
Home » Posts tagged 'organic chemistry' (Page 4)
Tag Archives: organic chemistry
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Rilpivirine

Rilpivirine
500287-72-9 cas no
4-{[4-({4-[(E)-2-cyanovinyl]-2,6-dimethylphenyl}amino)pyrimidin-2-yl]amino}benzonitrile

Rilpivirine (TMC278, trade name Edurant) is a pharmaceutical drug, developed byTibotec, for the treatment of HIV infection.[1][2] It is a second-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with higher potency, longer half-life and reducedside-effect profile compared with older NNRTIs, such as efavirenz.[3][4]
Rilpivirine entered phase III clinical trials in April 2008,[5][6] and was approved for use in the United States in May 2011.[7] A fixed-dose drug combining rilpivirine with emtricitabine andtenofovir, was approved by the U.S. Food and Drug Administration in August 2011 under the brand name Complera.[8]
Like etravirine, a second-generation NNRTI approved in 2008, rilpivirine is a diarylpyrimidine(DAPY). Rilpivirine in combination with emtricitabine and tenofovir has been shown to have higher rates of virologic failure than Atripla in patients with baseline HIV viral loads greater than 100,000 copies.
- Rilpivirine bound to proteins in the PDB
- “TMC278 – A new NNRTI”. Tibotec. Retrieved 2010-03-07.
- Stellbrink HJ (2007). “Antiviral drugs in the treatment of AIDS: what is in the pipeline ?”.Eur. J. Med. Res. 12 (9): 483–95. PMID 17933730.
- Goebel F, Yakovlev A, Pozniak AL, Vinogradova E, Boogaerts G, Hoetelmans R, de Béthune MP, Peeters M, Woodfall B (2006). “Short-term antiviral activity of TMC278–a novel NNRTI–in treatment-naive HIV-1-infected subjects”. AIDS 20 (13): 1721–6.doi:10.1097/01.aids.0000242818.65215.bd. PMID 16931936.
- Pozniak A, Morales-Ramirez J, Mohap L et al. 48-Week Primary Analysis of Trial TMC278-C204: TMC278 Demonstrates Potent and Sustained Efficacy in ART-naïve Patients. Oral abstract 144LB.
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-1 Patients Comparing TMC278 to Efavirenz in Combination With Tenofovir + Emtricitabine
- ClinicalTrials.gov A Clinical Trial in Treatment naïve HIV-Subjects Patients Comparing TMC278 to Efavirenz in Combination With 2 Nucleoside/Nucleotide Reverse Transcriptase Inhibitors
- “FDA approves new HIV treatment”. FDA. Retrieved 2011-05-20.
- “Approval of Complera: emtricitabine/rilpivirine/tenofovir DF fixed dose combination”. FDA. August 10, 2011.
FORMULATION
EDURANT (rilpivirine, Janssen Therapeutics) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of human immunodeficiency virus type 1 (HIV-1). EDURANT is available as a white to off-white, film-coated, round, biconvex, 6.4 mm tablet for oral administration. Each tablet contains 27.5 mg of rilpivirine hydrochloride, which is equivalent to 25 mg of rilpivirine.
The chemical name for rilpivirine hydrochloride is 4-[[4-[[4-[(E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]2-pyrimidinyl]amino]benzonitrile monohydrochloride. Its molecular formula is C22H18N6 • HCl and its molecular weight is 402.88. Rilpivirine hydrochloride has the following structural formula:
 |
Rilpivirine hydrochloride is a white to almost white powder. Rilpivirine hydrochloride is practically insoluble in water over a wide pH range.
Each EDURANT tablet also contains the inactive ingredients croscarmellose sodium, lactose monohydrate, magnesium stearate, polysorbate 20, povidone K30 and silicified microcrystalline cellulose. The tablet coating contains hypromellose 2910 6 mPa.s, lactose monohydrate, PEG 3000, titanium dioxide and triacetin.
…………………………….
papers
Sun, et al.: J. Med. Chem., 41, 4648 (1998),
Kashiwada, et al.: Bioorg. Med. Chem. Lett., 11, 183 (2001)
Journal of Medicinal Chemistry, 2005 , vol. 48, 6 , pg. 2072 – 2079
………………………………………………
patents
WO201356003, WO200635067,
WO2013038425
The following PCT Publications describe the synthesis of Rilpivirine:
WO03016306, WO2005021001, WO2006024667, WO2006024668, W02994916581, WO2009007441, WO2006125809, and WO2005123662. [0006] Crystalline Rilpivirine base Forms I and II are described in the US Patent
Publication: US2010189796. Crystalline Rilpivirine HC1, Forms A, B, C, and D, are described in the US Patent Publications: US2009/012108, and US2011/0008434. Rilpivirine fumarate and a synthesis thereof are disclosed in WO2006024667.
country……………….patent……………approved……………expiry
| United States | 6838464 | 2011-05-20 | 2021-02-26 |
| United States | 7067522 | 2011-05-20 | 2019-12-20 |
| United States | 7125879 | 2011-05-20 | 2014-04-14 |
| United States | 7638522 | 2011-05-20 | 2014-04-14 |
| United States | 8080551 | 2011-05-20 | 2023-04-11 |
| United States | 8101629 | 2011-05-20 | 2022-08-09 |
Rilpivirine and its hydrochloride salt were disclosed in U.S. patent no. 7,125,879.Process for the preparation of rilpivirine was disclosed in U.S. patent no. 7,399,856 (‘856 patent). According to the ‘856 patent, rilpivirine can be prepared by reacting the (E)-3-(4-amino-3,5-dimethylphenyI)acrylonitrile hydrochloride of formula II with 4-(4-chloropyrimidin-2-ylamino)benzonitrile of formula III-a in the presence of potassium carbonate and acetonitrile under reflux for 69 hours. The synthetic procedure is illustrated in scheme I, below:
 Scheme 1 Process for the preparation of rilpivirine was disclosed in U.S. patent no. 7,705,148 (Ί48 patent). According to the Ί48 patent, rilpivirine can be prepared by reacting the 4-[[4-[[4-bromo-2,6-dimethylphenyl]amino]-2- pyrimidinyl]amino]benzonitrile with acrylonitrile in the presence of palladium acetate, Ν,Ν-diethylethanamine and tris(2-methylphenyl)phosphine in acetonitrile. According to the Ί48 patent, rilpivirine can be prepared by reacting the compound of formula IV with 4-(4-chloropyrimidin-2-ylamino)benzonitrile formula Ill-a in the presence of hydrochloric acid and n-propanol to obtain a compound of formula Vll, and then the compound was treated with acetonitrile and potassium carbonate under reflux for 69 hours. The synthetic procedure is illustrated in scheme II, below:  Rilpivirine Scheme II U.S. patent no. 7,563,922 disclosed a process for the preparation of (E)-3-(4- amino-3,5-dimethylphenyl)acrylonitrile hydrochloride. According to the patent, (E)-3-(4- amino-3,5-dimethylphenyl)acrylonitrile hydrochloride can be prepared by reacting the 4- iodo-2,6-dimethyl-benzenamine in Ν,Ν-dimethylacetamide with acrylonitrile in the presence of sodium acetate and toluene, and then the solid thus obtained was reacted with hydrochloric acid in 2-propanol in the presence of ethanol and diisopropyl ether. U.S. patent no. 7,956,063 described a polymorphic Form A, Form B, Form C and Form D of rilpivirine hydrochloride. An unpublished application, IN 1415/CHE/201 1 assigned to Hetero Research Foundation discloses a process for the preparation of rilpivirine. According to the application, rilpivirine can be prepared by reacting the 4-(4-chloropyrimidin-2- ylamino)benzonitrile with (E)-3-(4-amino-3,5-dimethylphenyl)acrylonitrile hydrochloride in the presence of p-toluene sulfonic acid monohydrate and 1 ,4-dioxane. It has been found that the rilpivirine produced according to the prior art procedures results in low yields.
|
The synthesis is as follows:
………………
more info………………………..
Rilpivirine, which is chemically known as 4-{[4-({4-[(lE)-2-cyanoethenyl]-2,6- dimethylphenyl} amino) pyrimidin-2-yl]amino}benzonitrile, is a non-nucleoside reverse transcriptase inhibitor (NNRTI) and exhibits human immunodeficiency virus (HIV) replication inhibiting properties. Rilpivirine is used as its hydrochloride salt in the anti-HIV formulations.

Conventionally, various processes followed for the synthesis of Rilpivirine hydrochloride (I), generally involve preparation of the key intermediate, (E)-4-(2- cyanoemenyl)-2,6-dimethylphenylamine hydrochloride of formula (II).

(E)-4-(2-cyanoethenyl)-2,6-dimethylphenylamine hydrochloride (II)
WO 03/016306 first disclosed the synthesis of Rilpivirine involving different routes for synthesis of 4-(2-cyanoethenyl)-2,6-dimethylphenylamine. The first route involved protection of the amino group of 4-bromo-2,6-dimemylphenylarnine by converting to Ν,Ν-dimethylmethanimidamide, followed by formylation involving n- butyl lithium and dimethylformamide. The resulting formyl derivative was treated with diethyl(cyanomethyl) phosphonate to give the cyanoethenyl compound which was deprotected using zinc chloride to yield the cyanoethenylphenylamine intermediate having an undisclosed E/Z ratio. This route involved an elaborate sequence of synthesis comprising protection of amine by its conversion into imide, use of a highly moisture sensitive and pyrophoric base such as butyl lithium and a low yielding formylation reaction. All these factors made the process highly unviable on industrial scale.
The second route disclosed in WO 03/016306 employed 4-iodo-2,6- dimethylphenylamine as a starting material for synthesis of cyanoemenylphenylamine intermediate, which involved reaction of the dimethylphenylamine derivative with acrylonitrile for atleast 12 hours at 130 C in presence of sodium acetate and a heterogeneous catalyst such as palladium on carbon. Isolation of the desired compound involved solvent treatment with multiple solvents followed by evaporation. This route also does not give any details of the E/Z ratio of the unsaturated intermediate product. Although this route avoids use of phosphine ligands but lengthy reaction time and problem of availability of pure halo-phenylamine derivatives coupled with moderate yields hampers the commercial usefulness of this route.
The third route disclosed in WO 03/016306 involved reaction of 4-bromo-2,6- dimethylphenylamine with acrylamide in presence of palladium acetate, tris(2- methylphenyl)phosphine and N,N-diethylethanamine. The resulting amide was dehydrated using phosphoryl chloride to give 4-(2-cyanoethenyi)-2,6- dimethylphenylamine in a moderate yield of 67% without mentioning the E/Z ratio. Although the E/Z isomer ratio for the cyanoethenyl derivative obtained from these routes is not specifically disclosed in the patent, however, reproducibility of the abovementioned reactions were found to provide an E/Z ratio between 70/30 and 80/20. Various other methods have also been reported in the literature for introduction of the ‘ cyanoethenyl group in Rilpivirine. The Journal of Medicinal Chemistry (2005), 48, 2072-79 discloses Wittig or Wadsworth-Emmons reaction of the corresponding aldehyde with cyanomethyl triphenylphosphonium chloride to provide a product having an E/Z isomer ratio of 80/20. An alternate method of Heck reaction comprising reaction of aryl bromide with acrylonitrile in presence of tri-o- tolylphosphine and palladium acetate gave the same compound with a higher E/Z isomer ratio of 90/10. The method required further purification in view of the presence of a significant proportion of the Z isomer in the unsaturated intermediate. A similar method was disclosed in Organic Process Research and Development (2008), 12, 530-536. However, the E/Z ratio of 4-(2-cyanoethenyl)-2,6- dimethylphenylamine was found to be 80/20, which was found to improve to 98/2 (E/Z) after the compound was converted to its hydrochloride salt utilizing ethanol and isopropanol mixture.
It would be evident from the foregoing that prior art methods are associated with the following drawbacks:
a) High proportion of Z isomer, which requires elaborate purification by utilizing column chromatographic techniques, crystallization, or successive treatment with multiple solvents, which decreases the overall yield,
b) Introduction of cyanoethenyl group to the formylated benzenamine derivatives involves a moisture sensitive reagent like n-butyl lithium, which is not preferred on industrial scale. Further, the method utilizes cyanomethyl phosphonate esters and is silent about the proportion of the Z isomer and the higher percentage of impurities which requires elaborate purification and ultimately lowers the yield,
c) Prior art routes involve use of phosphine ligands which are expensive, environmentally toxic for large scale operations,
d) Prior art methods utilize phase transfer catalysts such as tetrabutyl ammonium bromide in stoichiometric amounts and the reactions are carried out at very high temperatures of upto 140-150°C.
Thus, there is a need to develop an improved, convenient and cost effective process for preparation of (E)-4-(2-cyanoethenyl)-2,6-dimethylphenylamine hydrochloride of formula (II) having Z-isomer less than 0.5%, without involving any purification and does not involve use of phosphine reagent and which subsequently provides Rilpivirine hydrochloride (I) conforming to regulatory specifications.
……………………………..
http://www.google.com/patents/EP2643294A2?cl=en
The present inventors have developed a process for stereoselective synthesis of the key Rilpivirine intermediate, (E)-4-(2-cyanoethenyl)-2,6-dimemylphenylarnine hydrochloride (II), comprising diazotization of 2,6-dimethyl-4-amino-l- carboxybenzyl phenylamine followed by treatment with alkali tetrafluoroborate to provide the tetrafluoroborate salt of the diazonium ion which is followed by reaction with acrylonitrile in presence of palladium (II) acetate and subsequent deprotection of the amino group with an acid followed by treatment with hydrochloric acid to give the desired E isomer of compound (II) having Z isomer content less than 0.5% and with a yield of 75-80%. The compound (II) was subsequently converted to Rilpivirine hydrochloride of formula (I) with Z isomer content less than 0.1%.



……………………………………

Chemical structures of selected NNRTIs
…………………………….
http://pubs.acs.org/doi/full/10.1021/jm040840e
Iloperidone (Fanapt)

Iloperidone (Fanapt), ILO-522, HP-873, Zomaril, 133454-47-4, antipsychotic
1-[4-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone; 1-[3-(4-Acetyl-2-methoxyphenoxy)propyl]-4-(6-fluoro-1,2-benzisoxazol-3-yl)piperidine; 4′-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3′-methoxyacetophenone
Aventis Pharma (Originator), Novartis (Licensee), Titan (Licensee) Vanda Pharmaceuticals (Licensee)
Vanda Pharmaceuticals (Licensee)
Iloperidone(Fanapt) is a monoamine directed towards acting upon and antagonizing specific neurotransmitters, particularly multiple dopamine and serotonin receptor subtypes.
Schizophrenia is a chronic, severe, and debilitating mental disorder that affects approximately 2.4 million Americans, around 1.1% of the population. The net cost of this disorder is staggering as estimates from 2002 reveal this disorder to cost $62.7 billion. A major issue with the treatment of schizophrenia is that patients show varying levels of response and tolerance to available therapies. Although the symptoms of the disease are very severe, estimates show that approximately 3 out of 4 patients discontinue medication prior to completing 18 months of treatment, many times due to the severe side effects of the approved medications.
Synthesis
J.T. Strupczewski, K.J. Bordeau, Y. Chiang, E.J. Glamkowski, P.G.
Conway, R. Corbett, H.B. Hartman, M.R. Szewczak, C.A. Wilmot andG.C. Helsley, J. Med. Chem., 38, 1119 (1995).
US 4355037
V. Miklos, WO Patent 031497 (2010).
J.T. Strupczewski, EP Patent 0402644 (1990)
The product is protected by the U.S. Pat. No. 5,364,866, U.S. Pat. No. RE 39198 E and EP 402644 B1.U.S. Pat. No. 5,364,866 and U.S. Pat. No. 5,663,449.EP 542136, EP 612318, EP 730452, JP 95501055, JP 97511215, US 5364866, US 5776963, WO 9309102, WO 9511680.US 4355037,EP 0542136; EP 0612318; EP 0730452; EP 0957102; EP 0959075; EP 0959076; EP 0963984; JP 1995501055; JP 1997511215; US 5364866; US 5776963; WO 9309102; WO 9511680
The first reported synthetic method for Iloperidone is described in patent EP 402644 A1.
In U.S. Patent US5776963 and patent family EP4 (^ 644, there is disclosed a method for preparing iloperidone,
The synthetic method reported(4, 5) for 1 involves two chemical steps: O-alkylation of acetovanillone (2) with 1-bromo-3-chloropropane (3) to obtain chloro derivative 4 followed byN-alkylation of piperidine intermediate 5 with 4. The reported process for 4 comprises O-alkylation of 2 with 3 in acetone in the presence of potassium carbonate for 20 h to provide 4as an oil after usual work up, which was then vacuum (0.1 mmHg) distilled to collect desired product 4 at 141–143 °C with around 85% yield (Scheme 1, Path A). Some of the drawbacks of this process are as follows: longer reaction time (around 20 h), formation of 6–7% of dimer impurity (10, Scheme 2), high-vacuum distillation to achieve the quality, which is always a cumbersome process at industrial scale, requiring special apparatus and skill set, and degradation and charring of some portion of product during high-vacuum distillation. Further, the next step comprises N-alkylation of 4 with 5 in N,N-dimethylformamide (DMF) in the presence of potassium carbonate to provide iloperidone (1) as a crude solid, which was purified by crystallization using ethanol to yield pure 1 with 58% yield (Scheme 1, Path A). Some of the lacunae observed with the above process includes the following: (a) low yields, (b) formation of carbamate impurity 13 (Scheme 2) in the range 15–20% due to the use of potassium carbonate, (c) ineffective purification by crystallization using ethanol to eliminate carbamate impurity below 0.15%, and (d) iloperidone obtained by the above synthetic process was beige in color.



The synthetic route is as follows:

The reaction of piperidine-4-carboxylic acid (I) with formic acid and acetic anhydride gives 1-formylpiperidine-4-carboxylic acid (II), which is treated with SOCl2 and acetic anhydride to yield the corresponding acyl chloride (III). The Friedel-Crafts condensation of (III) with refluxing 1,3-difluorobenzene (IV) by means of AlCl3 affords 4-(2,4-difluorobenzoyl)-1-formylpiperidine (V), which is treated with hydroxylamine in refluxing ethanol to give the corresponding oxime (VI). The cyclization of (VI) by means of NaH in hot THF/DMF yields 6-fluoro-3-(1-formylpiperidin-4-yl)-1,2-benzisoxazole (VII), which is treated with HCl in refluxing ethanol to afford 6-fluoro-3-(4-piperidyl)-1,2-benzisoxazole (VIII). Finally, this compound is condensed with 4-(3-chloropropoxy)-3-methoxyacetophenone (IX) by means of K2CO3 in hot DMF. The intermediate 4-(3-chloropropoxy)-3-methoxyacetophenone (IX) can be obtained by condensation of 4-hydroxy-3-methoxyacetophenone (IX) with 3-chcloropropyl bromide (X) by means of NaH or K2CO3 in DMF.

