
Home » Posts tagged 'NEW DRUGS' (Page 30)
Tag Archives: NEW DRUGS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Health Canada Approves ADCETRIS® (Brentuximab Vedotin) for the Treatment of Relapsed or Refractory Hodgkin Lymphoma (HL) and Systemic Anaplastic Large Cell Lymphoma (sALCL)


Structure of brentuximab vedotin
Brentuximab vedotin on track for BLA filing with FDA during first half of 2011. [© Sebastian Kaulitzki – Fotolia.com]
Brentuximab is a human antibody. The antibody portion of Brentuximab vedotin has the sequence of two copies of:
>Brentuximab vedotin - heavy chain QIQLQQSGPEVVKPGASVKISCKASGYTFTDYYITWVKQKPGQGLEWIGWIYPGSGNTKY NEKFKGKATLTVDTSSSTAFMQLSSLTSEDTAVYFCANYGNYWFAYWGQGTQVTVSAAST KGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLY SLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSV FLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTY RVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTK NQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQG NVFSCSVMHEALHNHYTQKSLSLSPG >Brentuximab vedotin - light chain DIVLTQSPASLAVSLGQRATISCKASQSVDFDGDSYMNWYQQKPGQPPKVLIYAASNLES GIPARFSGSGSGTDFTLNIHPVEEEDAATYYCQQSNEDPWTFGGGTKLEIKRTVAAPSVF IFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLS STLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
February 01, 2013
Seattle Genetics, Inc. today announced that Health Canada has issued a Notice of Compliance with conditions (NOC/c), authorizing marketing of ADCETRIS for two lymphoma indications: (1) the treatment of patients with Hodgkin lymphoma (HL) after failure of autologous stem cell transplant (ASCT) or after failure of at least two multi-agent chemotherapy regimens in patients who are not ASCT candidates, and (2) the treatment of patients with systemic anaplastic large cell lymphoma (sALCL) after failure of at least one multi-agent chemotherapy regimen. The indications for ADCETRIS were authorized based on promising response rates demonstrated in single-arm trials. No data demonstrate increased survival with ADCETRIS.
“they are focused on making ADCETRIS available globally to all eligible patients with relapsed HL and sALCL. The approval of ADCETRIS in Canada, as well as the recent approval in the European Union, are important milestones to accomplish this goal,” said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. “Now that Health Canada has approved ADCETRIS, we are committed to working closely with public and private insurers to secure reimbursement coverage for patients in Canada.”
“The approval of ADCETRIS in Canada marks a significant milestone for patients with relapsed HL or sALCL who have had few new treatment options in several decades,” Joseph M. Connors, M.D., FRCPC, Clinical Director, Center for Lymphoid Cancer at BC Cancer Agency in Vancouver, Canada.
Health Canada grants NOC/c, a form of market approval, on the basis of promising evidence of clinical effectiveness, for products intended for the treatment of serious, life-threatening or severely debilitating illnesses that meet a serious unmet medical need or demonstrate a significant improvement in the benefit/risk profile over existing therapies. Conditions associated with market authorization under the NOC/c policy include a requirement that Seattle Genetics conduct clinical trials designed to confirm the anticipated clinical benefit of ADCETRIS in these patients. Two confirmatory phase III clinical trials evaluating ADCETRIS in the front-line treatment setting of HL and mature T-cell lymphoma (MTCL), including sALCL, are currently underway and enrolling patients.
ADCETRIS (brentuximab vedotin) was issued marketing authorization under the NOC/c policy based on results from a single-arm, phase II pivotal trial in HL patients with relapsed or refractory disease following an ASCT and a single-arm, phase II pivotal trial in relapsed or refractory sALCL patients. ADCETRIS is administered in hospitals through IV infusion over 30 minutes every three weeks and patients who achieve stable disease or better should receive a minimum of 8 cycles and up to a maximum of 16 cycles (approximately one year).
ADCETRIS is the first in a new class of antibody-drug conjugates (ADCs) to be approved in Canada. Using Seattle Genetics’ proprietary technology, the ADC consists of a monoclonal antibody directed to an antigen called CD30. The monoclonal antibody is connected to a cell-killing agent by a linker system that is designed to be stable in the bloodstream but to release the cell-killing agent into CD30-expressing cells, resulting in target cell death. The CD30 antigen is known to be expressed on the Reed-Sternberg cells of HL and on sALCL, an aggressive type of T-cell non-Hodgkin lymphoma.
“Health Canada’s approval of ADCETRIS is the first step in getting patients access to this important therapy,” said Sue Robson, Executive Director of Lymphoma Foundation Canada. “The Lymphoma Foundation is committed to working with Canada provincial governments to ensure that appropriate patients have access to this new therapy.”
About Lymphoma
Lymphoma is a general term for a group of cancers that originate in the lymphatic system. There are two major categories of lymphoma: Hodgkin lymphoma and non-Hodgkin lymphoma. Hodgkin lymphoma is distinguished from other types of lymphoma by the presence of one characteristic type of cell, known as the Reed-Sternberg cell. The Reed-Sternberg cell generally expresses CD30. Systemic ALCL is an aggressive type of T-cell non-Hodgkin lymphoma that also expresses CD30.
Brentuximab vedotin (INN, codenamed SGN-35 and previously cAC10-vcMMAE) is an antibody-drug conjugate approved to treat anaplastic large cell lymphoma (ALCL) and Hodgkin lymphoma. The U.S. Food and Drug Administration granted the agent an accelerated approval on August 19, 2011 for use against these two diseases.[1] It is marketed as Adcetris.[2]
The compound consists of the chimeric monoclonal antibody brentuximab (which targets the cell-membrane protein CD30) linked to three to five units of the antimitotic agent monomethyl auristatin E (MMAE, reflected by the ‘vedotin’ in the drug’s name). The antibody portion of the drug attaches to CD30 on the surface of malignant cells, delivering MMAE which is responsible for the anti-tumour activity.[3][4] Hence it is an antibody-drug conjugate.
In a 2010 clinical trial,[5] 34% of patients with refractory Hodgkin Lymphoma achieved complete remission and another 40% had partial remission.[6] Tumor reductions were achieved in 94% of patients. In ALCL, 87% of patients had tumors shrink at least 50% and 97% of patients had some tumors shrinkage.[7]
On 28 February 2011 a Biologics License Application (BLA) was submitted to the U.S. Food and Drug Administration (FDA) for the use of brentuximab vedotin in relapsed or refractory Hodgkin’s lymphoma and relapsed or refractory systemic anaplastic large cell lymphoma.[8] Both indications were approved by the FDA in Aug 2011.[9]
For these same indications brentuximab vedotin received a conditional Marketing authorization from the European Medicines Agency in october 2012.[10]
- FDA: Brentuximab Vedotin
- Seattle Genetics to Present Brentuximab Vedotin and SGN-75 Clinical Data at the American Society of Clinical Oncology Annual Meeting
- Seattle Genetics: Brentuximab vedotin (SGN-35)
- Francisco, Joseph A; et al. (2003). “cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity”. Blood 102 (4): 1458–1465. doi:10.1182/blood-2003-01-0039. PMID 12714494.
- ClinicalTrials.gov NCT00848926 A Pivotal Open-Label Trial of SGN-35 for Hodgkin Lymphoma
- “Seattle Genetics and Millennium Report Positive Data from Pivotal Trial of Brentuximab Vedotin (SGN-35) in Relapsed or Refractory Hodgkin Lymphoma at ASH Annual Meeting”. Dec 2010.
- “Is Seattle Genetics the Next Big Thing?”. 2 Dec 2010.
- “Seattle Genetics Submits BLA to FDA for Brentuximab Vedotin in Relapsed or Refractory Hodgkin Lymphoma and Systemic ALCL”. 28 Feb 2011.
- Genetic Engineering & Biotechnology News: Seattle Genetics’ Antibody-Drug Conjugate Receives FDA Okay to Treat Lymphomas
- http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/002455/WC500135004.pdf
FDA Approves Ravicti (glycerol phenylbutyrate)for the Chronic Management of Some Urea Cycle Disorders

