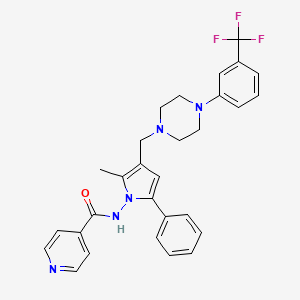

LL 3858, SUDOTERB
UNII-SK2537665A;
CAS 676266-31-2;
N-[2-methyl-5-phenyl-3-[[4-[3-(trifluoromethyl)phenyl]piperazin-1-yl]methyl]pyrrol-1-yl]pyridine-4-carboxamide;
N-[2-Methyl-5-phenyl-3-[[4-[3-(trifluoromethyl)phenyl]-1-piperazinyl]methyl]-1H-pyrrol-1-yl]-4-pyridinecarboxamide
Sudoterb(TM)
| Molecular Formula: |
C29H28F3N5O |
| Molecular Weight: |
519.572 g/mol |
- Originator Lupin
- Class Antituberculars; Isonicotinic acids; Pyrroles
- Mechanism of Action Undefined mechanism
- Orphan Drug Status No
- New Molecular Entity Yes
Highest Development Phases
- No development reported Tuberculosis
Most Recent Events
- 23 Jul 2015 No recent reports on development identified – Phase-II for Tuberculosis in India (unspecified route)
- 11 Dec 2013 Lupin completes a phase II trial in Tuberculosis in India prior to December 2013 (CTRI2009-091-000741)
- 31 Jul 2010 Lupin completes enrolment in its phase II trial for Tuberculosis in India (CTRI2009-091-000741)

Sudoterb HCl
CAS: 1044503-04-9 (2HCl)
Chemical Formula: C29H30Cl2F3N5O
Molecular Weight: 592.4882


SYNTHESIS
WO 2006109323

Tuberculosis (TB) is a contagious disease, which usually runs a protracted course, ending in death in majority of the cases, with relapse being a common feature of the disease. It is one of the most important causes of prolonged disability and chronic ill health. It is caused by the tubercle bacillus Mycobacterium tuberculosis, which is comparatively difficult to control. Drugs such as isoniazid, rifampicin, pyrazinamide, ethambutol streptomycin, para- aminosalisylic acid, ethionamide, cycloserine, capreomycin, kanamycin, thioacetazone etc. have been and are being currently used to treat TB. Amongst these, isoniazid, rifampicin, ethambutol and pyrazinamide are the first-line drugs of choice, which are administrated either as a single drug formulation or as a fixed-dose combination of two or more of the aforesaid drugs. Even though, each of the abovementioned first-line drug regimen is highly effective for treatment of TB, however, they are associated with shortcomings, such as unpleasant side- effects and relatively long course of treatment. The later one results in non-compliance of the patient to the treatment leading often to failure of the treatment and most importantly, development of drug resistance. The development of drug resistance has long constituted a principal difficulty in treating human tuberculosis. The second-line drugs, on the other hand are less effective, more expensive and more toxic.
It is estimated that in the next twenty years over one billion people would be newly infected with TB, with 35 million people succumbing to the disease (WHO Fact Sheet No. 104, Global
Alliance for TB Drug Development- Executive Summary of the Scientific Blueprint for TB
Development : http://www.who.int/inf-fs/en/factl04.hfaiil). With the emergence of HIV related
TB, the disease is assuming alarming proportions as one of the killer diseases in the world today.
A major thrust in research on antimycobacterials in the last decade has witnessed the development of new compounds for treatment of the disease, a) differing widely in structures, b) having different mode/mechanism of action, c) possessing favourable pharmacokinetic properties, d) which are safe and having low incidence of side-effects, and e) which provide a cost-effective dosage regimen.
Several new class of compounds have been synthesized and tested for activity against Mycobacterium tuberculosis, the details of chemistry and biology of which could be found in a recent review by B. N. Roy et. al. in J. Ind. Chem. Soc, April 2002, 79, 320-335 and the references cited therein.
Substituted pyrrole derivatives constitute another class of compounds, which hold promise as antimycobacterial agents. The pyrrole derivatives which have been synthesized and tested for antitubercular as well as non-tubercular activity has been disclosed by : a) D. Deidda et. al. in Antimicrob. Agents and Chemother., Nov 1998, 3035-3037. This article describes the inhibitory activity shown by one pyrrole compound, viz. BM 212 having the structure shown below, against both Mycobacterium tuberculosis including drug-resistant mycobacteria and some non-tuberculosis mycobacteria.