Iloperidone, also known as Fanapt, Fanapta, and previously known as Zomaril, is an atypical antipsychotic for the treatment ofschizophrenia.
Accordingly, 6-fluoro-3-(4-piperidyl)-1,2-benzoxazole 1 and 1-[4-(3-chloropropoxy)-3-methoxy-phenyl]ethanone 2 were heated in presence of potassium carbonate using dimethylformamide solvent to afford 1-[4-[3-[4-(6-fluoro-1,2-benzoxazol-3-yl)-1-piperidyl]propoxy]-3-methoxy-phenyl]ethanone also called Iloperidone
It was approved by the U.S. Food and Drug Administration (FDA) for use in the United States on May 6, 2009.
It’s not yet approved in India.
Hoechst Marion Roussel Inc. made initial inquiries into the drug; however, in May 1996, they discontinued research, and in June 1997 gave research rights to Titan Pharmaceuticals. Titan then handed over worldwide development, manufacturing and marketing rights to Novartis in August 1998. On June 9, 2004, Titan Pharmaceuticals announced that the Phase III development rights have been acquired by Vanda Pharmaceuticals. The original launch date was scheduled for 2002. On November 27, 2007, Vanda Pharmaceuticals announced that the U.S. Food and Drug Administration (FDA) had accepted their New Drug Application for iloperidone, confirming the application is ready for FDA review and approval. On July 28, 2008, the FDA issued a “Not Approvable” letter to Vanda Pharmaceuticals concerning the drug, stating that further trials are required before a decision can be made concerning marketed usage of iloperidone.
Chemically designated as 1-[4-[3-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone, is a second generation atypical antipsychotic agent. Iloperidone, also known as Fanapt, Fanapta, and Zomaril, was approved by the U.S. Food and Drug Administration (FDA) for use in the United States on May 6, 2009 and is indicated for the acute treatment of schizophrenia in adults. Iloperidone has been shown to act as an antagonist at all tested receptors. It was found to block the sites of noradrenalin (α2C), dopamine (D2A and D3), and serotonin (5-HT1A and 5-HT6) receptors.(2) In addition, pharmacogenomic studies identified single nucleotide polymorphisms associated with an enhanced response to iloperidone during acute treatment of schizophrenia. It is considered an “atypical” antipsychotic because it displays serotonin receptor antagonism, similar to other atypical antipsychotics. The older typical antipsychotics are primarily dopamine antagonists.(3)
Iloperidone won FDA approval for use treating schizophrenia in the United States on May 6, 2009
Iloperidone (1-[4-[3-[4-(6-fluoro-1,2-benzisoxazole-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone) is an atypical new-generation antipsychotic medicament belonging to the class of piperidinyl-benzisoxazole derivatives, which is used to treat schizophrenia, bipolar disorder and other psychiatric conditions. Iloperidone acts as a serotonin/dopamine receptor antagonist (5-HT2A/D2).
Iloperidone, also known as Fanapt, Fanapta, and previously known as Zomaril, is an atypical antipsychotic drug used for the treatment of schizophrenia. The chemical name of iloperidone is l-[4-[3-[4-(6-fluoro-l,2-benzisoxazol-3-yl)-l- piperidinyl]propoxy] -3-methoxyphenyl]ethanone.
EP 0402644 patent discloses first synthetic route of synthesis of iloperidone as shown in Scheme I, which consists of alkylation reaction between l-(4-(3-chloropropoxy-3- methoxyphenyl)ethanone of the formula (II) and 6-fluoro-3-piperidin-4-yl-l ,2 benzisoxazole hydrochloride of the formula (III) in presence of potassium carbonate in N,N dimethyl formamide. The reaction has been subsequently worked up and the compound of formula (I) is extracted from water using ethyl acetate. The compound of formula (I) is purified by crystallization using ethanol. The overall yield of compound of formula (I) is 58%.

Formula (I)
SCHEME 1 Further, we have analyzed the reported synthetic route for synthesis of iloperidone; following limitations have been observed and identified in the reported synthetic route:
a) The yield obtained using said synthetic route as reported in US RE39198 is 58%. Hence, this route of synthesis is not cost efficient at commercial scale due to low yield;
b) Use of potassium carbonate as a base in reaction leads to formation of carbon dioxide as one of the side products during the reaction, which further hinders in the manufacturing process by actively participating in manufacturing process and thereby leads to the formation o

Formula (IV)
which is in the range of 15-20%, and thereby resulting in low yield of iloperidone;
c) Purification by crystallization using ethanol as a solvent is not effective in eliminating or controlling carbamate impurity below 0.15% as per the ICH guide lines for the known impurities; and
d) Iloperidone obtained by the above synthetic process is beige in colour.
CN101768154 discloses the synthesis of iloperidone by N-alkylation reaction between l-(4-(3- chloropropoxy-3-methoxyphenyl)ethanone of the formula (II) and 6-fluoro-3-piperidin-4-yl-l,2 benzisoxazole hydrochloride of the formula (III) in inorganic alkaline solution, particularly; alkali metal carbonate solution. We have analyzed the reported synthetic route for synthesis of iloperidone and have observed and identified that the use of alkaline carbonate solution leads to the formation of carbamate impurity in the range of 1 to 1.5%.
Several patents were published after, describing essentially the same synthetic way such as US5364866 and US5663449.
The synthesis of iloperidone is described in USRE39198 (corresponding to EP 0 402 644 example 3) according to the following synthesis scheme:

In agreement with said patent, the intermediate isolated, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone, is reacted with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride in N,N-dimethyl formamide at 90° C. for 16 hours. When the reaction is complete, the mixture is poured into water and extracted with ethyl acetate. The crude product thus obtained is crystallised twice from ethanol to give crystallised iloperidone with a total yield of 58%.
The yield of this process is very low; moreover, the process begins with two isolated intermediates, and requires an aqueous extractive work-up step with an increase in volumes and consequent reduction in the productivity and efficiency of the process. Said process also requires a double crystallisation step to obtain a beige product. The quality levels obtained are not described in the text of the example, but a beige color does not suggest a high-quality product, as iloperidone is a white substance.
The synthesis of intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is disclosed in U.S. Pat. No. 4,366,162. Example 1 describes the preparation of said intermediate by reacting acetovanillone with 1-bromo-3-chloropropane in acetone with potassium carbonate. At the end of the reaction the resulting product is purified by distillation and obtained as an oily intermediate which is left to stand in order to obtain the solid intermediate.
The synthesis of intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is also disclosed in U.S. Pat. No. 4,810,713. Preparation 12 describes the synthesis of said intermediate from acetovanillone and 1-bromo-3-chloropropane in sodium hydroxide alkalinized water. At the end of the reaction the product obtained is extracted in toluene, the organic phases are washed with basic aqueous solutions and finally, the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone is crystallised with the aid of diisopropyl ether. The intermediate isolated is then recrystallised twice from cyclohexane and twice from petroleum ether.
An alternative process for the synthesis of iloperidone is reported in CN 102070626.
Scheme 2 shows the synthesis procedure:

The decision to alkylate acetovanillone with 1-chloro-3-propanol requires an extra synthesis step (to convert the OH group to an OR leaving group) compared with the procedure reported by the combination of patents USRE39198 (EP402644) and U.S. Pat. No. 4,366,162/U.S. Pat. No. 4,810,713, making said process less efficient from the economic standpoint.
WO2011061750 discloses an alternative iloperidone synthesis process as reported in Scheme 3:

Said process uses reagents such as methyl magnesium chloride to effect the Grignard reaction to convert the aldehyde group to a secondary alcohol group, which are much more complicated to manage on an industrial scale than the synthesis methods previously described. Moreover, the oxidation reaction of the next step uses reagents such as chromic acid or potassium permanganate, which have a very high environmental impact and very low industrial applicability.
WO2011055188 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride. The same patent application also gives preparation examples of the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone isolated as crystalline solid by procedures similar to those known in the literature.
CN 101824030 reports an iloperidone synthesis method similar to that of CN 102070626 which involves the same problems of inefficiency due to the additional step of inserting the leaving group required for alkylation with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
CN 101781243 discloses an alternative iloperidone synthesis process as reported in Scheme 4.

Said process is not advantageous compared with the preceding processes as the intermediate with the oxime group, due to the nature of this functional group, is particularly liable to degradation due to the action of numerous factors such as the presence of metals, acid pHs and basic pHs.
CN101768154 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
CN 101735208 describes a process for the synthesis of iloperidone comparable to the one reported in CN 101781243, namely through the intermediate with the functional oxime group.
IN 2007MU01980 discloses a process for the synthesis of iloperidone comparable to the one reported in USRE39198 from two isolated intermediates, 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone and 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride.
WO 2010031497 describes an alternative iloperidone synthesis process as reported in Scheme 5.

The considerable economic disadvantage of the process reported in WO2010031497 is based on the fact that by reversing the order of alkylation and performing that of intermediate 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride first, a greater loss of yield is generated on this intermediate which, according to the literature, is more difficult to synthesise and consequently more expensive than the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone, with a globally greater economic inefficiency of the iloperidone preparation process.
CN 102212063 discloses a process for the synthesis of iloperidone with the same arrangement of the synthesis steps as patent application WO 2010031497.
WO2011154860 describes a process for the synthesis of iloperidone wherein a phase transfer catalyst is used to prepare the intermediate 1-[4-(3-chloropropoxy)-3-methoxyphenyl]ethanone which, as in all the other preparation examples previously described, is crystallised, isolated and dried before use in the next step with 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole hydrochloride. Scheme 6 shows the synthesis scheme of the process of WO2011154860.

………………………………

……………………………………
EXAMPLE 1:
Tetrabutyl ammonium bromide (2.40 gm) was added to a stirred solution of Potassium hydroxide (0.724 kg) in mixture of Heptane (2.0L). and water (10.0L), followed by addition of 1- [4-(3-chloropropoxy)-3-methoxyphenyl]ethanone (2, 1.0kg) and 6-fluoro-3-piperidin-4-yl-l,2- benzisoxazole hydrochloride^, 1.1 1kg) at 30°C. This reaction mass was stirred for 15 to 20 min. The temperature of the reaction mass was raised to 70°C and was maintained for 8 to 10 hours. After completion of reaction (by TLC, Mobile Phase: Toluene/ Acetone/Ethyl acetate = 6:2:2 mL), the mixture was cooled to 30°C, diluted with dichloromethane (2.5 L) and stirred for 30 minutes. The dichloromethane layer was separated. The aqueous layer was re-extracted with dichloromethane (1.0L). The combined dichloromethane layer was washed with water (1.5L) and decolorized with activated charcoal (0.05 kg). The solvent was distilled off completely to obtain the residue. The residue obtained was dissolved in isopropyl alcohol (5.0L) at reflux temperature to obtain the clear solution. The clear solution obtained was cooled to 30°C followed by 0°C and stirred for 60 min to precipitate out crystals. The colorless crystals of compound (I) obtained were filtered. The crystalline solid was dried under vacuum (650-700 mm/Hg) to obtain pure compound (I) as a crystalline solid. HPLC analysis was performed for the crystalline solid obtained. The purity of Iloperidone, impurity profile and yield are shown in table 1 below.
Table 1 : Analysis data of iloperidone i.e. purity, yield and impurity profile.

EXAMPLE 2:
Tetrabutyl ammonium bromide (2.40 gm) was added to a stirred solution of Potassium hydroxide (0.724 kg) in mixture of Heptane (2.0L) and water (10.0L), followed by addition of 1- [4-(3-chloropropoxy)-3-methoxyphenyl]ethanone (2, 1.0kg) and 6-fluoro-3-piperidin-4-yl-l,2- benzisoxazole hydrochloride^, 1.1 1kg) at 30°C. This reaction mass was stirred for 15 to 20 min. The temperature of the reaction mass was raised to 70°C and maintained for 8 to 10 hours. After completion of reaction (by TLC, Mobile Phase: Toluene/ Acetone/Ethyl acetate = 6:2:2 mL), the mixture was cooled to 30°C, the reaction mixture was filtered to obtain wet crude iloperidone. Further, the obtained wet crude was dried at 60-65 °C under vacuum to furnish crude iloperidone (1.72 kg). The dried crude iloperidone was dissolved in isopropyl alcohol (5.0 L) at reflux temperature and decolorized with activated charcoal (0.05 kg). Obtained filtrate was cooled to 30°C followed by 0°C and stirred for 60 min to precipitate out crystals. The colorless crystals of compound (I) obtained were filtered. The crystalline solid was dried under vacuum (650-700 mm/Hg) to obtain pure compound (I) as a crystalline solid. HPLC analysis was performed for the crystalline solid obtained. The purity of Iloperidone, impurity profile and yield are shown in table 2 below.
Table 2: Analysis data of iloperidone i.e. purity, yield and impurity profile.

EXAMPLE-3:
……………………..
UPLC-MS [M+H+]=427
1H-NMR (in DMSO) (chemical shifts expressed in ppm with respect to the TMS signal): 2.06-1.78 (6H, m); 2.13 (2H, m); 2.49 (2H, t); 2.52 (2H, m); 2.97 (2H, m); 3.11 (1H, tt); 3.83 (3H, s); 4.12 (2H, t); 7.06 (1H, d); 7.22 (1H, m); 7.46 (1H, d); 7.61-7.58 (2H, m); 7.94 (1H, dd).
………………………………
.Improved and Efficient Process for the Production of Highly Pure Iloperidone: A Psychotropic Agent
http://pubs.acs.org/doi/full/10.1021/op400335p?prevSearch=iloperidone&searchHistoryKey=

The present work describes an improved and highly efficient process for the synthesis ofiloperidone (1), an antipsychotic agent, which is free from potential impurities. The synthesis comprises N-alkylation of 1-(4-(3-chloropropoxy)-3-methoxyphenyl)ethanone (4) with 6-fluoro-3-piperidin-4-yl-1,2-benzisoxazole hydrochloride (5) in a mixture of water and heptane as solvent and sodium hydroxide as a base in the presence of tetrabutylammonium bromide as a phase transfer catalyst to yield iloperidone (1) with a yield of around 95% and a purity of 99.80% by HPLC. The present work also describes the optimization details performed to achieve the process attributes responsible for high yield and purity.
FT-IR (KBr, λmax, cm–1): 3031, 2949, 2779, 2746, 2822, 1669, 1614, 1585, 1510, 1462, 1448, 1415, 1380, 1313, 1262, 1221, 1177, 1150, 1123, 1077, 1034, 997, 985, 955, 884, 876, 853, 812, 781, 643, 610, 569, 475.
1H NMR (CDCl3): δ 2.03–2.10 (m, 6H), 2.12–2.18 (m, 2H), 2.55–2.56 (s, 3H), 2.58–2.60 (t, 2H), 3.02–3.09 (m, 3H), 3.91 (s, 3H), 4.10–4.19 (t, 2H), 6.91–6.93 (d, 1H), 7.01–7.06 (dd, 1H), 7.21–7.24 (dd, 1H), 7.51–7.52 (d, 1H), 7.53–7.56 (dd, 1H), 7.69–7.65 (dd, 1H).
13C NMR (CDCl3): 26.02, 26.40, 30.36, 34.34, 53.36, 54.90, 55.80, 67.16, 97.04, 97.31, 110.20, 111.02, 111.98, 112.23, 117.12, 122.36, 122.46, 123.06, 130.11, 149.00, 152.66, 160.91, 162.60, 163.53, 163.66, 165.09, 198.59.
MS (ESI, m/z): 427.2 [M + H].+
Anal. Calcd (%) for C24H27FN2O4(426.48): C, 67.54; H, 6.33; found (%): C, 67.24; H, 6.18.
HPLC
HPLC analysis developed at Megafine India using a Hypersil BDS C18 column (250 mm × 4.6 mm, particle size 5 μm); mobile phase A comprising a mixture of 5.0 mM ammonium dihydrogen orthophosphate buffer and 0.1% triethylamine; mobile phase B comprising a mixture of acetonitrile/methanol in the ratio 80:20 v/v; gradient elution: time (min)/A (v/v): B (v/v); T0.01/65:35, T8.0/65:35, T25.0/35:65, T35.0/35:65, T37.0/65:35, T45.0/65:35; flow rate 1.0 mL/min; column temperature 30 °C; wavelength 225 nm. The observed retention time of iloperidone under these chromatographic conditions is about 17.0 min.
…….
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=25_10_2
N oxide impurity
m.p. 155-157 ºC;
FT-IR (KBr, νmax, cm-1):
3083, 2958, 2878, 1655, 1606, 1584, 1509, 1467, 1419, 1348,1273, 1223, 1182, 1143, 1121, 1032, 971, 957, 881, 857, 813,
802;
1H NMR (300 MHz, CDCl3)
δ 1.89-1.93 (m, 2H), 2.31-2.40 (m, 2H), 2.55 (s, 3H), 2.60-2.72 (m, 2H), 3.29-3.52 (m,
2H), 3.29-3.52 (m, 2H), 3.29-3.52 (m, 2H), 3.29-3.52 (m, 1H),3.85 (s, 3H), 4.23(t, 2H, J = 6.0 Hz), 7.11 (d, 1H, J = 8.4 Hz),7.30-7.36 (m, 1H), 7.62-7.65 (m, 1H), 7.71-7.74 (dd, J = 9.3and 2.0 Hz, 1H), 8.02-8.07 (dd, J = 8.7 and 5.4 Hz, 1H);
13CNMR (75 MHz, CDCl3)
δ 22.13, 24.70, 26.35, 31.49, 55.54,63.21, 67.07, 67.82, 97.51, 110.35, 111.86, 112.67, 123.11,
123.67, 129.95, 148.63, 152.22, 160.79, 163.10, 163.69,196.40;
MS (ESI, m/z): 443 [M + H]+.
Anal. calcd. (%) forC24H27N2O5F (442.19): C, 65.15; H, 6.15; N, 6.33; found (%):C, 65.11; H, 6.09; N, 6.29.
………………………
INTERMEDIATES

