
Home » Posts tagged 'OMARIGLIPTIN'
Tag Archives: OMARIGLIPTIN
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, New patent


(WO2016015596) PROCESS FOR PREPARING 2, 3-DISUBSTITUTED-5-OXOPYRAN COMPOUND
SUNSHINE LAKE PHARMA CO., LTD. [CN/CN]; Northern Industrial Area, Songshan Lake Dongguan, Guangdong 523000 (CN)
SUN, Guodong; (CN).
LIU, Yongjun; (CN).
WEI, Mingjie; (CN).
LAI, Cailang; (CN).
LI, Dasheng; (CN).
ZHANG, Shouhua; (CN).
WANG, Zhongqing; (CN)
To a mixture of methanol (42 mL) and tert-butyl ( (1R, 2S) -1- (2, 5-difluorophenyl) -1-hydroxypent-4-yn -2-yl) carbamate (7.0 g) cooled to-5℃ was added a solution of KOH (3.2 g) in methanol (28 mL) dropwise. After dropwise addition, the resulting mixture was stirred for 30 minutes, then iodine (5.7 g) was added to the mixture. The reaction mixture was stirred at 0 ℃ for 10 minutes, followed by 25 ℃ for 6 hours, and then quenched with water (140 mL) . Then the mixture was stirred at 25 ℃ for 2 hours. The precipitate was collected by filtration and washed sequentially with methanol/water (40 mL, v: v=1: 1) . The resulting solid was dried at 45 ℃ in vacuo to give the title compound as awhite solid (8.8 g, purity: 95.0%)

//////////WO 2016015596, Omarigliptin, Sunshine Lake Pharma Co Ltd, NEW PATENT
WO 2016014324, New Patent, Omarigliptin, MERCK SHARP & DOHME CORP
 Omarigliptin , MK-3102
Omarigliptin , MK-3102
WO2016014324, PROCESS FOR PREPARING CHIRAL DIPEPTIDYL PEPTIDASE-IV INHIBITORS
MERCK SHARP & DOHME CORP. [US/US]; 126 East Lincoln Avenue Rahway, New Jersey 07065-0907 (US).
CHUNG, John, Y. L.; (US).
PENG, Feng; (US).
CHEN, Yonggang; (US).
KASSIM, Amude Mahmoud; (US).
CHEN, Cheng-yi; (US).
MAUST, Mathew; (US).
MCLAUGHLIN, Mark; (US).
ZACUTO, Michael, J.; (US).
CHEN, Qinghao; (US).
TAN, Lushi; (US).
SONG, Zhiguo Jake; (US).
CAO, Yang; (US).
XU, Feng; (US)
![]()
A process for preparing a compound of structural Formula Ia: comprising Boc deprotection with TFA of, reductive amination of:.
The present invention is directed to a novel process for the preparation of omarigliptin, (2R,35,,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3 -amine, a dipeptidyl peptidase-IV (DPP-4) inhibitor, for the treatment of Type 2 diabetes, and related intermediates.
BACKGROUND OF THE INVENTION
Syntheses of omarigliptin have previously been described in PCT international patent applications numbers WO 2010/056708 and WO2013/003250. The process described in WO 2010/056708 does not result in a favorable yield of the compound of structural Formula la, as it results in a racemic mixture. WO2013/003250 describes the following scheme to make the compound of structural Formula la, an intermediate for synthesizing omarigliptin:

In WO2013/003250, synthesis of the compound of structural Formula la involves using benzenesulfonic acid (BSA) to remove the Boc protecting group of the compound of structural Formula 1, by first forming a BSA salt of the compound of structural Formula la. The BSA salt is then isolated and undergoes reductive amination with Boc -ketone of the compound of structural Formula 7, to produce the compound of structural Formula la, as a 19: 1 diastereomeric mixture. The BSA mediated Boc deprotection requires up to 72 h to reach full conversion.
An alternative process which eliminates the need to isolate the BSA salt of the compound of Formula la and reduces the overall reaction time of the process is desired. The inventors have now discovered a process for making the compound of structural Formula la which eliminates the step of isolating a salt of the compound of structural Formula la and reduces the overall reaction time. The present process also produces an end-of reaction homogeneous solution via reductive amination, which facilitates crystallization of the compound of structural Formula la. The described process also improves the diastereoselectivity, overall yield, cost and cycle time over the process described in WO2013/003250.
WO2013/003250 also describes the Boc deprotection of the compound of Formula la to produce omarigliptin (Formula I) shown below. As described in WO2013/003250, the Boc deprotection of the compound of Formula la involves aging the substrate in aqueous sulfuric acid in DMAc at 30 °C for 15-20 h, then working up with ammonium hydroxide. This work up produces large amounts of poorly soluble ammonium sulfate which co-crystallizes with the desired product. As a result, isolation of the desired product requires a long cycle time for filtration, washing and drying.

Formula I (omarigliptin)
Because the processes described herein use trifluoroacetic acid with or without a co-solvent for the transformation of the compound of Formula la to omarigliptin, which offers good solubility for the compound of Formula la, omarigliptin is achieved with fast reaction kinetics and good purity profiles.

the compound of structural Formula 1 is prepared by the following processes:

reagents
and,

or alternatively

10 R = Ms
X=OAc
SCHEME 3: Synthesis of the Boc Ketone

16 17 18 19
IPA, H2Q ,
1)956
Step 1 : As

A round bottom flask was charged with ligand L (0.829 g), Cu(II) propionate
monohydrate (0.402 g) (or Cu(II) acetate (0.31 g) or CuCl or CuCl2) and EtOH (350 ml) and agitated at room temperature for lh. 2,4-Difluorobenzaldehyde (100.0 g) was added followed by DABCO (2.368 g) (or 2,4-dimethylpiperizine) and the mixture was cooled to -5 – -15 °C. Cold (0°C) nitromethane (190 ml or 215 g) was added slowly to the cold solution and the solution was aged at -5 to -15 °C for 20-24 h and at 0 °C for 2-4h. 5 wt% EDTA»2Na (500 ml) followed by
water (200 mL) and MTBE (1.0 L) was added to the cold solution, and the temperature was raised to 20°C. The layers were separated and the organic layer was washed with additional 5 wt% EDTA»2Na (500 ml), followed by water (50 mL) and brine (250 mL). The organic layer, containing Compound 17, was concentrated to remove nitromethane, then the solvent was switched to THF.
Step 2: Michael-Lactolization – Nitro lactol