FDA Approves Ravicti (glycerol phenylbutyrate)for the Chronic Management of Some Urea Cycle Disorders
Ravicti is marketed by Hyperion Therapeutics, based in South San Francisco, Calif.
February 1, 2013 — The U.S. Food and Drug Administration today approved Ravicti (glycerol phenylbutyrate) for the chronic management of some urea cycle disorders (UCDs) in patients ages 2 years and older.
UCDs are genetic disorders that involve deficiencies of specific enzymes involved in the urea cycle, a series of biochemical steps normally required to remove ammonia from the blood. When protein is absorbed and broken down by the body, it produces nitrogen as a waste product. The urea cycle removes nitrogen from the blood and converts it to urea, which is removed from the body through urine. In people with UCDs, nitrogen accumulates and remains in the body as ammonia, which can travel to the brain and cause brain damage, coma or death.
Ravicti, a liquid taken three times a day with meals, helps dispose of ammonia in the body. It is intended for patients whose UCD cannot be managed by a protein-restricted diet or amino acid supplements alone. Ravicti must be used with a protein-restricted diet and, in some cases, dietary supplements.
“Ravicti provides another treatment for chronic management of urea cycle disorders, a group of life-threatening conditions,” said Donna Griebel, M.D., director of the Division of Gastrointestinal and Inborn Errors Products in the FDA’s Center for Drug Evaluation and Research. “The approval of this new therapeutic option demonstrates FDA’s commitment to providing treatments for patients suffering from rare diseases.”
Ravicti was reviewed under the agency’s fast track program, designed to facilitate the development and expedite the review of drugs to treat serious diseases, fill unmet medical needs, and get important new drugs to patients earlier. Ravicti also was granted orphan product designation because it is intended to treat a rare disease.
Glycerol phenylbutyrate (HPN-100) is a pro-drug of phenylbutryrate and a pre-pro-drug of phenylacetic acid (PAA), the active moiety of Buphenyl, the only therapy currently FDA-approved as adjunctive therapy for the chronic management of patients with the most prevalent urea cycle disorders — carbamylphosphate synthetase, ornithine transcarbamylase, and argininosuccinic acid synthetase. HPN-100, which is dosed orally in liquid form, provides an alternative pathway to the urea cycle for the disposal of waste nitrogen through the renal excretion of phenylacetylglutamine, which is formed from PAA and glutamine.
伯舒替尼 Bosutinib



Bosutinib Monohydrate (伯舒替尼)
(Bosulif®)
Approved sept4 2012 by FDA
PMDA SEPT26 2014
EMA MAR 27 2013
A kinase inhibitor indicated for the treatment of adult patients with Ph+ chronic myelogenous leukemia (CML).
WYETH INNOVATOR
PFIZER DEVELOPER

SKI-606; SK-606
CAS No.380843-75-4 (Free form)
CAS 918639-08-4(Bosutinib Monohydrate)
Bosutinib (rINN/USAN; codenamed SKI-606, marketed under the trade name Bosulif) is atyrosine kinase inhibitor undergoing research for use in the treatment of cancer. [1] [2]Originally synthesized by Wyeth, it is being developed by Pfizer.
。

Some commercial stocks of bosutinib (from sources other than the Pfizer material used for clinical trials) have recently been found to have the incorrect chemical structure, calling the biological results obtained with them into doubt.[3]
Bosutinib received US FDA approval on September 5, 2012 for the treatment of adult patients with chronic, accelerated, or blast phase Philadelphia chromosome-positive (Ph+)chronic myelogenous leukemia (CML) with resistance, or intolerance to prior therapy.[4][5][6]

Article
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) on January 17, 2013, adopts a positive opinion, recommending a conditional marketing authhorization for Pfizer’s orphan drug Bosulif (Bosutinib) for Chronic Leukemia (CML). Bosutinib receives orphan designation from the European Commission (EC) on August 4, 2010, for CML.
Pfizer receives FDA approval on September 4, 2012, for orphan drug Bosulif (Bosutinib) for CML. Pfizer receives on February 24, 2009, FDA Orphan Drug Designation (ODD) for Bosutinib for CML.