FANAPT is a psychotropic agent belonging to the chemical class of piperidinyl-benzisoxazole derivatives. Its chemical name is 4′-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)piperidino]propoxy]-3′-methoxyacetophenone. Its molecular formula is C24H27FN2O4 and its molecular weight is 426.48. The structural formula is:
 |
Iloperidone is a white to off-white finely crystalline powder. It is practically insoluble in water, very slightly soluble in 0.1 N HCl and freely soluble in chloroform, ethanol, methanol, and acetonitrile.
|
Title: Iloperidone
CAS Registry Number: 133454-47-4
CAS Name: 1-[4-[3-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]propoxy]-3-methoxyphenyl]ethanone
Manufacturers’ Codes: HP-873; ILO-522
Trademarks: Zomaril (Novartis)
Molecular Formula: C24H27FN2O4
Molecular Weight: 426.48
Percent Composition: C 67.59%, H 6.38%, F 4.45%, N 6.57%, O 15.01%
Literature References: Combined dopamine (D2) and serotonin (5HT2) receptor antagonist. Prepn: J. T. Strupczewski et al., EP402644; eidem, US 5364866 (1990, 1994 both to Hoechst-Roussel); eidem, J. Med. Chem. 38, 1119 (1995).
Pharmacology: M. R. Szewczak et al., J. Pharmacol. Exp. Ther. 274, 1404 (1995).
Clinical pharmacokinetics: S. M. Sainati et al., J. Clin. Pharmacol.35, 713 (1995).
HPLC determn in plasma: A. E. Mutlib, J. T. Strupczewski, J. Chromatogr. B 669, 237 (1995). Receptor binding study: S. Kongsamut et al., Eur. J. Pharmacol. 317, 417 (1996).
Review of pharmacology and therapeutic potential in schizophrenia: J. M. K. Hesselink, Curr. Opin. Cent. Peripher. Nerv. Syst. Invest. Drugs 2, 71-78 (2000); K. K. Jain, Expert Opin. Invest. Drugs 9, 2935-2943 (2000).
Properties: Crystals from ethanol, mp 118-120°.
Melting point: mp 118-120°
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzisoxazoles; Serotonin-Dopamine Antagonist.
|
..
-
Bjork, A. K. K.; Abramo, A. L.; Kjellberg, B. E. S. US 4366162, 1982.
-
Strupczewski, J. T.; Helsley, G. C.; Chiang, Y.; Bordeau, K. J. EP 0402644A1, 1990.
-
Ansari, S. A.; Hirpara, H. M.; Yadav, A. K.; Gianchandani, J. P. WO2012164516, 2012.
-
Azad, M. A. K.; Pandey, G.; Singh, K.; Prasad, M.; Arora, S. K. WO2012/090138 A1, 2012.
-
Dwivedi, S. D.; Patel, D. J.; Shah, A. P. WO2012/063269, 2012.
-
Athalye, S. S.; Parghi, K. D.; Ranbhan, K. J.; Sarjekar, P. B. WO2012/153341, 2012.
-
Raman, J. V.; Rane, D.; Kevat, J.; Patil, D. WO2011/154860, 2011.
-
Reguri, B. R.; Arunagiri, M.; Yarroju, P. C.; Kasiyappan, G. S.; Ponnapall, K. WO2011/055188, 2011.
-
Shiwei, Z.; Feng, J. US 2012/0172699A1, 2012.
-
Bettoni, P.; Roletto, J.; Paissoni, P. EP 2644608A1, 2013.
-
Mathad, V. T.; Solanki, P. V.; Pandit, B. S.; Uppelli, S. B. WO2012/123963 A2, 2012.
-
Strupczewski, J. T.; Allen, R. C.; Gardner, B. A.; Schmid, B. L.; Stache, U.; Glamkowski, E. J.; Jones, M. C.; Ellis, D. B.; Huger, F. P.; Dunn, R. W. J. Med. Chem. 1985, 28, 761–769
[ACS Full Text
 ], [PubMed], [CAS]
], [PubMed], [CAS]-
20 Mucke, H. A. M.; Castañer, J. Drugs Future 2000, 25(1), 29.
-
(21) Steiner, G.; Bach, A.; Bialojan, S.; Greger, G.; Hege, H.-G.;.Höger, T.; Jochims, K.; Munschauer, R.; Neumann, B.; Teschendorf, H.-J.; Traut, M.; Unger, L.; Gross, G. Drugs Future 1998 23(2), 191.
WO2003037337A1 * Oct 29, 2002 May 8, 2003 Markus Ahlheim Depot formulations of iloperidone and a star polymer WO2008027993A2 * Aug 29, 2007 Mar 6, 2008 Eurand Inc Drug delivery systems comprising solid solutions of weakly basic drugs CN101768154A Sep 19, 2009 Jul 7, 2010 浙江华海药业股份有限公司 New preparation method of iloperidone EP0402644A1 May 16, 1990 Dec 19, 1990 Hoechst-Roussel Pharmaceuticals Incorporated N-(aryloxyalkyl)heteroarylpiperidines and -heteroarylpiperazines,a process for their preparation and their use as medicaments EP0542136A1 * Nov 5, 1992 May 19, 1993 Hoechst-Roussel Pharmaceuticals Incorporated Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analgetics US5364866 Oct 30, 1992 Nov 15, 1994 Hoechst-Roussel Pharmaceuticals, Inc. Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analetics US5663449 Jun 6, 1995 Sep 2, 1997 Hoechst Marion Roussel, Inc. Intermediate compounds in the synthesis of heteroarylpiperidines, pyrrolidines and piperazines USRE39198 Nov 15, 2000 Jul 18, 2006 Aventis Pharmaceuticals Inc. Heteroarylpiperidines, pyrrolidines and piperazines and their use as antipsychotics and analgesics -
IRBESARTAN

IRBESARTAN, SR 47436, BMS-186295
Avapro® (Bristol-Myers Squibb) and Karvea®
(Sanofi-Winthrop)
2-butyl-3-({4-[2-(2H-1,2,3,4-tetrazol-5-yl)phenyl]phenyl}methyl)-1,3-diazaspiro[4.4]non-1-en-4-one
138402-11-6 CAS NO
U.S. Patents 5,270,317 and 5,352,788, 6,162,922
The compound prepared according to US 5270317 is polymorph A
-
Irbesartan is known by following chemical names:
- (a) 2-Butyl-3-[[2′-(1H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (b) 2-Butyl-3-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]-1,3-diazaspiro[4,4]non-1-en-4-one
- (c) 2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl) methyl]-2-imidazolin-5-one.
-
The structural formula of Irbesartan is represented below.

Irbesartan
-
The synthesis of irbesartan is first disclosed in US5270317 (equivalentEP0454511 ) and subsequently, several other patents disclose the synthesis of irbesartan by different methods. Basically the synthesis of this molecule involves two common intermediates namely spiroimidazole and substituted 4′-bromomethylbiphenyl.
-
US 5270317 describes preparation of irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin -5-one which is reacted with tributyltin azide in xylene at reflux temperature for 66 hours to give a product which is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol/THF mixture using 4N hydrochloric acid to get irbesartan.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
Irbesartan (INN) /ɜrbəˈsɑrtən/ is an angiotensin II receptor antagonist used mainly for the treatment of hypertension. Irbesartan was developed by Sanofi Research (now part ofsanofi-aventis). It is jointly marketed by sanofi-aventis and Bristol-Myers Squibb under thetrade names Aprovel, Karvea, and Avapro.
It is marketed in Brazil by Sanofi-Aventis under the trade name Aprovel .

As with all angiotensin II receptor antagonists, irbesartan is indicated for the treatment ofhypertension. Irbesartan may also delay progression of diabetic nephropathy and is also indicated for the reduction of renal disease progression in patients with type 2 diabetes,[1]hypertension and microalbuminuria (>30 mg/24 hours) or proteinuria (>900 mg/24 hours).[2]
Irbesartan is also available in a combination formulation with a low dose thiazide diuretic, invariably hydrochlorothiazide, to achieve an additive antihypertensive effect. Irbesartan/hydrochlorothiazide combination preparations are marketed under similar trade names to irbesartan preparations, including Irda, CoIrda, CoAprovel, Karvezide,Avalide and Avapro HCT.
A large randomized trial following 4100+ men and women with heart failure and normal ejection fraction (>=45%) over 4+ years found no improvement in study outcomes or survival with irbesartan as compared to placebo.[3]

BMS annual sales approx $1.3bn. Sanofi-aventis annual sales approx $2.1bn. In the United States, a generic version is available. Patent expired March 2012.
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. (2001). “Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes”. N Engl J Med 345 (12): 851–60. doi:10.1056/NEJMoa011303.PMID 11565517.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, Anderson S, Donovan M, Iverson E, Staiger C, Ptaszynska A (December 2008). “Irbesartan in patients with heart failure and preserved ejection fraction”. N. Engl. J. Med. 359 (23): 2456–67.doi:10.1056/NEJMoa0805450. PMID 19001508.
4……….C. A. Bernhart, P. M. Perreaut, B. P. Ferrari, Y. A. Muneaux,
J.-L. A. Assens, J. Clement, F. Haudricourt, C. F. Muneaux,
J. E. Taillades, M.-A. Vignal, J. Gougat, P. R. Guiraudou, C.
A. Lacour, A. Roccon, C. F. Cazaubon, J.-C. Brelihre, G. Le
Fur, D. Nisato, J. Med. Chem. 1993, 36, 3371–3380.
5…. K. F. Croom, M. P. Curran, K. L. Goa, Drugs 2004 64,
999–1028.
6… C. Bernhard, J.-C. Breliere, J. Clement, D. Nisato, P. M. Perreaut, C. F. Muneaux, (Elf Sanofi) US 5 270 317; Chem. Abstr. 1993, 119, 95560.
7. S. Chava, M. Bandari, K. S. Mathuresh, (Matrix Laboratories) WO 2005/122699; Chem. Abstr. 2005, 144, 88292.
5. S. Zupan~i~, A. Pe~avar, R. Zupet, (Krka) WO 2006/073376;
Chem. Abstr. 2006, 145, 124576.
8. C. V. Kavitha, S. L. Gaonkar, J. N. Chandra, S. Narendra, C.
T. Sadashiva, K. S. Rangappa, Bioorg. Med. Chem. 2007, 15,
7391–7398.
9. S. Rádl, J. Stach, O. Klecán, (Zentiva) WO 2005/021535;
Chem. Abstr. 2005, 142, 298118.
10. B. Satyanarayana, Y. Anjaneyulu, P. Veerasomaiah, P. P.
Reddy, Heterocycl. Commun. 2007, 13, 223–228.
11. V. V. Korrapati, P. Rao, R. Dandala, V. K. Handa, I. V. S. Rao,
A. Rani, A. Naidu, Synth. Commun. 2007, 37, 2897–2905.
12. J. Havlí~ek, Z. Mandelová, R. Weisemann, I. Strˇelec, S.
Rádl, Collect. Czech. Chem. Commun. 2009, 77, 347.
Irbesartan of formula (I).

The chemical name of Irbesartan is 2-Butyl-3-[[2′-(lH-tetrazol-5-yl)[l,l’-biphenyl]-4- yl]methyl]-l,3-diazaspiro[4,4]non-l-en-4-one and formula is C2SH2SN6O and molecular weight is 428.53. The current pharmaceutical product containing this drug is being sold by Sanofi Synthelabo using the tradename AVAPRO, in the form of tablets. Irbesartan is useful in the treatment of diabetic neuropathy, heart failure therapy and hypertension. Irbesartan is angiotension II type I (AΙIi)-receptor antagonist. Angiotension II is the principal pressor agent of the rennin-angiotension system and also stimulates aldosterone synthesis and secretion by adrenal cortex, cardiac contraction, renal resorption of sodium, activity of the sympathetic nervous system and smooth muscle cell growth. Irbesartan blocks the vasoconstrictor and aldosterone- secreting effects of angiotension II by selectively binding to the ATi angiotension II receptor. U.S. Pat. Nos. 5,270,317 and 5,559,233 describes a process for the preparation of N- substituted heterocyclic derivatives which involves reacting a heterocyclic compound of the formula

with a (biphenyl-4-yl)methyl derivative of the formula

wherein R1, R2, R3, R4, R5, and t, z and Hal have the meanings given in said U.S. Pat. No.
5,270,317, in the presence of an inert solvent such as DMF, DMSO or THF, with a basic reagent, for example KOH, a metal alcoholate, a metal hydride, calcium carbonate or triethylamine. The products of the reaction were purified by chromatography.
U.S. Pat. Nos. 5,352,788, and 5,559,233, and WO 91/14679 also describe identical alkylation of the nitrogen atom of the heterocyclic compound with the halo-biphenyl compound using the same inert solvent and the same basic reagents.
-
US5629331 describes a process for the preparation of irbesartan from 1-[(2′-cyanobiphenyl)4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one using sodium azide, TEA.HCl in N-methylpyrrolidone. The product is isolated from the alkaline reaction mass after acidification to pH 4.7 to 5.8 and the crude product is recrystallised from IPA/water to get Form A and ethanol/water to get Form B.
-
WO 2005/051943 A1 describes a process for the preparing irbesartan wherein 1-[(2′-cyanobiphenyl-4-yl)methyl]-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with tributyltin chloride, sodium azide and TBAB in toluene at reflux temperature for 20 hours. Product is isolated from the reaction mass as trityl irbesartan and then deprotected in methanol and formic acid to get irbesartan.
-
WO 2006/023889 describes a method for preparing irbesartan, wherein 1-(2′-cyanobiphenyl-4-yl)methyl)-2-n-butyl-4-spirocyclopentane-2-imidazolin-5-one is reacted with sodium azide and triethylamine hydrochloride in N-methyl-2-pyrrolidone to give irbesartan.
-
WO 2005/113518 describes a process for preparing irbesartan wherein cyano irbesartan in xylene, is reacted with tributyltin chloride and sodium azide at reflux temperature till reaction is completed followed by aqueous work-up and recrystallization to give irbesartaN
-
The process involving use of zinc salt for the transformation of nitrile to tetrazole is a safe and efficient process as reported in JOC (2001) 66, 7945-50. The use of zinc salt for transforming nitrile to tetrazole has also been published in WO9637481 and US5502191
Also Canadian Patent No. 2050769 describes the alkylation of the nitrogen atom of the heterocycle of the formula
with a compound of the formula

wherein X, R1, Z1 and Z6 have the meanings given therein, in the presence of N,N- dimethylformamide and a basic reagent, such as alkali metal hydrides for example sodium or potassium hydride.
All of the above identified patents describe alkylation in solvents, such as N5N- dimethylformamide or DMSO, etc. in the presence of a basic reagent, for example, a metal hydride or a metal alcoholate etc. The strong bases, such as metal hydride or a metal alcoholate require anhydrous reaction conditions. Since N,N-dimethylformamide is used as a solvent, its removal requires high temperature concentration by distillation, which can result in degradation of the final product. The product intermediate is also purified by chromatography which is commercially not feasible and cumbersome on large scale. Another process given in Canadian Patent No. 2050769 provides synthetic scheme as herein given below.

This process comprises the steps of protecting carboxylic group present on cyclopentane ring which is deprotected in consecutive step by vigourous hydrogenation condition in autoclave which is operationally difficult at a large scale.
US Patent No. 2004242894 also discloses the process of preparation of lrbesartan from 4- bromomethyl biphenyl 2′-(lH-tetrazol (2-triphenylmethyl) 5-yl) and Ethyl ester of 1- Valeramido cyclopentanecarboxylic acid in toluene in presence of base and PTC, and then hydrolyzing the protecting group. However this requires chromatographic purification.
This patent also discloses the process of preparation of tetrazolyl protected lrbesartan using 2,6 lutidine and oxalylchloride in toluene. However in this process the yield is as low as 30%.
US Patent No. 2004192713 discloses the process of preparation of lrbesartan by condensing the two intermediates via Suzuki coupling reaction. The reaction scheme is as given herein below.

However, this process has several disadvantages such as use of the reagents like butyl lithium and triisobutyl borate at low temp such as -20 to -30°C under Argon atmosphere condition which is difficult to maintain at commercial scale.
WO2005113518 discloses the process of preparation of Irbesartan by condensing n- pentanoyl cycloleucine (V) with 2-(4-aminomethyl phenyl) benzonitrile (VI) using dicyclocarbodiimide (DCC) and 1 -hydroxy benzotriazole as catalyst to give an open chain intermediate of formula (VIII) which is then cyclized in the presence of an acid, preferably trifluoro acetic acid to give cyano derivative of formula (VII) and which in turn is converted to Irbesartan by treating it with tributyl tin chloride and sodium azide.

In this application further describes another process comprising the steps of reacting 2- butyl-l,3-diazasρiro[4,4]non-l-en-4-one monohydrochloride (A) with 4-bromobenzyl bromide (B) in presence of base and solvent to give 3-[4-bromobenzyl]-2-butyl-l,3- diazaspiro[4,4]non-l-en-4-one (C) which is condensed with 2-[2′-(triphenylmethyl-2’H- tetrazol-5′-yl)phenyl boronic acid in the presence of tetrakis triphenyl phosphine palladium and base to give lrbesartan (I). However these processes suffer with several disadvantages such as it uses trifluoroacetic acid for the cyclization step which is highly corrosive material. The process requires an additional step of activation by DCC. This step not only increases number of steps but also create problem in handling DCC at an industrial scale as it is highly prone to hazard which makes the process least preferred on a large scale production of lrbesartan. Further it uses phenyl boronic acid derivative and triphenyl phosphine complex which are harmful for the skin and eye tissue and also harmful for respiratory system. Tetrakis triphenyl phosphine palladium is also a costly material which increases overall cost for the production of lrbesartan. Moreover the yield is as low as 22%. All the above patents/applications are incorporated herein as reference. In summary, prior art relating to the process for the preparation of lrbesartan suffers with several drawbacks such as i) It requires chromatographic purification of intermediates at various stages. ii) It requires specific autoclave conditions for a deprotection of protecting group. iii) It requires maintaining low temperature conditions such as -300C and requires special handling care and air and moisture tight condition with the reagents such as butyl lithium and triisobutyl borate. iv) It uses hazardous and highly corrosive reagents, v) It suffers low yield problem. vi) All the process is having more number of reaction steps.
- Irbesartan is described in Bernhart et al., U.S. Patent No. 5,270,317
-
Irbesartan, is a potent, long-acting angiotensin II receptor antagonist which is particularly useful in the treatment of cardiovascular ailments such as hypertension and heart failure. Its chemical name is2-n-butyl-4-spirocyclopentane-1-[(2′-(tetrazol-5-yl)biphenyl-4-yl)methyl]-2-imidazolin-5-one.
Irbesartan is an antihypertensive agent known from EP 454511. From EP 708103, which discloses their X-ray spectra, two polymorphs are known where form A can be produced form a solvent system containing less than 10% of water, while Form B from a system with more than 10% of water. The specific morphological variant of form A can be prepared having properties as disclosed in EP 1089994. Additional form has been disclosed in WO 04089938. Amorphous irbesartan is known from WO 03050110. It is said that Irbesartan produced as taught in EP 454511 is a fluffy material with relatively low bulk and tap densities and undesirable flow characteristics, which consequently has unadvantageous electrostatic properties, among them a high chargeability as measured by tribugeneration between -30 and -40 nanocoulomb/g (10‘9As/g). Alternativelyirbesartan could be prepared by complex process using sonifications and/or temperature oscillations according to EP 1089994 to exhibit a chargeability as measured by tribugeneration between -0 and -10 nanocoulomb/g.
According to EP 454511 a solid composition in form of tablets is prepared by mixing the active ingredient with a vehicle such as gelatine, starch, lactose, magnesium stearate, talc, gum Arabic or the like and can be optionally coated. The compositions containing from 20% to 70% by weight of irbesartan are known from EP 747050.
WO 04/007482 teaches the acidification to pH 2 – 3,5 of trityl irbesartan, which is sufficient to remove the protecting group, but not to convert into an acid addition salt; WO 04/065383 is likewise silent on hydrohalide acid addition salts. WO
06/011859 relates to the preparation of a hydrochloride salt of irbesartan in order to incorporate it into a pharmaceutical formulation. W099/38847 mentions optional conversion of irbesartan into hydrochloride, hydrobromide or hydrogen sulfate salts
……………………………………………
…………………