The MIC value (μg/ml) against the M. tuberculosis strain 103471 exhibited by BM 212 was 0.70 as against 0.25 found for isoniazid.
b) M. Biava et. al. in J. Med. Chem. Res., 1999, 19-34 have reported the synthesis of several analogues of BM 212, having the general formula (The compounds disclosed by M. Biava et. al. inJ. Med. Chem. Res., 1999, 19-34.) shown hereunder
wherein,
X is H, . CH2— (Oy-Cl ; CH2-(CH2)4-CH3
Z is H ; Y
and the in vitro antimicrobial activity of the compounds against Candida albicans, Candida sp, Cryptococcus neoforma s, Gram- positive or Gram-negative bacteria, isolates of pathogenic plant fungi, Herpes simplex virus, both HSV1 and HSN2, M. tuberculosis, M. smegmαtis, M. mαrinum and M. αvium.
However, the MIC values (μg/ml) of these compounds against the M. tuberculosis strain 103471 are found to be inferior to BM 212 and are in the range of 4-16.
M. Biava et. al. in Bioorg. & Med. Chem. Lett., 1999, 9, 2983-2988. This article describes the synthesis of pyrrole compounds of formula (: The compounds disclosed by M. Biava et. al. in Bioorg. & Med. Chem. Lett., 1999, 9, 2983-2988) shown hereunder
wherein,
X is H or Cl Y is H or Cl
R is N-methyl piperazinyl or thiomorphinyl
and their respective in vitro activity against M. tuberculosis and non-tuberculosis species of mycobacteria .
However, the MIC values (μg/ml) of these compounds against the M. tuberculosis strain 103471 are found to be inferior to BM 212 and are in the range of 2-4.
d) F. Cerreto et. al. in Eur. J. Med. Chem., 1992, 27, 701-708 have reported the synthesis of certain 3-amino-l,5-diary-2 -methyl pyrrole derivatives and their in vitro anti-fungal activity against Candida albicans and Candida sp. However, there is no report on the activity of such compounds against M. tuberculosis.
e) C. Gillet et. al. in Eur. J. Med. Chem.-Chimica Therapeutica, March- April 1976, ϋ(2), 173-181 report the synthesis of several pyrrole derivatives useful as anti-inflammatory agents and as anti-allergants.
f) R. Ragno et. al., Bioorg. & Med. Chem., 2000, 8, 1423-1432. This article reports the synthesis and biological activity of several pyrrole derivatives as well as describes a structure activity relationship between the said pyrrole compounds and antimycobacterial activity. The compounds (The compounds disclosed by R. Rango et. al., Bioorg. & Med. Chem., 2000, 8, 1423-1432)synthesized and tested by the authors is summarized hereunder
wherein,
X is COOH, COOEt, CONHNH2, CH2OH, CH(OH)C6H5, NO2
Y is H, CH3, OCH3, CH2, SO2, or a group of formula
wherein,
R is H, Cl, C2H5, or OCH3 and R1 is H, Cl, F, CH3, or NO2,
A is H or R
Z is a group of formula,
R2 is H, Cl, OH, or OCH3 and R3 is H or Cl
None of the abovementioned disclosures report or suggest the in vivo efficacy including toxicity of any of the compounds described therein against experimental tuberculosis in animal model. Moreover, the higher MIC values of the compounds reported suggest that they may not be very effective in inhibition of Mycobacterium tuberculosis.