Per a September 2012 article in FierceBioTech.com, a Pfizer spokesperson says that “the drug will cost less than $8,200/month”/patient in the US. In other words, treatment will cost approximately $98,400/patient/year. Also per FierceBiotech,“Bosulif is the 3rd new medicine from Pfizer Oncology’s pipeline to be approved by the FDA in just 13 months ….”.
ARTICLE
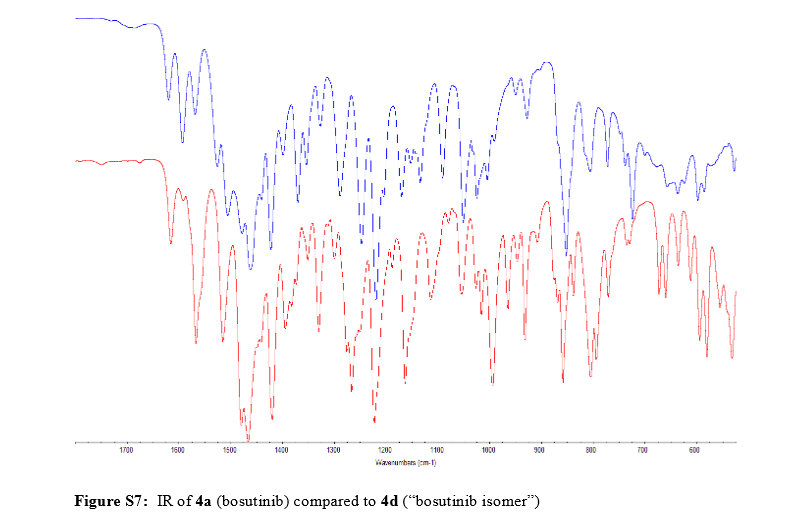
Pfizer’s response to compound fraud spotlights quality issues
read ………http://www.rsc.org/chemistryworld/2015/12/pfizer-bogus-bosutinib-isomer-fraud-leukaemia-drug

Synthesis
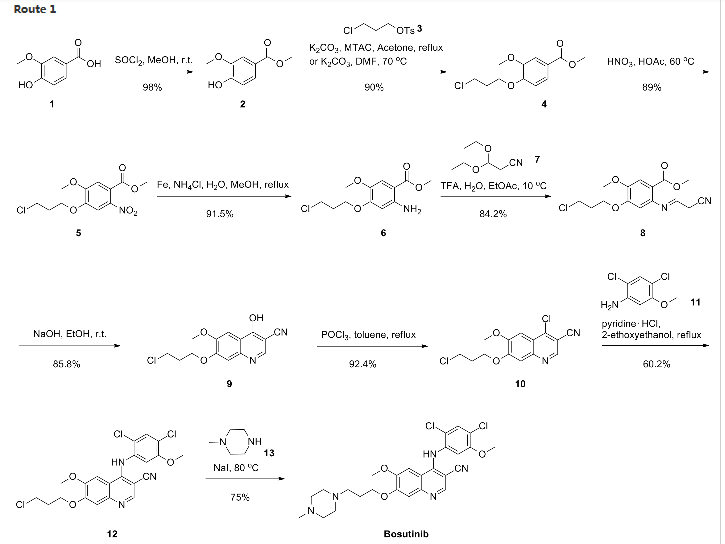

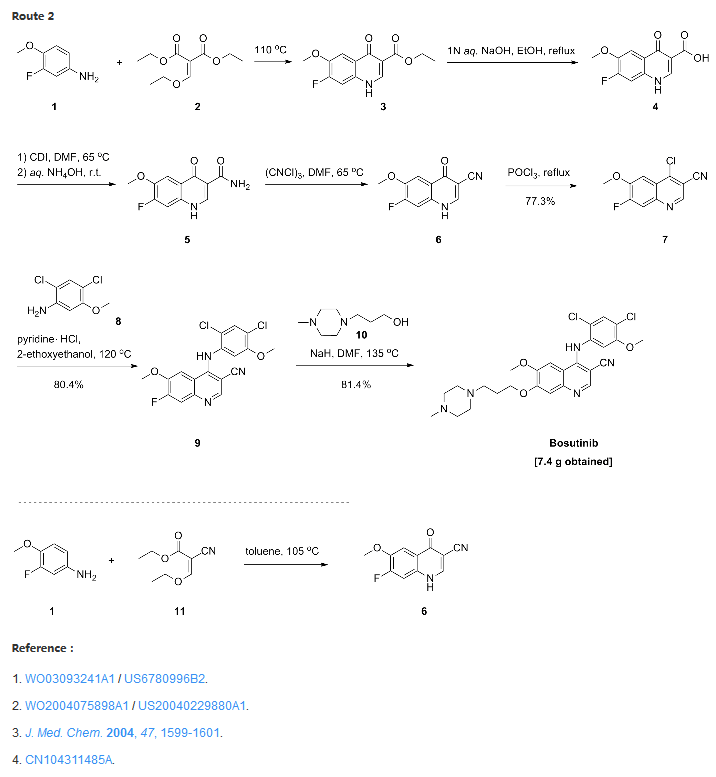
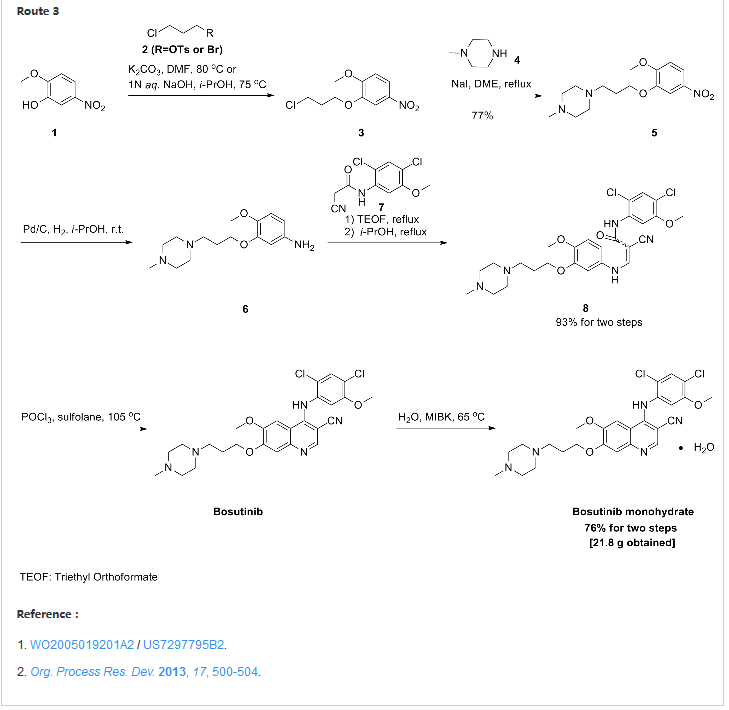



Confirmation of Bosutinib Structure; Demonstration of Controls To Ensure Product Quality

