
Home » Posts tagged 'Onc-01210'
Tag Archives: Onc-01210
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
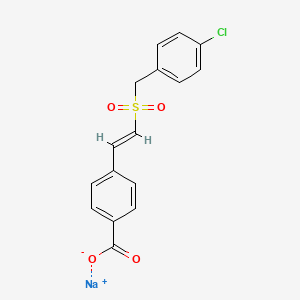
Recilisib Sodium, EX-RAD

Recilisib Sodium
Phase I
| C16H12ClNaO4S | |
| Molecular Weight: | 358.771849 g/mol |
|---|
A protein kinase inhibitor potentially for the treatment of acute radiation syndrome.
sodium;4-[(E)-2-[(4-chlorophenyl)methylsulfonyl]ethenyl]benzoate
![]()
Onc-01210; ON-01210.Na, Ex-RAD; ON 01210.Na; ON-01210; ON-01210-Na; Recilisib
CAS No. 334969-03-8(free)
CAS 922139-31-9(Recilisib sodium)
Benzoic acid, 4-[(1E)-2-[[(4-chlorophenyl)methyl]sulfonyl]ethenyl]-, sodium salt (1:1)
Onconova Therapeutics Inc, Univ Temple INNOVATOR
Stephen C Cosenza, Lawrence Helson,Premkumar E Reddy, Ramana M V Reddy INVENTORS
![]()
![]()

| Latest Stage of Development | Phase I |
| Standard Indication | Poisoning |
| Indication Details | Prevent radiation poisoning; Provide radation protection; Treat and prevent acute radiation syndrome (ARS) |
- Originator Onconova Therapeutics
- Class Radioprotectives; Small molecules; Sulfonamides
- Mechanism of Action Apoptosis inhibitors; Protein kinase inhibitors
- Orphan Drug Status Yes – Acute radiation syndrome
- Phase I Acute radiation syndrome
Most Recent Events
- 22 Apr 2016 Phase I development is ongoing in the US (PO & SC)
- 20 Mar 2014 Recilisib receives Orphan Drug status for Acute radiation syndrome in USA
- 03 Oct 2012 Phase-I clinical trials in Acute radiation syndrome in USA (PO)

Ex-Rad (or Ex-RAD), also known by the code name ON 01210.Na, or recilisib sodium (INN, USAN) is a drug developed by Onconova Therapeutics and the U.S. Department of Defense.[1][2] This newly developed compound is said to be a potent radiation protection agent. Chemically, it is the sodium salt of 4-carboxystyryl-4-chlorobenzylsulfone.[3]
Clinical trials
The results of two Phase I clinical studies in healthy human volunteers indicate that subcutaneously injected Ex-Rad is safe and well tolerated, with “no evidence of systemic side effects”.[4] A study in mice demonstrated the efficacy of Ex-Rad by increasing the survival rate of mice exposed to typically lethal whole-body irradiation. The study tested oral and parenteral administration of Ex-Rad for both pre- and post-exposure radiomitigation.[1]
Research on Ex-Rad has involved collaboration with the Armed Forces Radiobiology Research Institute (AFRRI), the Department of Biochemistry and Molecular & Cellular Biology at Georgetown University, Long Island University‘s Arnold & Marie Schwartz College of Pharmacy, and the Department of Oncological Sciences at the Mt. Sinai School of Medicine.[1]
Mechanism of action
Onconova suggests that Ex-Rad protects cells exposed to radiation against DNA damage, and that the drug’s mechanism of action does not involve scavenging free radicals or arresting the cell cycle. Instead, they claim it employs a “novel mechanism” involving “intracellular signaling, damage sensing, and DNA repair pathways”.[4] Ex-RAD is a chlorobenzylsulfone derivative that works after free radicals have damaged DNA. Onconova CEO Ramesh Kumar believes this is a better approach than trying to scavenge free radicals. “Free radicals are very short-lived, and so the window of opportunity to give a drug is very narrow,” he says. In cell and animal models, Ex-RAD protects hematopoieticand gastrointestinal tissues from radiation injury when given either before or after exposure.[5]