To Compound 17 in 2 volumes of THF (258 mL) from Step 1 under 2 and cooling at 0 °C, 1 equivalent of Hunig’s base was added. 1.15 equivalents of acrolein was added over 1 h via syringe pump at 0-5 °C. The reaction was stirred at -10-0 °C overnight. The resulting mixture was used directly in the next step.
Alternatively, the mixture was concentrated at 0-5 °C to remove excess acrolein, then the residue was flushed with acetonitrile until Hunig’s base and water are mostly removed. The residue was taken up in 8 volumes of acetonitrile and used directly in the next step.
Alternatively, at the end of the reaction the mixture was worked up by diluting with MTBE and washing with aqueous citric acid solution, and aqueous NaHCC solution, and the solvent was switched to acetonitrile. Alternatively, the end reaction mixture was taken forward directly to the next step.
Step 3: Dehydration – Nitro dihydropyrans

1.1 Equivalents of TEA was added to the acetonitrile solution of lactol 18 from Step 2 followed by 1.2 equivalents of mesyl chloride and 1.2 equivalents of S-collidine under < +10 °C . The reaction was aged at 10°C for 0.5-1 h. Alternatively, the end of the reaction mixture from Step 2 was cooled to between -20 °C to 0 °C. Two equivalents of S-collidine and 1.4 equivalents of mesyl chloride were then added. The mixture was heated to 36 °C and aged overnight. The mixture was cooled to room temperature. 15 volumes of MTBE was added and the solution was
washed with 3 volumes 10 wt% citric acid and 6 volumes water, 10 volumes water, then 3 volumes of 5% aHC03 solution and 6 volumes water. The organic was concentrated with 20 volumes of MTBE using 10 volumes MTBE. The organic solution was stirred with 20-30 wt% AQUAGUARD for 2 hours at room temperature. The mixture was filtered and washed with 2 volumes of MTBE.
Step 4: Dynamic Kinetic Resolution (DKR) crystallization – rraws-nitro-dihydropyran (19t)

The organic MTBE solution of Step 3 was solvent switched to 2 volumes of IPA and the final volume was -300 mL. 10 Mol% of TEA (or DAB CO or morpholine or DMAP) was added. Then water (1 15 mL) was slowly added over 3 hours. The slurry was filtered, washed with 80/20 IP A/water (2×100 mL) and vacuum dried under N2.
Step 5: Hydroboration/oxidation – Trans-nitro-pyranol

To a vessel charged with /raws-nitro-dihydropyran (10 g), MTBE (100 mL) was added under nitrogen. The mixture was stirred at room temperature to give a clear orange solution. The solution was cooled to +2 °C and borane dimethyl sulfide complex (9.55 ml) was added. The clear solution was aged for 2-5h until >99% conversion by HPLC analysis. The reaction was slowly quenched with water (7.25 ml) keeping at < +9 °C. After the solution was aged at 5°C for 5 min, water (78 mL) was added at < +13 °C. Solid sodium percarbonate (13.26 g, 84 mmol) was added. The suspension was stirred at 5 °C for 15h. The mixture was transferred to a separatory funnel with the aid of 60 mL MTBE and 20 mL water. The mixture was allowed to warm to room temperature. The aqueous phase was back-extracted with 40 mL MTBE. The combined organic phase was washed once with 30 mL half saturated sodium chloride solution, once with 15 mL brine and 15 mL 0.2N HC1, and once with 30 mL half-saturated sodium chloride solution. The organic layer was dried over a2S04. The organic was filtered, washed with 10 mL MTBE and concentrated to an oil. The oil was diluted to 200 mL for a 0.191M solution.
Step 6: Nitro Reduction/Boc protection – Pyranol

A 3 -neck jacketed round bottom flask equipped with overhead stirrier was charged with 0.191M (5R,6S)-5-nitro-pyran-3-ol (119 ml) (Compound 20) in ethanol and ethanol (32 ml). The solution was cooled to 1 1-12 °C. Cold 6N HC1 (19.55 ml, 1 17 mmol) was added at <
+17°C. Zinc dust (12.93 g) was added in five portions (5×2.59g) at < +26 °C. The mixture was stirred at 12 °C for 22 h. 1M K2C03 (76 mL) was added in one portion. MTBE (59 mL) was added then EDTA 2K 2H20 (22.55 g) was added over 10 min at < +14 °C. To the solution 45 wt% KOH (4.86 mL) solution was added. The solution was cooled to 5 °C, and 1.1 equivalents of B0C2O (5.46 g) was added. The solution was rinsed with MTBE (10 mL) and stirred at 5 °C for 2h, then at 12 °C for 16h, and then at 24 °C for lOh until >99.5% conversion. The solution was transferred to a separatory funnel with the aid of MTBE (30 mL) and water (5 mL). The organic layer was filtered and washed with MTBE (20 mL). The organic filtrate was concentrated. MTBE (60 mL), water (30 mL) and saturated sodium chloride solution (15 mL) were added. The mixture was warmed in a 30 °C bath to dissolve solid, and then concentrated. The concentrate was flushed with toluene in a 60 °C bath, then concentrated. Toluene (8.4 mL) was added and the mixture was heated to 80 °C. Heptane (70.8 mL) was added over lh at 80 °C, then cooled slowly to room temperature. The mixture was filtered and washed with 1 :2 toluene/heptane (23.55 mL), filterated and vacuum dried under nitrogen until a constant weight.
The purity could be further upgraded by the following procedure: a round bottom flask was charged with the product of Step 6 (7.069 g) from above. EtOH (21 mL) was added and the mixture was heated to 45 °C. Water (31.5 mL) was slowly added over 1 h at 45 °C. The mixture was aged for lh. Water (31.5 mL) was added in one portion, then cooled slowly to room temperature and aged overnight. The slurry was filtered and washed with 1 :3.5 EtOH/water (23.56 mL). Crystals were vacuum dried under nitrogen until a constant weight.
Alternatively, Compound 20 was reduced with 100 psi hydrogen in 20 volume wet THF in the presence of 10-30 wt% Raney nickel at 50 °C. Then the reaction mixture was basified with 2 equivalent of K2CO3 and a slight execess B0C2O to afford crude Compound 21 after aqueous work up.
Compound 7 was obtained from 21via oxidation as described in WO2013/003250.
S