Example 1Preparation of Compounds of formula IVa and IVb:
-

-
A jacketed 1,000 mL 3-neck flask was charged with 4′-methylbiphenyl-2-carbonitrile (Compound 1, 100.0 g) and CH2CI2 (500 mL) under nitrogen. To a 500 mL Erlenmeyer flask with magnetic stirrer, sodium bromate (NaBrO3; 31.2 g) was dissolved in water (170 mL). The NaBrO3 solution was transferred to the 1,000 mL flask and the reaction mixture was cooled to about 5 °C or less. Aqueous HBr solution (48 %, 105.0 g) was added to the 1,000 mL flask and the resulting reaction mixture was recycled though a UV lamp reactor. The reaction mixture was kept at 0-20 °C and the recycling was continued until the reaction was deemed complete by HPLC. Optionally, additional sodium bromate and hydrogen bromide may be added. The relative amounts of Compound 2 and Compound 3 were about 80-90% and about 10-20% respectively. Aqueous sodium metabisulfite solution (2.0 g of in 10 mL water) was added to the reaction mixture. Allow the phases to settle and the methylene chloride phase was washed with water and used in the next step without further purification.
Example 2Preparation of Compound II:
-

-
A 1L 3-neck flask was charged with Compound V (134.0 g), MTBAC (5.0 g) and CH2Cl2 (170 mL) and cool to -5 to 5 °C. An aqueous solution of KOH (182.6 g in 212 mL water) was added slowly to the 1L flask and the reaction temperature was kept at ≤ 5 °C. The methylene chloride solution of Compound IVa and Compound IVb from Example 1 was added to the reaction mixture slowly, while maintaining the temperature at 0-10 °C. Diethyl phosphite (39.66g) was added drop wise at 0-10 °C. Check the reaction mixture for completion of the reduction reaction, and additional diethyl phosphite may be added.
-
The reaction mixture was allowed to warm to ambient (20-30 °C) and agitated until the reaction was deemed complete by HPLC. Water (150 mL) was added and the phases were separated. The organic layer was extracted with water (230 mL) and polish filtered.
-
The methylene chloride (which contained the crude Compound II) was distilled off and exchanged with about 400 mL of methyl tert-butyl ether (MTBE) (optionally, the MTBE recycled from washing below can be used here). Upon cooling, crystallization occurred (optionally seeds were added) and after further cooling to below 25°C, crystals of Compound II were isolated, washed with MTBE and dried in vacuum at a temperature of less than 60°C. HPLC retention time: 18.126 min. Typically, the yield was about 85 to about 88%. Alternatively, IPA could be used as the crystallization and washing solvent
-
Optionally, the solvent (i.e., MTBE or IPA) used to wash the crystals of Compound II above can be recycled and used to crystallize the crude Compound II in the next batch. Since the washed solvent contains Compound II as well as impurities, it was surprisingly found that the washed solvent can be recovered and used again in crystallizing the crude compound of formula II in the next batch without sacrificing its purity while increasing its yield.
Example 3Preparation of Compound I:
-

-
A reactor was charged with Compound II (1 kg), triethylamine chlorhydrate (0.713 kg), sodium azide (0.337 kg) and N-methyl pyrrolidinone (2.07 kg), and the reaction mixture was heated to about 122°C under stirring. After completion of the reaction as determined by HPLC, the reaction mixture was cooled to about 45°C, and an aqueous solution of sodium hydroxide (35%, 5.99 kg) and water (3.0 kg) were added, the resulting mixture was stirred at a temperature between about 20 and about 40°C for about 0.5 hours. The aqueous phase was discarded and the organic phase was treated with toluene (1.73 kg) and water (5.0 kg), and stirred for about 0.5 hours at about 20 – about 30°C. The toluene phase was discarded and the aqueous phase was washed with ethyl acetate (1.8 kg) and treated with aqueous HCl until pH was adjusted to about 4.8 – about 5.2. Precipitation occurred and the resulting suspension was stirred for about 1 hour at about 20 – about 25°C. The precipitation was collected and washed with water three times (1.0 kg x 3). The crude wet product was recrystallized using a mixture of iso-propanol (0.393 kg) and water (4.5 kg). HPLC retention time: 11.725 min. The yield for Compound I was about 87%.
…………………………………………….
SPECTRAL DATA
The ESI mass spectrum of irbesartan showed a protonated molecular ion peak at m/z 429.3 confirming the molecular weight 428. The fragmentation pattern of parent ion 429.3 showed the fragment ions at m/z 385.9, 235.1, 207, 195.4, 192.1, 180.2 and 84
The FT-IR spectrum exhibited a characteristic stretching absorption band at 1732 cm-1 for the carbonyl group of amide functionality. The presence of this band at higher frequency was due to the ring stretching due to five member ring system. Another band at 1614cm-1 was due to C=N stretching vibrations
1H and 13C- NMR were recorded using DMSO-d6 as a solvent. In 1H-NMR the signal due to tetrazole NH proton was not detected may probably due to the tautomerism.
SEE
http://orgspectroscopyint.blogspot.in/2013/12/irbesartan-spectral-data.html
DP 1 IS IMPURITY
………………………………………….
NMR
1H-NMR (DMSO d6): δppm 0.78 (t, 3H); 1.17-1.30 (sex, 2H); 1.40-1.50 (quent, 2H); 1.64-1.66 (m, 2H); 1.80-1.82 (m, 6H); 2.22-2.29 (t, 2H); 4.67 (s, 2H); 7.07 (s, 4H); 7.50- 7.68 (m, 4H) M+: 429.6
,…………………..
m.p:181-182oC,
IR (KBr, cm-1) 1732 (C=O), 1616 (C=N); 1H NMR (DMSO-d6): δ 7.95–7.32 (m, 8 H), 4.80 –4.60 (s, 2 H), 3.60– 3.00 (br s, 1 H), 2.40– 2.20 (t, 2 H , J = 6.04 Hz), 2.00– 1.60 (m, 8 H),1.60–1.45 (quint, 2 H), 1.40– 1.20 (sext, 2 H), 0.91–0.70 (t, 3H, J = 7.41 Hz);
13C-NMR (DMSOd6): δ 186.5, 162.0,155.9, 141.9, 139.2, 137.2. 131.9, 131.4, 130.1, 128.7, 127.1, 124.3, 76.7, 43.1,
37.7, 28.3, 27.4, 26.3, 22.4, 14.5;
MS: m/z= 429 [M+1];
Anal. Calcd for C25H28N6O : C, 70.07; H,
6.59; N, 19.61. Found: C, 70.04; H, 6.57; N, 19.58.
http://www.acgpubs.org/OC/2011/Volume%204/Issue%201/13-OC-1106-199.pdf
………………………………………………..
1H NMR in DMSO-D6 : 7.68 (d. 2H, Ar-H), 7.52 (d, 2 H, Ar-H), 7.08 (s, 4 H, Ar-H), 4.68(s, 2H, -CH2), 2.69(t,2H,-CH2),2.18(m,2H,-CH2),1.83(m,2H,-CH2),1.81 (t, 2H, -CH2), 1.65 (t, 2H, -CH2), 1.45 (m, 2 H, -CH2), 1.24(m , 2H, -CH2), 0.77 (t, 3H, -CH3),
IR (KBR): 3061 (Aromatic C-H stretching), 2960 (Aliphatic C-H stretching), 3443 (N-H stretching), 1733 (C=0 stretching), 1617(CN stretching), 1337.99(CN stretching), 1407(N=N stretching) cm“1.
……………………….
HPLC condition:
Column: Alltima C18 (Alltech 88050) 15.0cm in length x 4.6mm in internal diameter and 5 micron particle size;
Column temperature: 40 C;
Solvent A: Buffer solution A 1.1 g of heptanesulfonic acid in 1 liter of water and adjust the pH to 2.5;
Solvent B: Methanol Flow rate: 1.2mL/min;
Gradient Elution Condition:
Time% A % %B
0 min 50 50
35 min 15 85
Detector: 240 nm;
Injection volume: 10 uL.
The chromatographic purity of
the compounds was analyzed using Agilent 1200 series HPLC instrument under the following conditions:
Column : Symmetry C18, 4.6 × 75 mm, 3.5 µm
Mobile phase : Eluent A: Deionized water, Eluent B: HPLC grade Methanol
Chromatographic Conditions
a. Column temperature : Ambient
b. Sample compartment : Ambient
c. Detector : 225 nm
d. Injection volume : 10 µL
e. Run time : 45 minutes
f. Flow rate :1.0 mL/min
g. Injector :Auto sampler with variable volume injector
h. Diluent : HPLC grade Acetonitrile
Argatroban

Argatroban
Molecular Formula: C23H36N6O5S
Formula Weight: 508.63
CAS No.: 74863-84-6
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl]
sulfonylamino]pentanoyl]-4-methyl-piperidine-2-
carboxylic acid
PATENT
US 7,589,106, 7,687,516, EP 0008746; US 4258192, US 4201863
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).

Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.
……………………………………………………….
Argatroban monohydrate

Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5-[(aminoiminomethyl) amino]-1-oxo2[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4-methyl-2- piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. cas 141396-28-3
Argipidine, Argatroban monohydrate, GN1600, DK-7419, MDI-805 Acova, Slonnon, Novastan
Mitsubishi Chemical (Originator), Encysive Pharmaceuticals (Licensee), Mitsubishi Pharma (Distributor), Daiichi Pharmaceutical (Codevelopment), GlaxoSmithKline (Codevelopment), Mitsubishi Pharma (Codevelopment), Sanofi-SynthLabo (Codevelopment)
Antithrombocytopenic, CARDIOVASCULAR DRUGS, Cerebrovascular Diseases, Treatment of, HEMATOLOGIC DRUGS, Hematopoiesis Disorders Therapy, Ischemic Stroke, Treatment of, NEUROLOGIC DRUGS, Peripheral Vascular Disease, Treatment of, Stroke, Treatment of, Treatment of Peripheral Obstructive Vascular Disease, Thrombin Inhibitors
Synthesis of argatroban on the method reported in the literature there are two synthetic routes, patent EP8746, US4258192, US4201863, JP8115267 relates to a route is: with 4 – methyl-piperidine as a starting material was prepared first intermediate body (2R, 4R) -4 – methyl-2 – ethyl-piperidine, and the first and a t-BOC protected amino nitro-L-arginine condensation, and then the 3 – methyl – 8 – quinoline sulfonyl chloride condensation after hydrolysis, hydrogenation, hydration be argatroban. This entry route synthesis process complicated procedure to be carried out under the protection of nitrogen, the raw material is highly toxic gas phosgene, the operation more difficult.
US4117127, JP02-212473, EP823430, EP8746, JC S Perk Transl 1981 (5), JP02-212473 relates to an alternative route is: nitro L-arginine prior to the 3 – methyl-8 – subsequent condensation quinoline sulfonyl chloride Intermediate (2R, 4R) -4 – methyl-2 – piperidinecarboxylate condensation, and then after hydrolysis, hydrogenation, hydration be argatroban. This synthetic route despite the relatively simple process method, to obtain raw materials, but this method using reagents such as phosphorus oxychloride, phosphorus trichloride has a pungent odor, easy to absorb moisture in humid air, intense smoke, environmental pollution, greater stimulation of the body’s respiratory tract, can cause eye and skin irritation and burning, and the use of this method, complex operation, low yield, high cost.
Patent CN100586946C Argatroban discloses a method for separating optically active isomeric compounds, the feedstock argatroban mixed solvent of alcohol and water was heated to reflux 5-10 hours, cooled and allowed to stand, and filtered to give White crystalline product, dried, repeated 2-6 times. [0008] Patent CN101033223A discloses a Argatroban is the main by-product (2R, 4R)-l_ [N2-(3_ methyl-8 – quinolinesulfonyl)-L-arginyl] -4 – methyl-2 – carboxylic acid, argatroban, and the byproducts are difficult to isolate, argatroban two diastereomeric isomer 21 (S) and 21 (R) separation of work attracted a lot of research persons. Because both physical and chemical properties are very similar, so separation is very difficult. 1993 Rawson, Thomas E.; VanGorp, Kimmie A.; Yang, Janet so first by high pressure liquid chromatography and column chromatography separation to obtain a single 21 (S) and 21 (R) argatroban [ Journal of Pharmaceutical Sciences vol. 82, No. 6,672]; Thibaudeau Karen et al. reported Protein A chromatographic separation [US6440417]. However, due to the separation of these methods a small amount of low efficiency, so there is no practical value industrialization. 2006 China Tianjin Weijie Technology Co., Ltd. Song Honghai et al. Reported using recrystallization Separation 21 (S) and 21 (R) argatroban way [0 With 951,936 it], so that the mass 21 (5) Aga music classes as possible, but the law of low yield, complicated operation, high cost, and a large amount of a small amount of 21 (S) of 21 (R)-product argatroban, from the viewpoint of industrial production, is still a ideal method. [0009] These methods can be effectively prepared argatroban, but the purity of the desired product is not high, poor color, content is low, affecting the quality of the results of its preparation.
U.S. Pat. No. 4,201,863 (6 May 1980) and EP 8746 (filed on 22 Aug. 1979 with priority based on the application for the cited US patent) describe a class of N2-arylsulphonyl-L-argininamide drugs, with anti-thrombotic activity, and the processes for obtaining them. Of these, the compound 4-methyl-1-[N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid (argatroban, isomers mixture) is described. The described process comprises the synthesis of an intermediate NG-substituted-N2-quinolinesulphonyl-L-argininamide from which the desired compound is obtained by catalyzed hydrogenolysis or acidolysis and catalyzed hydrogenation. The general conditions provided for the hydrogenolysis and hydrogenation reaction are: i) inert solvents (methanol, ethanol, tetrahydrofuran or dioxane); ii) presence of a catalyst (Raney nickel, palladium, platinum, ruthenium, rhodium); iii) hydrogen atmosphere at a pressure between 1 and 100 kg/cm2 and preferably between 5 and 50 kg/cm2; iv) temperature between 0° C. and 200° C. and preferably between 50° C. and 150° C.; v) reaction temperature from 2 hours to 120 hours. The crude product obtained is then purified by trituration or by re-crystallization from diethyl ether-tetrahydrofuran, diethyl ether-methanol or from water-methanol or by chromatography. No example is given of this purification step. In particular, both U.S. Pat. No. 4,210,863 and EP 8746 in example 1(E) describe the preparation of argatroban, isomers mixture. This compound is obtained in amorphous form by hydrogenation of [NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid in ethanol in the presence of Pd/C with hydrogen pressure of 10 kg/cm2 at 100° C. for 8 hours. The catalyst is removed by filtration of the ethanol solution which is then evaporated without further purification and/or re-crystallization steps. In the US patent at issue as indeed in patent application EP 8746, no mention is made of polymorphic forms of the compounds and, for the obtained compound, the following characteristics are reported: Amorphous solid, I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.01; H 6.98; N 16.61.
U.S. Pat. No. 4,258,192 (24 Mar. 1981) (continuation-in-part of the aforesaid patent application U.S. Pat. No. 4,201,863) and the same patent application EP 8746 describe the stereoisomers and the preparation thereof, including argatroban used as an active principle in medicaments, i.e. the stereoisomer (2R,4R)-4methyl-[4N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid, with the following characteristics: melting point (m.p.). 188-191° C.; I.R. (KBr) (cm−1) 3400, 1620, 1460, 1380; Molecular composition (%): theoretical C 54.31; H 7.13; N 16.52; found (%) C 54.05; H 6.94; N 16.65. The compound is prepared according to the description given in examples 1(E) in U.S. Pat. No. 4,258,192 and 2(E) and 3 in EP 8746 respectively by hydrogenation of (2R,4R) 1-[NG-nitro-N2-(3-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid in ethanol in presence of acetic acid catalyzed by Pd/C. After filtering the mass to remove the catalyst, the solvent is evaporated and the residue suspended in chloroform, the solution treated with a saturated sodium bicarbonate solution or 1N sodium hydroxide solution and after washing, the solvent is evaporated. The compound is then re-crystallized from ethanol. Again in this case, no reference is made to the obtainment of monohydrate polymorphic forms.
Said polymorphic forms are described instead in the publication Biochem. Biophys. Res. Comm. 1981, 101, 440-446 in the context of stereoisomer preparation. The monohydrate polymorph of the (2R,4R) stereoisomer is prepared by re-crystallization from ethanol/water and the reported characteristics are: m.p. 176-180° C.; [α]D 27 +76.1° (c 1, 0.2N HCl).
U.S. Pat. No. 5,925,760 (20 Jul. 1999) and EP 0823430 (filed 4 Aug. 1997) subsequently describe a new method for preparing argatroban by means of a new intermediate N2-(3-methyl-8-quinolinesulphonyl)-NG-nitro-L-arginine. In particular the patent makes reference to the preparation of a crystalline monohydrate form of argatroban, referring back to examples (D) and (E) of Japanese patent publication No. (Hei)-2-31055/1990 and generically to an I.R. spectrum identical to that of the commercially available argatroban compound. The relevant example in the cited patent publication is example (E), while example (D) concerns the preparation of (2R,4R)-1-[NG-nitro-N2-(3-methyl-8-quinolinesulphonyl)-L-arginyl]-4-methyl-2-piperidine carboxylic acid. This compound represents the starting compound for argatroban preparation by catalytic reduction in the presence of Pd/C. The crude argatroban obtained is then purified by extraction with chloroform, treatment with a saturated sodium bicarbonate solution and, after solvent evaporation, re-crystallization from ethanol or from 15% alcohol in water. It should be noted however that the Japanese patent makes no mention of the monohydrate form of argatroban being obtained and that for the compound the following characteristics are reported: m.p. 188-191° C.; molecular composition (theoretical/found) (%): C 54.31/54.01; H 7.13/6.98; N 16.52/16.61; I.R. (KBr) (cm−1) 3400; 1620; 1460; 1380. These analytical data, with the exception of the unreported melting point, are the same as those indicated in the cited patent documents describing a mixture of (2R,4R)-4methyl-[4N2-(3S-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid and (2R,4R)-4methyl-1-[N2-(3R-methyl-1,2,3,4-tetrahydro-8-quinolinesulphonyl)-L-arginyl]-2-piperidine carboxylic acid isomers of argatroban, but do not correspond to the melting point given in the publication, being the only document that identifies the monohydrate form of argatroban.
More recently, patent application CN 1,951,937 (filing date 10 Nov. 2006) described a method for preparing hydrated argatroban by treating argatroban with large quantities of water (more than 60 and up to 80 volumes of distilled water per gram of argatroban) at a temperature of 80-100° C. for a time of 0.5-1 hour and crystallization by cooling. The water content reported is comprised between 3.3 and 3.8% and the ratio of dextroisomer R to levoisomer S is R:S=63-67: 37-33.
Argatroban is a compound of wide therapeutic use, for which reason the need still exists to provide a compound of pharmaceutically acceptable quality obtained by easily industrialized and economically convenient methods. With regard to the monohydrate, this form is preferable for the applicative purpose since the anhydrous form is unstable and tends to become hydrated and/or wet. Moreover it crystallizes only with difficulty at the correct ratio between the diastereoisomers.