While anti-radiation suits or other protective gear may be effective at reducing radiation exposure, such gear is expensive, unwieldy, and generally not available to public. Moreover, radioprotective gear will not protect normal tissue adjacent to a tumor from stray radiation exposure during radiotherapy. Pharmaceutical radioprotectants offer a cost-efficient, effective and easily available alternative to radioprotective gear. However, previous attempts at radioprotection of normal cells with pharmaceutical compositions have not been entirely successful. For example, cytokines directed at mobilizing the peripheral blood progenitor cells confer a myeloprotective effect when given prior to radiation (Neta et al., Semin. Radiat. Oncol. 6:306-320, 1996), but do not confer systemic protection. Other chemical radioprotectors administered alone or in combination with biologic response modifiers have shown minor protective effects in mice, but application of these compounds to large mammals was less successful, and it was questioned whether chemical radioprotection was of any value (Maisin, J. R., Bacq and Alexander Award Lecture. “Chemical radioprotection: past, present, and future prospects”, Int J. Radiat Biol. 73:443-50, 1998). Pharmaceutical radiation sensitizers, which are known to preferentially enhance the effects of radiation in cancerous tissues, are clearly unsuited for the general systemic protection of normal tissues from exposure to ionizing radiation.
The major biological effects of radiation exposure are the destruction of bone marrow cells, gastrointestinal (GI) damage, lung pneumonitis, and central nervous system (CNS) damage. The long-term effects of radiation exposure include an increase in cancer rates. It has been estimated that the exposure of 100 rems (roentgen equivalent man: a measurement used to quantify the amount of radiation that would produce harmful biological effects) would produce ARS symptoms. Exposure levels above 300 rems would result in the death of approximately 50% of the exposed population.
The α,β-unsaturated aryl sulfones, in particular benzyl styryl sulfones, provide significant and selective systemic protection of normal cells from radiation-induced damage in animals. When used in radiotherapy techniques, these compounds also exhibit independent toxicity to cancer cells. These α,β-unsaturated aryl sulfones, in particular benzyl styryl sulfones, are described in U.S. Pat. Nos. 6,656,973 and 6,667,346, which are particularly incorporated herein by reference in their entirety. Although these compounds are stable in solid state their aqueous formulations for parenteral administration are pH sensitive and pose challenging hurdles to overcome physical stability. The most likely causative factor may be attributed to the reactive styryl sulfone conjugated double bond, which is prone to Michael addition by nucleophiles and eventual fallout of the conjugated addition product.
U.S. Patent No. 6,656,973, describes in vitro pharmacological effects of DMSO solubilization of a benzyl styryl sulfone (e.g. ON 01210.NA) but fails to disclose a composition comprising ON 01210. NA formulation and specifically, a shelf stable formulation which is suitable for administration to humans.
PCT Application WO 2007/016201 describes pharmaceutical solution compositions for parenteral administration for reducing toxic effects of ionizing radiation in a subject, comprising an effective amount of at least one radioprotective α,β-Unsaturated aryl sulfone, and at least one component selected from the group consisting of a) a water soluble polymer in an amount between about 0.5% and about 90% w/v, b) at least one chemically modified cyclodextrin in an amount between about 20% and about 60% w/v, and c) DMA in an amount between 10% and about 50% w/v.