Boc-mesyl-pyrazole solid 1 was added to 2.5 volumes of TFA at 0-2 °C, over 2-3 minutes under nitrogen, followed by 0.5 volume of TFA rinse. Conversion to TFA salt was complete within 0.5-lh at 1-2 °C. DMAc (14 vol) followed by triethylamine (5 equivalents or 2.3 volumes) were slowly added to the TFA reaction mixture at 0 °C maintaining < +20 °C. Boc-ketone 7 (0.89 equivalent) was then added at -15 °C followed by solid NaBH(OAc)3 (1.4 equivalents) which was added in three portions over lh. The reaction solution was aged at -15 °C overnight. The solution was then warmed to 22 °C, and after aging for 2-5 h. Diastereomeric ratio was > 96.5:3.5.
The solution was seeded with Boc amine 1 wt% at 22 °C and stirred at 22-40 °C for 2-4 h. 0.36 volume 28% ammonium hydroxide was added over 2-4 h, then, 3.64 volumes 28% ammonium hydroxide was added over 4-10h at 22-60 °C. After cooling to 22 °C, the batch was filtered, washed with 5: 1 DMAc/water, then water. The wet cake was vacuum dried under nitrogen at ambient affording the product. Diastereoselectivity was > 30: 1.
Boc Deprotection of Formula la

A reactor was charged with 2.5 X (by volume) of trifluoroacetic acid. The batch was cooled to 5-10 °C. The reactor was then charged with 0.4 X (by volume) water. The batch was cooled to 0-5 °C. The reactor was then charged with 1 equivalent (1 kg) of the compound of Formula la over 0.5-lh while maintaining the temperature between 0 -5°C. The reactor was then charged with 0.5 X (by volume) trifluoroacetic acid to reactor while maintaining the temperature between 0-5°C. The batch was then heated between 15-20°C and aged for 2-2.5 h. The batch was then cooled to between 5-10°C. A crystallizer was charged with water 5.0 X (by volume) and 0.1 X (by volume) of ammonia water and adjusted to between 3-13°C. To generate a seed bed, Compound I seed (lwt% vs la) was added and the temperature as adjusted to between 3-13°C. A solution of ammonia water 3.8 X (by volume) and of the compound of Formula la was added simultaneously to the seed bed over 2.5 – 3.5 hours while maintaining temperature at 3-13°C and pH -9-10. The batch was aged for at least 30 minutes and then filtered. The resulting crystals were washed with 3. OX (by volume) water at 3 – 13°C twice and vacuum dried at < 50°C to afford the compound of formula I.
//////WO 2016014324, New Patent, Omarigliptin, MERCK SHARP & DOHME CORP, MK-3102
MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan

MARIZEV® (Omarigliptin), Merck’s Once-Weekly DPP-4 Inhibitor for Type 2 Diabetes, Approved in Japan
KENILWORTH, N.J.–(BUSINESS WIRE)–Merck (NYSE:MRK), known as MSD outside the United States and Canada, today announced that the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) has approved MARIZEV® (omarigliptin) 25 mg and 12.5 mg tablets, an oral, once-weekly DPP-4 inhibitor indicated for the treatment of adults with type 2 diabetes. Japan is the first country to have approved omarigliptin……….http://www.mercknewsroom.com/news-release/prescription-medicine-news/marizev-omarigliptin-mercks-once-weekly-dpp-4-inhibitor-type
syn…….https://newdrugapprovals.org/2014/04/18/omarigliptin-mk-3102-in-phase-3-for-type-2-diabetes/

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HEREJoin me on Facebook 

 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
/////////////MARIZEV, (Omarigliptin), Merck’s, Once-Weekly, DPP-4 Inhibitor, Type 2 Diabetes, Approved, Japan
OMARIGLIPTIN. MK 3102 IN PHASE 3 FOR TYPE 2 DIABETES
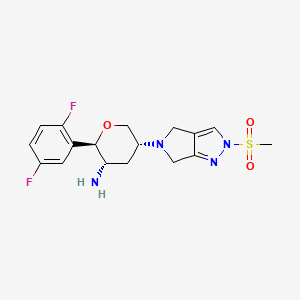
OMARIGLIPTIN. MK 3102
cas 1226781-44-7
Approved in japan SEPT 28 2015
(2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
(2R,3S,5R)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)oxan-3-amine
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
IN PHASE 3

CLICK ON IMAGES FOR CLARITY VIEW


PAPER

In our effort to discover DPP-4 inhibitors with added benefits over currently commercially available DPP-4 inhibitors, MK-3102 (omarigliptin), was identified as a potent and selective dipeptidyl peptidase 4 (DPP-4) inhibitor with an excellent pharmacokinetic profile amenable for once-weekly human dosing and selected as a clinical development candidate. This manuscript summarizes the mechanism of action, scientific rationale, medicinal chemistry, pharmacokinetic properties, and human efficacy data for omarigliptin, which is currently in phase 3 clinical development.
Omarigliptin (MK-3102) (2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, is a 2,3,5-substituted tetrahydropyran analogue currently in phase 3 clinical trial for type 2 diabetes mellitus (T2DM).
is a 2,3,5-substituted tetrahydropyran analogue currently in phase 3 clinical trial for type 2 diabetes mellitus (T2DM).
(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, Omarigliptin
Crystallization from ethyl acetate gave a compound with greater than 99% purity.
Optical rotation [α]D20 −12.0° (c 1.0, CH3OH).
1H NMR (CD3OD, 500 MHz) δ = 1.71 (q, 1H, J = 12 Hz), 2.56–2.61 (m, 1H), 3.11–3.18 (m, 1H), 3.36–3.40 (m, 1H), 3.48 (t, 1H, J = 12 Hz), 3.88–3.94 (m, 4H), 4.30–4.35 (m, 1H), 4.53 (d, 1H, J = 12 Hz), 7.14–7.23 (m, 2H), 7.26–7.30 (m, 1H), 7.88 (s, 1H).
LC–MS: 399.04 (M + 1).
PATENT
http://www.google.com.tr/patents/US20100120863?hl=tr&cl=ja
Example 1

