| Trade names | Ninlaro |
| AHFS/Drugs.com | entry |
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 58%[1] |
| Protein binding | 99% |
| Metabolism | hepatic, CYP3A4 (42%),CYP1A2 (26%) and others |
| Biological half-life | 9.5 days |
| Excretion | urine (62%), feces (22%) |
| Identifiers | |
| CAS Number | 1072833-77-2 |
| ATC code | L01XX50 |
| PubChem | CID 25183872 |
| ChemSpider | 25027391 |
| UNII | 71050168A2 |
| KEGG | D10130 |
| ChEBI | CHEBI:90942 |
| Synonyms | MLN2238 |
| Chemical data | |
| Formula | C14H19BCl2N2O4 |
| Molar mass | 361.03 g·mol−1 |

//////////
see….http://apisynthesisint.blogspot.in/2016/02/takedas-ixazomib-multiple-myeloma-drug.html

Simeprevir
03/28/2013| Medivir AB announced that a new drug application (NDA) has been filed with the U.S. Food and Drug Administration (FDA) seeking approval for simeprevir. The filing is based on phase III data in treatment-naïve and treatment-experienced patients with compensated liver disease.
The filing of a regulatory application in the US triggers a milestone payment of ?10m to Medivir.
Simeprevir is jointly developed by Medivir and Janssen Pharmaceuticals, Inc. (Janssen), and is an investigational NS3/4A protease inhibitor, administered as a 150 mg capsule once daily with pegylated interferon and ribavirin for the treatment of genotype 1 chronic hepatitis C in adult patients.
“The filing in the U.S. is a very important milestone for simeprevir, the hepatitis C patients and for Medivir as a company. In addition it triggers a ? 10m milestone payment to us, which strengthens our solid financial situation even more.” comments Maris Hartmanis, CEO of Medivir.
The regulatory submission for simeprevir is supported in part by data from three pivotal phase III studies: QUEST-1 and QUEST-2 in treatment-naïve patients and PROMISE in patients who have relapsed after prior interferon-based treatment. In each study, participants were treated with one 150 mg simeprevir capsule once daily for 12 weeks plus pegylated interferon and ribavirin for 24 or 48 weeks. Primary efficacy data from the phase III studies will be presented at different upcoming medical meetings.
About Simeprevir
Simeprevir, an investigational next generation NS3/4A protease inhibitor jointly developed by Janssen R&D Ireland and Medivir AB, is currently in late phase III studies as a once-daily capsule (150 mg) taken in combination with pegylated interferon and ribavirin for the treatment of genotypes 1 and 4 HCV.
Global phase III studies of simeprevir include QUEST-1 and QUEST-2 in treatment-naïve patients, PROMISE in patients who have relapsed after prior interferon-based treatment and ATTAIN in null-responder patients. In parallel to these trials, phase III studies for simeprevir are ongoing in treatment-naïve and treatment-experienced HIV-HCV co-infected patients, HCV genotype 4 patients and Japanese HCV genotype 1 patients. Janssen recently announced the submission of a new drug application for simeprevir in Japan for the treatment of genotype 1 hepatitis C.
Simeprevir is being studied in phase II interferon-free trials with and without ribavirin in combination with:
In addition, Janssen has a non-exclusive collaboration with Vertex Pharmaceuticals to evaluate in a phase II study the safety and efficacy of an all-oral regimen of simeprevir and Vertex’s investigational nucleotide analogue polymerase inhibitor VX-135 for the treatment of HCV. As a first step, Janssen Pharmaceutical Inc. will conduct a drug-drug interaction (DDI) study with simeprevir and VX-135.
We also recently announced plans to initiate a phase II trial of an investigational interferon-free regimen with simeprevir, TMC647055 and Idenix’s IDX719, a once-daily, pan-genotypic NS5A inhibitor, with and without ribavirin.
About Hepatitis C
Hepatitis C, a blood-borne infectious disease of the liver and a leading cause of chronic liver disease and liver transplants, is a rapidly evolving treatment area with a clear need for innovative treatments. Approximately 150 million people are infected with hepatitis C worldwide, and 350,000 people per year die from the disease.
About Medivir AB
Medivir is an emerging research-based pharmaceutical company focused on infectious diseases. Medivir has world class expertise in polymerase and protease drug targets and drug development which has resulted in a strong infectious disease R&D portfolio. The Company’s key pipeline asset is simeprevir, a novel protease inhibitor in late phase III clinical development for hepatitis C that is being developed in collaboration with Janssen R&D Ireland.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed by Medivir and Johnson & Johnson’s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Lesinurad
Acetic acid, 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]-,
sodium salt (1:1)
Sodium 2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-
yl]sulfanyl}acetate
2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid
MOLECULAR FORMULA C17H13BrN3NaO2S
MOLECULAR WEIGHT 426.3
http://clinicaltrials.gov/show/NCT01508702
http://www.ama-assn.org/resources/doc/usan/lesinurad.pdf
Ardea Biosciences, Inc.
Gout phase 3
Gout is associated with elevated levels of uric acid that crystallize and deposit in joints, tendons, and surrounding tissues. Gout is marked by recurrent attacks of red, tender, hot, and/or swollen joints.
This study will assess the serum uric acid lowering effects and safety of lesinurad compared to placebo in patients who are intolerant or have a contraindication to allopurinol or febuxostat.
http://euroscan.org.uk/technologies/technology/view/2386
Lesinurad (RDEA-594, lesinurad sodium) is a selective urate transporter-1 (URAT-1) inhibitor, which blocks the reabsorption of urate within the renal proximal tubule. It is intended for the treatment of gout after failure of first line therapy and is administered orally at 400mg once daily
A Phase 3 Randomized, Double-Blind, Multicenter, Placebo- Controlled Study to Assess the Efficacy and Safety of Lesinurad Monotherapy Compared to Placebo in Subjects With Gout and an Intolerance or Contraindication to a Xanthine Oxidase Inhibitor
AstraZeneca’s lesinurad (formerly known as RDEA-594) is a selective oral Uric Acid Transporter URAT1 inhibitor currently in Phase III development for the treatment of of gout. The regulatory filings for lesinurad in the US and Europe are expected for the first half of 2014.