U.S. Patent Application 20090247624, and corresponding PCT Application WO 2008/105808, are directed to aqueous solutions, which comprise between about 20 mg/ml to about 100 mg/ml of at least one α,β-unsaturated aryl sulfone (e.g., the compound ON 01210. Na ((E)-4-Carboxystyryl-4-chlorobenzylsulfone sodium salt, a cosolvent in an amount between about 25% and about 90% w/v (e.g., about 50% PEG 400), wherein the composition is buffered and exists within the range of about pH 7.0 to about pHIO (e.g., 0.2M Tris-EDTA, pH about 8.5). The aforementioned solution formulations have exhibited a sub-optimal shelf life and lack a preferred degree of solubility and/or stability. These formulations evolved progressively as a result of addressing the most challenging aspects in the formulation and drug development field, namely, solubility and stability parameters that defined the long term viability of these formulations. There seems to be a delicate balance between pH, solubility and stability of the active moiety in aqueous milieu, wherein achieving such balance and development of a shelf stable aqueous formulation has presented a formidable challenge. Therefore, a shelf stable effective solution formulation that prevents the breakdown of the therapeutically active entity and keeps the drug in the solution at the desired pH was most desired and significant effort was directed towards this goal.
What is needed therefore, is a shelf stable effective solution formulation of radioprotective α,β-unsaturated aryl sulfones that prevents the breakdown of the therapeutically active entity and keeps the drug in the solution at the desired pH. This invention solves these and other long felt needs by providing improved solution formulation of radioprotective α,β- unsaturated aryl sulfones having improved physical and chemical stability and enhanced shelf life.
SYNTHESIS BY WORLDDRUGTRACKER
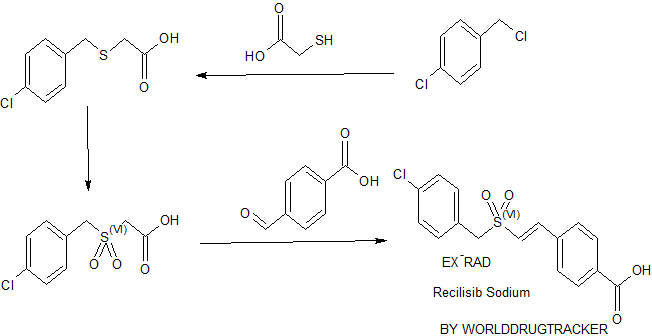
PATENT
WO 2011119863
An exemplary species of a radioprotective α,β-unsaturated aryl sulfone is ON 01210.Na. ON 01210.Na is a derivative of chlorobenzylsulfone. This compound is described in U.S. Pat. Nos. 6,656,973 and 6,667,346 as exhibiting valuable prophylactic properties which mitigate the effects of accidental and intentional exposure to life-threatening levels of irradiation. Hence, a systematic development of this compound is described with the objective of developing a shelf stable formulation.
Table 1 describes the general physical properties of ON. 1210. Na. The exemplary compound is a sodium salt of (E)-4-Carboxystyryl-4-chlorobenzylsulfone.
TABLE 1
Physical Properties of ON.1210.Na
Chemical Structure

Chemical Name (E)-4-Carboxystyryl-4-chlorobenzylsulfone,
Sodium Salt
Empirical Formula C16H12ClNa04S
Molecular Weight 358.79
Physical Nature White crystalline flakes
Melting Point 354-356° C.
Solubility Soluble in water at 8-10 mg/ml
The compound ON 01210. Na appears to form at least one polymorph. X-ray diffraction pattern, for example, of precipitated ON 01210. Na is different from that of the originally synthesized compound. Polymorphs of ON 01210.Na are intended to be within the scope of the claims appended hereto.
EXAMPLE 1
Preparation of ON 01210. Na
4-Chlorobenzyl-4-carboxystyryl sulfone (ON 01210) (49 g; 0.145 mol) was taken in a one-liter conical flask and 500 ml of distilled water was added. Sodium hydroxide solution (16 ml: 10 M stock) (0.150 mol.) was added to the conical flask. The contents of the flask were then boiled with stirring till ON 01210 was completely dissolved. The solution was then cooled to room temperature and shining crystals separated were filtered through a fluted filter paper. The crystalline material was dried under vacuum to yield (48 g) (92% yield) of pure ON 1210. Na.
