
Home » Posts tagged 'NEW DRUGS CHINA'
Tag Archives: NEW DRUGS CHINA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
DRUG DISCOVERY PRESENTATION BY DR ANTHONY CRASTO

Please wait and allow slideshare to load…………..
Merck KGaA has entered a collaboration with China-based biotech BeiGene to research a new treatment for cancer.

Merck KGaA has entered a collaboration with China-based biotech BeiGene to research a new treatment for cancer.
The compound, currently known as BeiGene-283, BGB-283 , is a second-generation BRAF inhibitor and is expected to enter clinical development in 2014.
It is designed to work by hindering the action of the BRAF protein, which is thought to play a part in the promotion of cancer cell growth and has been found to be mutated in some cancer patients.
read all at
http://www.pmlive.com/pharma_news/merck_kgaa_to_research_cancer_drug_with_chinese_biotech_480521


TLC388 (Lipotecan®) Taiwan Liposome Company Hepatic cancer drug candidate gets fast track approval status from SFDA
TLC388 (Lipotecan®) structure can be figured out from a link below of a poster
http://www.tlcbio.com/files/news/2011111701580783.pdf
IT IS A CAMPOTHECIN ANALOGUE
The str can be concluded from above picture from a poster by TLC BIO
TLC388 (Lipotecan) is a potent Topoisomerase-1 inhibitor and it can disrupt both Sonic Hedgehog and HIF1-α pathways to overcome cancer drug resistance and inhibit angiogenesis induced by tumor hypoxia. This phase I first-in-human study of Lipotecan examined the MTD, safety, anti-tumor activity and pharmacokinetic profiles of TLC388 in patients with advanced incurable solid tumors.
Methods: Lipotecan was administered intravenously on day 1, 8 and 15 of a 28-day cycle. Patients underwent safety assessments regularly and tumor assessments every other cycle. Pharmacokinetic samples were drawn on days 1, 8 and 15 of cycles 1 and 2 for all treated patients.
http://mct.aacrjournals.org/cgi/content/meeting_abstract/10/11_MeetingAbstracts/A89
http://clinicaltrials.gov/show/NCT00747474
MAR19 2013
China SFDA has granted fast track approval status to Taiwan Liposome company hepatic cancer drug Lipotecan, shortening the review period. The drug will enter Phase 2 clinical trials in China in the second half of this year. Lipotecan has been granted orphan drug status by US FDA and EU EMEA for the treatment of hepatocellular carcinoma (HCC)
Nexavar is the standard of care in first line advanced liver cancer patients. Lipotecan as a second-line treatment allows patients who have failed prior treatment with Nexavar to maintain a six month course of the disease without progressing
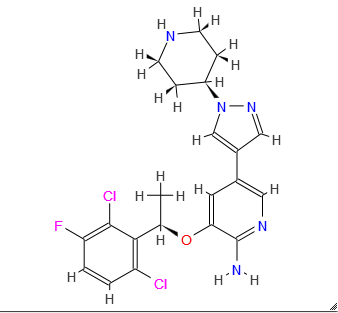
Pfizer Gains China Approval of Kinase-Specific Lung Cancer Drug, Xalkori (crizotinib)