NO PIC
Sudershan Kumar Arora
sudershan arora, Formerly: President R&D, Ranbaxy Lab Limited,
Experience
-

Semi Retired,
Company NameSelf Employed, Dates EmployedAug 2015 – Present
-
Head Business development,
Company NameNIPER, Chandigarh, Dates EmployedJul 2014 – Jun 2015
-
President-R&D,
Company NameRanbaxy Lab Limited, Dates EmployedMay 2008 – Jun 2014,
Employment Duration6 yrs 2 mos
-
President– New Chemical research and Development,
Company NameLupin limited, R&D Center, Dates Employed2006 – 2008,
Employment Duration2 yrs, LocationPune Area, India
-

Global Head-API Research,
Company NameSandoz, Dates Employed2005 – 2006
PATENT
WO 2004026828
https://www.google.com/patents/WO2004026828A1?cl=en
PATENT
US 20050256128
PATENT
https://encrypted.google.com/patents/WO2005107809A2?cl=en
Thus the invention relates to an antimycobacterial combination comprising a therapeutically effective amount of N-(3-[[4-(3-trifluoromethylphenyl)piperazinyl]methyl]-2- methy 1-5 -phenyl- pyrrolyl)-4-pyridylcarboxamide of formula (I) or a pharmaceutically acceptable non- toxic salt thereof
and a therapeutically effective amount of one or more first line antitubercular drugs selected from the group consisting of isoniazid, rifampicin, ethambutol and pyrazinamide. Further according to the invention there is provided a process for preparation of an antimycobacterial pharmaceutical composition comprising combining a compound of formula I or a pharmaceutically acceptable salt thereof
and one or more of the first line antitubercular drugs using a dry granulation method, a wet granulation method or a direct compression method. The present invention further provides an antimycobacterial combination comprising a therapeutically effective amount of N-(3-[[4-(3-trifluoromethylphenyl)piperazinyl]methyl]-2- methyl-5-phenyl-pyrrolyl)-4-pyridylcarboxamide of formula (I) the compound of formula (I) or a pharmaceutically acceptable non-toxic salt thereof
and a therapeutically effective amount of one or more first line antitubercular drugs selected firom isoniazid, rifampicin, ethambutol and pyrazinamide for treatment of multi-drug resistant tuberculosis including latent tuberculosis. The present invention provides an antimycobacterial combination comprising a therapeutically effective amount of N-(3-[[4-(3-trifluoromethylphenyl)piperazinyl]methyl]-2- methyl-5-phenyl-pyrrolyl)-4-pyridylcarboxamide of formula (I) or a pharmaceutically acceptable non-toxic salt thereof
and a therapeutically effective amount of one or more first line antitubercular drugs selected from isoniazid, rifampicin, ethambutol and pyrazinamide for treatment and/or inhibition of one or more mycobacterial conditions/ cells including but not limited to sensitive and multi- drug resistant strains of Mycobacterium tuberculosis, Mycobacterium avium – intracellular complex, M. fortutium, M. kansasaii and other related mycobacterial species.
ynthesis of Compound of Formula (I) The compound of formula (I) and the pharmaceutically acceptable salts thereof can be synthesized by any known method including but not limited to the methods disclosed in our PCT Application No. PCT/IN02/00189 (WO 04/026828 Al), which is incorporated herein by reference. An example of the preparation of N-(3-[[4-(3-trifluoromethylphenyl) piperazinyl]methyl]-2-methyl-5-phenyl-pyrrolyl)-4-pyridylcarboxamide is as follows:
Preparation of N-(3 ~[[4-(3 -trifluoromethylphenyl)piperazinyl]methyl)] -2-methyl-5 – phenylpyrrolyl)-4-pyridylcarboxamide
Step l 1 -(4-chlorophenyl)pentane- 1 ,4-dione To a well stirred suspension of anhydrous aluminium chloride (27.0gm, 205.9mmol) in
126ml. of chlorobenzene was added oxopentanoylchloride (23.0gm, 171.6 mmol) drop-wise, over a period of 30-35 minutes at room temperature (25-30EC). The reaction mixture was stirred at the same temperature for 1 hour. After decomposition of the reaction mixture by the addition of solid ice and hydrochloric acid (10ml) the precipitated solid was filtered and the filtrate evaporated on a rotary evaporator to remove all the solvents. The residue was dissolved in ethyl acetate (400 ml), washed with water (2 x 100ml.), brine (100 ml.) and dried over anhydrous sodium sulfate and the solvent evaporated off. The crude product so obtained was chromatographed over silica gel (100-200 mesh) using chloroform as eluent to give 8.6gm (24.07%) of the title compound.
Step 2 N-(5-methyl-2-phenylpyrrolyl)-4 pyridylcarboxamide
A mixture of 1- (chlorophenyl)pentane-l,4-dione (6.0g, 28.50 mmol, as obtained in Step-1) and isonicotinic hydrazide (4.30gm, 31.35 mmol) in benzene (6.0 ml.) was refluxed by over molecular sieves. After two hours, benzene was removed under reduced pressure and the residue dissolved in ethyl acetate, washed with water (2 x 100 ml.) and brine (1 x 50 ml.). The ethyl acetate layer was dried over anhydrous sodium sulfate and the solvent evaporated off. The crude product so obtained as purified by column chromatography over silica gel (100-200 mesh) using 0.2% methanol in chloroform as eluent to give 3.50gm (39.42%) of the title compound.