EXAMPLE II
Preparation of ON 01210. Na Formulation A (Without Vitamin E TPGS)
TRIS (968.0 mg), EDTA (233.8 mg), and deionized (DI) water (24 ml) were combined in a beaker equipped with a Teflon coated stirring bar. The mixture was stirred until complete dissolution occurred, and the resulting solution was covered with aluminum foil and allowed to stir gently overnight at room temperature. The following morning, PEG 400 NF (40.0 ml) was added to the TRIS/EDTA aqueous solution with continued stirring. The vessel containing PEG 400 NF was rinsed with DI water (2 x 3.2 ml), and the rinsate added to the formulation mixture. After stirring the mixture to homogeneity (approx. 10 minutes), the pH was measured to be 9.46 using a calibrated electronic pH meter. The pH was adjusted to 8.37 (target pH = 8.40) by the careful addition of 98 pipet drops of 1.0 M HCl (aq) with stirring and allowed to fully equilibrate over a 10-15 minute period. Once the pH steadied at 8.37, ON 01210. Na (4.0 g) was added to the stirring formulation mixture. Complete dissolution required vigorous stirring and brief periodic sonication to break up ON 01210.Na clumps over a two hour period. After complete dissolution of ON 01210. Na, DI water (approx. 5 ml) was added to bring the final volume to approximately 80 milliliters. The pH of the resulting solution was determined to be 8.31, and thus 20 pipet drops of 1.0N NaOH(aq) were added to adjust the final formulation batch (defined as ON 01210.Na Formulation A) pH to 8.41-8.42. Formulation A was 0.22 micron filtered using a 100 ml Gastight Syringe equipped with a Millex®GP filter unit (Millipore Express® PES Membrane; Lot No R8KN13888).
PATENT
PATENT
PATENT
The α,β unsaturated aryl sulfones are characterized by cis-trans isomerism resulting from the presence of one or more double bonds. The compounds are named according to the Cahn-Ingold-Prelog system, the IUPAC 1974 Recommendations, Section E: Stereochemistry, in Nomenclature of Organic Chemistry, John Wiley & Sons, Inc., New York, NY, 4th ed., 1992, p.
127-138. Stearic relations around a double bond are designated as “Z” or “E”.
(E)-α,β unsaturated aryl sulfones may be prepared by Knoevenagel condensation of aromatic aldehydes with benzylsulfonyl acetic acids or arylsulfonyl acetic acids. The procedure is described by Reddy et al, Ada. Chim. Hung. 115:269-71 (1984); Reddy et al, Sulfur Letters 13:83-90 (1991); Reddy et al, Synthesis No. 4, 322-23 (1984); and Reddy et al, Sulfur Letters 7:43-48 (1987), the entire disclosures of which are incorporated herein by reference.
According to the Scheme 1 below, Ra and Rb each represent from zero to five substituents on the depicted aromatic nucleus. For purposes of illustration, and not limitation, the aryl groups are represented as phenyl groups, that is, the synthesis is exemplified by the preparation of styryl benzylsulfones. Accordingly, the benzyl thioacetic acid B is formed by the reaction of sodium thioglycollate and a benzyl chloride A. The benzyl thioacetic acid B is then oxidized with 30% hydrogen peroxide to give a corresponding benzylsulfonyl acetic acid C. Condensation of the benzylsulfonyl acetic acid C with an aromatic aldehyde D via a Knoevenagel reaction in the presence of benzylamine and glacial acetic acid yields the desired (E)-styryl benzylsulfone E.

Scheme 1
The following is a more detailed two-part synthesis procedure for preparing (E)-styryl benzylsulfones according to the above scheme.
General Procedure 1: Synthesis (E)-Styryl Benzylsulfones
Part A. To a solution of (8g, 0.2 mol) sodium hydroxide in methanol (200 ml), thioglycollic acid (0.1 mol) is added slowly and the precipitate formed is dissolved by stirring the contents of the flask. Then an appropriately substituted benzyl chloride (0.1 mol) is added stepwise and the reaction mixture is refluxed for 2-3 hours. The cooled contents are poured onto crushed ice and neutralized with dilute hydrochloric acid (200 ml). The resulting corresponding benzylthioacetic acid (0.1 mol) is subjected to oxidation with 30% hydrogen peroxide (0.12 mol) in glacial acetic acid (125 ml) by refluxing for 1 hour. The contents are cooled and poured onto crushed ice. The separated solid is recrystalized from hot water to give the corresponding pure benzylsulfonylacetic acid.