(2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amineStep A: tert-Butyl {(2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5 (4H)-yl]tetrahydro-2H-pyran-3-yl}carbamate
A mixture of Intermediate 2 (26.3 g, 80 mmol) and 2-(methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole (Intermediate 5) (15.07 g, 80 mmol) in anhydrous methanol (1.5 L) was stirred at room temperature for 2 h. To the resulting white suspension was added decaborane (2.95 g, 24.15 mmol) and the mixture was stirred at room temperature overnight. Methanol was removed and the residue was purified on two 65i Biotage™ columns eluting with 5-50% ethyl acetate in dichloromethane to afford the title compound as a white solid. LC-MS: 499.10 (M+1).
Step B: (2R,3S,5R)-2-(2,5-Difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine
Removal of the BOC group in the product from Step A (13.78 g, 27.67 mmol) was accomplished with trifluoroacetic acid (100 ml) in dichloromethane (200 mL) at room temperature. After stirring for 2 h, the reaction was concentrated and neutralized with 25% MeOH and 2.5% ammonium hydroxide in dichloromethane. Solvents were removed under reduced pressure and the resulting crude material was purified on a 65i Biotage™ column eluting with 1.25-5% MeOH and 0.125-0.5% ammonium hydroxide in dichloromethane. The isolated material was further purified by recrystallization from 5:1 EtOAc/CH2Cl2 at 60° C. The crystalline product was washed with cold 2:1EtOAc/hexanes to give the title compound as a light brown solid. 1H NMR (500 MHz, CD3OD): 1.71 (q, 1H, J=12 Hz), 2.56-2.61 (m, 1H), 3.11-3.18 (m, 1H), 3.36-3.40 (m, 1H), 3.48 (t, 1H, J=12 Hz), 3.88-3.94 (m, 4H), 4.30-4.35 (m, 1H), 4.53 (d, 1H, J=12 Hz), 7.14-7.23 (m, 2H), 7.26-7.30 (m, 1H), 7.88 (s, 1H). LC-MS: 399.04 (M+1).
Intermediate 2

tert-Butyl[(2R,3S)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate Step A: 1-(2,5-Difluorophenyl)-2-nitroethanol
To sodium hydroxide (1N, 3L) and methanol (1500 mL) at 5° C. was added a solution of 2,5-difluorobenzaldehyde (350 g, 2.46 mol) and nitromethane (157 mL, 2.9 mol) in methanol (350 mL) dropwise over a period of 1 h. The reaction mixture was then neutralized with glacial acetic acid (165 mL). Diethyl ether (1500 mL) was added and the layers separated. The organic layer was washed successively with saturated aqueous sodium carbonate solution (1000 mL), and saturated aqueous brine (1000 mL). The organic layer was dried over anhydrous magnesium sulfate, filtered and concentrated to afford 1-(2,5-difluorophenyl)-2-nitroethanol that was used without further purification in Step B.
Step B: 2-Nitro-1-(2,5-difluorophenyl)ethanone
A solution of Dess-Martin periodinane (125 g) in dichloromethane (600 mL) was added to a solution of the nitroalcohol made in Step A (46.3 g) at 10° C. over a period of 30 min. Stirring was continued for 2 h, and the reaction mixture was then poured onto a mixture of sodium bicarbonate (300 g) and sodium thiosulfate (333 g) in water (3 L). The desired product was extracted with methyl t-butyl ether (MTBE) (2 L). The aqueous layer was neutralized with HCl (2N, 1.5 L) and extracted with MTBE (3 L). The combined organic layers were dried over anhydrous magnesium sulfate, filtered, evaporated and the residue was purified by chromatography (silica gel, eluting with dichloromethane) to yield the desired nitroketone.
Step C: 3-Iodo-2-(iodomethyl)prop-1-ene
A mixture of 3-chloro-2-(chloromethyl)prop-1-ene (1.0 g, 8 mmol) and sodium iodide (6.6 g, 44 mmol) in acetone (60 mL) was stirred at room temperature for 20 h, evaporated under reduced pressure and partitioned between dichloromethane (150 mL) and water (50 mL). The organic layer was dried over sodium sulfate, filtered and evaporated to yield 3-iodo-2-(iodomethyl)prop-1-ene as a reddish oil.
Step D: 3-Methylene-5-nitro-6-(2,5-difluorophenyl)-3,4-dihydro-2H-pyran
N,N-diisopropylethylamine (184 mL) was added to a solution of 2-nitro-1-(2,5-difluorophenyl)ethanone (92.7 g, 461 mmol) in N,N-dimethylformamide (1000 mL) and 3-iodo-2-(iodomethyl)prop-1-ene (156 g, 507 mmol). The mixture was heated at 60° C. for 2 h, evaporated and purified by chromatography (silica gel, gradient 0-30% dichloromethane in hexane) to yield 3-methylene-5-nitro-6-(2,5-difluorophenyl)-3,4-dihydro-2H-pyran.
Step E: (2R,3S)-5-Methylene-3-nitro-2-(2,5-difluorophenyl)tetrahydro-2H-pyran
This compound was made by following the same method described in Intermediate 1, Step D by using 3-methylene-5-nitro-6-(2,5-trifluorophenyl)-3,4-dihydro-2H-pyran.
Step F: (2R,3S)-5-Methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-amine
This compound was made by following the same method described in Intermediate 1, Step E by using (2R,3S)-5-Methylene-3-nitro-2-(2,5-difluorophenyl)tetrahydro-2H-pyran.
Step G: tert-Butyl[(2R,3S)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
This compound was made by following the same method described in Intermediate 1, Step F by using (2R,35)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-amine.
Step H: tert-Butyl[(2R,3S)-5-hydroxy-5-(hydroxymethyl)-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
This compound was made by following the same method described in Intermediate 1, Step G by using tert-butyl[(2R,35)-5-methylene-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate.
Step I: tert-Butyl[(2R,3S)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate
To a solution of tert-butyl[(2R,3S)-5-hydroxy-5-(hydroxymethyl)-2-(2,5-trifluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate (10.5 g) in methanol (100 mL) at 0° C. was added pyridine (7.8 mL) and lead tetraacetate (21.7 g). The reaction mixture was stirred for 20 min. Aqueous work-up with ethyl acetate gave crude product which was purified by chromatography (silica, 0-50% ethyl acetate/heptane) to yield tert-butyl[(2R,35)-5-oxo-2-(2,5-difluorophenyl)tetrahydro-2H-pyran-3-yl]carbamate as white solid.
Intermediate 3