Step 3 N-(3 – { [4-(3-trifuoromethylphenyl)piperazinyl]methyl} -2-methyl-5 -phenylpyrrolyl)-4- pyridylcarboxamide
To a stirred solution of N-(5-methyl-2-phenylpyrrolyl)-4-pyridylcarboxamide (0.300gm, 1.083 mmol, as obtained in Step-2) in acetonitrile (5.0 ml.) was added a mixture of l-(3-trifluoromethylphenyl)piperazine hydrochloride (0.288gm, 1.083mmol), 40% formaldehyde (0.032gm, 1.083 mmol) and acetic acid (0.09 ml), drop-wise. After the completion of addition, the reaction mixture was stirred at room temperature for 4 hours. The reaction mixture was neutralized with sodium hydroxide (20% aq. Soln.) and extracted with ethyl acetate (2 x 50 ml.). The combined ethyl acetate extract was washed with water (2 x 25 ml.), brine (1-χ 20 ml.), and dried over anhydrous sodium sulfate and the solvent evaporated off. TLC of the crude product indicated two spots, which were separated by column chromatography over silica gel (100-200mesh). The more polar compound a eluted out using 80% ethyl acetate- hexane mixture was obtained in 24.34 % (0.130 gm) and was identified as N-(3-{[4-(3- trifluoromethylphenyl)piperazinyl]methyl}-2-methyl-5-phenylpyrrolyl)-4- pyridylcarboxamide m.p.80-82°C, MS: m/z 520 (M+l)
1HNMR(CDC13, *): 2:13 (s, 3H,CH3), 2.60 (bs, 4H, 2xN-CH2), 3.18 (bs, 4H, 2xN-CH2), 3.41 (s, 2H, N-CH2), 6.24 (s, lH,H-4), 6.97-7.03 (4H, m, ArH), 7.22-7.29 (m, 5H,AιΗ), 7.53 (d, 2H, J=6Hz, pyridyl ring), 8.50 (bs, 1H,NH D2O exchangeable), 8.70 (d, 2H, J=6Hz, pyridyl ring).
PATENT
WO 2006109323
Compounds of Formula I are known from PCT International Patent Application WO 2004026828, and were screened for antimycobacterial activity, in various in vitro and in vivo models in mice and guinea pigs. Several compounds exhibited strong antimycobacterial activity against sensitive and MDR strains of Mycobacterium tuberculosis in the in vitro and in vivo experiments. Further the compounds of Formula I were also found to be bioavailable, less toxic and safe compared to available anti TB drugs in various animal models.
Thus compounds of Formula I are useful for the effective treatment of Mycobacterium tuberculosis infection caused by sensitive/MDR strains. PCT International Patent Application WO 2004026828 also discloses the synthesis of compounds of Formula I,
wherein,
Ri is phenyl or substituted phenyl
R2 is selected from a group consisting of i) phenyl which is unsubstituted or substituted with 1 or 2 substituents, each independently selected from Cl, F, or, ii) pyridine, or iii) naphthalene, or iv) NHCOR4 wherein R4 is aryl, unsubstituted or substituted heteroaryl, unsubstituted or substituted heterocyclyl. R3 is selected from a group of formula
/~-\ /-Un
— N N-R5 and — N X
wherein R5 is phenyl which is unsubstituted or substituted with 1 or 2 substituents each independently selected from the group consisting of halogen, Ci-C4 alkyl, Ci-C4 alkoxy, nitro, amino, haloalkyl, haloalkoxy etc.; unsubstituted or substituted benzyl; unsubstituted or substituted heteroaryl; unsubstituted or substituted heteroaroyl; unsubstituted or substituted diphenylmethyl,
n = 0-2 and X = -NCH3, CH2, S, SO, or SO2
Such that when R2 is phenyl, which is unsubstituted or substituted with 1 or 2 substituents, each independently selected from Cl, F; R5 is not Ci-C4 alkyl, or X is not -NCH3, CH2, S, SO, or SO2, when n = 1, or X is not -CH2 when n = 0 which comprises reacting the compound of Formula Il
»o-i >-CH, (H)
O O
with thionyl chloride, followed by reaction with RiH (wherein Ri is phenyl or substituted phenyl) in presence of aluminium chloride, and then condensation with R2NH2 (wherein R2 is as described above) in presence of p-toluenesulphonic acid to yield the corresponding unsubstituted pyrrole derivatives of Formula V,
which on further treatment with suitable secondary amines in the presence of formaldehyde and acetic acid afforded the desired pyrrole derivatives of Formula I,
which, on reacting with hydrochloric acid give a hydrochloride salt of compound of Formula Ia. wherein m = 1-2, Ri, R2 and R3 are the same as defined earlier. The above-mentioned methods in the prior art for the synthesis of compound of the Formula I suffer from the limitations,
1. In methods described in PCT International Patent Application WO 2004026828 for the synthesis of compounds of Formula I, positional isomers, the compound of Formula I’, are formed. The necessity of their removal through column chromatography decreases the yield of final pure product.