Part B. A mixture of the benzylsulfonyl acetic acid (10 mmol), an appropriately substituted aromatic aldehyde (10 mmol), and benzylamine (0.2 ml) in glacial acetic acid (12 ml) is refluxed for 2-3 hours. The contents are cooled and treated with cold ether (50 ml). Any product precipitated out is separated by filtration. The filtrate is diluted with more ether and washed successively with a saturated solution of sodium bicarbonate (20 ml), sodium bisulfite (20 ml), dilute hydrochloric acid (20 ml) and finally with water (35 ml). Evaporation of the dried ethereal layer yields styryl benzylsulfones as a solid material.
According to an alternative to Part A, the appropriate benzylsulfonylacetic acids may be generated by substituting a thioglycollate
HSCH2COOR for thioglycollic acid, where R is an alkyl group, typically C1-C6 alkyl. This leads to the formation of the alkylbenzylthioacetate intermediate (F),
![]()
which is then converted to the corresponding benzyl thioacetic acid B by alkaline or acid hydrolysis.
(E)-styryl phenyl sulfones (formula I: n=zero; Qls Q2 = substituted or unsubstituted phenyl) are prepared according to the method of General Procedure 1, replacing the benzylsulfonyl acetic acid in Part B with the appropriate substituted or unsubstituted phenylsulfonyl acetic acid.
(Z)-Styryl benzylsulfones are prepared by the nucleophilic addition of the appropriate thiols to substituted phenylacetylene with subsequent oxidation of the resulting sulfide by hydrogen peroxide to yield the (Z)-styryl benzylsulfone. The procedure is generally described by Reddy et al., Sulfur Letters 13:83-90 (1991), the entire disclosure of which is incorporated herein as a reference.
In the first step of the (Z)-styryl benzylsulfones synthesis, the sodium salt of benzyl mercaptan or the appropriate substituted benzyl mercaptan is allowed to react with phenylacetylene or the appropriate substituted phenylacetylene forming the pure (Z)-isomer of the corresponding styryl benzylsulfide in good yield.
In the second step of the synthesis, the (Z)-styryl benzylsulfide intermediate is oxidized to the corresponding sulfone in the pure (Z)-isomeric form by treatment with hydrogen peroxide.
The following is a more detailed two-part synthesis procedure for preparing (Z)-styryl benzylsulfones:
Procedure 2: Synthesis of (Z)-Styryl Benzylsulfones
Part A. To a refluxing methanolic solution of substituted or unsubstituted sodium benzylthiolate prepared from 460 mg (0.02g atom) of (i) sodium, (ii) substituted or unsubstituted benzyl mercaptan (0.02 mol) and (iii) 80 ml of absolute methanol, is added freshly distilled substituted or unsubstituted phenylacetylene. The mixture is refluxed for 20 hours, cooled and then poured on crushed ice. The crude product is filtered, dried and recrystalized from methanol or aqueous methanol to yield a pure (Z)- styryl benzylsulfide.
Part B. An ice cold solution of the (Z)- styryl benzylsulfide (3.0g) in 30 ml of glacial acetic acid is treated with 7.5 ml of 30% hydrogen peroxide. The reaction mixture is refluxed for 1 hour and then poured on crushed ice. The separated solid is filtered, dried, and recrystalized from 2-propanol to yield the pure (Z)-styryl benzylsulfone. The purity of the compounds is ascertained by thin layer chromatography and geometrical configuration is assigned by analysis of infrared and nuclear magnetic resonance spectral data.