Step A: tert-Butyl (3Z)-3-[(dimethylamino)methylene]-4-oxopyrrolidine-1-carboxylate
A solution of tert-butyl 3-oxopyrrolidine-1-carboxylate (40 g, 216 mmol) was treated with DMF-DMA (267 g, 2241 mmol) and heated at 105° C. for 40 min. The solution was cooled and evaporated under reduced pressure and the resulting orange solid was treated with hexane (200 mL) and cooled in a refrigerator for 3 days. The resulting brownish-yellow solid obtained as such was collected by filtration, dried and used in the next step without further purification.
Step B: 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazole
A solution of hydrazine (3 mL) and tert-butyl (3Z)-3-[(dimethylamino)methylene]-4-oxopyrrolidine-1-carboxylate (19.22 g) in ethanol (40 mL) was heated at 85° C. in a sealed tube for 4 h. Solvent was removed under reduced pressure, and the residue was triturated with dichloromethane (160 mL) and ethyl acetate (15 mL). The resulting solid was filtered. The filtrate was concentrated and the resulting solid was triturated again and filtered. The combined solids were treated with 4N hydrochloric acid (250 mL) in methanol and stirred for 6 h. The reaction mixture was concentrated and dried. The resulting solid was treated again for 6 h with 4N hydrochloric acid (250 mL) in methanol. After concentration and drying, the resulting hydrochloride salt was treated with ammonia in methanol (2N, 300 mL) and ammonium hydroxide solution in water (28%, 30 mL) and concentrated to dryness. The solid obtained was treated with methanol (70 mL) and water (5 mL) and purified in three batches on Biotage Horizon® system (silica, gradient 5-17% methanol containing 10% concentrated ammonium hydroxide in ethyl acetate) to yield 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole. 1H NMR (500 MHz, CD3OD): δ 4.04 (d, 4H); 7.39 (s, 1H).
Intermediate 5

2-(Methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole Step A: tert-Butyl 1-(methylsulfonyl)]-4,6-dihydropyrrolo[3,4-c]pyrazole-5(1H)-carboxylate (A) and tent-butyl 2-(methylsulfonyl)]-2,6-dihydropyrrolo[3,4-c]pyrazole-5(4H)-carboxylate (B)
A suspension of N-Boc-pyrazolopyrrolidine (Intermediate 3, Step B) (27.16 g, 130 mmol) in anhydrous acetonitrile (1.0 L) was charged in a 2.0 L three-neck flask fitted with a thermometer and an addition funnel and then treated with sodium hydride (60% dispersion in oil, 6.23 g, 156 mmol) while under nitrogen atmosphere in one portion. The reaction mixture was stirred at room temperature for 2 h. The resulting white suspension was then cooled in an ice bath and methanesulfonyl chloride (25.2 mL, 324 mmol) was slowly added via addition funnel The ice bath was then removed and the mixture was stirred 1 h at room temperature. The reaction mixture was quenched with water (500 mL) and the layers were separated. The aqueous layer was then extracted with 2×500 mL of dichloromethane. The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure to give a mixture of products A and B as colorless syrups. NMR in CD3OD indicated a 1:1 mixture of two products, in which the proton on the pyrazole ring in product A appeared at 7.70 ppm while the proton in product B appeared at 7.95 pm. LC-MS: 288.08 (M+1).
Step B: 2-(Methylsulfonyl)-2,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole
Trifluoroacetic acid (200 mL) was added slowly to a solution containing intermediates A and B prepared in the previous step (48.4 g, 168 mmol) in dichloromethane (400 mL) at 0° C. After addition, the cooling bath was removed and the reaction was allowed to stir at room temperature for 2 h. Solvent was removed under reduced pressure and the resulting trifluoroacetate salt was then neutralized with 500 mL of 25% methanol and 2.5% ammonium hydroxide in dichloromethane. After removal of solvent, the desired Intermediate 5 was obtained after chromatography on a Biotage™ column (2×340 g) eluting with 2.5-12.5% methanol and 0.25-1.25% ammonium hydroxide in dichloromethane. LC-MS: 109.85 (M+1).
PATENT
below patent caution…………….similar not same….examples below will help you in synthesis similarities
http://www.google.com/patents/WO2014018355A1?cl=en