2. The synthesis of oxopentanoyl chloride (compound of Formula III) for the synthesis of compound of Formula I has been described in J. Org. Chem.
1960, 25, 390-392. It comprises reaction of levulinic acid with thionyl chloride at 50 0C for 1h, which results in poor yield.
3. In method described in PCT International Patent Application WO 2004026828 for the synthesis of 1-aryl-pentane-1,4-dione (compound of Formula IV), impurities are formed and purification involves column chromatography which decreases the yield of the product. 4. The synthesis of the intermediate of Formula V requires the use of benzene and high temperature conditions, which involves the formation of undesired by- products.
5. The above-mentioned methods in prior art for the synthesis of all the intermediates and final compounds of Formula I involves column chromatography for purification, which is cumbersome, tedious and not practicable on an industrial scale.
Example 1: Preparation of /V-(2-methyl-5-phenyl-3-f4-C3-trifluoromethyl-phenyl)- piperazin-1-ylmethyli-pyrrol-i-ylHsonicotinamide hydrochloride
Step (a): Preparation of 4-oxo-pentanoyl chloride
To a stirred mixture of levulinic acid (340.23 g, 2.93 mol) and Λ/./V- dimethylformamide (6.8 mL, catalytic amount) was added thionyl chloride (367.36 g, 3.087 mol, 1.05 equivalent) drop-wise at 20-30 0C in 1.5-2.0 h. After the complete addition of thionyl chloride, the reaction mixture was stirred at same temperature for 0.5 h (completion of reaction or formation of acid chloride was monitored by GC). After the completion of reaction, thionyl chloride was distilled off under reduced pressure at 20-30 0C. Traces of thionyl chloride were removed by adding benzene (136 mL) under reduced pressure at 30-35 0C and residue was dried at reduced pressure (1-2 mm) at 20-30 0C for 30-60 min to yield 370 g (93.8%) of 4-oxo-pentanoyl chloride as light orange oil. Step (b): Preparation of 1-phenyl-pentane-1,4-dione
(B) (A)
To a stirred suspension of benzene (3700 mL, 10 T w/v of acid chloride) and anhydrous aluminium chloride (440.02 g, 3.30 mol, 1.20 equivalent) was added A- oxo-pentanoyl chloride (370 g, 2.75 mol) drop-wise; the rate of addition was regulated so that the addition required 1.5-2 h and the temperature of the reaction mixture was kept at 25-35 0C. The reaction was completed in 2 h and monitored by GC. After completion of reaction, the reaction mixture was added slowly into cold (5-10 0C) 5% HCI (3700 mL) solution maintaining the temperature below 30 0C. The layers were separated; aqueous layer was extracted with ethyl acetate (1×1850 mL). The combined organic phase was washed with water (1 *1850 mL), 5% NaHCO3 solution (1×1850 mL), water (1×1850 mL), 5% NaCI solution (1×1850 mL), dried (Na2SO4), filtered and concentrated under reduced pressure at 35-40 0C, which was finally dried under reduced pressure (1-2 mm) at 35-400C to yield 185.6 g (38.3%) of 1-phenyl-pentane-1,4-dione as thick oil.