The bis(styryl) sulfones of formula IN are prepared according to Procedure 3:
Procedure 3
Synthesis of (E)(E)- and (E)(Z)-bis(Styryl) Sulfones
To freshly distilled phenyl acetylene (51.07 g, 0.5 mol) is added sodium thioglycollate prepared from thioglycollic acid (46 g, 0.5 mol) and sodium hydroxide (40 g, 1 mol) in methanol (250 ml). The mixture is refluxed for 24 hours and poured onto crushed ice (500 ml) after cooling. The styrylthioacetic acid, formed after neutralization with dilute hydrochloric acid (250 ml), is filtered and dried; yield 88 g (90%); m.p. 84-86°C.
The styrylthioacetic acid is then oxidized to styrylsulfonylacetic acid as follows. A mixture of styrylthioacetic acid (5 g, 25 mmol) in glacial acetic acid (35 ml) and 30% hydrogen peroxide (15 ml) is heated under reflux for 60 minutes and the mixture is poured onto crushed ice (200 ml) after cooling. The compound separated is filtered and recrystalized from hot water to give white crystalline flakes of (Z)-styrylsulfonylacetic acid; yield 2.4 g (41%); m.p. 150-51°C.
A solution of (Z)-styrylsulfonylacetic acid (2.263 g, 10 m mol) in glacial acetic acid (6 ml) is mixed with an aromatic aldehyde (10 mmol) and benzylamine (0.2 ml) and refluxed for 3 hours. The reaction mixture is cooled, treated with dry ether (50 ml), and any product separated is collected by filtration. The filtrate is diluted with more ether and washed successively with a saturated solution of sodium hydrogen carbonate (15 ml), sodium bisulfite (15 ml), dilute hydrochloric acid (20 ml) and finally with water (30 ml). Evaporation of the dried ethereal layer yields (E)(Z)-bis(styryl)sulfones.
(E),(E)-bis(styryl)sulfones are prepared following the same procedure as described above with exception that sulfonyldiacetic acid is used in place of (Z)-styrylsulfonylacetic acid, and twice the amount of aromatic aldehyde (20 mmol) is used.
The styryl sulfones of formula N, which are systematically identified as 2-(phenylsulfonyl)-l-phenyl-3-phenyl-2-propen-l-ones, may be prepared according to either Method A or Method B of Procedure 4:
Procedure 4
Synthesis of 2-(Phenylsulfonyl)-l-phenyl-3-phenyl-2-propen-l-ones
These compounds are synthesized by two methods which employ different reaction conditions, solvents and catalysts.
Method A: Phenacyl aryl sulfones are made by refluxing α-bromoacetophenones (0.05 mol) and sodium arylsulfinates (0.05 mol) in absolute ethanol (200 ml) for 6-8 hours. The product which separates on cooling is filtered and washed several times with water to remove sodium bromide. The product is then recrystalized from ethanol: phenacyl-phenyl sulfone, m.p. 90-91°C; phenacyl-p-fluorophenyl sulfone, m.p. 148-149°C; phenacyl-p-bromophenyl sulfone, m.p. 121-122°C; phenacyl-p-methoxyphenyl sulfone, m.p. 104-105°C; p-nitrophenacyl-phenyl sulfone, m.p. 136-137°C.
A solution of phenacyl aryl sulfone (0.01 mol) in acetic acid (10 ml) is mixed with an araldehyde (0.01 mol) and benzylamine (0.02 ml) and refluxed for 3 hours. The solution is cooled and dry ether (50 ml) is added. The ethereal solution is washed successively with dilute hydrochloric acid, aqueous 10% NaOH, saturated NaHSO3 solution and water. Evaporation of the dried ethereal layer gives a solid product which is purified by recrystallization.