Xalkori, crizotinib,
(PF-02341066)
3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine
Crizotinib; 877399-52-5; Xalkori; PF-2341066; PF-02341066; (R)-crizotinib; 877399-52-5
| Molecular Formula: | C21H22Cl2FN5O |
|---|---|
| Molecular Weight: | 450.336683 g/mol |
Crizotinib an inhibitor of receptor tyrosine kinase for the treatment of non-small cell lung cancer (NSCLC). Verification of the presence of ALK fusion gene is done by Abbott Molecular’s Vysis ALK Break Apart FISH Probe Kit. This verification is used to select for patients suitable for treatment. FDA approved in August 26, 2011.
Crizotinib (1), an anaplastic lymphoma kinase (ALK) receptor tyrosine kinase inhibitor approved by the U.S. Food and Drug Administration in 2011, is efficacious in ALK and ROS positive patients
Feb 25, 2013
Pfizer has been granted China approval for Xalkori (crizotinib), an innovative treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK) positive. The ALK-positive variation, which comprises between 3% and 5% of all NSCLC tumors, must be proved by a biomarker test. Pfizer said China’s approval came just eleven months after it submitted a new drug application to the SFDA for Xalkori
Crizotinib (trade name Xalkori,[1] Pfizer), is an anti-cancer drug acting as an ALK (anaplastic lymphoma kinase) and ROS1 (c-ros oncogene 1) inhibitor, approved for treatment of some non-small cell lung carcinoma (NSCLC) in the US and some other countries, and undergoing clinical trials testing its safety and efficacy in anaplastic large cell lymphoma, neuroblastoma, and other advanced solid tumors in both adults and children.[2]
- FDA approves Xalkori with companion diagnostic for a type of late-stage lung cancer. U.S. Food and Drug Administration.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm269856.htm
- ClinicalTrials.gov NCT00932451 An Investigational Drug, PF-02341066, Is Being Studied In Patients With Advanced Non-Small Cell Lung Cancer With A Specific Gene Profile Involving The Anaplastic Lymphoma Kinase (ALK) Gene
Crizotinib the core structure is a substituted pyridine, the 3 – position of the ether as a chiral center adjacent, so with Mitsunobu reaction to complete, as is a typical Mitsunobu SN2 reaction, the reaction chiral center occurs in reverse, so easy to control, no racemization occurs. Pyridine substituted at position 5 by Suzuki reaction constructed.
Compound 1 The activation of the hydroxyl groups of methanesulfonyl chloride, and then with a 4 – iodopyrazole reaction 2 , 2 to 4 Suzuki reaction conversion can be used, but will generate a large quantity of the reaction product of their coupling, the first 2 converted to a Grignard reagent, and then with a boronic acid ester of 3 reaction 4 .

……………………
http://www.specchemonline.com/articles/view/biocatalyst-breakthroughs#.VTcW9yxabEs
http://www.google.com/patents/WO2014020467A2?cl=en
(R)-3-[l-(2,6-Dichloro-3-fluoro-phenyl)-ethoxy]-5-(l-piperidin-4-yl-lH-py- razol-4-yl)-pyridin-2-ylamine, also known as Crizotinib, is represented by the Formula (I):

Formula (I)
Crizotinib is a potent small-molecule inhibitor of c-Met/HGFR (hepatocyte growth factor receptor) kinase and ALK (anaplastic lymphoma kinase) activity. Enantiomerically pure compound of formula I was first disclosed in US Patent No. 7,858,643. Additionally, the racemate of compound of formula I was disclosed in U.S. patent application 2006/0128724, both of these references discloses similar methods for the synthesis of Compound of Formula I.
Conventionally, the compounds of formula I are prepared by reacting Bis(pinacolato)diboron with protected 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-pyridin-2-ylamine in the presence of Pd catalyst. The obtained product after deprotection is reacted with N- protected 4-(4-bromo-pyrazol-l-yl)-piperidine in the presence of Pd Catalyst. The obtained product is filtered through celite pad and purified by Column Chromatography. The final product of formula I was obtained by deprotection of the purified compound by using HCl/dioxane. US Patent No. 7,858,643 provides enantiomerically pure aminoheteroaryl compounds, particularly aminopyridines and aminopyrazines, having protein tyrosine kinase activity. More particularly, US 7,858,643 describes process for the preparation of 3-[(lR)-l-(2,6- dichloro-3-fluorophenyl)ethoxy]-5-(l-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine. The Scheme is summarized below in Scheme- 1 :