…..

the protection of 4-methylpiperidine (I) with (Boc)2O gives the carbamate (II), which is condensed with benzyl chloroformate by means of sec-butyl lithium and TMEDA in ethyl ether to yield (?-trans-1-(tert-butoxycarbonyl)-4-methylpiperidine-2-carboxylic acid benzyl ester (III). Deprotection of the NH group of (III) with HCl in ethyl acetate affords (?-trans-4-methylpiperidine-2-carboxylic acid benzyl ester (IV), which is condensed with the protected arginine derivative (V) by means of isobutyl chloroformate and TEA to provide the corresponding amide as a diastereomeric mixture. Resolution of this mixture by flash chromatography furnishes the desired diastereomer (VI), which is treated with HCl in ethyl acetate in order to remove the Boc-protecting group to yield compound (VII). Condensation of compound (VII) with 3-methylquinoline-8-sulfonyl chloride (VIII) by means of TEA in dichloromethane affords the expected sulfonamide (IX). Finally, this compound is submitted to hydrogenation with H2 over Pd/C in AcOH/ethanol in order to produce debenzylation, cleavage of the NO2 group and hydrogenation of the pyridine ring to yield argatroban.
………….
Argatroban, i.e., (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino) pentyl)-4-methyl-2-piperidine carboxylic acid, has two diastereoisomers: 21(R) and 21(S). Usually the ratio of 21(R) to 21(S) is 64-65: 36-35 (U.S. Pat. No. 6,440,417, Cossy. J., et al, Bioorganic & Medicine Chemistry Letters, 11 (2001), 1989-1992, Journal of pharmaceutical Sciences, Vol. 82, No. 6, 672 (1993)).
The structure formula of Argatroban is reported below:

-
- 21(S) Argatroban, X=CH3, Y═H;
- 21(R) Argatroban, X=H, Y=CH3;
- Argatroban, 21(S): 21 (R)=35:65.
The chemical names of the two diastereoisomers mentioned above are:
- 21(S) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3S)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-72-6); and
- 21(R) Argatroban: (2R,4R)-1-((2S)-5-((Aminoiminomethyl)amino)-1-oxo-2-((((3R)-1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl)amino)pentyl)-4-methyl-2-piperidine carboxylic acid (121785-71-5).
In 1978, S. Akamoto et al from Japanese Mitsubishi Chemical Corporation first disclosed the anti-thrombin activity of Argatroban monohydrate (U.S. Pat. No. 4,101,653). In the next 20 years, numerous researchers had in-depth studies on Argatroban about its biological activity and medicine values. In 1981, S. Akamoto compared Argatroban with heparin in vivo (Okamoto, S. et al., Biochem. Biophys. Res. Commun. 101, 440 (1981)); T. Kumoto disclosed its three-dimensional selective activity (Kumada, T. et al., Thromb. Res. 24, 285 (1981)). In 1984, R. Kumato made a clinical evaluation of hemodialysis of Argatroban (Kikumoto, R. et al., Biochemistry 23, 85 (1984)), and in 1986, he further disclosed that Argatroban can inhibit the thrombin activity of mammals, and can be used as active ingredient to treat and prevent thrombosis and as an inhibitor of platelet aggregation. Argatroban monohydrate can be used as a selective anti-thrombosis agent for treatment of chronic arterial blockage and cerebral thrombosis, etc (JP 61-48829). In 1992 and 1993, Taparelli and Jakubowski separately disclosed the reversibility of Argatroban in anti-thrombin (Taparelli, C., Trends Pharmacol. Sci., 1993, 14, 366, Jakubowski, J. A. et al, Rep. Med. Chem., 1992, 27, 99). In 1990s, many researchers such as L. R. Buch reported other related research (Buch, L. R., Cadiosvasc. Drug Rev., 1991, 9, 247, Strupcnewski, J. D. et al., Academic: San Diego, 1991; Vol. 26, p 299, Brundish, D. et al., J. Med. Chem. 1999; 42, 4584, Shebuski, R. J., Academic: San Diego, 1999; Vol. 26, p 98). In 1992, Argatroban monohydrate was first approved as an anti-thrombin medicine in Japan (Hijikata-Okunomiya, A., et al, Thromb. Hemostasis, 1992, 18, 135).
Updated in sept 2015, reader there may be duplication
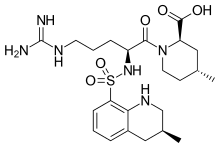
| Argatroban |
| AC1L99H9; AC-15185; TL8005144; C04931; | |
| Molecular Formula: | C23H36N6O5S |
|---|---|
| Molecular Weight: | 508.63414 g/mol |
(2R,4R)-1-[5-(diaminomethylideneamino)-2-[(3-methyl-1,2,3,4-tetrahydroquinolin-8-yl)sulfonylamino]pentanoyl]-4-methylpiperidine-2-carboxylic acid, cas 74863-84-6
| Patent | Submitted | Granted |
|---|---|---|
| Prodrugs of (2R)-2-Propyloctanoic Acid For the Treatment of Stroke [US7495029] | 2008-06-05 | 2009-02-24 |
Tomiya Mano, Jin Shiomura, “Argatroban preparations for ophthalmic use.” U.S. Patent US5506241, issued October, 1986.
Argatroban is an anticoagulant that is a small molecule direct thrombin inhibitor.[1] In 2000, argatroban was licensed by the Food and Drug Administration (FDA) for prophylaxis or treatment of thrombosis in patients with heparin-induced thrombocytopenia (HIT). In 2002, it was approved for use during percutaneous coronary interventions in patients who have HIT or are at risk for developing it. In 2012, it was approved by the MHRA in the UK for anticoagulation in patients with Heparin-Induced Thrombocytopenia Type II (HIT) who require parenteral antithrombotic therapy.[2]
Argatroban is given intravenously and drug plasma concentrations reach steady state in 1-3 hours.[3] Argatroban is metabolized in the liver and has a half-life of about 50 minutes. It is monitored by PTT. Because of its hepatic metabolism, it may be used in patients with renal dysfunction. (This is in contrast to lepirudin, a direct thrombin inhibitor that is primarily renally cleared).
Argatroban is a direct, selective thrombin inhibitor. The American College of Cardiologists (ACC) recommend using bivalirudin or argatroban in patients who have had, or at risk for, heparin induced thrombocytopenia (HIT) and are undergoing percutaneous coronary intervention. Argatroban is a non-heparin anticoagulant shown to both normalize platelet count in patients with HIT and prevent the formation of thrombi. Parental anticoagulants must be stopped and a baseline activated partial thromboplastin time must be obtained prior to administering argatroban.

Transitioning to warfarin in individuals with heparin induced thrombocytopenia
Argatroban is used as an anticoagulant in individuals with thrombosis and heparin induced thrombocytopenia. Often these individuals require long term anticoagulation. If warfarin is chosen as the long term anticoagulant, this poses particular challenges due to the falsely elevated prothrombin time and INR caused by argatroban. The combination of argatroban and warfarin may raise the INR to greater than 5.0 without a significant increased risk of bleeding complications.[4] One solution to this problem is to measure the chromogenic factor X level. A level < 40-45% typically indicates that the INR will be therapeutic (2-3) when the argatroban is discontinued.

http://www.google.com/patents/WO2009124906A2?cl=en
………….
NMR paper
Complete 1H and 13C assignments of (21R) and (21S) diastereomers of argatroban
Article first published online: 20 DEC 2007
DOI: 10.1002/mrc.2122, http://onlinelibrary.wiley.com/doi/10.1002/mrc.2122/abstract



click on image for clear view
1H NMR PREDICT


13C NMR PREDICT
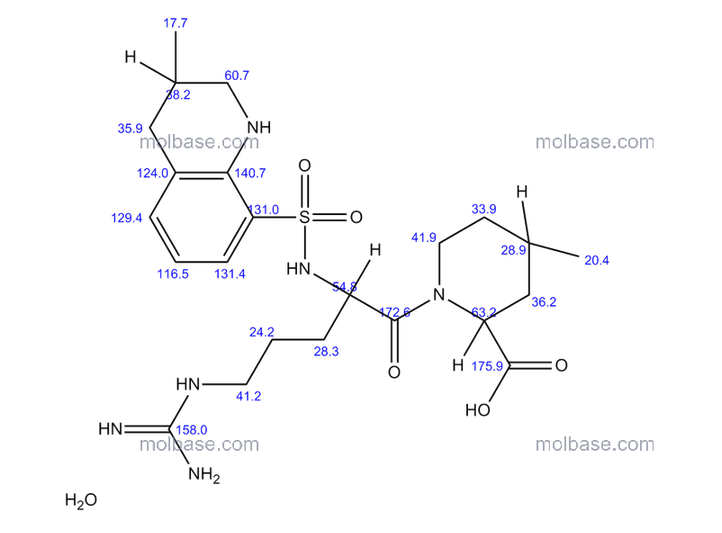
References
1 Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005;353:1028-40. PMID 16148288
2http://www.pharmatimes.com/Article/12-07-03/UK_launch_for_Mitsubishi_s_blood_thinner_Exembol.aspx
3Dhillon S. Argatroban: A Review of its Use in the Management of Heparin-Induced Thrombocytopenia. Am J Cardiovasc Drugs 2009; 9 (4): 261-82. Link text
- Hursting MJ, Lewis BE, Macfarlane DE. (2005). “Transitioning from argatroban to warfarin therapy in patients with heparin-induced thrombocytopenia.”. Clin Appl Thromb Hemost 11 (3): 279–87. doi:10.1177/107602960501100306. PMID 16015413.
External links
| EP0823430A1 * | Aug 4, 1997 | Feb 11, 1998 | Mitsubishi Chemical Corporation | Method for preparing n2-arylsulfonyl-l-argininamides |
| US4201863 * | Aug 31, 1978 | May 6, 1980 | Mitsubishi Chemical Industries, Limited | N2 -Arylsulfonyl-L-argininamides and the pharmaceutically acceptable salts thereof |
| Reference | ||||
|---|---|---|---|---|
| 1 | None | |||
| 2 | * | OKAMOTO S ET AL: “Potent inhibition of thrombin by the newly synthesized arginine derivative No. 805. The importance of stereo-structure of its hydrophobic carboxamide portion” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 101, no. 2, 30 July 1981 (1981-07-30), pages 440-446, XP024844713 ISSN: 0006-291X [retrieved on 1981-07-30] cited in the application | ||
| 3 | * | SONG H: “Method for preparing argatroban monohydrate in pure water” CASREACT,, 25 April 2007 (2007-04-25), XP002493272 & CN 1 951 937 A (TIANJIN WEIJIE TECHNOLOGY CO L [CN]) 25 April 2007 (2007-04-25) | ||
| Citing Patent | Filing date | Publication date | Applicant | Title |
| WO2012136504A1 | Mar 26, 2012 | Oct 11, 2012 | Lundbeck Pharmaceuticals Italy S.P.A. | Method for the preparation of process intermediates for the synthesis of argatroban monohydrate |
| CN102408468A * | Sep 20, 2011 | Apr 11, 2012 | 海南灵康制药有限公司 | Argatroban compound and preparation method thereof |
| EP2752412A1 | Mar 26, 2012 | Jul 9, 2014 | Lundbeck Pharmaceuticals Italy S.p.A. | Intermediates for the synthesis of Argatroban monohydrate |
Argatroban is a synthetic direct thrombin inhibitor and the chemical name is 1-[5[(aminoiminomethyl)amino]1-oxo-2-[[(1,2,3,4-tetrahydro-3-methyl-8-quinolinyl)sulfonyl]amino]pentyl]-4methyl-2-piperidinecarboxylic acid, monohydrate. Argatroban has 4 asymmetric carbons. One of the asymmetric carbons has an R configuration (stereoisomer Type I) and an S configuration (stereoisomer Type II). Argatroban consists of a mixture of R and S stereoisomers at a ratio of approximately 65:35.
The molecular formula of argatroban is C23H36N6O5S•H2O. Its molecular weight is 526.66 g/mol. The structural formula is:
|
Argatroban Injection is a sterile, non-pyrogenic, clear, colorless to pale yellow isotonic solution. It is supplied in a single use polyolefin bag containing 250 mg of argatroban in 250 mL sodium chloride solution (1 mg/mL). Each mL contains 1 mg argatroban, 9 mg sodium chloride, USP, and 3 mg sorbitol, NF in water for injection, USP. The pH of the solution is between 3.2 to 7.5.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
(2R,4R)-1-[(2S)-5-(diaminomethylideneamino)-2-
[[(3R)-3-methyl-1,2,3,4-tetrahydroquinolin-8-yl] sulfonylamino]pentanoyl]-4-methyl-piperidine-2- carboxylic acid |
|
| Clinical data | |
| Trade names | Argatroban |
| AHFS/Drugs.com | monograph |
| Routes of administration |
intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (intravenous) |
| Protein binding | 54% |
| Metabolism | hepatic |
| Biological half-life | 39 and 51 minutes |
| Identifiers | |
| CAS Registry Number | 74863-84-6 |
| ATC code | B01AE03 |
| PubChem | CID: 440542 |
| DrugBank | DB00278 |
| ChemSpider | 389444 |
| UNII | OCY3U280Y3 |
| KEGG | C04931 |
| ChEMBL | CHEMBL1166 |
| Chemical data | |
| Formula | C23H36N6O5S |
| Molecular mass | 508.635 g/mol |
//////
Drug spotlight- Zafirlukast

ZAFIRLIKAST 
cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate 107753-78-6
Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 (1990);
U. S. Patent No. 4,859,692;
U. S. Patent No. 5,993,859;
http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020547s031lbl.pdf
Zafirlukast is an oral leukotriene receptor antagonist (LTRA) for the maintenance treatment of asthma, often used in conjunction with an inhaled steroid and/or long-acting bronchodilator. It is available as a tablet and is usually dosed twice daily. Another leukotriene receptor antagonist is montelukast (Singulair), taken once daily. Zileuton (Zyflo), also used in the treatment of asthma via its inhibition of 5-lipoxygenase, is taken four times per day.
Zafirlukast blocks the action of the cysteinyl leukotrienes on the CysLT1 receptors, thus reducing constriction of the airways, build-up of mucus in the lungs andinflammation of the breathing passages.
Zafirlukast is marketed by Astra Zeneca with the brand names Accolate, Accoleit, and Vanticon. It was the first LTRA to be marketed in the USA and is now approved in over 60 countries, including the UK, Japan, Taiwan, Italy, Spain, Canada, Brazil, China and Turkey
Healthy young men who received a single oral 40 mg dose attained peak plasma zafirlukast concentrations that averaged 607 μg/L at 3.4 hours. The elimination half-life ranged from 12 to 20 hours. In another study involving a 20 mg single oral dose in healthy men, the elimination half-life averaged 5.6 hours.[1][2]
A letter was submitted to the FDA by Zeneca Pharmaceuticals on July 22, 1997, notifying them of a change in product labeling that includes the following potential reaction in patients undergoing a dosage reduction of oral steroids who are currently taking zafirlukast:
PRECAUTIONS-Eosinophilic Conditions: The reduction of the oral steroid dose, in some patients on ACCOLATE therapy, has been followed in rare cases by the occurrence of eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy sometimes presenting as Churg–Strauss syndrome, a systemic eosinophilic vasculitis. Although a causal relationship with ACCOLATE has not been established, caution is required when oral steroid reduction is being considered.1
NDA..020547 26/09/1996, ACCOLATE, ASTRAZENECA, 20MG TABLET
| US Patent No | Expirey Date | patent use code |
|---|---|---|
| 5482963 | Jan 9, 2013 | |
| 5612367 | Mar 18, 2014 | U-189 |
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | R03DC01 | C 31 H 33 N 3 O 6 S | 575.69 g / mol | 107753-78-6 |
| monohydrate | R03DC01 | C 31 H 33 N 3 O 6 S · H 2 O | 593.70 g / mol | 143052-93-1 |
| calcium (2: 1) | R03DC01 | C 62 H 64 CaN 6 O 12 S 2 | 1189.43 g / mol | 107753-86-6 |
Application
-
antihistamine effect
-
LTD4-antagonist
Classes of substances
-
Benzenesulfonamide (s -imidy), as well as their derivatives
-
Esters of carbamic acid
-
Cyclopentanes
-
Hydroxybenzoic acid amides, and hydroxy acids alkoksibenzoynyh
-
Indoles
-
-
-
-
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), with the chemical name 4(5-cyclopentyloxy-carbonylamino-1-methyl-indol-3ylmethyl)-3-methoxy-N-o-tolylsulfonylbenzamide. The molecular weight of zafirlukast is 575.7 and the structural formula is:

Zafirlukast, a fine white to pale yellow amorphous powder, is practically insoluble in water. It is slightly soluble in methanol and freely soluble in tetrahydrofuran, dimethylsulfoxide, and acetone.The empirical formula is: C31H33N3O6S
- Fischer JD, Song MH, Suttle AB, Heizer WD, Burns CB, Vargo DL, Brouwer KL. Comparison of zafirlukast (Accolate) absorption after oral and colonic administration in humans. Pharmaceut. Res. 17: 154-159, 2000.
- Bharathi DV, Naidu A, Jagadeesh B, Laxmi KN, Laxmi PR, Reddy PR, Mullangi R. Development and validation of a sensitive LC-MS/MS method with electrospray ionization for quantitation of zafirlukast, a selective leukotriene antagonist in human plasma: application to a clinical pharmacokinetic study. Biomed. Chromatogr. 22: 645-653, 2008.
- Zafirlukast (U.S. National Library of Medicine)
- Zafirlukast (patient information)