Step 1 2 Step 2

Example 1
Synthesis of 3: (Step 1 & 2)
Dimethyldisulfide 1 (5 g, 53 mmol) and acetic acid (6 mL, 106 mmol) were mixed under nitrogen atmosphere and cooled to – 20 °C. Sulfuryl chloride (13 mL, 159 mmol) was added dropwise with stirring. The mixture was then stirred for 1 hour at -20 °C and afterwards allowed to come to room temperature and continued for another two hours. Acetyl chloride was distilled off from the reaction mixture. Crude methanesulfinyl chloride 2 obtained was used in the next step without further purification.
To a solution of chloramine T (14.95 g, 53 mmol) in dry toluene (220 mL) was added a solution of methanesulfinyl chloride 2 (5.2 g, 53 mmol) in dry toluene (10 mL) at 0 °C. The resulting suspension was heated at 80 °C for 2 hours with stirring. After cooling, the solid was filtered off and washed with dry toluene (100 mL). The filtrate was evaporated in vacuo and the crude mixture was purified through silica gel chromatography to obtain 3 as off white solid. XH NMR (300 MHz, CDC13): δ 7.85 – 7.91 (m, J= 8.42 Hz, 2H), 7.31 – 7.38 (m, J= 8.23 Hz, 2H), 3.78 (s, 3H), 2.45 (s, 3H).
Synthesis of 4: (Step 3)
To a solution of Ml (1.0 g, 2.2 mmol) in THF (10 mL) and DMF (10 mL) under nitrogen atmosphere at 0 °C was added Et3N (0.92 mL, 6.6 mmol) followed by B0C2O (0.48 g, 2.2 mmol). The reaction mixture was allowed to come to room temperature and continued the stirring for over night. The reaction mixture was diluted with water (100 mL) and extracted with CH2CI2 (3 x 100 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 4 as a off white solid.
XH NMR (400 MHz, CDC13): δ 7.27 – 7.35 (m, 1H), 4.44 – 4.54 (m, 4H), 1.52 (s, 9H).
Synthesis of 5: (Step 4)
To a suspension of NaH (0.30 g, 7.5 mmol) in dry THF (5 mL) under nitrogen atmosphere at 0 °C was added a solution of 4 (0.78 g, 3.7 mmol) in dry THF (30 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Reaction mixture was again cooled to 0 °C. A solution of 3 (2.0 g, 7.4 mmol) in THF (25 mL) was added to the reaction mixture and continued the stirring for another 1 hour. The reaction mixture was quenched with water (100 mL) and extracted with EtOAc (3 x 200 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 5 as an off-white solid.
XH NMR (400 MHz, CDC13): δ 7.84 – 7.88 (m, 1H), 7.78 (t, J= 8.27 Hz, 2H), 7.23 – 7.30 (m, 2H), 4.39 – 4.49 (m, 4H), 3.53 (d, J= 2.40 Hz, 3H), 2.42 (s, 3H), 1.53 (s, 9H).; Molecular Formula: Ci8H24N405S2; LCMS purity: 98.18%; Expected: 440.1 ; Observed: 341.0 (M-99).
Synthesis of 6: (Step 5)
To a solution of 5 (0.47 g, 1.06 mmol) in dry CH2CI2 (1 1 mL) under nitrogen atmosphere at 0 °C was added TFA (3 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Solvent was removed under vacuum and solid mass was washed with Et20 (3 x 10 mL) to get amine TFA salt as white solid.
XH NMR (300 MHz, CD3OD): δ 7.78 (s, 1H), 7.63 – 7.70 (m, J= 8.11 Hz, 2H), 7.26 – 7.35 (m, J = 8.33 Hz, 2H), 3.93 (s, 2H), 3.86 (s, 2H), 3.34 (s, 3H), 2.42 (s, 3H).
The amine TFA salt was dissolved in minimum volume of MeOH:CHCi3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (N¾)] using MeOH as eluent.
Organics were concentrated under vacuum to get free 6.
Synthesis of 7: (Step 6) To a stirred solution of 6 (0.34 g, 0.95 mmol) and M2 (0.26 g, 0.79 mmol) in DMAc (6.78 mL) under nitrogen atmosphere for 10 minutes was added AcOH (0.067 mL, 1.19 mmol). The reaction mixture was stirred for further 5 minutes and cooled to 0 °C. NaBH(OAc)3 (0.20 g, 0.95 mmol) was added to the reaction mixture and allowed to stirrer at room temperature for overnight. NH4OH (2 mL) was added to the reaction mixture and heated at 50 °C for 1 hour followed by water (3.39 mL) and again heated at 50 °C for another hour. Reaction mixture was cooled to room temperature and filtered. The solid residue was washed with water (4 x 100 mL) and the crude residue was purified by silica gel chromatography to afford 7.
XH NMR (300 MHz, CDC13): δ 7.80 (d, J= 6.95 Hz, 3H), 7.25 – 7.29 (m, 2H), 7.22 (br. s., 1H), 6.92 – 7.02 (m, 2H), 4.52 (d, J= 9.33 Hz, 1H), 4.24 – 4.40 (m, 2H), 3.85 (br. s., 5H), 3.48 (s, 3H), 3.39 – 3.47 (m, 1H), 3.07 (br. s., 1H), 2.52 (d, J= 10.25 Hz, 1H), 2.44 (s, 3H), 1.61 (br. s., 1H), 1.28 (s, 9H).; Molecular Formula: C29H35F2N506S2; LCMS purity: 99.08%; Expected: 651.2; Observed: 652.0 (M+l). Synthesis of Example 1: (Step 7)
To a solution of 7 (20 mg, 0.03 mmol) in dry CH2CI2 (2 mL) under nitrogen atmosphere at 0 °C was added TFA (0.5 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Solvent was removed under vacuum and solid mass was washed with Et20 to get amine di-TFA salt Example 1 as white solid. Unless otherwise noted the IC50 values were determined using the assay discussed earlier.
XH NMR (400 MHz, CD3OD): δ 8.05 (s, 1H), 7.73 (d, J= 8.03 Hz, 2H), 7.36 (d, J= 8.28 Hz, 2H), 7.29 – 7.34 (m, 1H), 7.20 – 7.27 (m, 2H), 4.71 (d, J= 10.04 Hz, 1H), 4.40 – 4.53 (m, 5H), 3.72 – 3.82 (m, 2H), 3.68 (s, 3H), 3.59 – 3.65 (m, 1H), 2.77 – 2.85 (m, 1H), 2.44 (s, 3H), 2.00 – 2.14 (m, 1H).; Molecular Formula: C24H27F2 504S2; HPLC purity: 99.74%; LCMS Expected: 551.2; Observed: 552.2 (M+l).
SCHEME 2