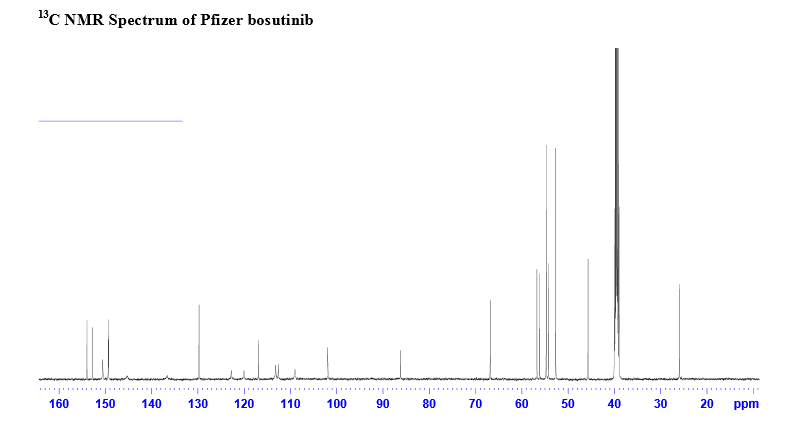
Nonbranded/unauthorized vendors had been manufacturing/selling what they described as bosutinib, while the material supplied was actually an isomer of bosutinib. This raised concerns within the worldwide research community around the established control strategies for bosutinib. This manuscript summarizes that the appropriate testing was in place to ensure product quality, along with additional experimentation that was performed to confirm that testing (methods) can differentiate the potential isomeric compounds. Testing includes the use of IR for identity confirmation of raw materials, material characterization by NMR, single crystal X-ray to confirm structure, and evaluation of several potential isomers by HPLC, melting point, and IR, thus demonstrating the control strategy needed to ensure the product controls.




REFERENCES
- Puttini M, Coluccia AM, Boschelli F, Cleris L, Marchesi E, Donella-Deana A, Ahmed S, Redaelli S, Piazza R, Magistroni V, Andreoni F, Scapozza L, Formelli F, Gambacorti-Passerini C. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006 Dec 1;66(23):11314-22. Epub 2006 Nov 17.
- Vultur A, Buettner R, Kowolik C, et al. (May 2008). “SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells”.Mol. Cancer Ther. 7 (5): 1185–94. doi:10.1158/1535-7163.MCT-08-0126.PMC 2794837. PMID 18483306.
- Derek Lowe, In The Pipeline (blog), “Bosutinib: Don’t Believe the Label!”
- Cortes JE, Kantarjian HM, Brümmendorf TH, Kim DW, Turkina AG, Shen ZX, Pasquini R, Khoury HJ, Arkin S, Volkert A, Besson N, Abbas R, Wang J, Leip E, Gambacorti-Passerini C. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011 Oct 27;118(17):4567-76. Epub 2011 Aug 24.
- Cortes JE, Kim DW, Kantarjian HM, Brümmendorf TH, Dyagil I, Griskevicus L, Malhotra H, Powell C, Gogat K, Countouriotis AM, Gambacorti-Passerini C. Bosutinib Versus Imatinib in Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia: Results From the BELA Trial. J Clin Oncol. 2012 Sep 4. [Epub ahead of print]
- “Bosulif Approved for Previously Treated Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia”. 05 Sep 2012.
-
P Bowles et al, Org. Process Res. Dev., 2015, DOI: 10.1021/acs.oprd.5b00244
8 N M Levinson and S G Boxer, PLoS One, 2012, 7, e29828 (DOI: 10.1371/journal.pone.0029828)
9 N Beeharry et al, Cell Cycle, 2014, 13, 2172 (DOI: 10.4161/cc.29214)
update…………….
file:///C:/Users/Inspiron/Downloads/molecules-15-04261%20(1).pdf
Synthesis of Bosutinib from 3-Methoxy-4-hydroxybenzoic Acid
Jun 11, 2010 – Abstract: This paper reports a novel synthesis of bosutinib starting from … C-NMR, MS and the purities of all the compounds were determined …
4-(2,4-Dichloro-5-methoxyphenylamino]-6-methoxy-7-[3-(4-methylpiperazin-1-yl)propoxy]quinoline- 3-carbonitrile (10). A mixture of 7-(3-chloropropoxy)-4-(2,4-dichloro-5-methoxyphenylamino)-6-methoxyquinoline-3-carbonitrile (9, 0.328 g, 0.7 mmol) and sodium iodide (0.11 g, 0.70 mmol) in N-methylpiperazine (4 mL) was heated at 80 ºC for 12 h. The reaction mixture was concentrated in vacuo and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography, eluting with 30% methanol in dichloromethane. The fractions containing product were collected and concentrated in vacuo. Diethyl ether was added to the residue, and the light pink solid was collected by filtration (0.28 g, 75% yield, 98.7% HPLC purity): m.p. 116–120 ºC;
1H-NMR (DMSO-d6): 1.92–1.97 (m, 2H), 2.15 (s, 3H), 2.32–2.46 (m, 10H), 3.84 (s, 3H), 3.93 (s, 3H), 4.19 (t, J = 6.3 Hz, 2H), 7.31 (br s, 2H), 7.43 (s, 1H), 7.64 (s, 1H), 8.52 (s, 1H), 9.51 (s, 1H);
13C-NMR (CDCl3): 25.96, 45.68, 52.67, 52.67, 54.24, 54.72, 54.72, 56.01, 60.71, 66.87, 89.10, 101.66, 101.66, 109.12, 113.95, 117.17, 122.99, 122.99, 128.27, 137.88, 146.15, 148.13, 148.51, 149.50, 150.43, 153.03;
MS (ES) m/z 530.2, 532.2 (M+1).
…………………..
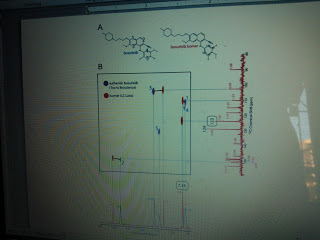
Structural and spectroscopic analysis of the kinase inhibitor …
files.figshare.com/337292/Figure_S2.doc
by NM Levinson – Cited by 39 – Related articles
NMR spectroscopy on bosutinib and the bosutinib isomer. As described in the main text, the 1H NMR spectra of the compounds we purchased from LC Labs and …
Figure S2. NMR experiments on bosutinib and the bosutinib isomer. A) The structure of bosutinib and a putative structure for the bosutinib isomer are shown. The blue numbers on the bosutinib structure represent the five aromatic proton-carbon pairs. The numbers on the aniline ring of the bosutinib isomer are 13C chemical shifts. B) NMR spectra. In the top left panel, 1H-13C HSQC spectra of bosutinib and the bosutinib isomer are shown. The thick black lines connect the peaks that arise from the equivalent proton-carbon pairs in the two compounds. The thin gray lines are intended to guide the eye to the corresponding peaks in the 1-dimensional spectra. The peaks for the five aromatic proton-carbon pairs in authentic bosutinib are indicated with large blue numbers. These putative assignments are based on 13C chemical shift predictions. The bottom panel shows the 1H NMR spectra of both compounds. The peak located at 7.34 ppm in the bosutinib isomer sample, which integrates to 2, is indicated. The colored numbers directly next to the peaks are the peak integrations. The panel on the upper right shows the aromatic region of the 13C NMR spectrum of the bosutinib isomer. The peak located at 123 ppm, which displays an integrated intensity of 2, is indicated.
//////////////Clc1c(OC)cc(c(c1)Cl)Nc4c(C#N)cnc3cc(OCCCN2CCN(CC2)C)c(OC)cc34
BOTOX® (onabotulinumtoxinA) Receives U.S. Food and Drug Administration Approval for the Treatment of Overactive Bladder for Adults Who Have an Inadequate Response to or Are Intolerant of an Anticholinergic Medication