Gout (also known as podagra when it involves the big toe), while not life-threatening, is an excruciatingly painful condition caused by a buildup of a waste product in the blood called uric acid, which is normally eliminated from the body through urine. Excess Uric acid crystallizes and get deposited in the joints (usually the big toes), creating symptoms similar to an acute arthritis flare. Gout has seen a recent gradual resurgence as a result of rising obesity rates and poor diet according to a study in the journal Annals of the Rheumatic Diseases.
The current Standard treatment for gout works by inhibiting a protein called xanthine oxidase that helps in the formation of the uric acid. These therapies, some of which have been used for more than 50 years, are not effective in all patients. One is a generic drug called allopurinol that was approved in the U.S. in 1966. The other is febuxostat, marketed by Takeda Pharmaceutical Co. in the U.S. asUloric and by Ipsen SA and others in Europe as Adenuric and approved in the U.S. in 2009.
AstraZeneca’s new product Lesinurad, a selective uric acid re-absorption inhibitor (SURI), tackles gout by blocking a protein called Uric acid trasporter 1 (URAT1) that otherwise would cause the body to reabsorb the uric acid. AstraZeneca acquired lesinurad (aka RDEA-594) as part of its $1.26 billion takeouver of San Diego-based Ardea Biosciences in 2012. RDEA594 is a metabolite of RDEA806, a non-nucleoside reverse transcriptase inhibitor originally developed for HIV.

In top-line results from a Phase III LIGHT study released by AstraZeneca in December 2013 on gout patients who get no benefit from Zyloprim (allopurinol) and febuxostat, lesinurad alone significantly reduced serum levels of uric acid. The company has three other phase III studies ongoing that are testing the use of the drug alongside allopurinol and febuxostat, and these should generate results in the middle of 2014. Analysts at JPMorgan Chase forecast lesinurad alone may have peak sales of $1 billion a year. AstraZeneca also has a second, more potent drug called RDEA3179 to treat elevated levels of uric acid or hyperuricemia. Pfizer’s KUX-1151, licensed from Japan’s Kissei Phmarceuticals, is in early stage development.
Gout is not an automatic success indication of drugmakers. Savient Pharmaceuticals filed for Chapter 11 bankruptcy in October 2013 in the face of a severe cash crisis, having spent hundreds of millions of dollars on its would-be flagship — the gout-fighting drug Krystexxa (pegloticase) — with limited results. Krystexxa (pegloticase), a twice-monthly infusion designed to treat severe chronic gout that doesn’t respond to conventional therapy, was approved by the U.S. Food and Drug Administration in September 2010. Crealta Pharmaceuticals acquired Savient for $120.4 million in December 2013.
Lesinurad
RDEA-594
2-{[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]sulfanyl}acetic acid
CAS number: 878672-00-5 (Lesinurad), 1151516-14-1 (Lesinurad sodium)
Mechanism of Action:once-daily inhibitor of URAT1, a transporter in the kidney that regulates uric acid excretion from the body
US patents:US8242154 , US8173690, US808448
Indication: Gout
Developmental Status: Phase III (US, UK, EU)
Originator: Ardea Biosciences (Acquired by AstraZeneca for $1.26 billion in 2012)
Developer: AstraZeneca
…………………………
http://www.google.co.in/patents/US8242154
Example 8 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)-N-(2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume, to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 mins and the organic layer removed. The aqueous layer was cooled to 0° C. and acidified by treatment with HCl (1N) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3×) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
Example 102 Methyl 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate


Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0° C., and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%).

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).

A solution of 1-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropylnaphthalene (3.1 g, 73%).

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of 1-amino-4-cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) at 0° C. The reaction mixture was stirred for 5 min at 0° C. and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanatonaphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50° C. for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.0 g, 49%).

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 mins to a suspension of 5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
Example 104 Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate

Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 mins to a solution of 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10° C. The mixture was stirred at 10° C. for a further 10 mins. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 103 2-(5-Bromo-4-(1-cyclopropylnapthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 mins to a solution of methyl 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetate (prepared as described in example 1 above; 1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0° C. The mixture was stirred at 0° C. for a further 45 mins and then neutralized to pH 7 by the addition of 0.5N HCl solution at 0° C. The resulting mixture was concentrated in vacuo to ⅕th of its original volume, then diluted with water (˜20 mL) and acidified to pH 2-3 by the addition of 0.5N HCl to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with DCM is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2-(5-bromo-4-(1-cyclopropylnaphthalen-4-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid (1.02 g, 93%).
 |
1H NMR (400 MHz, DMSO-d6) δ ppm 0.84-0.91 (m, 2 H) 1.12-1.19 (m, 2 H) 2.54-2.61 (m, 1 H) 3.99 (d, J = 1.45 Hz, 2 H) 7.16 (d, J = 7.88 Hz, 1 H) 7.44 (d, J = 7.46 Hz, 1 H) 7.59-7.70 (m, 2 H) 7.75 (td, J = 7.62, 1.14 Hz, 1 H) 8.59 (d, J = 8.50 Hz, 1 H) 12.94 (br. s., 1 H) | Mass found: 404.5 (M + 1) | B |
Described herein are various polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate which decreases uric acid levels, (see for example US patent publication 2009/0197825, US patent publication 2010/0056464 and US patent publication 2010/0056465). Details of clinical studies involving sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate have been described in International patent application
PCT/US2010/052958.
Polymorph Form A
In one embodiment, sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H- l,2,4-triazol-3-ylthio)acetate polymorph Form A exhibits an x-ray powder diffraction pattern characterized by the diffraction pattern summarized in Table 1 A or Table IB. In some embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4- cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 3 peaks of (±0.1°2Θ) of Table 1A or IB. In certain embodiments, provided herein is a polymorph of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetate comprising at least 4 peaks of (±0.1°2Θ) of Table 1A or IB, at least 5 peaks of (±0.1°2Θ) of Table 1A or IB, at least 6 peaks of (±0.1°2Θ) of Table 1A or IB, at least 8 peaks of
(±0. Γ2Θ) of Table 1A or IB, at least 10 peaks of (±0. Γ2Θ) of Table 1A, at least 15 peaks of (±0. Γ2Θ) of Table 1A, at least 20 peaks of (±0. Γ2Θ) of Table 1A, at least 25 peaks of (±0.1 °2Θ) of Table 1A, or at least 30 peaks of (±0.1 °2Θ) of Table 1A.


Examples
I Preparation of compounds
Example 1: Preparation of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate
Sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetate was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.

[00103] Aqueous sodium hydroxide solution (1M, 2.0 mL, 2.0 mmol) was added dropwise over 5 min to a solution of 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H- l,2,4-triazol-3-ylthio)acetic acid (810 mg, 2.0 mmol) in ethanol (10 mL) at 10 °C. The mixture was stirred at 10 °C for a further 10 min. Volatile solvents were removed in vacuo to dryness to provide sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4- triazol-3-ylthio)acetate as a solid (850 mg, 100%).
Example 2: Preparation of 2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol- 3-ylthio)acetic acid
2-(5-Bromo-4-(4-cyclopropylnaphthalen- 1 -yl)-4H- 1 ,2,4-triazol-3-ylthio)acetic acid was prepared according to previously described procedures (see US patent publication
2009/0197825) and as outlined below.
[00104] Route i:

Sodium hydroxide solution (2M aqueous, 33.7 mL, 67 mmol, 2 eq) was added to a suspension of 2-(5-bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)-N- (2-chloro-4-sulfamoylphenyl)acetamide (prepared by previously published procedures, see US 2009/0197825; 20 g, 34 mmol) in ethanol (200 mL) and the mixture heated at reflux for 4 hours. Charcoal (10 g) was added, the mixture stirred at room temperature for 12 hours and the charcoal removed by filtration. The charcoal was washed several times with ethanol and the filtrate then concentrated. Water (200 mL) was added and then concentrated to approx. one third volume to remove all ethanol. Water (200 mL) and ethyl acetate (250 mL) were added, the mixture stirred vigorously for 15 min and the organic layer removed. The aqueous layer was cooled to 0 °C and acidified by treatment with HCl (IN) resulting in the formation of a cloudy oily precipitate. The mixture was extracted with ethyl acetate (3x) and the combined organic extracts dried over sodium sulfate and concentrated to give 2-(5- bromo-4-(4-cyclopropylnaphthalen-l-yl)-4H-l,2,4-triazol-3-ylthio)acetic acid as an off white solid (11.2 g, 82%).
[00105] Route ii:

STEP A: 1-Cyclopropylnaphthalene

Cyclopropylmagnesium bromide (150 mL, 0.5M in tetrahydrofuran) was slowly added to a solution of 1-bromonaphthalene (10 g, 50 mmol) and [l,3-bis(diphenylphosphino)propane] dichloro nickel (II) in tetrahydrofuran (10 mL) stirred at 0 °C, and the reaction mixture stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and ethyl acetate and aqueous ammonium chloride were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropylnaphthalene (6.4 g, 76%). ] STEP B: l-Cyclopropyl-4-nitronaphthalene

Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropylnaphthalene (6.4 g, 38 mmol) stirred at 0 °C. The reaction mixture was stirred at 0 °C for an extra 30 min and then slowly poured into ice. Water was added, followed by ethyl acetate. After extraction, the organic layer was washed with aqueous sodium hydroxide (1%) and water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-cyclopropyl-4-nitronaphthalene (5.2 g, 64%).
[00108] STEP C: l-Amino-4-cyclopropylnaphthalene

A solution of l-cyclopropyl-4-nitronaphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, filtered over celite, and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield l-amino-4-cyclopropylnaphthalene (3.1 g, 73%).
STEP D: l-Cyclopropyl-4-isothiocvanatonaphthalene

Thiophosgene (1.1 g, 9.7 mmol) was added to a stirred solution of l-amino-4- cyclopropylnaphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in
dichloromethane (50 mL) at 0 °C. The reaction mixture was stirred for 5 min at 0 °C and then aqueous HCl (1% solution) was added. The organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent removed under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield l-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%>).
[00110] STEP E: 5-Amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol

A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), l-cyclopropyl-4- isothiocyanato naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50 °C for 15 hours. The solvent was removed under reduced pressure, toluene added, and the solvent was evaporated again. Sodium hydroxide solution (2M, 30 mL) was added and the reaction mixture heated at 50 °C for 60 hours. The reaction mixture was filtered and the filtrate neutralized with aqueous HCl (2M). The mixture was re-filtered and the solvent removed under reduced pressure. The residue was purified by silica gel chromatography to yield 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3- thiol (2.0 g, 49%). [00111] STEP F: Methyl 2-(5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- -3 -ylthio)acetate

Methyl 2-chloroacetate (0.73 mL, 8.3 mmol) was added dropwise over 5 min to a suspension of 5-amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazole-3-thiol (2.24 g, 7.9 mmol) and potassium carbonate (1.21 g, 8.7 mmol) in DMF (40 mL) at room
temperature. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the presence of P2O5 to yield methyl 2-(5- amino-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (2.24 g, 80%).
[00112] STEP G: Methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3 -ylthio)acetate

Sodium nitrite (2.76 g, 40 mmol) was added to a solution of methyl 2-(5-amino-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (0.71 g, 2 mmol) and benzyltriethylammonium chloride (1.63 g, 6 mmol) in bromoform (10 mL). Dichloroacetic acid (0.33 mL, 4 mmol) was then added and the reaction mixture stirred at room
temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel, packed with dichloromethane (DCM). The column was first eluted with DCM until all bromoform eluted, then eluted with acetone/DCM (5:95) to give methyl 2-(5-bromo-4-(l- cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3-ylthio)acetate (713 mg, 85%).
[00113] STEP H: 2-(5-Bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- )acetic acid

A solution of lithium hydroxide (98 mg, 4.1 mmol) in water (10 mL) was added dropwise over 5 min to a solution of methyl 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4- triazol-3-ylthio)acetate (1.14 g, 2.7 mmol) in ethanol (10 mL) and THF (10 mL) at 0 °C. The mixture was stirred at 0 °C for a further 45 min and then neutralized to pH 7 by the addition of 0.5N HC1 solution at 0 °C. The resulting mixture was concentrated in vacuo to l/5th of its original volume, then diluted with water (~20 mL) and acidified to pH 2-3 by the addition of 0.5N HC1 to produce a sticky solid. (If the product comes out as an oil during acidification, extraction with dichloromethane is recommended.) The tan solid was
collected by vacuum filtration and dried under high vacuum at 50 °C for 16 h in the
presence of P2O5 to yield 2-(5-bromo-4-(l-cyclopropylnaphthalen-4-yl)-4H-l,2,4-triazol-3- ylthio)acetic acid (1.02 g, 93%).
………………………….
EXAMPLES
The following experiments are provided only by way of example, and should not be understood as limiting the scope of the invention.
COMPOUNDS OF THE INVENTION 2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method A)

1-Cyclopropyl-naphthalene
Cyclopropylmagnesium bromide (150 mL, 0.5 M in tetrahydrofuran) was slowly added to a solution of 1-bromo-naphthalene (10 g, 50 mmol) and [1,3-bis(diphenylphosphino)propane]dichloronickel(II) in tetrahydrofuran (10 mL) stirred at 0° C. The reaction mixture was stirred at room temperature for 16 hours and the solvent was evaporated under reduced pressure. EtOAc and ammonium chloride in water were added. After extraction, the organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-naphthalene (6.4 g, 76%).
1-Cyclopropyl-4-nitro-naphthalene
Sodium nitrite (30 mL) was slowly added (over 2 hours) to 1-cyclopropyl-naphthalene (6.4 g, 38 mmol) stirred at 0° C. The reaction mixture was stirred at 0° C. for an extra 30 min and then was slowly poured into ice. Water was added, followed by EtOAc. After extraction, the organic layer was washed with a 1% aqueous solution of NaOH, then washed with water, dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 1-cyclopropyl-4-nitro-naphthalene (5.2 g, 64%).
1-Amino-4-cyclopropyl-naphthalene
A solution of 1-cyclopropyl-4-nitro-naphthalene (5 g, 23 mmol) in ethanol (200 mL) was stirred under hydrogen in the presence of Pd/C (10% net, 1.8 g). The reaction mixture was shaken overnight, then filtered over celite. The solvent was evaporated, and the residue was purified by silica gel chromatography to yield 1-amino-4-cyclopropyl-naphthalene (3.1 g, 73%).
1-Cyclopropyl-4-isothiocyanato-naphthalene
Thiophosgene (1.1 g, 9.7 mmol) was added to a solution of 1-amino-4-cyclopropyl-naphthalene (1.8 g, 9.7 mmol) and diisopropylethylamine (2 eq) in dichloromethane (50 mL) stirred at 0° C. The reaction mixture was stirred for 5 min at this temperature, then a 1% solution of HCl in water was added and the organic layer was separated, washed with brine, dried over sodium sulfate, filtered and the solvent was evaporated under reduced pressure. Hexane was added, and the resulting precipitate was filtered. The solvent was evaporated to yield 1-cyclopropyl-4-isothiocyanatonaphthalene (1.88 g, 86%).
5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol
A mixture of aminoguanidine hydrochloride (3.18 g, 29 mmol), 1-cyclopropyl-4-isothiocyanato-naphthalene (3.24 g, 14 mmol) and diisopropylethylamine (3 eq) in DMF (20 mL) was stirred at 50° C. for 15 hours. The solvent was evaporated, toluene was added, and the solvent was evaporated again. A 2.0 M aqueous solution of sodium hydroxide (30 mL) was added and the reaction mixture was heated at 50° C. for 60 hours. The reaction mixture was filtered, and the filtrate was neutralized with a 2.0 M aqueous solution of HCl. New filtration, then evaporation of solvent and purification of the residue by silica gel chromatography to yield 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (2.0 g, 49%).
2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)Acetamide
In a solution of 5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-thiol (708 mg, 2.5 mmol), K2CO3 (380 mg, 2.5 mmol) in DMF (20 mL) was added 2-chloro-N-(2-chloro-4-sulfamoylphenyl)acetamide (710 mg, 2.5 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion of the reaction, the solvent was evaporated. The residue was purified by silica gel chromatography to yield 2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (1.26 g, 95%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide
Dichloroacetic acid (180 uL, 2.2 mmol) was added to a suspension of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (0.59 g, 1.1 mmol), sodium nitrite (1.5 g, 22 mmol) and BTEABr (0.91 g, 3.3 mmol) in dibromomethane (30 mL). The reaction mixture was stirred at room temperature for 4 hours, then extracted with dichloromethane and sodium bicarbonate in water. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography to yield 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (224 mg, 31%).
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazole-3-ylsulfanyl]-N-(2-chloro-4-sulfamoylphenyl)acetamide (Method B)