Scheme-1
wherein, “Boc” means tert-butoxycarbonyl; and a) (Boc)2, DMF, Dimethylaminopyridine b) Pd(dppf)Cl2, KOAc, Dichloromethane; c) HC1, Dioxane, Dichloromethane; d) Pd(PPh3)2Cl2, Na2C03, DME/H20; e) 4M HCl/Dioxane, Dichloromethane
A similar process has been disclosed in the U.S. patent application 2006/0128724 for the preparation of Crizotinib. J. Jean Cui et. al. in J. Med. Chem. 2011, 54, 6342-6363, also provides a similar process for the preparation of Crizotinib and its derivatives.
However, above mentioned synthetic process requires stringent operational conditions such as filtration at several steps through celite pad. Also column chromatography is required at various steps which is not only tedious but also results in significant yield loss. Another disadvantage of above process involves extensive use of palladium catalysts, hence metal scavengers are required to remove palladium content from the desired product at various steps which makes this process inefficient for commercial scale.
Yet another disadvantage of above process is the cost of Bis(pinacolato)diboron. This reagent is used in excess in the reaction mixture resulting in considerable cost, especially during large-scale syntheses.
US Patent No. 7,825,137 also discloses a process for the preparation of Crizotinib where Boc protected 4-(4-iodo-pyrazol-l-yl)-piperidine is first reacted with Bis(pinacolato)diboron in the presence of Pd catalyst. The reaction mixture is filtered through a bed of celite and the obtained filtrate is concentrated and purified by silica gel chromatography to give to form tert-butyl-4-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl]piperidine-l- carboxylate. To this compound, 5-bromo-3-[l-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]- pyridin-2-ylamine is added in the presence of a Pd catalyst. The reaction mixture is stirred for 16h at 87°C. The reaction mixture is filtered through celite pad and the concentrated filtrate is purified on silica gel column to obtain (4-{6-amino-5-[(R)-l-(2,6-dichloro-3-fluoro- phenyl)-ethoxy]-pyri- din-3-yl}-pyrazol-l-yl)-piperidine-l-carboxylic acid tert-butyl ester of 95% purity. To the solution of resulting compound in dichloromethane 4N HCl/Dioxane is added and thereby getting the reaction suspension is filtered in Buchner funnel lined with filter paper. The obtained solid is dissolved in HPLC water and pH is adjusted to 10 with the addition of Na2C03 Compound is extracted using dichloroform and is purified on a silica gel column by eluting with CH2Cl2 MeOH/NEt3 system to obtain Crizotinib. The scheme is summarized below in scheme 2:

Formula (i) Formula (ii)

Formula (iii) Formula (ii) ula (iv)

Formula (v) Formula (I)
Scheme-2
Preparation of Crizotinib:
To a stirred solution of Tert-butyl 4-(4-{ 6-amino-5-[(li?)-l-(2,6-dichloro-3- fluorophenyl)ethoxy]pyridin-3 -yl } – lH-pyrazol- 1 -yl)piperidine- 1 -carboxylate (material obtained in Example 3) (l.Og, 0.00181 moles) in dichloromethane (-13 ml) at 0°C was added 4.0 M dioxane HQ (6.7 ml, 0.0272 moles). Reaction mixture was stirred at room temperature for 4h. After the completion of reaction monitored by TLC, solid was filtered and washed with dichloromethane (10 ml). The obtained solid was dissolved in water (20 ml); aqueous layer was extracted with dichloromethane (10×2). The pH of aqueous layer was adjusted to 9-10 with Na2C03 and compound was extracted with dichloromethane (10 x 3), combined organic layers were washed with water (20 ml), evaporated under vacuum to get solid product. The solid was stirred with ether (10 ml), filtered off, washed well with ether, dried under vacuum to get Crizotinib.
Yield: 0.45g (55 %)
HPLC Purity: 99.35 %
1HNMR (400 MHz, CDC13) δ: 7.76 (d, J = 1.6 Hz, 1H), 7.56 (s, 1H), 7.49 (s, 1H), 7.30 (dd, J = 9.2 Hz), 7.0 (m, 1H), 6.86 (d, J = 1.6 Hz, 1H), 6.09 ( q, J= 6.8 Hz, 1H), 4.75 (brs, 1H), 4.19 (m, 1H), 3.25 (m, 2H), 2.76 (m, 2H), 2.16 (m, 2H), 1.92 (m, 2H), 1.85 (d, J= 6.8 Hz, 3H), 1.67 (brs, 1H)
…………………………
http://www.sciencedirect.com/science/article/pii/S0040403914000872
Abstract
A novel approach for the synthesis of Crizotinib (1) is described. In addition, new efficient procedures have been developed for the preparation of (S)-1-(2,6-dichloro-3-fluorophenyl)ethanol (2) and tert-butyl 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazol-1-yl)piperidine-1-carboxylate (4), the key intermediates required for the synthesis of Crizotinib.
Graphical abstract