BOTOX® (onabotulinumtoxinA) Receives U.S. Food and Drug Administration Approval for the Treatment of Overactive Bladder for Adults Who Have an Inadequate Response to or Are Intolerant of an Anticholinergic Medication
Allergan, Inc. (NYSE:AGN) announced today that the U.S. Food and Drug Administration (FDA) has approved BOTOX® (onabotulinumtoxinA) for the treatment of overactive bladder (OAB) with symptoms of urge urinary incontinence, urgency and frequency in adults who have had an inadequate response to or are intolerant of an anticholinergic medication. In two double-blind, randomized, multi-center, placebo-controlled 24-week clinical trials among adults with overactive bladder who had not been adequately managed with anticholinergic treatments, BOTOX® reduced daily urinary incontinence (leakage) episodes as compared to placebo by 50 percent or more by week 12 (reduction of 2.5 episodes from baseline of 5.5 episodes in one study and reduction of 3 episodes from baseline of 5.5 episodes in the second study for those treated with BOTOX® vs. a reduction of 0.9 episodes from a baseline of 5.1 episodes in one study and a reduction of 1.1 episodes from a baseline of 5.7 episodes in the second study for those treated with placebo).1
Allergan has a long-standing commitment to study the potential of BOTOX® to treat a number of different medical conditions
“Allergan has a long-standing commitment to study the potential of BOTOX® to treat a number of different medical conditions,” said Scott Whitcup, M.D., Allergan’s Executive Vice President, Research and Development, Chief Scientific Officer. “With today’s approval, BOTOX® is now approved for 26 different indications in more than 85 countries. Most importantly, today’s FDA approval is a milestone in the treatment of this burdensome condition and will provide a novel option for urologists and their OAB patients.”
While the exact cause is often unknown, OAB is a medical condition that results in an uncontrolled urge to urinate, frequent urination and, in many patients, uncontrollable leakage of urine. In the United States, an estimated 14.7 million adults experience symptoms of OAB with urinary incontinence (unexpected leakage of urine).2 Anticholinergics, which are often prescribed as pills, are used by approximately 3.3 million Americans with OAB, with or without urinary incontinence, to manage their condition.3 It is estimated, however, that greater than 50 percent of these patients stop taking at least one oral medication within 12 months, likely due to an inadequate response to, or intolerance of, the medication.4
“Overactive bladder can be a difficult condition to treat as there have been limited options for patients when currently available medications have failed to provide them with adequate relief,” said Dr. Victor Nitti*, Vice Chairman, Department of Urology and Director of Female Pelvic Medicine and Reconstructive Surgery at NYU Langone Medical Center. “With the approval of BOTOX®, we have a new treatment option to offer these patients that has demonstrated efficacy in reducing urinary leakage and other symptoms of OAB with the effect lasting up to six months.”
The median duration for efficacy with BOTOX® at reducing urinary leakage and other symptoms of OAB in the two clinical studies was 135-168 days compared to 88-92 days with placebo based on qualification for retreatment. To qualify for retreatment, at least 12 weeks must have passed since the prior treatment, post-void residual urine volume must have been less than 200 mL and patients must have reported at least two urinary incontinence episodes over three days. BOTOX® treatment relieves OAB symptoms by temporarily calming muscle contractions by blocking the transmission of nerve impulses to the bladder muscle.

BOTOX Cosmetic (onabotulinum toxin A) For Injection, is a sterile, vacuum-dried purified botulinum toxin type A, produced from fermentation of Hall strain Clostridium botulinum type A grown in a medium containing casein hydrolysate, glucose, and yeast extract, intended for intramuscular use. It is purified from the culture solution bydialysis and a series of acid precipitations to a complex consisting of the neurotoxin, and several accessory proteins. The complex is dissolved in sterile sodium chloride solution containing Albumin Human and is sterile filtered (0.2 microns) prior to filling and vacuum-drying.
The primary release procedure for BOTOX Cosmetic uses a cell-based potency assay to determine the potency relative to a reference standard. The assay is specific to Allergan’s products BOTOX and BOTOX Cosmetic. One Unit of BOTOX Cosmetic corresponds to the calculated median intraperitoneal lethal dose (LD50) in mice. Due to specific details of this assay such as the vehicle, dilution scheme and laboratory protocols, Units of biological activity of BOTOX Cosmetic cannot be compared to nor converted into Units of any other botulinum toxin or any toxin assessed with any other specific assay method. The specific activity of BOTOX Cosmetic is approximately 20 Units/nanogram of neurotoxin protein complex.
Each vial of BOTOX Cosmetic contains either 50 Units of Clostridium botulinum type A neurotoxin complex, 0.25 mg of Albumin Human, and 0.45 mg of sodium chloride; or 100 Units of Clostridium botulinum type A neurotoxin complex, 0.5 mg of Albumin Human, and 0.9 mg of sodium chloride in a sterile, vacuum-dried form without a preservative.
 COCK WILL TEACH YOU NMR
COCK WILL TEACH YOU NMR COCK SAYS MOM CAN TEACH YOU NMR
COCK SAYS MOM CAN TEACH YOU NMR
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 amcrasto@gmail.com
amcrasto@gmail.com
TAKE A TOUR
KISUMU, KENYA

Kisumu – Wikipedia, the free encyclopedia
Kisumu is a port city in Kisumu County, Kenya 1,131 m (3,711 ft), with a population of 409,928 (2009 census). It is the third largest city in Kenya, the principal city …
Kisumu CountyKisumu County is one of the new devolved counties of Kenya. Its …
|
Kisumu International AirportKisumu International Airport is an airport in Kisumu, Kenya (IATA …
|