2-[5-Amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole | 2.24 | g | 282.36 | 7.9 |
| methyl chloroacetate | 0.73 | ml | 108.52 | 8.3 (1.05 eq) |
| potassium carbonate | 1.21 | g | 138.21 | 8.7 (1.1 eq) |
| dimethylformamide | 40 | ml | (5 mL/mmol) | |
Procedure:
To a suspension of thiotriazole and potassium carbonate in DMF was added methyl chloroacetate dropwise at room temperature for 5 min. The reaction was stirred at room temperature for 24 h and slowly poured into a stirred ice-cold water solution. The tan precipitate was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 2.24 g (80%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester
| Materials | Amount | Mol. Wt. | mmoles | ||
| thiotriazole L10183-58 | 709 | mg | 354.43 | 2.0 | |
| bromoform | 10 | ml | (5 ml/mmol) | ||
| sodium nitrite | 2.76 | g | 69.00 | 40 | (20 eq) |
| benzyltriethylammonium | 1.63 | g | 272.24 | 6.0 | (3 eq) |
| bromide | |||||
| dichloroacetic acid | 0.33 | ml | 128.94 | 4.0 | (2 eq) |
Procedure:
To a solution of 2-[5-amino-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester and benzyltriethylammonium chloride in bromoform was added sodium nitrite. To the mixture was added dichloroacetic acid and the reaction mixture was stirred at room temperature for 3 h. The mixture was directly loaded onto a 7-inch column of silica gel that was packed with CH2Cl2. The column was first eluted with CH2Cl2 until all CHBr3 eluted, and was then eluted with acetone/CH2Cl2 (5:95) to give 713 mg (85%) of the title compound.
2-[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid
| Materials | Amount | Mol. Wt. | mmoles | |
| thiotriazole methyl ester | 1.14 | g | 418.31 | 2.7 |
| tetrahydrofuran | 10 | ml | (~3 ml/mmol) | |
| ethanol | 10 | ml | (~3 ml/mmol) | |
| water | 10 | ml | (~3 ml/mmol) | |
| lithium hydroxide | 98 | mg | 23.95 | 4.1 (1.5 eq) |
Procedure:
To a solution of 2-[5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-[1,2,4]triazol-3-ylsulfanyl]acetic acid methyl ester, in a mixture of THF and EtOH at 0° C., was added a solution of LiOH in H2O dropwise over 5 min. The reaction was complete after stirring at 0° C. for an additional 45 min. The reaction was neutralized to pH 7 by the addition of 0.5 N HCl solution at 0° C., and the resulting mixture was concentrated in vacuo to ⅕th of its original volume. The mixture was diluted with H2O (˜20 mL) and acidified to pH 2-3 by the addition of 0.5 N HCl to produce sticky solid. (If the product comes out as an oil during acidification, extraction with CH2Cl2 is recommended.) The tan solid was collected by vacuum filtration and dried under high vacuum at 50° C. for 16 h in the presence of P2O5 to yield 1.02 g (93%) of the title compound.
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8242154 B2 ;Also published as US20100056465, US20130040907;Original Assignee: Ardea Biosciences, Inc
Esmir Gunic, Jean-Luc Girardet, Jean-Michel Vernier, Martina E. Tedder, David A. Paisner;Compounds, compositions and methods of using same for modulating uric acid levels;US patent number US8173690 B2;Also published as US20100056464;Original Assignee: Ardea Biosciences, Inc
Barry D. Quart, Jean-Luc Girardet, Esmir Gunic, Li-Tain Yeh;Compounds and compositions and methods of use;US patent number US8084483 B2; Also published as CA2706858A1, CA2706858C, CN101918377A, CN102643241A, CN103058944A, EP2217577A2, EP2217577A4, US8283369, US8357713, US8546437, US20090197825, US20110268801, US20110293719, US20120164222, US20140005136, WO2009070740A2, WO2009070740A3;Original Assignee:Ardea Biosciences, Inc.
Gunic, Esmir; Galvin, Gabriel;Manufacture of 2-[5-bromo-4-(cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio]acetic acid and related compounds;PCT Int. Appl., WO2014008295 A1
Zamansky, Irina et al;Process for preparation of polymorphic, crystalline, and mesophase forms of 2-[[5-bromo-4-(4-cyclopropyl-1-naphthalenyl)-4H-1,2,4-triazol-3-yl]thio]acetic acid sodium salt; PCT Int. Appl., WO2011085009
Gunic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels; U.S. Pat. Appl. Publ., 20100056465
unic, Esmir et al;Preparation of naphthalene thio triazole derivatives and their use for modulating uric acid levels;U.S. Pat. Appl. Publ., 20100056464
Quart, Barry D. et al;Preparation of azole carboxylates as modulators of blood uric acid levels;PCT Int. Appl., 2009070740, 04 Jun 2009
Girardet, Jean-Luc et al;Preparation of S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase;PCT Int. Appl., WO2006026356
US20100056465 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
US20100056542 * Sep 4, 2009 Mar 4, 2010 Ardea Biosciences Compounds, compositions and methods of using same for modulating uric acid levels
WO2009070740A2 * Nov 26, 2008 Jun 4, 2009 Ardea Biosciences Inc Novel compounds and compositions and methods of use
WO2011085009A2 * Jan 5, 2011 Jul 14, 2011 Ardea Biosciences, Inc. Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof
| WO2011159732A1 * | Jun 14, 2011 | Dec 22, 2011 | Ardea Biosciences,Inc. | Treatment of gout and hyperuricemia |
| WO2012092395A2 * | Dec 28, 2011 | Jul 5, 2012 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio) acetic acid and uses thereof |
| EP2560642A2 * | Mar 29, 2011 | Feb 27, 2013 | Ardea Biosciences, Inc. | Treatment of gout |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US20100056465 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20100056542 * | Sep 4, 2009 | Mar 4, 2010 | Ardea Biosciences | Compounds, compositions and methods of using same for modulating uric acid levels |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| WO2011085009A2 * | Jan 5, 2011 | Jul 14, 2011 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetate, and uses thereof |
| US8283369 | 30 Jun 2011 | 9 Oct 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | 30 Jun 2011 | 22 Jan 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8546437 | 30 Jun 2011 | 1 Oct 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8633232 * | 4 May 2012 | 21 Jan 2014 | Ardea Biosciences, Inc. | Compounds, compositions and methods of using same for modulating uric acid levels |
| US20130040907 * | 4 May 2012 | 14 Feb 2013 | Ardea Biosciences Inc. | Compounds, Compositions and Methods of Using Same for Modulating Uric Acid Levels |
| WO2006026356A2 * | Aug 25, 2005 | Mar 9, 2006 | La Rosa Martha De | S-TRIAZOLYL α-MERCAPTOACETANILDES AS INHIBITORS OF HIV REVERSE TRANSCRIPTASE |
| WO2009070740A2 * | Nov 26, 2008 | Jun 4, 2009 | Ardea Biosciences Inc | Novel compounds and compositions and methods of use |
| US20090197825 * | Nov 26, 2008 | Aug 6, 2009 | Ardea Biosciences, Inc. | Novel compounds and compositions and methods of use |
| US7947721 | Aug 25, 2005 | May 24, 2011 | Ardes Biosciences Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8084483 | Nov 26, 2008 | Dec 27, 2011 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |
| US8106205 | Feb 2, 2010 | Jan 31, 2012 | Ardea Biosciences, Inc. | N[S(4-aryl-triazol-3-yl)α-mercaptoacetyl]p-amino benzoic acids as HIV reverse transcriptase inhibitors |
| US8252828 | Jun 30, 2011 | Aug 28, 2012 | Ardea Biosciences, Inc. | S-triazolyl α-mercapto acetanilides as inhibitors of HIV reverse transcriptase |
| US8283369 | Jun 30, 2011 | Oct 9, 2012 | Ardea Biosciences. Inc. | Compounds and compositions and methods of use |
| US8357713 | Jun 30, 2011 | Jan 22, 2013 | Ardea Biosciences Inc. | Compounds and compositions and methods of use |
| US8372807 | May 20, 2010 | Feb 12, 2013 | Ardea Biosciences, Inc. | Methods of modulating uric acid levels |
| US8481581 | Jul 18, 2012 | Jul 9, 2013 | Ardea Biosciences, Inc. | S-triazolyl α-mercaptoacetanilides as inhibitors of HIV reverse transcriptase |
| US8524754 | Jan 5, 2011 | Sep 3, 2013 | Ardea Biosciences, Inc. | Polymorphic, crystalline and mesophase forms of sodium 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio) acetate, and uses thereof |
| US8546436 | Dec 28, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US8546437 | Jun 30, 2011 | Oct 1, 2013 | Ardea Biosciences, Inc. | Compounds and compositions and methods of use |