Method B: Dry tetrahydrofuran (200 ml) is taken in a 500 ml conical flask flushed with nitrogen. To this, a solution of titanium (IN) chloride (11 ml, 0.01 mol) in absolute carbon tetrachloride is added dropwise with continuous stirring. The contents of the flask are maintained at -20°C throughout the course of the addition. A mixture of phenacyl aryl sulfone (0.01 mol) and aromatic aldehyde (0.01 mol) is added to the reaction mixture and pyridine (4 ml, 0.04 mol) in tetrahydrofuran (8 ml) is added slowly over a period of 1 hour. The contents are stirred for 10-12 hours, treated with water (50 ml) and then ether (50 ml) is added. The ethereal layer is separated and washed with 15 ml of saturated solutions of 10% sodium hydroxide, sodium bisulfite and brine. The evaporation of the dried ethereal layer yields 2-(phenylsulfonyl)-l-phenyl-3-phenyl-2 propen-l-ones.
PATENT
https://www.google.com/patents/CN104817488A?cl=en
The structure of this medicine formula (I) shown below,

Wherein, R1 is absent or is halogen, C1-3 alkyl, alkoxy and -CF3; R2 is absent or is halogen, C1-3 alkyl, alkoxy and -cf3; structural formula (I) The method for the preparation of compounds as follows:

| WO2007016201A2 | Jul 28, 2006 | Feb 8, 2007 | Onconova Therapeutics, Inc. | FORMULATION OF RADIOPROTECTIVE α, β UNSATURATED ARYL SULFONES |
| WO2008105808A2 | Jul 27, 2007 | Sep 4, 2008 | Onconova Therapeutics, Inc. | FORMULATIONS OF RADIOPROTECTIVE α, β UNSATURATED ARYL SULFONES |
| US6656973 | Nov 27, 2002 | Dec 2, 2003 | Temple University – Of The Commonwealth System Of Higher Education | (E)-4-carboxystyrl-4-chlorobenzyl sulfone and pharmaceutical compositions thereof |
| US6667346 | Feb 28, 2002 | Dec 23, 2003 | Temple University – Of The Commonwealth System Of Higher Education | Method for protecting cells and tissues from ionizing radiation toxicity with α, β unsaturated aryl sulfones |
| US6982282 * | May 17, 2002 | Jan 3, 2006 | Sonus Pharmaceuticals, Inc. | Emulsion vehicle for poorly soluble drugs |
| US20090247624 | Jul 27, 2007 | Oct 1, 2009 | Onconova Therapeutics Inc. | Formulations of radioprotective alpha beta unsaturated aryl sulfones |
References
- “Onconova Therapeutics presents new data demonstrating radioprotection by Ex-RAD at RRS annual meeting” (Press release). EurekAlert. 2010-09-27. Archived from the originalon 2011-03-22. Retrieved 2011-03-22.
- Hipp, Van (2011-03-16). “Ex-Rad, the U.S. Military’s Radiation Wonder Drug”. FoxNews.com (FOX News Network). Archived from the original on 2011-03-26. Retrieved 2011-03-26.
- Ghosh, Sanchita P.; Perkins, Michael W.; Hieber, Kevin; Kulkarni, Shilpa; Kao, Tzu-Cheg; Reddy, E. Premkumar; Reddy, M. V Ramana; Maniar, Manoj; Seed, Thomas; Kumar, K. Sree (2009). “Radiation Protection by a New Chemical Entity, Ex-Rad™: Efficacy and Mechanisms”. Radiation Research 171 (2): 173–9. doi:10.1667/RR1367.1. PMID 19267542.
- “Ex-RAD® for Protection from Radiation Injury”. Onconova Therapeutics. 2009. Archived from the original on 2011-03-22. Retrieved 2011-03-22.
- http://cen.acs.org/articles/90/i26/Drugs-Never-Used.html[full citation needed]
- Kouvaris, J. R.; Kouloulias, V. E.; Vlahos, L. J. (2007). “Amifostine: The First Selective-Target and Broad-Spectrum Radioprotector”. The Oncologist 12 (6): 738–47.doi:10.1634/theoncologist.12-6-738. PMID 17602063.