Boat riding in Kisumu

Local inhabitants near Kisumu, 1911


Clockwise: Lake Victoria Panorama, Kisumu Panorama, sunset at Oginga Odinga street, Downtown, Kiboko Point, Nighttime in Kisumu and Jomo Kenyatta Stadium.
|
|

Kisumu
|
|
Coordinates:  0°6′S 34°45′E 0°6′S 34°45′E |
|
| Country | |
|---|---|
| County | Kisumu County |

Kisumu panorama, viewed from Lake Victoria

Jomo Kenyatta Stadium

Kisumu Harbour. The green vegetation is water hyacinth

Nairobi University Kisumu Campus





DR ANTHONY MELVIN CRASTO Ph.D

Shenzhen Neptunus Bioengineering submits first ever application for conducting Phase 2 trials in the US for Traditional Chinese Medicine ( Polydatin Injection) in the class of innovative drugs
polydatin
Resveratrol 3-beta-mono-D-glucoside
trans-piceid
3,5,4′-trihydroxystilbene-3-O-β-D-glucopyranoside
Shenzhen Neptunus Bioengineering submits first ever application for conducting Phase 2 trials in the US for Traditional Chinese Medicine ( Polydatin Injection) in the class of innovative drugs

Piceid is a stilbenoid glucoside and is a major resveratrol derivative in grape juices.[1] It can be found in the bark of Picea sitchensis.[2] It can also be isolated from Polygonum cuspidatum,[3] the Japanese knotweed (syn. Fallopia japonica).
Resveratrol can be produced from piceid fermented by Aspergillus oryzae.[3] This latter species produces a piceid-b-D-glucosidase.[4]
Trans-piceid is the glucoside formed with trans-resveratrol, while cis-piceid is formed with cis-resveratrol.
Trans-resveratrol-3-O-glucuronide is one of the two metabolites of trans-piceid in rat.[5]
Resveratrol glucoside from transgenic alfalfa has been used for the prevention of aberrant crypt foci in mice.[6]
- Romero-Pérez, A. I.; Ibern-Gómez, M.; Lamuela-Raventós, R. M.; De La Torre-Boronat, M. C. (1999). “Piceid, the Major Resveratrol Derivative in Grape Juices”. Journal of Agricultural and Food Chemistry 47 (4): 1533–1536. doi:10.1021/jf981024g.PMID 10564012. edit
- Aritomi, M.; Donnelly, D. M. X. (1976). “Stilbene glucosides in the bark of Picea sitchensis”. Phytochemistry 15 (12): 2006. doi:10.1016/S0031-9422(00)88881-0. edit
- Wang, H.; Liu, L.; Guo, Y. -X.; Dong, Y. -S.; Zhang, D. -J.; Xiu, Z. -L. (2007). “Biotransformation of piceid in Polygonum cuspidatum to resveratrol by Aspergillus oryzae”. Applied Microbiology and Biotechnology 75 (4): 763–768. doi:10.1007/s00253-007-0874-3. PMID 17333175. edit
- Purification and characterization of piceid-b-D-glucosidase from Aspergillus oryzae. Chunzhi Zhang, Dai Li, Hongshan Yu, Bo Zhang and Fengxie Jin, Process Biochemistry, 2007, 42, pages 83–88, doi:10.1016/j.procbio.2006.07.019
- Zhou, M.; Chen, X.; Zhong, D. (2007). “Simultaneous determination of trans-resveratrol-3-O-glucoside and its two metabolites in rat plasma using liquid chromatography with ultraviolet detection”. Journal of Chromatography B 854: 219.doi:10.1016/j.jchromb.2007.04.025. edit
- RKineman, B. D.; Brummer, E. C.; Paiva, N. L.; Birt, D. F. (2010). “Resveratrol from Transgenic Alfalfa for Prevention of Aberrant Crypt Foci in Mice”. Nutrition and Cancer 62(3): 351–361. doi:10.1080/01635580903407213. PMID 20358473
Novartis gets European approval for first Meningitis B vaccine