- …………………
- http://www.sciencedirect.com/science/article/pii/S0040403911021745
-
Abstract
4-(4-Iodo-1H-pyrazol-1-yl)piperidine is a key intermediate in the synthesis of Crizotinib. We report a robust three-step synthesis that has successfully delivered multi-kilogram quantities of the key intermediate. The process includes nucleophilic aromatic substitution of 4-chloropyridine with pyrazole, followed by hydrogenation of the pyridine moiety and subsequent iodination of the pyrazole which all required optimization to ensure successful scale-up.
Graphical abstract

……………………

A robust six-step process for the synthesis of crizotinib, a novel c-Met/ALK inhibitor currently in phase III clinical trials, has been developed and used to deliver over 100 kg of API. The process includes a Mitsunobu reaction, a chemoselective reduction of an arylnitro group, and a Suzuki coupling, all of which required optimization to ensure successful scale-up. Conducting the Mitsunobu reaction in toluene and then crystallizing the product from ethanol efficiently purged the reaction byproduct. A chemoselective arylnitro reduction and subsequent bromination reaction afforded the key intermediate 6. A highly selective Suzuki reaction between 6 and pinacol boronate 8, followed by Boc deprotection, completed the synthesis of crizotinib 1.
3-[(1R)-1-(2,6-Dichloro-3-fluorophenyl)ethoxy]-5-[1-(piperidin-4-yl)-1H-pyrazol-4-yl]pyridin-2-amine 1
crizotinib1 (20.7 kg, 80%) as a white solid.
Mp 192 °C;
1H NMR (400 MHz, CDCl3) δ: 7.78 (d, J = 1.8 Hz, 1H), 7.58 (s, 1H), 7.52 (s, 1H), 7.31 (dd, J = 9.0, 4.9 Hz, 1H), 7.06 (m, 1H), 6.89 (d, J = 1.7 Hz, 1H), 6.09 (q, 1H), 4.79 (br s, 2H), 4.21 (m, 1H), 3.26 (m, 2H), 2.78 (m, 2H), 2.17 (m, 2H), 1.90 (m, 2H), 1.87 (d, J = 6.7 Hz, 3H), 1.63 (br s, 1H).
13C NMR (100.6 MHz, CDCl3) δ: 157.5 (d, J = 250.7 Hz), 148.9, 139.8, 137.0, 135.7, 135.6, 129.9, 129.0 (d, J = 3.7 Hz), 122.4, 122.1 (d, J = 19.0 Hz), 119.9, 119.3, 116.7 (d, J = 23.3 Hz), 115.0, 72.4, 59.9, 45.7, 34.0, 18.9.
LC-MS: found m/z 450.0, 451.0, 452.0, 453.0, 454.0, 455.0.
Anal. Calcd for C21H22Cl2FN5O: C, 56.01; H, 4.92; N, 15.55. Found: C, 56.08; H, 4.94; N, 15.80.
Cui, J. J.; Botrous, I.; Shen, H.; Tran-Dube, M. B.; Nambu, M. D.; Kung, P.-P.; Funk, L. A.; Jia, L.; Meng, J. J.; Pairish, M. A.; McTigue, M.; Grodsky, N.; Ryan, K.; Alton, G.; Yamazaki, S.; Zou, H.; Christensen, J. G.; Mroczkowski, B.Abstracts of Papers; 235th ACS National Meeting, New Orleans, LA, United States, April 6–10, 2008.