 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| cyclopentyl 3-{2-methoxy-4-[(o-tolylsulfonyl)carbamoyl]benzyl}-1-methyl-1H-indol-5-ylcarbamate | |
| Clinical data | |
| Trade names | Accolate |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a697007 |
| Pregnancy cat. | B1 (Australia), B (United States) |
| Legal status | POM (UK) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | Unknown |
| Protein binding | 99% |
| Metabolism | Hepatic (CYP2C9-mediated) |
| Half-life | 10 hours |
| Excretion | Biliary |
| Identifiers | |
| CAS number | 107753-78-6 |
| ATC code | R03DC01 |
| PubChem | CID 5717 |
| IUPHAR ligand | 3322 |
| DrugBank | DB00549 |
| ChemSpider | 5515 |
| UNII | XZ629S5L50 |
| KEGG | D00411 |
| ChEBI | CHEBI:10100 |
| ChEMBL | CHEMBL603 |
| Chemical data | |
| Formula | C31H33N3O6S |
| Mol. mass | 575.676 g/mol |
Trade Names
| Country | Trade name | Manufacturer |
|---|---|---|
| United Kingdom | Akkolat | AstraZeneca |
| Italy | Akkoleit | – “- |
| Zafirst | Chiesi | |
| Japan | Akkolat | AstraZeneca |
| USA | – “- | Zeneca |
| Ukraine | No | No |
Formulations
-
Tablets of 20 mg, 40 mg

is a first anti-asthmatic leukotriene antagonist (Matassa, V.G. et al, J. Med. Chem., v. 33, 1781 ‘(1990); U. S. Patent No. 4,859,692 and The Merck Index, 12th Edition, 10241). Methods for the preparation of Zafirlukast are described in J. Med. Chem., v. 33, 1781 (1990), U. S. Patent 4,859,692 and U.S. Patent 5,993,859 starting from methyl 3-methoxy-4-(l-methyl-5-nitroindol-3-ylmethyl)benzoate [la]



in the presence of an equivalent quantity of silver(I) oxide,

The above process has serious disadvantages in the isolation of the product [4] in step (b) which is due to the fact that alkylation of indole, that is unsubstituted at positions 1-, 2- and 3-, at the 3-position, is accompanied by the undesired process of poly alkylation, to form polysubstituted indoles of formula [7] and/or formula [8] :

while at the same time some quantity of the starting unreacted indole remains in the reaction mixture. Most common methods for the separation of alkyl (indol-3-ylmethyl)benzoate of formula [4] from by-products of polyalkylation and starting unreacted indole, which are all covalent compounds with similar physical properties, include column chromatography that is an unpractical method for industrial scale applications.
Formula (I) compound for the synthesis of an important intermediate of zafirlukast.Reported in the patent EP199543 synthesized compound (I) of the conventional method, the following formula:

(A) (I)
In this method, Intermediate A and 5 – nitro-indole silver oxide in the presence of a catalyst, for docking composite formula (I) compound. Reported only 45% of the reaction yield, the reaction is difficult to complete the reaction and post-treatment using chromatographic methods, resulting in product purification more difficult. And the use of more expensive silver oxide catalysts, high cost.
W00246153 reported a catalyst for the above reaction to zinc bromide, Compound (I), after treatment of the compound (I) with sodium hydroxide hydrolysis of the intermediate (B), separating the product and raw materials purification products.

The method reported in the literature a yield of 60%, but the actual operation is repeated only about 30% yield, and the operation is complicated, cumbersome and costly.
zaafirlukast is a selective and competitive receptor antagonist of leukotriene D4 and E4 (LTD4 and LTE4), components of slow-reacting substance of anaphylaxis (SRSA). Cysteinyl leukotriene production and receptor occupation have been correlated with the pathophysiology of asthma, including airway edema, smooth muscle constriction, and altered cellular activity associated with the inflammatory process, which contribute to the signs and symptoms of asthma.
The cysteinyl leukotrienes (LTC4 LTD4, LTE4) are the products of arachidonic acid metabolism and are various cells, including mast cells and eosinophills, these eicosinoids bind to cysteinyl leukotriene (CysLT) receptors. The CysLT type-1 (CysLT1) receptor is found in human airway and other pro-inflammatory cells. CysLTs have been correlated with the pathophysiology of asthma.
Zafirlukast is a synthetic, selective peptide leukotriene receptor antagonist (LTRA), useful for the treatment of asthma and is commercially available in products sold under the brand name ACCOLATE™ as 10 and 20 mg tablets for oral administration. ACCOLATE™ is indicated for the prophylaxis and treatment of asthma in adults and children 5 years of age and older.
ACCOLATE™ film coated tablets contain amorphous zafirlukast as the active ingredient and the excipients croscarmellose sodium, lactose, magnesium stearate, microcrystalline cellulose, povidone, hypromellose, and titanium dioxide.
The greatest prevalence of asthma is in preschool children; however, the clinical utility of asthma therapy for this age group is limited by a narrow therapeutic index, long-term tolerability, and frequency and/or difficulty of administration. Asthma treatment requires an immediate perceivable effect. Inhalation therapy is a very common therapy prescribed for young children; inhalation therapy has the disadvantage of high dose variability.
 Process for the preparation of zafirlukast
Process for the preparation of zafirlukastUS 20040186300 A1



An Improved and Scalable Process for Zafirlukast: An Asthma Drug
Melting range: 142−145 °C; MS (m/z): 576 (M+ + H); IR (KBr, cm−1): 3326 (NH), 1679 (−C═O), 1H NMR (CDCl3) δ 7.0−8.0 (m, 11H), 3.7 (s, 3H), 4.0 (s, 2H), 3.9 (s, 3H), 2.6 (s, 3H), 1.45−1.8 (s, 9H). ……………………………………………………………….. US 20040186300 A1 http://www.google.com/patents/US20040186300 zafirlukast ethanolate as white powder with mp 132-133° C. (dec.) and 99.8% purity by HPLC. 1H NMR (CDCl3, δ, ppm): 1.22 (t, J 7.05 Hz, 3H), 1.45-1.87 (m, 8H), 2.66 (s, 3H), 3.67 (s, 3H), 3.73 (q, J 7.05 Hz, 4H), 3.79 (s, 3H), 3.98 (s, 2H), 5.08-5.23 (m, 1H), 6.58 (s, 1H), 6.73 (s, 1H), 7.01-7.51 (m, 9H), 8.23 (d, J 7.52 Hz, 1H), 9.67 (s, 1H).
Synthesis pathway
| Synthesis a) |
|---|
      |
| Synthesis of b) |
   |
-
Synthesis a)
-
US 4,859,692 (ICI; 08/22/1989; GB -prior. 4/17/1985; 17.10.1985).
-
EP 199 543 (ICI, Zeneca; appl. 16.4.1986; GB -prior. 4/17/1985).
-
-
Synthesis of b)
-
EP 490 649 (ICI, Zeneca; 11.12.1991; GB -prior. 12.12.1990).
-
Matassa, G. et al .: J. Med. Chem. (JMCMAR) 33, 1781 (1990).
-
Srinivas, K. et al .: Org. Process Res. Dev. (OPRDFK) 8 (6), 952 (2004).
-
added info Asthma is a disease that causes swelling and narrowing the airways of the lungs. Airways are air carriers to and from lungs. Swollen and narrower airways affect the air flow to and from the lungs and this lead to tightness of chest, wheezing, shortness of breath and cough. These symptoms are often occurs in early morning and in night. Asthma is caused by genetic and environmental factors, it was not curable completely but this can be controlled with good medical care. Leukotriene antagonists also known as leukast are the medicaments that are used to reduce leukotrienes, which are produced by several types of cells and causes inflammation in asthma and bronchitis. Leukotriene antagonists that are available in market are Montelukast, Zafirlukast and Pranlukast. Zafirlukast is the first leukast compound approved for management of Asthma. US FDA approved zafirlukast in the form of 10 mg and 20 mg tablet with the brand name of Accolate®.1 Subsequently this was approved and launched by innovator in few other countries. There are many synthetic routes for the preparation of Zafirlukast 4 is well documented in literature. Some of the key approaches are discussed here under. Scientists from ICI Americas Inc2 have reported process for the synthesis of 4, which starts with esterification of 3-methoxy-4-methyl benzoic acid 53 using methanol in presence of acetyl chloride PRODUCT PATENT ROUTE Allylic bromination of methyl ester 54 using bromine in presence of CCl4 resulted bromo compound 55, which was reacted with 5-nitro indole 124 using silver oxide as catalyst to obtain condensed compound 125. N-methylation of 125 utilizing methyl iodide in presence of NaH afforded N-methyl indole derivative 57. Thus obtained 57 was subjected to reduction using palladium carbon (Pd/C) in methanol followed by reacted with cyclopentyl chloroformate to obtain compound 59. Hydrolysis of 59 using LiOH.H2O subsequently reaction with o-toluene sulfonamide (OTSA) in presence of 1-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (DMAPEC) and DMAP furnished zafirlukast 4. Matassa et al3 also reported similar procedure for the synthesis of Zafirlukast 4. 


FDA okays Vifor Fresenius phosphate binder Velphoro
THERAPEUTIC CLAIM Oral phosphate binder, treatement of elevated
phosphate levels in patients undergoing dialysis
CHEMICAL DESCRIPTIONS
1. Ferric hydroxide oxide
2. Mixture of iron(III) oxyhydroxide, sucrose, starches
3. Polynuclear iron(III) oxyhydroxide stabilized with sucrose and starches
structure
O =Fe -OH
MOLECULAR FORMULA FeHO2•xC12H22O11•y(C6H10O5)n
SPONSOR Vifor (International) Inc.
CODE DESIGNATIONS PA21
CAS REGISTRY NUMBER 12134-57-5
sucroferric oxyhydroxide
Sucroferric oxyhydroxide nonproprietary drug name
1. February 27, 2013. N13/36. STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL. USAN (ZZ-19). SUCROFERRIC …
The US Food and Drug Administration has given the green light to Vifor Fresenius Medical Care Renal Pharma’s hyperphosphatemia drug Velphoro.
The approval for Velphoro (sucroferric oxyhydroxide), formerly known as PA21, is based on Phase III data demonstrated that the drug successfully controls the accumulation of phosphorus in the blood with the advantage of a much lower pill burden than the current standard of care in patients with chronic kidney disease on dialysis, namely Sanofi’s Renvela (sevelamer carbonate). read this at
http://www.pharmatimes.com/Article/13-11-28/FDA_okays_Vifor_Fresenius_phosphate_binder_Velphoro.aspx
Velphoro (PA21) receives US FDA approval for the treatment of hyperphosphatemia in Chronic Kidney Disease Patients on dialysis
Velphoro (sucroferric oxyhydroxide) has received US Food and Drug Administration (FDA) approval for the control of serum phosphorus levels in patients with Chronic Kidney Disease (CKD) on dialysis. Velphoro will be launched in the US by Fresenius Medical Care North America in 2014.
Velphoro (previously known as PA21) is an iron-based, calcium-free, chewable phosphate binder. US approval was based on a pivotal Phase III study, which met its primary and secondary endpoints. The study demonstrated that Velphoro® successfully controls hyperphosphatemia with fewer pills than sevelamer carbonate, the current standard of care in patients with CKD on dialysis. The average daily dose to control hyperphosphatemia was 3.3 pills per day after 52 weeks.
Velphoro was developed by Vifor Pharma. In 2011, all rights were transferred to Vifor Fresenius Medical Care Renal Pharma, a common company of Galenica and Fresenius Medical Care. In the US, Velphorowill be marketed by Fresenius Medical Care North America, a company with a strong marketing and sales organization, and expertise in dialysis care. The active ingredient of Velphoro is produced by Vifor Pharma in Switzerland.
Hyperphosphatemia, an abnormal elevation of phosphorus levels in the blood, is a common and serious condition in CKD patients on dialysis. Most dialysis patients are treated with phosphate binders. However, despite the availability of a number of different phosphate binders, up to 50% of patients depending on the region are still unable to achieve and maintain their target serum phosphorus levels. In some patients, noncompliance due to the high pill burden and poor tolerability appear to be key factors in the lack of control of serum phosphorus levels. On average, dialysis patients take approximately 19 pills per day with phosphate binders comprising approximately 50% of the total daily pill burden. The recommended starting dose of Velphoro is 3 tablets per day (1 tablet per meal).
Full results from the pivotal Phase III study involving more than 1,000 patients were presented at both the 50th ERA-EDTA (European Renal Association European Dialysis and Transplant Association) Congress in Istanbul, Turkey, in May 2013, and the American Society of Nephrology (ASN) Kidney Week in Atlanta, Georgia, in November 2013. Velphorowas shown to be a potent phosphate binder, with lower pill burden and a good safety profile.
Based on these data, Vifor Fresenius Medical Care Renal Pharma believes that Velphoro offers a new and effective therapeutic option for the control of serum phosphorus levels in patients with chronic kidney disease on dialysis.
The regulatory processes in Europe, Switzerland and Singapore are ongoing and decisions are expected in the first half 2014. Further submissions for approval are being prepared.
Teva Gets Orphan Drug Designation for Treanda
Teva Announces Additional Regulatory Exclusivity for TREANDA® (Bendamustine HCI) for Injection
Orphan Designation combined with pediatric extension provides regulatory exclusivity through April 2016 for indolent B-cell non-Hodgkin lymphoma indication
JERUSALEM, November 27, 2013 –(BUSINESS WIRE)–Teva Pharmaceutical Industries Ltd. (NYSE: TEVA) today announced that the U.S. Food and Drug Administration (FDA) has granted orphan drug exclusivity for TREANDA through October 2015 for indolent B-cell non-Hodgkin lymphoma (iNHL) that has progressed during or within six months of treatment with rituximab or a rituximab-containing regimen.http://www.pharmalive.com/teva-announces-additional-regulatory-exclusivity-for-treanda
read my old post, contains synthesis
https://newdrugapprovals.wordpress.com/2013/09/19/fda-oks-tevas-injectable-treanda/
VBL Therapeutics announced FDA has granted Fast Track designation to its lead oncology drug VB-111

VB-111
VBL Therapeutics announced today that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to its lead oncology drug VB-111, for prolongation of survival in patients with recurrent glioblastoma multiforme (rGBM).
VB-111 – highly targeted anti-angiogenic agent for the specific inhibition of tumor vascular growth
VB-111 is the first highly targeted anti-angiogenic agent for the specific inhibition of tumor vascular growth to use VTS™™, our proprietary platform technology, for cancer therapy. VB-111 is an IV-administered anti angiogenic agent that works in a manner akin to a “biological knife” to destroy tumor vasculature, thus cutting off blood vessels feeding the tumor.
Preclinical Insights
VB-111 has shown significant promise as a targeted cancer treatment with the potential to work synergistically in combination with conventional chemotherapy treatments to provide an effective treatment regimen for cancer patients. Pharmacological and toxicology studies of VB-111 have showed tissue specificity for the tumor tissue, no significant damage to normal non-cancerous tissues or to the normal vasculatures in the body and more than 90 percent tumor burden reduction in a metastatic lung cancer model with only one injection. Similar efficacy was shown in other tumor models.
Completed Clinical Trials
Phase 1 Clinical Trial – in a Phase 1 “all comers” dose escalation study in 33 patients with advanced metastatic cancer, therapeutic doses of VB-111 demonstrated antitumor activity and was found to be safe and well tolerated with no effect on liver function or major changes in complete blood count. Findings have been presented at the American Association of Cancer Research (AACR) and the American Society of Clinical Oncology (ASCO) annual meetings.
GSK obtains FDA approval for bird flu vaccine
GlaxoSmithKline (GSK) has received approval from the US Food and Drug Administration (FDA) for the first adjuvanted vaccine to prevent H5N1 influenza, also known as bird flu.
GSK obtains FDA approval for bird flu vaccine http://www.pharmaceutical-technology.com/news/newsgsk-obtains-fda-approval-bird-flu-vaccine?WT.mc_id=DN_News
26 November 2013

GlaxoSmithKline (GSK) has received approval from the US Food and Drug Administration (FDA) for the first adjuvanted vaccine to prevent H5N1 influenza, also known as bird flu.
The FDA cleared the pandemic Influenza A (H5N1) virus monovalent vaccine, adjuvanted (also referred to as Q-Pan H5N1 influenza vaccine), for use in people aged 18 and older who are at increased risk of exposure to the virus.
The vaccine is composed of monovalent, inactivated, split A/H5N1 influenza virus antigen and GSK’s AS03 adjuvant.
The company said that in clinical studies, the adjuvanted formulation stimulated the required immune response while using a smaller amount of antigen as compared with a formulation without adjuvant.
FDA Approves Olysio (simeprevir) for Hepatitis C Virus

Simeprevir
Inhibits HCV NS3/4A protease.
MEDIVIR … originator
launched 2013
923604-59-5 CAS
C38H47N5O7S MF
749.93908 MW
IUPAC standard name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – ({7-methoxy-8-methyl-2-[4 – (propan-2-yl) -1,3-thiazol-2 -yl] quinolin-4-yl} oxy)-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
IUPAC traditional name
(1R, 4R, 6S, 15R, 17R)-N-(cyclopropanesulfonyl) -17 – {[2 – (4-isopropyl-1 ,3-thiazol-2-yl)-7-methoxy-8-methylquinolin-4- yl] oxy}-13-methyl-2 ,14-dioxo-3 ,13-diazatricyclo [13.3.0.0 4 , 6 ] octadec-7-ene-4-carboxamide
- Olysio
- Simeprevir
- TMC 435
- TMC 435350
- TMC-435
- TMC435
- TMC435350
- UNII-9WS5RD66HZ
November 22, 2013 — The U.S. Food and Drug Administration approved Olysio (simeprevir), a new therapy to treat chronic hepatitis C virus infection.
OLYSIO™ is the first once-daily protease inhibitor approved for the treatment of chronic hepatitis C in a combination antiviral regimen for adults with compensated liver disease
Hepatitis C is a viral disease that causes inflammation of the liver that can lead to diminished liver function or liver failure. Most people infected with the hepatitis C virus have no symptoms of the disease until liver damage becomes apparent, which may take several years. Most of these people then go on to develop chronic hepatitis C. Some will also develop scarring and poor liver function (cirrhosis) over many years, which can lead to complications such as bleeding, jaundice (yellowish eyes or skin), fluid accumulation in the abdomen, infections or liver cancer. According to the Centers for Disease Control and Prevention, about 3.2 million Americans are infected with the hepatitis C virus