Bexero is a vaccine indicated for the treatment of the meningococal gp B disease
Novartis has received approval from the European Commission for the first vaccine to protect children against Meningitis B.
Bexsero (4CMenB) will be used in Europe to prevent meningococcal B meningitis (MenB), one of the most common and deadly forms of the disease in babies and infants under five years of age.
There is currently no approved vaccine offering protection against this particular type of meningitis.
Novartis has committed to making the Bexsero available as soon as possible, the firm said in a statement on Tuesday.
Meningitis UK is today urging the government to introduce the vaccine into the UK, which has one of the highest incidence rates for MenB in the world.
Meningitis UK Founder Steve Dayman said; “The Government must introduce the Meningitis B vaccine into the immunisation schedule as soon as possible – it will save thousands of lives and spare families so much suffering.
“Any delay means lives will be lost.”
The Joint Committee on Vaccination and Immunisation (JCVI), which advises the government, plans to meet in June 2013 to discuss the vaccine.
MenB is caused by bacteria, leading to inflammation of the lining around the brain and spinal cord. It can kill within 24 hours.
In December 2010, Novartis submitted a Marketing Authorisation Application (MAA) to the European Medicines Agency (EMA) for bexsero based on positive results from Phase III trials.
In November 2012, the Committee for Medicinal Products for Human Use (CHMP) of EMA adopted a positive opinion for approval of the MAA.
Novartis plans to submit marketing applications in Asia, Latin America and North America.
Meningococcal is a life threatening disease which can lead to death within 24 to 48 hours of the first symptoms. The disease manifests in the form of bacterial meningitis, which leads to an infection of the membrane around the brain and spine and a bloodstream infection called sepsis.
The bacteria which causes meningococcal disease is called meningococcus and is divided into five main groups, called serogroups, namely A, B, C, W135 and Y. MenB is the most common type of bacteria causing meningococcal disease.
MenB strains can mutate making it very difficult to diagnose and treat. It has led to several outbreaks across the world. The highest rates of the disease occur in the semi-arid and sub-Saharan Africa region.
Most of the MenB cases occur in healthy patients. A person can carry the bacteria for up to six months. It is easily transmitted through physical contact, coughing and sneezing. Infants and adolescents are the most vulnerable groups of the disease.
Initial symptoms of the disease are flu-like and hence difficult to diagnose. The main symptoms such as neck stiffness and rashes appear at a later stage of the illness. Existing treatments for the disease include hospitalisation and antimicrobial therapy. The disease, however, is difficult to treat due to its rapid rate of progression.
An estimated 20,000 to 80,000 cases of MenB are reported every year. About 5-10% of the people die even after being diagnosed and treated. Those who survive the disease suffer from severe complications such as brain damage, learning disabilities, behavioural problems and hearing loss.
DR ANTHONY MELVIN CRASTO Ph.D
FDA Approves Exjade to Remove Excess Iron in Patients with genetic blood disorder
Exjade (deferasirox) is an iron chelating agent. Exjade tablets for oral suspension contain 125 mg, 250 mg, or 500 mg deferasirox. Deferasirox is designated chemically as 4-[3,5-Bis (2-hydroxyphenyl)-1H-1,2,4-triazol-1yl]-benzoic acid and its structural formula is:
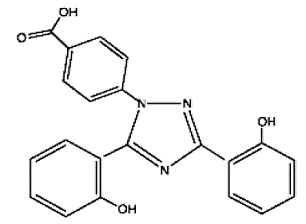 |
Deferasirox is a white to slightly yellow powder. Its molecular formula is C21H15N3O4 and its molecular weight is 373.4.
FDA Approves Exjade to Remove Excess Iron in Patients with genetic blood disorder
January 23, 2013 — The U.S. Food and Drug Administration today expanded the approved use of Exjade (deferasirox) to treat patients ages 10 years and older who have chronic iron overload resulting from a genetic blood disorder called non-transfusion-dependent thalassemia (NTDT).
NTDT is a milder form of thalassemia that does not require individuals to get frequent red blood cell transfusions. However, over time, some patients with NTDT are still at risk for iron overload that can lead to damage to vital organs.
The FDA is also authorizing marketing of FerriScan as an imaging companion diagnostic for Exjade. The agency previously cleared FerriScan for measuring liver iron concentration (LIC), but its use in Exjade clinical studies to select patients for therapy, and to manage therapy, defined its role as an imaging companion diagnostic necessary for Exjade’s safe and effective use. FerriScan measures LIC non-invasively using magnetic resonance imaging.
An estimated 1,000 people in the United States have thalassemia, according to the National Heart, Lung, and Blood Institute. Thalassemia conditions can cause the body to make fewer healthy red blood cells and less hemoglobin, a protein that carries oxygen to all parts of the body and returns carbon dioxide to the lungs so it can be exhaled. Some patients with thalassemia require frequent transfusions of red blood cells to maintain an acceptable level of hemoglobin. Iron overload is common in these patients.
Exjade was previously approved for treatment of chronic iron overload due to blood transfusions in patients ages 2 years and older, and this approval extends its use to treat patients with NTDT who show iron overload. Exjade should be used in patients with NTDT who have an LIC of at least 5 milligrams of iron per gram of dry liver tissue weight.
Exjade’s new indication is being approved under the FDA’s accelerated approval program, which provides patients earlier access to promising new drugs intended to treat serious or life-threatening illnesses while the company conducts additional studies to confirm the drug’s clinical benefit. Exjade was approved based on clinical data showing it can reduce LIC to less than 5 mg/g dry weight, a surrogate endpoint that is judged reasonably likely to predict a clinical benefit to patients.
“Using our accelerated approval process, FDA is able to expedite the availability of this drug to patients who need to reduce excess iron,” said Richard Pazdur, M.D., director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Exjade is the first drug approved to treat patients with NTDT who show iron overload.”
The safety and effectiveness of Exjade to treat chronic iron overload in patients with NTDT were established in two clinical trials designed to measure the number of patients whose LIC was reduced to less than 5 mg/g dry weight after 52 weeks of treatment. In the first trial, 166 patients were randomly assigned to receive 5 mg/kg of Exjade, 10 mg/kg of Exjade, or a placebo daily. Results showed 15 percent and 27 percent of Exjade-treated patients achieved the target LIC, respectively, compared with 4 percent in placebo-treated patients. The second trial contained 133 patients from the first study who received an additional year of Exjade treatment or switched from placebo to Exjade treatment. Thirty-five percent of the evaluable patients in this extension trial achieved the target LIC.
The FDA reviewed data for the FerriScan through the de novo classification process, a regulatory pathway for medical devices that are generally moderate-risk but are not comparable to an already legally marketed device. The FDA’s granting of the de novo request for FerriScan was based largely on data from the Exjade clinical studies that used FerriScan LIC results as the primary outcome measure. Additionally, investigators conducted a 230-patient study that found FerriScan results were as accurate as liver biopsy for measuring LIC.
“The FerriScan device is a non-invasive test that helps physicians to select appropriate patients for Exjade therapy as well as monitor their response to the drug, and discontinue therapy when LIC reaches safe levels,” said Alberto Gutierrez, Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health.
Exjade is marketed by East Hanover, N.J.-based Novartis. FerriScan is marketed by Resonance Health, based in Australia.
structure
http://www.rxlist.com/exjade-drug.htm
http://en.wikipedia.org/wiki/Deferasirox
http://www.us.exjade.com/index.jsp?usertrack.filter_applied=true&NovaId=4029462066891750194
http://www.novartisoncology.com/index.jsp?lightbox=global-patient
Sihuan’s Drug Nalmefene Hydrochloride Receives Approval for Pharmaceutical Registration from State Food and Drug Administration
 |
|
|---|---|
| Nalmefene | |
| 17-cyclopropylmethyl-4,5α-epoxy-6-methylenemorphinan-3,14-diol |
REVEX (nalmefene hydrochloride injection), an opioid antagonist, is a 6-methylene analogue of naltrexone. The chemical structure is shown below:
 |
Molecular Formula: C21H25NO3•HCl
Molecular Weight: 375.9, CAS # 58895-64-0
Chemical Name: 17-(Cyclopropylmethyl)-4,5a-epoxy-6-methylenemorphinan-3,14-diol, hydrochloride salt.
Nalmefene hydrochloride is a white to off-white crystalline powder which is freely soluble in water up to 130 mg/mL and slightly soluble in chloroform up to 0.13 mg/mL, with a pKa of 7.6.
REVEX is available as a sterile solution for intravenous, intramuscular, and subcutaneous administration in two concentrations, containing 100 µg or 1.0 mg of nalmefene free base per mL. The 100 µg/mL concentration contains 110.8 µg of nalmefene hydrochloride and the 1.0 mg/mL concentration contains 1.108 mg of nalmefene hydrochloride per mL. Both concentrations contain 9.0 mg of sodium chloride per mL and the pH is adjusted to 3.9 with hydrochloric acid.
Concentrations and dosages of REVEX are expressed as the free base equivalent of nalmefene.
Nalmefene (Revex), originally known as nalmetrene, is an opioid receptor antagonistdeveloped in the early 1970s,[1] and used primarily in the management of alcoholdependence, and also has been investigated for the treatment of other addictions such aspathological gambling and addiction to shopping.
Nalmefene is an opiate derivative similar in both structure and activity to the opiate antagonist naltrexone. Advantages of nalmefene relative to naltrexone include longer half-life, greater oral bioavailability and no observed dose-dependent liver toxicity. As with other drugs of this type, nalmefene can precipitate acute withdrawal symptoms in patients who are dependent on opioid drugs, or more rarely when used post-operatively to counteract the effects of strong opioids used in surgery.
Nalmefene differs from naltrexone by substitution of the ketone group at the 6-position of naltrexone with a methylene (CH2) group, which considerably increases binding affinity to the μ-opioid receptor. Nalmefene also has high affinity for the other opioid receptors, and is known as a “universal antagonist” for its ability to block all three.
- US patent 3814768, Jack Fishman et al, “6-METHYLENE-6-DESOXY DIHYDRO MORPHINE AND CODEINE DERIVATIVES AND PHARMACEUTICALLY ACCEPTABLE SALTS”, published 1971-11-26, issued 1974-06-04
- Barbara J. Mason, Fernando R. Salvato, Lauren D. Williams, Eva C. Ritvo, Robert B. Cutler (August 1999). “A Double-blind, Placebo-Controlled Study of Oral Nalmefene for Alcohol Dependence”. Arch Gen Psychiatry 56 (8): 719.
- Clinical Trial Of Nalmefene In The Treatment Of Pathological Gambling
- http://www.fda.gov/cder/foi/label/2000/20459S2lbl.pdf
- “Efficacy of Nalmefene in Patients With Alcohol Dependence (ESENSE1)”.
- “Lundbeck submits Selincro in EU; Novo Nordisk files Degludec in Japan”. thepharmaletter. 22 December 2011.
- Nalmefene Hydrochloride Drug Information, Professional
Takeda Receives FDA Approval For Three New Type 2 Diabetes Therapies