- http://www.news-medical.net/news/20110323/Cellerant-commences-CLT-008-Phase-III-trial-in-patients-with-leukemia.aspx
- Reliene, Ramune; Pollard, Julianne M.; Sobol, Zhanna; Trouiller, Benedicte; Gatti, Richard A.; Schiestl, Robert H. (2009). “N-acetyl cysteine protects against ionizing radiation-induced DNA damage but not against cell killing in yeast and mammals”. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 665: 37. doi:10.1016/j.mrfmmm.2009.02.016.
- Mansour, Heba H.; Hafez, Hafez F.; Fahmy, Nadia M.; Hanafi, Nemat (2008). “Protective effect of N-acetylcysteine against radiation induced DNA damage and hepatic toxicity in rats”.Biochemical Pharmacology 75 (3): 773–80. doi:10.1016/j.bcp.2007.09.018. PMID 18028880.
- Demirel, C; Kilçiksiz, S; Ay, OI; Gürgül, S; Ay, ME; Erdal, N (2009). “Effect of N-acetylcysteine on radiation-induced genotoxicity and cytotoxicity in rat bone marrow”. Journal of radiation research 50 (1): 43–50. doi:10.1269/jrr.08066. PMID 19218780.
- Demirel, C; Kilciksiz, S; Evirgen-Ayhan, S; Gurgul, S; Erdal, N (2010). “The preventive effect of N-acetylcysteine on radiation-induced dermatitis in a rat model”. Journal of the Balkan Union of Oncology 15 (3): 577–82. PMID 20941831.
- Geiger, Hartmut; Pawar, Snehalata A; Kerschen, Edward J; Nattamai, Kalpana J; Hernandez, Irene; Liang, Hai Po H; Fernández, Jose Á; Cancelas, Jose A; Ryan, Marnie A; Kustikova, Olga; Schambach, Axel; Fu, Qiang; Wang, Junru; Fink, Louis M; Petersen, Karl-Uwe; Zhou, Daohong; Griffin, John H; Baum, Christopher; Weiler, Hartmut; Hauer-Jensen, Martin (2012).“Pharmacological targeting of the thrombomodulin–activated protein C pathway mitigates radiation toxicity”. Nature Medicine 18 (7): 1123–9. doi:10.1038/nm.2813. PMC 3491776.PMID 22729286.
External links
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015265549 | 2015-09-24 | STABLE AQUEOUS FORMULATION OF (E)-4-CARBOXYSTYRYL-4-CHLOROBENZYL SULFONE |
| US2015238448 | 2015-08-27 | FORMULATION OF RADIOPROTECTIVE ALPHA, BETA UNSATURATED ARYL SULFONES |
| US2013012588 | 2013-01-10 | COMPOSITIONS AND METHODS FOR PREVENTION AND TREATEMENT OF WOUNDS |
| US2013012589 | 2013-01-10 | STABLE AQUEOUS FORMULATION OF (E)-4-CARBOXYSTYRYL-4-CHLOROBENZYL SULFONE |
| US2011250184 | 2011-10-13 | METHODS FOR DETERMINING EFFICACY OF A THERAPEUTIC REGIMEN AGAINST DELETERIOUS EFFECTS OF CYTOTOXIC AGENTS IN HUMAN |
| US2011028504 | 2011-02-03 | Formulation of radioprotective alpha beta unsaturated aryl sulfones |
| US2009247624 | 2009-10-01 | FORMULATIONS OF RADIOPROTECTIVE ALPHA BETA UNSATURATED ARYL SULFONES |
 |
|
| Identifiers | |
|---|---|
| 922139-31-9 |
|
| PubChem | 23668369 |
| Properties | |
| C16H12ClNaO4S | |
| Molar mass | 358.77 g·mol−1 |
//////////Onc-01210, ON-01210.Na, 334969-03-8, 922139-31-9, Recilisib Sodium, Phase I , A protein kinase inhibitor, treatment of acute radiation syndrome, Orphan Drug Status, Ex-RAD
C1=CC(=CC=C1CS(=O)(=O)C=CC2=CC=C(C=C2)C(=O)[O-])Cl.[Na+]