An unexpected phenomenon concerning an otherwise common impurity of lesinurad has been observed in the context of synthetic process development. A new industrial process was designed as a chlorine-free process, but the critical chlorinated impurity 10 was surprisingly detected in the isolated product. Because of the structural similarity of the impurity and the product, no efficient separation of 10 by conventional methods (e.g., crystallization) was discovered. The formation of the impurity was explained by a chlorine impurity in a commercial brominating agent. This communication also describes control of the critical impurity.
Identification of an Unexpected Impurity in a New Improved Synthesis of Lesinurad
2-[[5-Bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-yl]thio]acetic Acid (1)
1H NMR (DMSO-d6), δ (ppm): 0.87 (m, 2H); 1.15 (m, 2H); 2.56 (m, 1H); 4.00 (m, 2H); 7.16 (d, J = 8.5 Hz, 1H); 7.44 (d, J = 7.3, 1H); 7.64 (d, J = 7.3 Hz, 1H); 7.66 (t, J= 7.6 Hz, 1H); 7.74 (t, J = 7.6 Hz, 1H); 8.58 (d, J = 8.5 Hz, 1H); 12.99 (s, COOH). 13C NMR (DMSO-d6), δ (ppm): 7.3; 7.4; 12.9; 34.1; 121.8; 122.7; 125.2; 126.6; 126.8; 127.3; 128.1; 128.6; 131.4; 133.5; 143.2; 153.5; 169.0.