Hepatitis C virus (HCV) infections affect approximately 3 percent of the worldwide population and often lead to cirrhosis and hepatocellular carcinoma. The standard therapy of pegylated- interferon and ribavirin induces serious side effects and provides viral eradication in less than 50% of patients. Combination therapy of HCV including ribavirin and interferonare currently is the approved therapy for HCV. Unfortunately, such combination therapy also produces side effects and is often poorly tolerated, resulting in major clinical challenges in a significant proportion of patients. Numerous direct acting agents (DAAs) have been or are being developed for treatment of HCV, such as telaprevir and boceprevir (both received MA approved in 2011 for use with interferon and ribavirin based therapy), however direct acting agents are linked to increased toxicity of treatment, the emergence of resistance, and to date do not provide a standard of care which is interferon free. The combination of direct acting agents can also result in drug-drug interactions. To date, no HCV therapy has been approved which is interferon free. There is therefore a need for new combination therapies which have reduced side effects, and interferon free, have a reduced emergence of resistance, reduced treatment periods and/or and enhanced cure rates.
Simeprevir (formerly TMC435) is an experimental drug candidate for the treatment of hepatitis C. It is being developed byMedivir and Johnson & Johnson‘s pharmaceutical division Janssen Pharmaceutica and is currently in Phase III clinical trials.[1]
Simeprevir is a hepatitis C virus protease inhibitor.[2]
Simeprevir is being tested in combination regimens with pegylated interferon alfa-2a and ribavirin,[3] and in interferon-free regimens with other direct-acting antiviral agents including daclatasvir[4] and sofosbuvir [5]
Simeprevir has been launched in 2013 in Japan by Janssen Pharmaceutical (JP) for use in combination with pegylated interferon (Peg-IFN) and ribavirin for the treatment of genotype 1 chronic hepatitis C virus (HCV) patients who are treatment naïve, prior non responders or relapsed following treatment with Peg-IFN with or without ribavirin. In 2013, the product has also been approved in the U.S. by Medivir and Janssen R&D Ireland for the oral treatment of chronic hepatitis C genotype 1 infection, in combination with peginterferon alfa and ribavirin in adults with compensated liver disease, including cirrhosis, who are treatment-naïve or who have failed previous interferon therapy (pegylated or non-pegylated) with ribavirin.
The drug candidate was originally developed at Medivir, which was acquired by Janssen R&D Ireland in 2012. In November 2004, Medivir entered into a license and research collaboration agreement with Tibotec, a Johnson & Johnson subsidiary, for the discovery and development of orally active protease inhibitors of the NS3/4A protease of HCV. In 2011, a codevelopment agreement between Pharmasset (now Gilead Sciences) and Tibotec was signed for the treatment of chronic hepatitis C (HCV) in combination with PSI-7977. Also in 2011, fast track designation was received in the U.S. for the treatment of chronic hepatitis C (CHC) genotype-1 infection.
In 2011, Tibotec Therapeutics, Division of Centocor Ortho Biotech Products, L.P. announced that it had changed its name to Janssen Therapeutics, Division of Janssen Products, LP.
“Hepatitis C is a complex disease and Janssen is committed to working with the HCV community, caregivers, and health care systems to address this global epidemic,” said Gaston Picchio, Hepatitis Disease Area Leader, Janssen Research & Development. “We are pleased that the FDA has granted simeprevir Priority Review, as it is a significant step forward in making this therapy available to physicians and their hepatitis C patients.”

Hepatitis C virus (HCV) is the leading cause of chronic liver disease worldwide.
Following initial acute infection, a majority of infected individuals develop chronic hepatitis because HCV replicates preferentially in hepatocytes but is not directly cytopathic. Chronic hepatitis can progress to liver fibrosis leading to cirrhosis, end- stage liver disease, and HCC (hepatocellular carcinoma), making it the leading cause of liver transplantations. This and the number of patients involved, has made HCV the focus of considerable medical research. Replication of the genome of HCV is mediated by a number of enzymes, amongst which is HCV NS3 serine protease and its associated cofactor, NS4A. NS3 serine protease is considered to be essential for viral replication and has become an attractive target for drug discovery.
Current anti-HCV therapy is based on (pegylated) interferon-alpha (IFN-α) in combination with ribavirin. Not only does this therapy result in a limited efficacy in that only part of the patients are treated successfully, but it also faces significant side effects and is poorly tolerated in many patients. Hence there is a need for further HCV inhibitors that overcome the disadvantages of current HCV therapy such as side effects, limited efficacy, poor tolerance, the emergence of resistance, as well as compliance failures.
Various agents have been described that inhibit HCV NS3 serine protease. WO05/073195 discloses linear and macrocyclic NS3 serine protease inhibitors with a central substituted proline moiety and WO 05/073216 with a central cyclopentyl moiety. Amongst these, the macrocyclic derivatives are attractive by overcoming one or more of the disadvantages of current anti-HCV therapy

(I) simeprevir
The compound of formula (I) is an inhibitor of the Hepatitis C virus (HCV) serine protease and is described in WO 2007/014926, published on 8 February 2007. This compound overcomes several of the disadvantages of current anti-HCV therapy and in particular shows pronounced activity against HCV, has an attractive pharmacokinetic profile, and is well-tolerated. Following the synthesis procedure described in Example 5 of WO 2007/014926, an amorphous solid form is obtained.
It now has been found that the compound of formula (I) can be converted into crystalline forms, which can advantageously be used as active ingredients in anti-HCV therapy. To that purpose, these crystalline forms are converted into pharmaceutical formulations.
………………………………………………………………………………………….
SIMEPREVIR

…………………………

OLYSIO (simeprevir) is an inhibitor of the HCV NS3/4A protease.
The chemical name for simeprevir is (2R,3aR,10Z,11aS,12aR,14aR)-N-(cyclopropylsulfonyl)-2[[2-(4-isopropyl-1,3-thiazol-2-yl)-7-methoxy-8-methyl-4-quinolinyl]oxy]-5-methyl-4,14-dioxo2,3,3a,4,5,6,7,8,9,11a,12,13,14,14atetradecahydrocyclopenta[c]cyclopropa[g][1,6]diazacyclotetradecine-12a(1H)-carboxamide. Its molecular formula is C38H47N5O7S2 and its molecular weight is 749.94. Simeprevir has the following structural formula:
 |
Simeprevir drug substance is a white to almost white powder. Simeprevir is practically insoluble in water over a wide pH range. It is practically insoluble in propylene glycol, very slightly soluble in ethanol, and slightly soluble inacetone. It is soluble in dichloromethane and freely soluble in some organic solvents (e.g., tetrahydrofuran and N,N-dimethylformamide).
OLYSIO (simeprevir) for oral administration is available as 150 mg strength hard gelatin capsules. Each capsule contains 154.4 mg of simeprevir sodium salt, which is equivalent to 150 mg of simeprevir. OLYSIO (simeprevir) capsules contain the following inactive ingredients: colloidal anhydrous silica, croscarmellose sodium, lactose monohydrate, magnesium stearate and sodium lauryl sulphate. The white capsule contains gelatin and titanium dioxide (E171) and is printed with ink containing iron oxide black (E172) and shellac (E904).
……………..
Synthesis
Example 1 : preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04‘6]octadec-7-ene- 4-carboxylic acid (16)
Synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinoline (6) Step 1 : synthesis of Λ/-(tert-butyloxycarbonyl)-3-methoxy-2-methylaniline (2)

1 2
Triethylamine (42.4 mL, 302 mmol) was added to a suspension of 3-methoxy-2- methylbenzoic acid (45.6 g, 274 mmol) in dry toluene (800 mL). A clear solution was obtained. Then, dppa (65.4 mL, 302 mmol) in toluene (100 mL) was slowly added. After 1 h at room temperature, the reaction mixture was successively heated at 500C for 0.5 h, at 700C for 0.5 h then at 1000C for 1 h. To this solution, t-BuOH (30.5 g, 411 mmol) in toluene (40 mL) was added at 1000C and the resulting mixture was refluxed for 7h. The solution was cooled to room temperature then successively washed with water, 0.5 N HCl, 0.5 N NaOH and brine, dried (Na2SO4), and evaporated to give 67 g of the target product: m/z = 237 (M)+.
_2: synthesis of 3-methoxy-2-methylaniline (3)

TFA (40.7 mL, 548 mmol) was added to a solution of jV-(teτt-butyloxycarbonyl)- 3-methoxy-2-methylaniline, in dichloro methane (500 mL). After 2 h at room temperature, TFA (40.7 mL, 548 mmol) was added and the resulting mixture was stirred at room temperature overnight. Then, volatiles were evaporated. The residue was triturated with toluene (100 mL) and diisopropylether (250 mL), filtered off and washed with diisopropyl ether (100 mL) to give 56.3 g of the title product as a TFA salt: m/z = 138 (M+H)+. The TFA salt was transformed to the free aniline by treatment with NaHCO3.
Step 3: synthesis of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (4)

A solution Of BCl3 (1.0 M, 200 mL, 200 mmol) in CH2Cl2 was slowly added under nitrogen to a solution of 3-methoxy-2-methylaniline (26.0 g, 190 mmol) in xylene (400 mL). The temperature was monitored during the addition and was kept below 100C. The reaction mixture was stirred at 5°C for 0.5 h. Then, dry acetonitrile (13 mL, 246 mmol) was added at 5°C. After 0.5 h at 5°C, the solution was transferred into a dropping funnel and slowly added at 5°C to a suspension OfAlCl3 (26.7 g, 200 mmol) in CH2Cl2 (150 mL). After 45 min at 5°C, the reaction mixture was heated at 700C under a nitrogen stream. After evaporation Of CH2Cl2, the temperature of the reaction mixture reached 65°C. After 12 h at 65°C, the reaction mixture was cooled at 00C, poured onto ice (300 g), and slowly heated to reflux for 7h. After 2 days at room temperature, 6 N NaOH (50 mL) was added. The pH of the resulting solution was 2-3. The xylene layer was decanted. The organic layer was extracted with CH2Cl2. The xylene and CH2Cl2 layers were combined, successively washed with water, IN NaOH, and brine, dried (Na2SO4) and evaporated. The residue was triturated in diisopropyl ether at O0C, filtered off and washed with diisopropylether to give 13.6 g (40 %) of the title product as a yellowish solid: m/z = 180 (M+H)+.
Step 4: synthesis of 2′-[[(4-isopropylthiazole-2-yl)(oxo)methyl]amino]-4′-methoxy-3 ‘- methylacetophenone (5)

A solution of the compound 4 (18.6 g, 104 mmol) in dioxane (50 rnL) was added under nitrogen to a suspension of 4-isopropylthiazole-2-carbonyl chloride in dioxane (250 rnL). After 2 h at room temperature, the reaction mixture was concentrated to dryness. Then, the residue was partitioned between an aqueous solution of NaHCOs and AcOEt, organic layer was washed with brine, dried (Na2SO4), and evaporated. The residue was triturated in diisopropyl ether, filtered off and washed with diisopropyl ether to give 30.8 g (90 %) of the title product 5.
Step 5: synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinoline (6)

Potassium tert-butoxide (21.8 g, 195 mmol) was added to a suspension of the compound 5 (30.8 g, 92.7 mmol) in tert-butanol. The resulting reaction mixtures was heated at 1000C overnight. Then, the reaction mixture was cooled at room temperature and diluted with ether (100 mL). The precipitate was filtered off and washed with Et2O to give a powder (fraction A). The mother liquor was concentrated in vacuo, triturated in ether, filtered off, and washed with ether to give a powder (fraction 2). Fractions 1 and 2 were mixed and poured into water (250 mL). The pH of the resulting solution was adjusted to 6-7 (control with pH paper) with HCl IN. The precipitate was filtered off, washed with water and dried. Then, the solid was triturated in diisopropyl ether, fϊltered off and dried to give 26 g (88%) of the compound 6 as a brownish solid: m/z = 315 (M+H)+.
Synthesis of (hex-5-enyl)(methyl)amine (8)
O CF,
FX N Br’ N O NH
H 7
(a) Sodium hydride (1.05 eq) was slowly added at 00C to a solution of JV-methyl- trifluoro-acetamide (25 g) in DMF (140 mL). The mixture was stirred for Ih at room temperature under nitrogen. Then, a solution of bromohexene (32,1 g) in DMF
(25 mL) was added dropwise and the mixture was heated to 700C for 12 hours. The reaction mixture was poured on water (200 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and evaporated to give 35 g of the target product 7 as a yellowish oil which was used without further purification in the next step.
(b) A solution of KOH (187.7 g) in water (130 mL) was added dropwise to a solution of 7 (35 g) in methanol (200 mL). The mixture was stirred at room temperature for
12 hours. Then, the reaction mixture was poured on water (100 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and the ether was distilled under atmospheric pressure. The resulting oil was purified by distillation under vacuum (13 mm Hg pressure, 500C) to give 7,4 g (34 %) of the title product 8 as a colourless oil: 1H-NMR (CDCl3): δ 5.8 (m, IH), 5 (ddd, J = Yl 2 Hz, 3.5 Hz, 1.8 Hz, IH), 4.95 (m, IH), 2.5 (t, J = 7.0 Hz, 2H), 2.43 (s, 3H), 2.08 (q, J= 7.0 Hz, 2H), 1.4 (m, 4H), 1.3 (br s, IH).
Preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxyl- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04‘6loctadec-7-ene-4-carboxylic acid (16)

3-Oxo-2-oxa-bicyclo[2.2.1]heptane-5-carboxylic acid 9 (500 mg, 3.2 mmol) in 4 mL DMF was added at 00C to HATU (1.34 g, 3.52 mmol) and JV-methylhex-5-enylamine (435 mg, 3.84 mmol) in DMF (3 mL), followed by DIPEA. After stirring for 40 min at 00C, the mixture was stirred at room temperature for 5 h. Then, the solvent was evaporated, the residue dissolved in EtOAc (70 rnL) and washed with saturated NaHCOs (IO mL). The aqueous layer was extracted with EtOAc (2 x 25 mL). The organic phases were combined, washed with saturated NaCl (20 mL), dried (Na2SO4), and evaporated. Purification by flash chromatography (EtO Ac/petroleum ether, 2:1) afforded 550 mg (68%) of the target product 10 as a colorless oil: m/z = 252 (M+H)+.

A solution of LiOH (105 mg in 4 mlof water) was added at 00C to the lactone amide 10. After Ih, the conversion was completed (HPLC). The mixture was acidified to pH 2 – 3 with IN HCl, extracted with AcOEt, dried (MgSO4), evaporated, co-evaporated with toluene several times, and dried under high vacuum overnight to give 520 mg (88%) of the target product 11: m/z = 270 (M+H)+.

The l-(amino)-2-(vinyl)cyclopropanecarboxylic acid ethyl ester hydrochloride 12
(4.92 g, 31.7 mmol) and HATU (12.6 g, 33.2 mmol) were added to 11 (8.14 g,
30.2 mmol). The mixture was cooled in an ice bath under argon, and then DMF (100 mL) and DIPEA (12.5 mL, 11.5 mmol) were successively added. After 30 min at 00C, the solution was stirred at room temperature for an additional 3 h. Then, the reaction mixture was partitioned between EtOAc and water, washed successively with 0.5 N HCl (20 mL) and saturated NaCl (2 x 20 mL), and dried (Na2SO4). Purification by flash chromatography (AcOEt/CH2Cl2/Petroleum ether, 1 :1 :1) afforded 7.41 g (60%) of the target product 13 as a colorless oil: m/z = 407 (M+H)+.

DIAD (1.02 niL, 5.17 mmol) was added at -15°C under nitrogen atmosphere to a solution of 13 (1.5 g, 3.69 mmol), quinoline 6 (1.39 g, 4.43 mmol) and triphenyl- phosphine (1.26 g, 4.80 mmol) in dry THF (40 mL). After 4.5 h, at -15°C, the reaction mixture was partitioned between ice-cold water and AcOEt, dried (Na2SO4) and evaporated. The crude material was purified by flash column chromatography (gradient of petroleum AcOEt/CH2Cl2, 1 :9 to 2:8) to give 1.45 g (56 %) of the target product 14: m/z = 703 (M+H)+.

A solution of 14 (1.07 g, 1.524 mmol) and Hoveyda-Grubbs 1st generation catalyst (33 mg, 0.03 eq) in dried and degassed 1 ,2-dichloroethane (900 mL) was heated at 75°C under nitrogen for 12 h. Then, the solvent was evaporated and the residue purified by silica gel chromatography (25% EtOAc in CH2Cl2). 620 mg (60%) of pure macrocycle 15 were obtained, m/z = 674 (M+H)+. 1H NMR (CDCl3): 1.18-1.39 (m, 12H), 1.59 (m, IH), 1.70-2.08 (m, 5H), 2.28 (m, IH), 2.38 (m, IH), 2.62 (m, 2H), 2.68 (s, 3H), 2.83 (m, IH), 3.06 (s, 3H), 3.19 (sept, J= 6.7 Hz, IH), 3.36 (m, IH), 3.83 (m, IH), 3.97 (s, 3H), 4.09 (m, 2H), 4.65 (td, J= 4 Hz, 14 Hz, IH), 5.19 (dd, J= 4 Hz,
10 Hz, IH), 5.31 (m, IH), 5.65 (td, J= 4 Hz, 8 Hz, IH), 7.00 (s, IH), 7.18 (s, IH), 7.46
(d, J= 9 Hz, IH), 7.48 (s, IH), 8.03 (d, J= 9 Hz, IH).