![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-1h.png)
![3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine NMR spectra analysis, Chemical CAS NO. 877399-52-5 NMR spectral analysis, 3-[(1R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy]-5-(1-piperidin-4-ylpyrazol-4-yl)pyridin-2-amine C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-07-29/000/437/336/877399-52-5-13c.png)
| WO2006021881A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors |
| WO2006021884A2 * | 15 Aug 2005 | 2 Mar 2006 | Pfizer | Enantiomerically pure aminoheteroaryl compounds as protein kinase inhibitors |
| WO2013181251A1 * | 29 May 2013 | 5 Dec 2013 | Ratiopharm Gmbh | Crizotinib hydrochloride salt in crystalline |
| EP2620140A1 * | 26 Jan 2012 | 31 Jul 2013 | ratiopharm GmbH | Crizotinib containing compositions |
-
WO2010048131A1 * Oct 20, 2009 Apr 29, 2010 Vertex Pharmaceuticals Incorporated C-met protein kinase inhibitors WO2011042389A2 * Oct 4, 2010 Apr 14, 2011 Bayer Cropscience Ag Phenylpyri(mi)dinylazoles US7825137 Nov 23, 2006 Nov 2, 2010 Pfizer Inc. Method of treating abnormal cell growth US7858643 Aug 26, 2005 Dec 28, 2010 Agouron Pharmaceuticals, Inc. Crizotinib, a c-Met protein kinase inhibitor anticancer agent; 3-[(R)-1-(2,6-dichloro-3-fluoro-phenyl)-ethoxy]-5-(1-piperidin-4-yl-1H-pyrazol-4-yl)-pyridin-2-ylamine is crizotinib US20060128724 Aug 26, 2005 Jun 15, 2006 Agouron Pharmaceuticals, Inc. Pyrazole-substituted aminoheteroaryl compounds as protein kinase inhibitors 1 J. JEAN CUI J. MED. CHEM. vol. 54, 2011, pages 6342 – 6363 2 ORG. PROCESS RES. DEV. vol. 15, 2011, pages 1018 – 1026 3 * PIETER D. DE KONING ET AL: “Fit-for-Purpose Development of the Enabling Route to Crizotinib (PF-02341066)“, ORGANIC PROCESS RESEARCH & DEVELOPMENT, vol. 15, no. 5, 16 September 2011 (2011-09-16), pages 1018-1026, XP055078841, ISSN: 1083-6160, DOI: 10.1021/op200131n
Celgene gains SFDA China, approval for marketing Revlimid (lenalidomide) to treat multiple myeloma

(RS)-3-(4-amino-1-oxo 1,3-dihydro-2H-isoindol- 2-yl)piperidine-2,6-dione
Lenalidomide
REVLIMID® is an oral immunomodulatory drug marketed in the United States and many international markets, in combination with dexamethasone, for treatment of patients with multiple myeloma who have received at least one prior therapy. It is also marketed in the United States and certain international markets for the treatment of transfusion-dependent anemia due to low- or intermediate-1-risk myelodysplastic syndromes, or MDS, associated with a deletion 5q cytogenetic abnormality with or without additional cytogenetic abnormalitie.Revlimid Worldwide annual sales in 2011 was $3.2bLenalidomide (Revlimid) is a derivative of thalidomideintroduced in 2004.It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Lenalidomide has significantly improved overall survival in myeloma (which generally carries a poor prognosis), although toxicity remains an issue for users. [1]It costs $163,381 per year for the average patient.[2]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past 10 years. There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[3] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of the microenvironment support for tumor cells, and immunomodulatory role. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
- McCarthy; Philip L. McCarthy, Kouros Owzar, Craig C. Hofmeister, et al. (May 10, 2012). “Lenalidomide after Stem-Cell Transplantation for Multiple Myeloma”. N Engl J Med 366 (19): 1770–1781. doi:10.1056/NEJMoa1114083. PMID 22571201.
- Badros, Ashraf Z. Badros (May 10, 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- Vallet S, Palumbo A, Raje N, Boccadoro M, Anderson KC (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