Example 2: Synthesis of Compound 1 & 2 (Step 1):
To a suspension of M2 (0.95 g, 2.8 mmol) in water (8.67 mL) was added sodium metabisulfite (0.55 g, 2.8 mmol) and stirred a room temperature for lhour. A solution of M3* (0.52 g, 2.8 mmol) in ethanol (8.67 mL) was added to the above reaction mixture and continued the stirring for further 4 hours. Neat aCN (0.14 g, 2.8 mmol) was added to the above reaction mixture in one portion and heated the reaction mixture at 50 °C for 2 days. Reaction mixture was concentrated under vacuum to remove most of the ethanol. The crude mixture was extracted with CHCI3 (50 x 3 mL). The combined organic layer was washed with water, dried over a2S04, filtered, concentrated and purified by flash chromatography to obtain 1 and 2 as solids.
Compound 1: ‘H NMR (300 MHz, CDC13): δ 7.77 (s, 1H), 7.26 – 7.35 (m, 1H), 7.00 (t, J= 5.76 Hz, 2H), 4.57 (t, J= 9.88 Hz, 2H), 4.32 – 4.39 (m, 1H), 3.85 – 4.09 (m, 5H), 3.60 (d, J= 11.34 Hz, 1H), 3.34 (s, 3H), 2.63 – 2.74 (m, 1H), 2.02 – 2.15 (m, 1H), 1.31 (s, 9H).
Compound 2: XH NMR (300 MHz, CDC13): δ 7.28 – 7.36 (m, 2H), 7.00 (t, J= 5.85 Hz, 2H), 4.55 (d, J= 8.97 Hz, 2H), 4.37 (dd, J= 2.65, 11.25 Hz, 1H), 3.88 – 4.07 (m, 5H), 3.60 (d, J = 1 1.34 Hz, 1H), 2.71 (td, J= 3.45, 12.49 Hz, 1H), 1.97 – 2.12 (m, 1H), 1.31 (s, 9H).; Molecular Formula: C22H25F2 503; LCMS purity: 94.48%; Expected: 445.2; Observed: 446.0 (M+l). (*Preparation of M3: M3.PI1SO3H (1.0 g, 2.8 mmol) was dissolved in minimum volume of MeOH:CHCl3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (NH2)] using MeOH as eluent. Organics were concentrated under vacuum to get free M3, which was used directly without further purification.) Synthesis of compound 3 (Step 2):
To a solution of compound 2 (0.40 g, 0.89 mmol) in THF (5 mL) under 2 atmosphere at -78 °C was added a solution of MeMgBr (0.89 mL, 2.6 mmol, 3M in Et20). The reaction mixture was allowed to attain room temperature over 1 hour. TLC shows complete conversion. The reaction mixture was again cooled to -10 °C and quenched with saturated aq. NH4CI solution (10 mL). The reaction mixture was extracted with CH2CI2 (50 x 3 mL).
Combined organics were dried over Na2S04, filtered, concentrated and purified by reversed phase chromatography to obtain 3 as di-TFA salt.
Molecular Formula: C22H28F2 4O3; LCMS purity: 88.82%; Expected: 434.2; Observed: 435.2 (M+l).
Synthesis of Example 2 (Step 3):
To a solution of compound 3 (35 mg, 0.053 mmol) in CH2CI2 (2 mL) was added TFA (0.5 mL) dropwise at 0 °C. Reaction mixture was allowed to attain room temperature over 2 hours time. TLC shows complete conversion. Reaction mixture was concentrated to dryness. The solid residue was washed with Et20 (10 x 3 mL) and dried under vacuum to obtain Example 2 as tri-TFA salt.
XH NMR (400 MHz, CD3OD): δ 7.60 (s, 1H), 7.37 (dd, J= 5.02, 8.03 Hz, 1H), 7.22 – 7.31 (m, 2H), 4.70 (d, J= 10.04 Hz, 1H), 4.48 – 4.61 (m, 4H), 4.17 (dd, J= 2.26, 11.29 Hz, 1H), 3.91 (d, J = 11.04 Hz, 1H), 3.73 – 3.83 (m, 1H), 2.54 – 2.62 (m, 1H), 2.22 (t, J= 12.05 Hz, 1H), 1.71 (s, 3H).; Molecular Formula: C17H20F2 4O; HPLC purity: 94.98%; Expected: 334.2; Observed: 335.2 (M+l).
SCHEME 3

Example 3
Synthesis of 1 & 2: (Step 1)
To a suspension of M2 (0.95 g, 2.8 mmol) in water (8.67 mL) was added sodium metabisulfite (0.55 g, 2.8 mmol) and stirred a room temperature for lhour. A solution of M3* (0.52 g, 2.8 mmol) in ethanol (8.67 mL) was added to the above reaction mixture and continued the stirring for further 4 hours. Neat aCN (0.14 g, 2.8 mmol) was added to the above reaction mixture in one portion and heated the reaction mixture at 50 °C for 2 days. Reaction mixture was concentrated under vacuum to remove most of the ethanol. The crude mixture was extracted with CHCI3 (50 x 3 mL). The combined organic layer was washed with water, dried over a2S04, filtered, concentrated and purified by flash chromatography to obtain 1 and 2 as solids.
Compound 1: ‘H NMR (300 MHz, CDC13): δ 7.77 (s, 1H), 7.35 – 7.26 (m, 1H), 7.00 (t, J= 5.76 Hz, 2H), 4.57 (t, J= 9.88 Hz, 2H), 4.39 – 4.32 (m, 1H), 4.09 – 3.85 (m, 5H), 3.60 (d, J= 1 1.34 Hz, 1H), 3.34 (s, 3H), 2.74 – 2.63 (m, 1H), 2.15 – 2.02 (m, 1H), 1.31 (s, 9H).
Compound 2: XH NMR (300 MHz, CDC13): δ 7.36 – 7.28 (m, 2H), 7.00 (t, J= 5.85 Hz, 2H), 4.55 (d, J= 8.97 Hz, 2H), 4.37 (dd, J= 2.65, 11.25 Hz, 1H), 4.07 – 3.88 (m, 5H), 3.60 (d, J= 1 1.34 Hz, 1H), 2.71 (td, J= 3.45, 12.49 Hz, 1H), 2.12 – 1.97 (m, 1H), 1.31 (s, 9H).; Molecular Formula: C22H25F2 503; LCMS purity: 94.48%; Expected: 445.2; Observed: 446.0 (M+l).
(*Preparation of M3: M3.PI1SO3H (1.0 g, 2.8 mmol) was dissolved in minimum volume of MeOH:CHCl3 (1 : 1) and passed through a column [Orochem 5 g, 10 ml, Amino (NH2)] using MeOH as eluent. Organics were concentrated under vacuum to get free M3, which was used directly without further purification.) Synthesis of compound 3 (Step 2):
To a solution of 2 (0.40 g, 0.89 mmol) in THF (5 niL) under 2 atmosphere at -78 °C was added a solution of MeMgBr (0.89 mL, 2.6 mmol, 3M in Et20). The reaction mixture was allowed to attain room temperature over 1 hour. TLC shows complete conversion. The reaction mixture was again cooled to -10 °C and quenched with saturated aq. NH4CI solution (10 mL). The reaction mixture was extracted with CH2CI2 (50 x 3 mL). Combined organics were dried over Na2S04, filtered, concentrated and purified by reversed phase chromatography to obtain 3 (0.05 g, 8.4%) as di-TFA salt.
Molecular Formula: C22H28F2 4O3; LCMS purity: 88.82%; Expected: 434.2; Observed: 435.2 (M+l).
Synthesis of compound 4 (Step 3):
To a suspension of NaH (22 mg, 0.55 mmol) in dry THF (0.1 mL) under nitrogen atmosphere at 0 °C was added a solution of 3 (120 mg, 0.27 mmol) in dry THF (4.8 mL). The reaction mixture was allowed to come to room temperature and continued the stirring for 2 hours. Reaction mixture was again cooled to 0 °C. Methanesulfonyl chloride (0.42 mL, 0.55 mmol) was added to the reaction mixture and continued the stirring for another 1 hour. The reaction mixture was quenched with water and extracted with EtOAc (3 x 50 mL). Combined organics were dried over Na2S04, filtered, concentrated under vacuum and purified by silica gel chromatography afforded 4 as off white solid.
Molecular Formula: C23H30F2N4O5S; LCMS purity: 95.64%; Expected: 512.2; Observed: 513.2 (M+l). Synthesis of Example 3: (Step 4)
To a stirred solution of compound 4 (9.0 mg, 0.017 mmol) in CH2CI2 (2.0 mL) was added TFA (0.2 mL) dropwise at 0 °C. Reaction mixture was allowed to attain room temperature over 2 hours time. TLC shows complete conversion. Reaction mixture was concentrated to dryness. The solid residue was washed with Et20 (2 x 10 mL) and dried under vacuum. The solids were once again washed with a mixture of CH2CI2 (0.1 mL) and Et20 (5.0 mL) to obtain Example 3 (8.0 mg, 72.7%) as di-TFA salt. The IC50 value of Example 3 is 4nM. ¾ NMR (400MHz ,CD3OD): δ 7.96 (s, 1 H), 7.41 – 7.31 (m, 1 H), 7.30 – 7.19 (m, 2 H), 4.68 – 4.60 (m, 1 H), 4.22 – 4.07 (m, 4 H), 4.01 (d, J= 11.0 Hz, 1 H), 3.77 (d, J= 11.0 Hz, 1 H), 3.74 – 3.63 (m, 1 H), 3.39 (s, 3 H), 2.43 (d, J= 10.8 Hz, 1 H), 2.04 (t, J= 11.9 Hz, 1 H), 1.51 (s, 3 H).; Molecular Formula: C18H22F2 4O3S; HPLC purity: 95.01%; LCMS mass Expected: 412.2;
Observed: 413.0 (M+l).