A solution of lithium hydroxide (1.65 g, 38.53 mmol) in water (15 rnL) was added to a stirred solution of ester 15 (620 mg, 0.920 mmol) in THF (30 mL) and MeOH (20 mL). After 16 h at room temperature, the reaction mixture was quenched with NH4Cl sat., concentrated under reduced pressure, acidified to pH 3 with HCl IN and extracted with CH2Cl2, dried (MgSO4) and evaporated to give 560 mg (88%) of carboxylic acid 16. m/z = 647 (M+H)+. 1H NMR (CDCl3): 1.11-1.40 (m, 8H), 1.42-1.57 (m, 2H), 1.74 (m, 2H), 1.88-2.00 (m, 2H), 2.13 (m, IH), 2.28 (m, IH), 2.40 (m, IH), 2.59 (m, 2H), 2.67 (s, 3H), 2.81 (m, IH), 2.97 (s, 3H), 3.19 (m, IH), 3.31 (m, IH), 3.71 (m, IH), 3.96 (s, 3H), 4.56 (dt, J= 4 Hz, 12 Hz, IH), 5.23 (m, 2H), 5.66 (m, IH), 7.01 (s, IH), 7.10 (s, IH), 7.22 (d, J= IO Hz, IH), 7.45 (s, IH), 8.00 (d, J= 10 Hz, IH).
Example 2: Preparation of Λ/-[17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[ 13.3.0.04‘6]octadec-7-ene- 4-carbonyll(cvclopropyl)sulfonamide (17) SIMEPREVIR

A solution of the compound 16 (560mg, 0.867 mmol) prepared according to Example 4, and carbonyldiimidazole (308 mg, 1.90 mmol) in dry THF (10 mL) was stirred at reflux under nitrogen for 2h. The reaction mixture was cooled to room temperature and cyclopropylsulfonamide (400 mg, 3.301 mmol) and DBU (286 mg, 1.881 mmol) were added. This solution was heated at 500C for 15 h. Then, the reaction mixture was cooled down at room temperature and concentrated under reduced pressure. The residue was partitioned between CH2Cl2 and HCl 1 N, the organic layer was washed with brine, dried (MgSO4) and evaporated. Purification by flash chromatography (gradient of EtOAc (0 to 25%) in CH2Cl2) afforded 314 mg of an off-white solid which was further washed with water, then isopropylether, and dried in the vacuum oven to deliver 282 mg (40%) of the pure title product 17, which is the compound of formula (I) SIMEPREVIR , as a white powder: m/z = 750 (M+H)+.
1H NMR (CDCl3): 0.99-1.52 (m, 14H), 1.64-2.05 (m, 4H), 2.77 (m, IH), 2.41 (m, 2H), 2.59 (m, 2H), 2.69 (s, 3H), 2.92 (m, 2H), 3.04 (s, 3H), 3.19 (m, IH), 3.40 (m, 2H), 3.98 (s, 3H), 4.60 (t, J= 13 Hz, IH), 5.04 (t, J= 11 Hz, IH), 5.37 (m, IH), 5.66 (m, IH), 6.21 (s, IH), 7.02 (s, IH), 7.22 (d, J= IO Hz, IH), 7.45 (s, IH), 7.99 (d, J= 10 Hz, IH), 10.82 (broad s, IH).
…………………
SYNTHESIS
Example 4: preparation of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methyl- quinolin-4-yloxy] – 13 -methyl-2, 14-dioxo-3 , 13 -diazatricyclo[ 13.3.0.04‘6]octadec-7-ene- 4-carboxylic acid (46) FREE ACID
Synthesis of 4-hvdroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinoline (36) Step 1: synthesis of iV-(tert-butyloxycarbonyl)-3-methoxy-2-methylaniline (32)

31 32
Triethylamine (42.4 mL, 302 mmol) was added to a suspension of 3-methoxy-2- methylbenzoic acid (45.6 g, 274 mmol) in dry toluene (800 mL). A clear solution was obtained. Then, dppa (65.4 mL, 302 mmol) in toluene (100 mL) was slowly added. After 1 h at room temperature, the reaction mixture was successively heated at 50°C for 0.5 h, at 70°C for 0.5 h then at 100°C for 1 h. To this solution, t-BuOH (30.5 g, 411 mmol) in toluene (40 mL) was added at 100°C and the resulting mixture was refluxed for 7h. The solution was cooled to room temperature then successively washed with water, 0.5 N HCl, 0.5 N NaOH and brine, dried (Na2SO4), and evaporated to give 67 g of the target product: m/z = 237 (M)+.
Step 2: synthesis of 3-methoxy-2-methylaniline (33)

TFA (40.7 mL, 548 mmol) was added to a solution of iV-(tert-butyloxycarbonyl)-3- methoxy-2-methylaniline, in dichloromethane (500 mL). After 2 h at room temperature, TFA (40.7 mL, 548 mmol) was added and the resulting mixture was stirred at room temperature overnight. Then, volatiles were evaporated. The residue was triturated with toluene (100 mL) and diisopropylether (250 mL), filtered off and washed with diisopropyl ether (100 mL) to give 56.3 g of the title product as a TFA salt: m/z = 138 (M+H)+. The TFA salt was transformed to the free aniline by treatment with NaHCO3.
Step 3: synthesis of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (34)

A solution OfBCl3 (1.0 M, 200 mL, 200 mmol) in CH2Cl2 was slowly added under nitrogen to a solution of 3-methoxy-2-methylaniline (26.0 g, 190 mmol) in xylene (400 mL). The temperature was monitored during the addition and was kept below 10°C. The reaction mixture was stirred at 5°C for 0.5 h. Then, dry acetonitrile (13 mL, 246 mmol) was added at 5°C. After 0.5 h at 5°C, the solution was transferred into a dropping funnel and slowly added at 5°C to a suspension OfAlCl3 (26.7 g, 200 mmol) in CH2Cl2 (150 mL). After 45 min at 5°C, the reaction mixture was heated at 70°C under a nitrogen stream. After evaporation Of CH2Cl2, the temperature of the reaction mixture reached 65°C. After 12 h at 65°C, the reaction mixture was cooled at 0°C, poured onto ice (300 g), and slowly heated to reflux for 7h. After 2 days at room temperature, 6 N NaOH (50 mL) was added. The pH of the resulting solution was 2-3. The xylene layer was decanted. The organic layer was extracted with CH2Cl2. The xylene and CH2Cl2 layers were combined, successively washed with water, IN NaOH, and brine, dried (Na2SO4) and evaporated. The residue was triturated in diisopropyl ether at O0C, filtered off and washed with diisopropylether to give 13.6 g (40 %) of the title product as a yellowish solid: m/z = 180 (M+H)+.
Step 4: synthesis of 2′-[[(4-isopropylthiazole-2-yl)(oxo)methyl]amino]-4′-methoxy-3 ‘- methylacetophenone (35)

A solution of (2-amino-4-methoxy-3-methylphenyl)(methyl)ketone (18.6 g, 104 mmol) in dioxane (50 mL) was added under nitrogen to a suspension of 4-isopropylthiazole-2- carbonyl chloride in dioxane (250 mL). After 2 h at room temperature, the reaction mixture was concentrated to dryness. Then, the residue was partitioned between an aqueous solution OfNaHCO3and AcOEt, organic layer was washed with brine, dried (Na2SO4), and evaporated. The residue was triturated in diisopropyl ether, filtered off and washed with diisopropyl ether to give 30.8 g (90 %) of the title product 35.
Step 5: synthesis of 4-hydroxy-2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinoline (36)

Potassium tert-butoxide (21.8 g, 195 mmol) was added to a suspension of 2′-[[(4-iso- propylthiazole-2-yl)(oxo)methyl]amino]-4′-methoxy-3′-methylacetophenone (35, 30.8 g, 92.7 mmol) in tert-butanol. The resulting reaction mixtures was heated at 100°C overnight. Then, the reaction mixture was cooled at room temperature and diluted with ether (100 mL). The precipitate was filtered off and washed with Et2O to give a powder (fraction A). The mother liquor was concentrated in vacuo, triturated in ether, filtered off, and washed with ether to give a powder (fraction 2). Fractions 1 and 2 were mixed and poured into water (250 mL). The pH of the resulting solution was adjusted to 6-7 (control with pH paper) with HCl IN. The precipitate was filtered off, washed with water and dried. Then, the solid was triturated in diisopropyl ether, filtered off and dried to give 26 g (88%) of the title product 36 as a brownish solid: m/z = 315 (M+H)+.
Synthesis of (hex-5-enyl)(methyl)amine (38)

Sodium hydride (1.05 eq) was slowly added at 0°C to a solution of iV-methyltrifluoro- acetamide (25 g) in DMF (140 mL). The mixture was stirred for Ih at room temperature under nitrogen. Then, a solution of bromohexene (32,1 g) in DMF (25 mL) was added dropwise and the mixture was heated to 70°C for 12 hours. The reaction mixture was poured on water (200 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and evaporated to give 35 g of the target product 37 as a yellowish oil which was used without further purification in the next step.
Step B:
A solution of potassium hydroxide (187.7 g) in water (130 mL) was added dropwise to a solution of 37 (35 g) in methanol (200 mL). The mixture was stirred at room temperature for 12 hours. Then, the reaction mixture was poured on water (100 mL) and extracted with ether (4 x 50 mL), dried (MgSO4), filtered and the ether was distilled under atmospheric pressure. The resulting oil was purified by distillation under vacuum (13 mm Hg pressure, 50°C) to give 7,4 g (34 %) of the title product 38 as a colourless oil: 1H-NMR (CDCl3): δ 5.8 (m, IH), 5 (ddd, J= 17.2 Hz, 3.5 Hz, 1.8 Hz, IH), 4.95 (m, IH), 2.5 (t, J= 7.0 Hz, 2H), 2.43 (s, 3H), 2.08 (q, J= 7.0 Hz, 2H), 1.4 (m, 4H), 1.3 (br s, IH).
Preparation of 17-r2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxyl-
13-methyl-2,14-dioxo-3,13-diazatricvclori3.3.0.04‘6loctadec-7-ene-4-carboxylic acid
£46}

3-Oxo-2-oxa-bicyclo[2.2.1]heptane-5-carboxylic acid 39 (500 mg, 3.2 mmol) in 4 mlDMF was added at 0°C to HATU (1.34 g, 3.52 mmol) and iV-methylhex-5- enylamine (435 mg, 3.84 mmol) in DMF (3 mL), followed by DIPEA. After stirring for 40 min at 0°C, the mixture was stirred at room temperature for 5 h. Then, the solvent was evaporated, the residue dissolved in EtOAc (70 mL) and washed with saturated NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (2 x 25 mL). The organic phases were combined, washed with saturated NaCl (20 mL), dried (Na2SO4), and evaporated. Purification by flash chromatography (EtOAc/petroleum ether, 2:1) afforded 550 mg (68%) of the target product 40 as a colorless oil: m/z = 252 (M+H)+.

A solution of LiOH (105 mg in 4 mlof water) was added at 0°C to the lactone amide 40. After Ih, the conversion was completed (HPLC). The mixture was acidified to pH 2 – 3 with IN HCl, extracted with AcOEt, dried (MgSO4), evaporated, co-evaporated with toluene several times, and dried under high vacuum overnight to give 520 mg (88%) of the target product 41: m/z = 270 (M+H)+.

The l-(amino)-2-(vinyl)cyclopropanecarboxylic acid ethyl ester hydrochloride 42 (4.92 g, 31.7 mmol) and HATU (12.6 g, 33.2 mmol) were added to 41 (8.14 g, 30.2 mmol). The mixture was cooled in an ice bath under argon, and then DMF (100 mL) and DIPEA (12.5 mL, 11.5 mmol) were successively added. After 30 min at 0°C, the solution was stirred at room temperature for an additional 3 h. Then, the reaction mixture was partitioned between EtOAc and water, washed successively with 0.5 N HCl (20 mL) and saturated NaCl (2 x 20 mL), and dried (Na2SO4). Purification by flash chromatography (AcOEt/CH2Cl2/Petroleum ether, 1:1:1) afforded 7.41 g (60%) of the target product 43 as a colorless oil: m/z = 407 (M+H)+.

DIAD (1.02 mL, 5.17 mmol) was added at -15°C under nitrogen atmosphere to a solution of 43 (1.5 g, 3.69 mmol), quinoline 36 (1.39 g, 4.43 mmol) and triphenyl- phosphine (1.26 g, 4.80 mmol) in dry THF (40 mL). After 4.5 h, at -15°C, the reaction mixture was partitioned between ice-cold water and AcOEt, dried (Na2SO4) and evaporated. The crude material was purified by flash column chromatography (gradient of petroleum AcOEt/CH2Cl2, 1 :9 to 2:8) to give 1.45 g (56 %) of the target product 44: m/z = 703 (M+H)+.

A solution of 44 (1.07 g, 1.524 mmol) and Hoveyda-Grubbs 1st generation catalyst (33 mg, 0.03 eq) in dried and degassed 1,2-dichloroethane (900 mL) was heated at 75°C under nitrogen for 12 h. Then, the solvent was evaporated and the residue purified by silica gel chromatography (25% EtOAc in CH2Cl2). 620 mg (60%) of pure macrocycle 45 were obtained, m/z = 674 (M+H)+. 1H NMR (CDCl3): 1.18-1.39 (m, 12H), 1.59 (m, IH), 1.70-2.08 (m, 5H), 2.28 (m, IH), 2.38 (m, IH), 2.62 (m, 2H), 2.68 (s, 3H), 2.83 (m, IH), 3.06 (s, 3H), 3.19 (sept, J= 6.7 Hz, IH), 3.36 (m, IH), 3.83 (m, IH), 3.97 (s, 3H), 4.09 (m, 2H), 4.65 (td, J= 4 Hz, 14 Hz, IH), 5.19 (dd, J= 4 Hz, 10 Hz, IH), 5.31 (m, IH), 5.65 (td, J= 4 Hz, 8 Hz, IH), 7.00 (s, IH), 7.18 (s, IH), 7.46 (d, J= 9 Hz, IH), 7.48 (s, IH), 8.03 (d, J= 9 Hz, IH).
Step F

A solution of lithium hydroxide (1.65 g, 38.53 mmol) in water (15 mL) was added to a stirred solution of ester 45 (620 mg, 0.920 mmol) in THF (30 mL) and MeOH (20 mL). After 16 h at room temperature, the reaction mixture was quenched with NH4Cl sat., concentrated under reduced pressure, acidified to pH 3 with HCl IN and extracted with CH2Cl2, dried (MgSO4) and evaporated to give 560 mg (88%) of carboxylic acid 46. m/z = 647 (M+H)+. 1H NMR (CDCl3): 1.11-1.40 (m, 8H), 1.42-1.57 (m, 2H), 1.74 (m, 2H), 1.88-2.00 (m, 2H), 2.13 (m, IH), 2.28 (m, IH), 2.40 (m, IH), 2.59 (m, 2H), 2.67 (s, 3H), 2.81 (m, IH), 2.97 (s, 3H), 3.19 (m, IH), 3.31 (m, IH), 3.71 (m, IH), 3.96 (s, 3H), 4.56 (dt, J= 4 Hz, 12 Hz, IH), 5.23 (m, 2H), 5.66 (m, IH), 7.01 (s, IH), 7.10 (s, IH), 7.22 (d, J= 10 Hz, IH), 7.45 (s, IH), 8.00 (d, J= 10 Hz, IH).
Example 5: Preparation of JV-ri7-r2-(4-isopropylthiazole-2-yl)-7-methoxy-8- methylquinolin-4- yloxyl – 13 -methyl-2, 14-dioxo-3 , 13 -diazatricyclol“ 13.3.0.04‘6loctadec- 7-ene-4-carbonyll (cvclopropyPsulfonamide (47) SIMEPREVIR

A solution of 17-[2-(4-isopropylthiazole-2-yl)-7-methoxy-8-methylquinolin-4-yloxy]- 13-methyl-2, 14-dioxo-3, 13-diazatricyclo[l 3.3.0.04,6]octadec-7-ene-4-carboxylic acid 46 (560mg, 0.867 mmol) prepared according to Example 4, and carbonyldiimidazole (308 mg, 1.90 mmol) in dry THF (10 mL) was stirred at reflux under nitrogen for 2h. The reaction mixture was cooled to room temperature and cyclopropylsulfonamide (400 mg, 3.301 mmol) and DBU (286 mg, 1.881 mmol) were added. This solution was heated at 50°C for 15 h. Then, the reaction mixture was cooled down at room temperature and concentrated under reduced pressure. The residue was partitioned between CH2CI2 and HCl 1 N, the organic layer was washed with brine, dried (MgSO4) and evaporated. Purification by flash chromatography (gradient of EtOAc (0 to 25%) in CH2CI2) afforded 314 mg of an off-white solid which was further washed with water, then isopropylether, and dried in the vacuum oven to deliver 282 mg (40%) of the pure title product 47 SIMEPREVIR as a white powder: m/z = 750 (M+H)+.
1H NMR (CDCl3): 0.99-1.52 (m, 14H), 1.64-2.05 (m, 4H), 2.77 (m, IH), 2.41 (m, 2H), 2.59 (m, 2H), 2.69 (s, 3H), 2.92 (m, 2H), 3.04 (s, 3H), 3.19 (m, IH), 3.40 (m, 2H), 3.98 (s, 3H), 4.60 (t, J= 13 Hz, IH), 5.04 (t, J= 11 Hz, IH), 5.37 (m, IH), 5.66 (m, IH), 6.21 (s, IH), 7.02 (s, IH), 7.22 (d, J= 10 Hz, IH), 7.45 (s, IH), 7.99 (d, J= 10 Hz, IH), 10.82 (broad s, IH).
…………………..
REFERENCES
- “Medivir Announces That Simeprevir (TMC435) Data Will Be Presented at the Upcoming AASLD Meeting”. Yahoo News. October 1, 2012. Retrieved November 6, 2012.
- Lin, TI; Lenz, O; Fanning, G; Verbinnen, T; Delouvroy, F; Scholliers, A; Vermeiren, K; Rosenquist, A et al. (2009). “In vitro activity and preclinical profile of TMC435350, a potent hepatitis C virus protease inhibitor”. Antimicrobial agents and chemotherapy 53 (4): 1377–85. doi:10.1128/AAC.01058-08. PMC 2663092. PMID 19171797.
|displayauthors=suggested (help) - “Phase 3 Studies Show Simeprevir plus Interferon/Ribavirin Cures Most Patients in 24 Weeks”. hivandhepatitis.com. December 27, 2012.
- Medivir announces TMC435 in an expanded clinical collaboration. Medivir. 18 April 2012.
- Results from a phase IIa study evaluating Simeprevir and Sofosbuvir in prior null responder Hepatitis C patients have been presented at CROI. 6 March 2013.
- TMC-435350
Drugs Fut 2009, 34(7): 545 - Structure-activity relationship study on a novel series of cyclopentane-containing macrocyclic inhibitors of the hepatitis C virus NS3/4A protease leading to the discovery of TMC435350
Bioorg Med Chem Lett 2008, 18(17): 4853 - Synthesis of enantiomerically pure trans-3,4-substituted cyclopentanols by enzymatic resolution
Acta Chem Scand (1989) 1992, 46: 1127
PATENTS
- WO 2008092954
- WO 2007014926
- WO 2008092955
- WO 2000009543
- CN 102531932
- WO 2013061285
- WO 2011113859
- WO 2013041655
| WO2010097229A2 * | 26 Feb 2010 | 2 Sep 2010 | Ortho-Mcneil-Janssen Pharmaceuticals Inc | Amorphous salt of a macrocyclic inhibitor of hcv |
| WO2013037705A2 * | 7 Sep 2012 | 21 Mar 2013 | Fovea Pharmaceuticals | Aniline derivatives,their preparation and their therapeutic application |
| WO2005073195A2 * | 28 Jan 2005 | 11 Aug 2005 | Per-Ola Johansson | Hcv ns-3 serine protease inhibitors |
| WO2007014926A1 * | 28 Jul 2006 | 8 Feb 2007 | Tibotec Pharm Ltd | Macrocyclic inhibitors of hepatitis c virus |

The compound ritonavir, and pharmaceutically acceptable salts thereof, and methods for its preparation are described in WO94/14436. For preferred dosage forms of ritonavir, see US6,037, 157, and the documents cited therein: US5,484, 801, US08/402,690, and WO95/07696 and WO95/09614. Ritonavir has the following formula:

