

| Patent ID
|
Title
|
Submitted Date
|
Granted Date
|
|---|---|---|---|
| US9790208 | CRYSTALLINE SALT FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1-YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AS OREXIN RECEPTOR ANTAGONIST |
2014-12-02
|
|
| US2016368901 | CRYSTALLINE FORM OF (S)-(2-(6-CHLORO-7-METHYL-1H-BENZO[D]IMIDAZOL-2-YL)-2-METHYLPYRROLIDIN-1 -YL)(5-METHOXY-2-(2H-1, 2, 3-TRIAZOL-2-YL)PHENYL)METHANONE AND ITS USE AS OREXIN RECEPTOR ANTAGONISTS |
2014-12-02
|
Home » Posts tagged 'Nemorexant'
Tag Archives: Nemorexant
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nemorexant
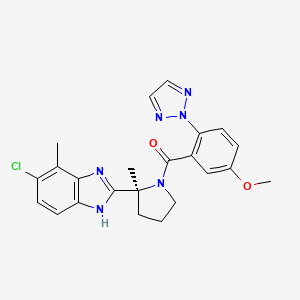
Nemorexant
ACT-541468, UNII LMQ24G57E9
[(2S)-2-(5-Chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]methanone
1505484-82-1 [RN]
LMQ24G57E9
Methanone, [(2S)-2-(5-chloro-4-methyl-1H-benzimidazol-2-yl)-2-methyl-1-pyrrolidinyl][5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl]-
- Originator Actelion Pharmaceuticals
- Developer Idorsia Pharmaceuticals
- Class Sleep disorder therapies
- Mechanism of Action Orexin receptor type 1 antagonists; Orexin receptor type 2 antagonists
- Phase III Insomnia
- 19 Oct 2018 Idorsia Pharmaceuticals plans a phase I trial for Liver disorders (Hepatic impairment) in November 2018 (PO) (NCT03713242)
- 09 Oct 2018 Idorsia Pharmaceuticals completes a phase I trial in Insomnia (In volunteers) in Netherlands (PO) (NCT03609775)
- 27 Sep 2018 Idorsia Pharmaceuticals plans a phase I trial for Hepatic impairment in November 2018 , (NCT03686995)
Nemorexant (developmental code name ACT-541468) is a dual orexin receptor antagonist (DORA) which was originated by Actelion Pharmaceuticals and is under development by Idorsia Pharmaceuticals for the treatment of insomnia.[1][2] It acts as a selective dual antagonist of the orexin receptors OX1 and OX2.[1][2] As of June 2018, nemorexant is in phase III clinical trials for the treatment of insomnia.[1]
Idorsia is developing nemorexant, a dual orexin receptor antagonist (DORA), for the oral treatment of insomnia and investigating the program for the treatment of COPD. In May 2018, a phase III study was initiated in subjects with insomnia disorder and in September 2018, a phase I trial was initiated in COPD.
SCHEME
SEE AT END OF PAGE
PATENT
WO2013182972 ,
PATENT
WO2015083094 ,
Patent
WO 2015083070

Synthesis of nemorexant, using 2-methyl-L-proline hydrochloride as the starting material
N-Protection of 2-methyl-L-proline hydrochloride with Boc2O gives N-Boc-2-methyl-L-proline,
Which upon condensation with 4-chloro-3-methylbenzene-1,2-diamine using HATU and DIEA in CH2Cl2 affords the corresponding amide.
Cyclization of diamine in the presence of AcOH at 100 °C provides imidazole derivative,
Whose Boc moiety is removed by means of HCl in dioxane to yield 5-chloro-4-methyl-2-[2(S)-methylpyrrolidin-2-yl]benzimidazole hydrochloride.
N-Acylation of pyrrolidine derivative with 5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid using HATU and DIEA in CH2Cl2 produces Nemorexant
5-methoxy-2-(1,2,3-triazol-2-yl)benzoic acid (prepared by the coupling of 2-iodo-5-methoxybenzoic acid with 1,2,3-triazole using CuI and Cs2CO3 in DMF)
PATENT
WO 2016020403
PATENT
WO 2015083071
Reference Example 1
1) Synthesis of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid
2-lodo-5-methoxy benzoic acid (15.0 g; 53.9 mmol) is dissolved in anhydrous DMF (45 ml) followed by the addition of 1 H-1 ,2,3-triazole (7.452 g; 108 mmol) and cesium carbonate (35.155 g; 108 mmol). By the addition of cesium carbonate the temperature of the reaction mixture increases to 40°C and gas evolved from the reaction mixture. Copper(l)iodide (514 mg; 2.7 mmol) is added. This triggers a strongly exothermic reaction and the temperature of the reaction mixture reaches 70°C within a few seconds. Stirring is continued for 30 minutes. Then the DMF is evaporated under reduced pressure followed by the addition of water (170 ml) and EtOAc (90 ml). The mixture is vigorously stirred and by the addition of citric acid monohydrate the pH is adjusted to 3-4. The precipitate is filtered off and washed with water and EtOAc and discarded. The filtrate is poured into a separation funnel and the phases are separated. The water phase is extracted again with EtOAc. The combined organic layers are dried over MgS04, filtered and the solvent is evaporated to give 7.1 g of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid as a white powder of 94% purity (6 % impurity is the regioisomerically N1-linked triazolo-derivative); tR [min] = 0.60; [M+H]+ = 220.21
2) Synthesis of (S)-1 -(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid
2-Methyl-L-proline hydrochloride (99.7 g; 602 mmol) is dissolved in a 1/1-mixture of MeCN and water (800 ml) and triethylamine (254 ml; 1810 mmol) is added. The temperature of the reaction mixture slightly rises. The reaction mixture is cooled to 10°C to 15°C followed by careful addition of a solution of Boc20 (145 g; 662 mmol) in MeCN (200 ml) over 10 minutes.
Stirring at RT is continued for 2 hours. The MeCN is evaporated under reduced pressure and aq. NaOH solution (2M; 250 ml) is added to the residual aq. part of the reaction mixture. The water layer is washed with Et20 (2x 300 ml) then cooled to 0°C followed by slow and careful addition of aq. HCI (25%) to adjust the pH to 2. During this procedure a suspension forms.
The precipitate is filtered off and dried at HV to give 1 10.9 g of the title compound as a beige powder; tR [min] = 0.68; [M+H]+ = 230.14
3) Synthesis of (S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-
(S)-1-(tert-butoxycarbonyl)-2-methylpyrrolidine-2-carboxylic acid (60 g; 262 mmol) and HATU (100 g; 264 mmol) is suspended in DCM (600 ml) followed by the addition of DIPEA (84.6 g; 654 mmol) and 6-chloro-2,3-diaminotoluene (41 g; 262 mmol). The reaction mixture is stirred at rt for 14 hours then concentrated under reduced pressure and to the residue is added water followed by the extraction of the product with EtOAc (3x). The combined organic layers are washed with brine, dried over MgS04, filtered and the solvent is evaporated under
reduced pressure to give 185 g of the title compound as a dark brownish oil, which is used in the next step without further purification; tR [min] = 0.89; [M+H]+ = 368.01
4) Synthesis of (S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1 -carboxylate
(S)-tert-butyl 2-((2-amino-4-chloro-3-methylphenyl)carbamoyl)-2-methylpyrrolidine-1-carboxylate (185 g; 427 mmol) are dissolved in AcOH (100%; 611 ml), heated to 100°C and stirring continued for 90 minutes. The AcOH is evaporated under reduced pressure and the residue is dissolved in DCM followed by careful addition of saturated sodium bicarbonate solution. The phases are separated, the aq. phase is extracted once more with DCM, the combined aq. phases are dried over MgS04, filtered and the solvent is evaporated under reduced pressure to give 142.92 g of the title compound as a dark brown oil which is used in the next step without further purification; tR [min] = 0.69; [M+H]+ = 350.04
5) Synthesis of (S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride
(S)-tert-butyl 2-(5-chloro-4-methyl-1 H-benzo[d]imidazol-2-yl)-2-methylpyrrolidine-1-carboxylate (355.53 g; 1.02 mol) are dissolved in dioxane (750 ml) followed by careful addition of HCI solution in dioxane (4M; 750 ml; 3.05 mol). The reaction mixture is stirred for 3 hours followed by the addition of Et20 (800 ml) which triggered precipitation of the product. The solid is filtered off and dried at high vacuum to give 298.84 g of the title compound as a redish powder; tR [min] = 0.59; [M+H]+ = 250.23
6) Synthesis of [(S)-2-(5-chloro-4-methyl-1 H-benzoimidazol-2-yl)-2-methyl-pyrrolidin-1- -(5-methoxy-2-[1,2,3]triazol-2-yl-phenyl)-methanone
(S)-5-chloro-4-methyl-2-(2-methylpyrrolidin-2-yl)-1 H-benzo[d]imidazole hydrochloride (62.8 g; 121 mmol) is dissolved in DCM (750 ml) followed by the addition of 5-methoxy-2-(2H-1 ,2,3-triazol-2-yl)benzoic acid (62.8 g; 121 mmol) and DIPEA (103 ml; 603 mmol). Stirring is continued for 10 minutes followed by the addition of HATU (47 g; 124 mmol). The reaction mixture is stirred for 16 hours at RT. The solvents are evaporated under reduced pressure and the residue is dissolved in EtOAc (1000 ml) and washed with water (3x 750 ml). The organic phase is dried over MgS04, filtered and the solvent is evaporated under reduced pressure. The residue is purified by CC with EtOAc / hexane = 2 / 1to give 36.68 g of the title compound as an amorphous white powder. tR [min] = 0.73; [M+H]+ = 450.96
Table 1 : Characterisation data for COMPOUND as free base in amorphous form
II. Preparation of crystalline forms of COMPOUND
Example 1 :
Preparation of seeding material of COMPOUND hydrochloride in crystalline Form 1
10 mg COMPOUND is mixed with 0.2 mL 0.1 M aq. HCI and 0.8 mL EtOH. The solvent is fully evaporated and 0.05 mL isopropanol is added. Alternatively 0.05 mL methyl-isobutylketone can be added. The sample is stored closed at room temperature for 4 days and crystalline material of COMPOUND hydrochloride in crystalline Form 1 is obtained. This material can be used as seeding material for further crystallization of COMPOUND hydrochloride in crystalline Form 1.
Example 2: Preparation and characterization of COMPOUND hydrochloride in crystalline form 1
5g COMPOUND is mixed with 0.9 mL 1 M aq. HCI and 20 mL EtOH. The solvent is evaporated and 25 mL isopropanol is added. Seeds of COMPOUND hydrochloride are added and the sample is allowed to stand at room temperature. After about 2 days the suspension is filtered and the solid residue is dried at reduced pressure (2 mbar for 1 hour) and allowed to equilibrate open for 2 hours at 24°C/46% relative humidity. The obtained solid is COMPOUND hydrochloride in crystalline Form 1
Table 2: Characterisation data for COMPOUND hydrochloride in crystalline form 1
PATENT
WO-2018202689
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018202689&tab=PCTDESCRIPTION&maxRec=1000
Process for the preparation of a crystalline potassium salt of a 2-(2H-[1,2,3]triazol-2-yl)-benzoic acid derivatives is claimed. Compound is disclosed to be useful for the preparation of pharmaceuticals, especially certain orexin receptor antagonists such as nemorexant .
References
- ^ Jump up to:a b c https://adisinsight.springer.com/drugs/800044843
- ^ Jump up to:a b Equihua-Benítez AC, Guzmán-Vásquez K, Drucker-Colín R (July 2017). “Understanding sleep-wake mechanisms and drug discovery”. Expert Opin Drug Discov. 12 (7): 643–657. doi:10.1080/17460441.2017.1329818. PMID 28511597.
- Muehlan, C.; Heuberger, J.; Juif, P.E.; Croft, M.; van Gerven, J.; Dingemanse, J.
Accelerated development of the dual orexin receptor antagonist ACT-541468: Integration of a microtracer in a first-in-human study
Clin Pharmacol Ther 2018, 104(5): 1022 - A Study to Evaluate the Pharmacokinetics of ACT-541468 in Subjects With Mild, Moderate and Severe Hepatic Impairment (NCT03713242)
ClinicalTrials.gov Web Site 2018, October 24 - Boof, M.-.L.; Ufer, M.; Halabi, A.; Dingemanse, J.
Impact of the dual orexin receptor antagonist ACT-541468 on the pharmacokinetics of the CYP3A4 probe drug midazolam and assessment of the effect of food on ACT-541468
119th Annu Meet Am Soc Clin Pharmacol Ther (ASCPT) (March 21-24, Orlando) 2018, Abst PI-043 - Muehlan, C.; Brooks, S.; Zuiker, R.; van Gerven, J.; Dingemanse, J.
Night-time administration of ACT-541468, a novel dual orexin receptor antagonist: Characterization of its pharmacokinetics, next-day residual effects, safety, and tolerability
32nd Annu Meet Assoc Sleep Soc (SLEEP) (June 2-6, Baltimore) 2018, Abst 0008 - Proposed international nonproprietary names (Prop. INN): List 118
WHO Drug Inf 2017, 31(4): 635
External links
from PubChem
 |
|
| Clinical data | |
|---|---|
| Synonyms | ACT-541468 |
| Routes of administration |
By mouth |
| Drug class | Orexin antagonist |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C23H23ClN6O2 |
| Molar mass | 450.927 g/mol |
| 3D model (JSmol) | |
///////////////Nemorexant, ACT-541468, Phase III, Insomnia