HY A0023, ALOGLIPTIN BENZOATE, NESINA, SYR 322
Takeda Receives FDA Approval For Three New Type 2 Diabetes Therapies
Furiex Pharmaceuticals Inc. Friday 25 jan 2013,confirmed that Takeda Pharmaceutical Company Limited has received approval from the U.S. Food and Drug Administration of three new type 2 diabetes therapies, NESINA (alogliptin) and the fixed-dose combination therapies, OSENI (alogliptin and pioglitazone) and KAZANO (alogliptin and metformin HCl), for the treatment of type 2 diabetes in adults as adjuncts to diet and exercise.
Under its agreement with Takeda, Furiex is entitled to receive a $25 million milestone payment as a result of this approval, as well as royalties on sales in the United States and potential sales-based milestones. Furiex has already been receiving royalty payments from Takeda for the sale of NESINA and LIOVEL in Japan.
“Receiving regulatory approvals for NESINA, OSENI and KAZANO in the U.S. marks an important milestone for Furiex,” said Fred Eshelman, chairman of Furiex.
“These approvals should enable Takeda to build on the success of NESINA in Japan and leverage its more than 20 years of clinical and patient experience in the type 2 diabetes therapeutic area.”
Type 2 diabetes is the most common form of diabetes and has reached epidemic proportions globally. The global health care expenditures to treat and prevent diabetes and its complications were estimated at $471 billion in 2012. In addition to diet and exercise, patients often need to take multiple medications to help manage blood glucose. Because of the chronic nature of this disease, combination therapy is often required to maintain diabetic control over many years of therapy.
NESINA is a DPP-4 inhibitor for the treatment of type 2 diabetes as an adjunct to diet and exercise. DPP-4 inhibitors address insulin deficiency by slowing the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide).
OSENI is a fixed dose combination therapy that combines alogliptin and pioglitazone in a single tablet for the treatment of type 2 diabetes in adults as an adjunct to diet and exercise. Pioglitazone is a thiazolidinedione that directly targets insulin resistance, a condition in which the body does not efficiently use the insulin it produces to control blood glucose levels. It is currently approved for use in adults for the treatment of type 2 diabetes as an adjunct to diet and exercise.
KAZANO is a fixed dose combination therapy for the treatment of type 2 diabetes that combines alogliptin and metformin in a single tablet. Metformin is a widely-used diabetes medication that acts primarily by reducing the amount of glucose produced by the liver. These medications work in combination to help patients with type 2 diabetes manage their blood glucose levels.
Nesina® (alogliptin) is a member of a new class of drugs for the oral treatment of type 2 diabetes (T2D). Nesina is being developed and marketed by Takeda Pharmaceuticals. In April 2010, Takeda received regulatory approval from Japan’s Ministry of Health, Labour and Welfare for Nesina and it is now being sold in Japan.
Takeda has resubmitted a new drug application (NDA) with the U.S. Food and Drug Administration (FDA), and has submitted a Marketing Authorization Application (MAA) with the European Medicines Agency (EMA).
As a result of their collaboration, Furiex has rights to royalties and sales-based milestones from Takeda for the sale of Nesina in Japan. Furiex will be entitled to receive regulatory milestones, royalties and sales-based milestones upon marketing approval of Nesina in other countries.
Alogliptin is a DPP-4 inhibitor that slows the inactivation of incretin hormones GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulinotropic peptide), which play a major role in regulating blood glucose levels and have the potential to improve pancreatic beta-cell function.
Alogliptin has been studied in 12 Phase III trials including more than 8,000 patients. Pivotal trials demonstrated alogliptin was well-tolerated and it significantly improved glycemic control in T2D patients without raising the incidence of hypoglycemia. Additionally, alogliptin has been shown to enhance glycemic control when used in combination with other commonly prescribed diabetes drugs.
Alogliptin was tested in 329 drug-naive patients with inadequately controlled T2D in a double-blind, placebo-controlled, multicenter study. Patients were randomized to once-daily treatment with 12.5 mg or 25 mg alogliptin or placebo for 26 weeks. The primary efficacy end point was HbA(1c). Alogliptin was well-tolerated and significantly improved glycemic control in these patients with T2D without raising the incidence of hypoglycemia.
DR ANTHONY CRASTO, PhD, ICT Organic chemistry, Currently working with GLENMARK GENERICS LTD research centre as Principal Scientist, process research (bulk actives) at Mahape, Navi Mumbai, India, helping millions, million hits on google on all organic chemistry websites, Hands on experience in developing novel routes for drug molecules and implementation on commercial scale. several international patents published.pushing boundaries, one lakh connections on all networking sites