Isopropyl (2S)-2-[[[(2R,3R,4R,5R)-5-(2,4-dioxopyrimidin-1-yl)-4-fluoro-3-hydroxy-4-methyl-tetrahydrofuran-2-yl]methoxy-phenoxy-phosphoryl]amino]propanoate

The Foster City, CA-based Gilead said that its experimental drug GS-7977, originally known as PSI-7977 before the acquisition, when combined with ribavirin, cured a group of genotype 1 hepatitis C patients after four weeks of treatment. The clinical study involved hepatitis C patients who either failed to respond to previous therapies or had not been treated before. The genotype 1 is the most common form of HCV in the United States. It affects 70 to 90 percent of the people in this country who have hepatitis C.
Norbert Bischofberger, chief scientific officer at Gilead said patients with genotype 1 hepatitis C had no detectable signs of the virus after treated with GS-7977 combination therapy for a course of close to a month. Previous study showed the drug candidate could also cure patients with genotype 2 and 3 HCV.
Gilead gained rights to GS-7977 through the $11 billion Pharmasset acquisition deal, which enable the company to be in an advanced position to compete with a few pharma companies seeking to develop an all-oral regimen for hepatitis C. The 100 percent cure rate data suggested that GS-7977 may be one of the most promising therapies for hepatitis C.
Last year, GS-7977, an oral uridine nucleotide analog polymerase inhibitor of HCV, received fast track designation from the U.S. FDA for the treatment of HCV infection.
The World Health Organization estimated that 3–4 million people are infected with HCV each year. Some 130–170 million people are chronically infected with HCV and at risk of developing liver cirrhosis and/or liver cancer, and more than 350,000 people die yearly from hepatitis C-related diseases.
Sofosbuvir (formerly PSI-7977 or GS-7977) is an experimental drug candidate for the treatment of hepatitis C.[1] It was discovered at Pharmasset and then acquired for development by Gilead Sciences. It is currently in Phase III clinical trials.[2]
Sofosbuvir is a prodrug that is metabolized to the active antiviral agent 2′-deoxy-2′-α-fluoro-β-C-methyluridine-5′-monophosphate.[3]
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus (HCV) polymerase.[4] The HCV polymerase or NS5B protein is a RNA-dependent RNA polymerase critical for the viral cycle.
Sofosbuvir is being studied in combination with pegylated interferon and ribavirin, with ribavirin alone, and with other direct-acting antiviral agents.[5] It has shown excellent clinical efficacy when used either with pegylated interferon/ribavirin or in interferon-free combinations. In particular, combinations of sofosbuvir with NS5A inhibitors, such as daclatasvir or GS-5885, have shown sustained virological response rates of up to 100% in people infected with HCV.[6]

TRIFLURIDINE

TIPIRACIL
TAS-102 is an anti-cancer drug under development for colorectal cancer.[1]
| Combination of | |
|---|---|
| Trifluridine | cytotoxin |
| Tipiracil | thymidine phosphorylase inhibitor |
A phase II trial reported in 2011[2] and a phase III trial is due to end in 2014.[1][3]
TAS-102 consists of the cytotoxin trifluridine and the thymidine phosphorylase inhibitor (TPI) tipiracil.[4] Trifluridine is incorporated into DNA during DNA synthesis and inhibits tumor cell growth. Tipiracil protects trifluridine from being broken down when taken orally.
| February 28, 2013, |
|
Taiho Pharmaceutical Co., Ltd. has submitted an application to the Japanese Ministry of Health, Labour and Welfare for approval of the manufacture and marketing of the novel oral nucleoside anti-tumour agent TAS-102 (combination of trifluorothymidine [FTD] and tipiracil hydrochloride [TPI]). Taiho is seeking approval of TAS-102 for the indication of unresectable, advanced, recurrent colorectal cancer. The application for approval is based on the results of a phase II clinical trial (Study 10040030) conducted at 20 facilities throughout Japan. It was a randomized, double-blind comparative study of TAS-102 and a placebo involving 172 patients with unresectable, advanced, recurrent colorectal cancer that was refractory to the standard chemotherapy of at least two or more regimens containing fluoropyrimidine, irinotecan, and oxaliplatin. The results indicated that the group administered TAS-102 had improved overall survival rates (median overall survival: 9.0 months vs. 6.6 months) and a significantly reduced risk of mortality (HR: 0.56, p=0.0011). The most frequently reported adverse drug reaction with a CTCAE grade of 3 or higher was neutropenia. Grade 3 or higher diarrhea, fatigue, nausea, and other adverse reactions were no more than 10 per cent. Taiho Pharmaceutical is currently proceeding with a global phase III clinical trial of TAS-102 in a similar colorectal cancer population (RECOURSE) with the ultimate goal of global registration and commercialization of the agent. Taiho Pharmaceutical believes that TAS-102 will make a significant contribution to cancer patients and will continue its development efforts to broaden its use. TAS-102 is an anti-tumour agent composed of a combination of trifluorothymidine (FTD), a nucleoside that incorporates into DNA and inhibits a variety of genetic functions required for the proliferation of cancer cells, and tipiracil hydrochloride (TPI), an inhibitor of thymidine phosphorylase (which degrades FTD) that maintains an effective blood concentration of FTD. TAS-102 is administered twice daily to achieve a total daily dose of 70mg/m2 for five days followed by two days of rest and then repeated a second time. This is followed by a 14-day rest period to make a 28-day schedule for one course. |
|
|
|---|---|
| TRIFLURIDINE | |
| 1-[4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5- (trifluoromethyl) pyrimidine-2,4-dione |
Trifluridine (also called trifluorothymidine or TFT) is an anti-herpesvirus antiviral drug, used primarily on the eye. It was sold under the trade name, Viroptic, by Glaxo Wellcome, now merged into GlaxoSmithKline. The brand is now owned by Monarch Pharmaceuticals, which is wholly owned by King Pharmaceuticals.
It is a nucleoside analogue, a modified form of deoxyuridine, similar enough to be incorporated into viral DNA replication, but the -CF3 group added to the uracil component blocks base pairing.
It is a component of the experimental anti-cancer drug TAS-102.
TIPIRACIL
| NAME | 5-chloro-6-[(2-iminopyrrolidin-1-yl)methyl]pyrimidine-2,4-(1H,3H)-dione | |||
| CAS | 183204-74-2 | |||
| MOL F | C9H11ClN4O2 | |||
| STR | |
|||
| USE | potentiator of antineoplastics; | |||
Taiho Pharmaceutical, a subsidiary of Otsuka Holdings Co., Ltd., is an R&D-driven specialty pharma focusing on the three fields of oncology, allergies and immunology, and urology.