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.5b00267

Development of a convergent synthesis of omarigliptin (MK-3102) suitable for commercial manufacture is described. The target molecule is assembled through a diastereoselective reductive amination of a highly functionalized pyranone with a mesylated pyrazole followed by deprotection of a Boc group. The synthesis of the pyranone relies on three Ru-catalyzed reactions: (1) a DKR reduction of a rac-α-aminoketone to set the two contiguous stereogenic centers, (2) a cycloisomerization of a bis-homopropargylic alcohol to a dihydropyran, and, finally, (3) a Ru-catalyzed oxidation of a pyranol to the desired pyranone. The regioselective synthesis of a N-Boc-1-mesyl pyrazole fragment was achieved via base-promoted mesyl group isomerization to afford 30:1 selectivity. A highlight of the endgame process development is telescoping a Boc deprotection and reductive amination followed by direct crystallization of the penultimate from the reaction mixture. This avoids handling of an unstable, mutagenic 1-mesylpyrazole BSA salt used in the earlier multikilogram deliveries and improves the overall diastereoselectivity and efficiency of the route.


Tesfaye Biftu et al, Omarigliptin (MK-3102): A Novel Long-Acting DPP-4 Inhibitor for Once-Weekly Treatment of Type 2 Diabetes;Journal of Medicinal Chemistry, Articles ASAP, March 24, 2014,DOI: 10.1021/jm401992e
Zacuto, Michael J. et al, Process for preparing chiral dipeptidyl peptidase-IV inhibitors;PCT Int. Appl., WO2013003250
Biftu, Tesfaye et al, Novel tetrahydropyran analogs as dipeptidyl peptidase IV inhibitors: Profile of clinical candidate (2R,3S,5R)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4H)-yl]tetrahydro-2H-pyran-3-amine, Bioorganic & Medicinal Chemistry Letters, 23(19), 5361-5366; 2013
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for the treatment or prevention of diabetes,PCT Int. Appl., WO2011028455
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for treatment or prevention of diabetes,U.S. Pat. Appl. Publ., US20100120863
Biftu, Tesfaye et al, Preparation of aminotetrahydropyrans as dipeptidyl peptidase IV inhibitors for treatment or prevention of diabetes,U.S. Pat. Appl. Publ., US20100120863
Xu, Feng et al, Process for preparation of chiral trans-2,3-disubstituted 5-oxotetrahydropyrans from ethyl N-(diphenylmethylene)glycinate and propargyl besylate, U.S. Pat. Appl. Publ., US20090187028
Ru(p-cymene)-N-sulfonyl-l,2-diphenylethylenediamine (DPEN) catalyst
R. Noyori, et al., J. Org. Chem., 66: 7931-7944 (2001)
B. Mohar, et al., Chem. Commun., 2572-2573 (2001)
The rhodium-catalyzed cycloisomerization
B. Trost etal., J.Amer. Chem.Soc., 125:7482-7483 (2003).
The ruthenium-catalyzed cycloisomerization
B. Trost, et al., J. Amer. Chem. Soc., 124: 2528-2533 (2002)
Gantz, I.; Chen, M.; Mirza, A.; Suryawanshi, S.; Davies, M. J.; Goldstein, B. J. Effect of MK-3102, a novel once-weekly DPP-4 inhibitor, over 12 weeks in patients with type 2 diabetes mellitus. Presented at the 48th Annual Meeting of the European Association for the Study of Diabetes (EASD), Berlin, Germany, October 2012; Abstract 101 (Clinical Research, Metabolism, Merck Research Laboratories).