3-{6-{[1-(2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonyl]amino}-3-methylpyridin-2-yl}benzoic acid
26,FEB 2013
syn at >>>>>>>https://newdrugapprovals.org/2013/07/28/3274/
Vertex Pharmaceuticals announced Tuesday night the design of two phase III studies for its combination therapy to treat the most common form of cystic fibrosis. The studies will each run for six months, so results could be ready as early as the end of 2013 or during first half of 2014.
The studies announced Tuesday will evaluate the two different doses of an experimental medicine VX-809 in combination with Kalydeco. Each study will enroll 500 cystic fibrosis patients randomized to either the VX-809/Kalydeco arms or a placebo for six months of treatment. The studies’ primary endpoint will be the relative improvement in lung function of VX-809/Kalydeco compared to placebo.
Last fall, Vertex presented data from a phase II study demonstrating that a 600 mg dose of VX-809 and Kalydeco worked synergistically to improve lung function in cystic fibrosis patients with the F508del mutation compared to placebo. This same dose combination will be tested in the phase III study along with a higher 800 mg (actually, 400 mg given twice a day) dose of VX-809 plus Kalydeco.
Vertex also announced new data from this phase II study on Tuesday night showing similar lung function improvements between the 800 mg and 600 mg doses of VX-809. For this reason, the higher dose was included in the phase III studies.
Along with the two phase III studies in adult patients, Vertex will also conduct a six-month study of the combination therapy in pediatric patients ages 6 to 11. This study, along with the data from the adult studies, may be used to expand the combination therapy’s approval into younger patients.
In January, FDA anointed Kalydeco and VX-809 with Breakthrough Therapy Designation as part of the agency’s efforts to accelerate the development and approval of drugs for serious and life-threatening disease. Vertex did not say whether Breakthrough Designation played a specific role in the VX-809/Kalydeco phase III program but the relatively short six-month duration of the studies plus the ability to test the combination in children at the same time does accelerate the development of the combination therapy. If the data from the studies are positive, the drugs could be approved sooner than expected and for more patients.
Lumacaftor (USAN, codenamed VX-809) is an experimental drug for the treatment of cystic fibrosis, being developed by Vertex Pharmaceuticals. The drug is designed to be effective in patients that have the F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR), the defective protein that causes the disease. F508del, meaning that the amino acid phenylalanine in position 508 is missing, is found in about 60% of cystic fibrosis patients.[1]
Interim results from a Phase II clinical trial indicate that patients with the most common form of genetic mutation causing cystic fibrosis homozygous F508del had an 8.5% increase in lung function (FEV1) after 56 days on a combination of lumacaftor and ivacaftor (Kalydeco).[2]

Chemical structure of ganetespib and its co-crystal structure with Hsp90 N-terminal.
A, chemical structure of ganetespib.
B, crystallographic complex of ganetespib in the Hsp90 N-terminal.
C, hydrogen bond interactions between ganetespib with amino acid residues in the Hsp90 N-terminal ATP-binding pocket.
Synta Pharmaceuticals has opened a ClinicalTrials.gov listing for their much discussed GALAXY-2 phase 3 trial of their HSP90 inhibitor ganetespib in second-line non-small cell lung cancer (NSCLC), in combination with docetaxel. For the moment at least, the phase 2b GALAXY-1 trial is still listed as enrolling patients, but that would be expected to complete soon.
http://clinicaltrials.gov/ct2/show/NCT01798485
![Molecular Structure of 888216-25-9 (3H-1,2,4-Triazol-3-one, 5-[2,4-dihydroxy-5-(1-methylethyl)phenyl]-2,4-dihydro-4-(1-methyl-1H-indol-5-yl)-)](http://www.lookchem.com/300w/2012-2/e100939d-148c-4f54-9b7c-83632fb0d675.gif)

Hsp90, a chaperone essential for the folding of JAK2, is the target of Synta Pharmaceuticals’ ganetespib.