Step (c): Preparation of /V-(2-methyl-5-phenyl-pyrrol-1-yI)-isonicotinamide
A mixture of 1-(phenyl)-pentane-1,4-dione (185 g, 1.05 mol), isonicotinic hydrazide (158.4 g, 1.155 mol, 1.1 equivalent), p-toluenesulphonic acid (1.85 g, 1% w/w) and dichloromethane (1850 ml_) was heated under reflux at 40-50 0C under azeotropic distillation for 2-3 h (water was collected in dean stark apparatus). The completion of reaction was monitored by HPLC. After cooling to 25-30 0C the resulting mixture was washed with saturated NaHCO3 solution (1×925 mL), aqueous layer was back extracted with EtOAc (1×925 ml_). The combined organic layers were washed with water (1×925 mL), 5% brine solution (1×925 mL), dried (Na2SO4) and filtered. The filtrate was concentrated under reduced pressure to obtain the solid product, which was further dried under reduced pressure (1-2 mm) at 35-40 0C. To this, cyclohexane (925 mL) was added and stirred for 25-30 min, solid separated out was filtered washed with cyclohexane (370 mL). This process was repeated two times more with the same amount of cyclohexane and finally solid was dried under reduced pressure (1-2 mm) at 40-500C; yield 162.23 g (55.7%). White solid, mp 177-179 0C. 1H NMR (CDCI3): δ 2.10 (s, 3H), 5.98 (d, J = 3.4 Hz, 1H), 6.22 (d, J = 3.7 Hz, 1H), 7.237.28 (m, 5H), 7.50 (d, J = 5.6 Hz, 2H), 8.55 (d, J = 5.0 Hz, 2H), 9.82 (s, 1H). MS: m/z (%) 278 (100) [M+1]. Anal. Calcd for C17H15N3O (277.32): C, 73.63; H, 5.45; N, 15.15. Found: C, 73.92; H, 5.67; N, 15.29.
Step (d): Preparation of /V-{2-methyl-5-phenyl-3-[4-(3-trifluoromethyl- phenyl)-piperazin-1-ylmethyl]-pyrrol-1-yl}-isonicotinamide
To a stirred solution of Λ/-(2-methyl-5-phenyl-pyrrol-1-yl)-isonicotinamide (160 g, 0.577 mol) in acetonitrile (1600 mil), was added drop-wise through pressure equalizing funnel a mixture of 1-(3-trifluoromethyl-phenyl)-piperazine monohydrochloride (153.75 g, 0.667 mol, 1.155 equivalent), formaldehyde (17.34 g, 0.577 mol, 1.0 equivalent) and acetic acid (480 mL) at 25-30 0C over a period of 60-90 min. The resulting reaction mixture was stirred for 14-16 h at same temperature and completion of reaction was monitored by TLC. After the completion of reaction, reaction mixture was treated with 20% aqueous NaOH solution (2600 mL). Layers were separated, EtOAc (4000 mL) was added to organic layer, washed with water (2×2000 mL), brine (2×1250 mL), dried (Na2SO4), and filtered. The filtrate was concentrated under reduced pressure at 35-38 0C and then dried under reduced pressure (1-2 mm) to yield the mixture of Λ/-{5-methyl-2-phenyl-3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-ylmethyl]-pyrrol- 1-yl}-isonicotinamide (A) and Λ/-{2-methyl-5-phenyl-3-[4-(3-trifluoromethyl- phenyl)-piperazin-1-ylmethyl]-pyrrol-1-yl}-isonicotinamide (B), yield 289 g (97.8%). The ratio of A and B was determined by reverse phase HPLC, which was found to be 19.4% and 76.7%, respectively.
Step (e): Purification of yV-{2-methyl-5-phenyl-3-[4-(3-trifluoromethyl-phenyl)- piperazin-1-ylmethyl]-pyrrol-1-yl}-isonicotinamide i) The mixture of A and B obtained from Step (d) (279 g) was dissolved in EtOAc (1960 ml_, 7 times) by heating at 50-60 0C. To this activated charcoal (14 g) was added and stirred for 10 min at the same temperature, filtered the activated charcoal through celite bed at 50-60 0C, washed with EtOAc (560 mL). After cooled to 25-30 0C, cyclohexane (2800 mL) was added to the filtrate and stirred the reaction mixture for 14-15 h at 20-35 0C. Solid separated out was filtered, washed with cyclohexane (3500 mL) and dried under reduced pressure (1-2 mm) for 4-5 hours. Yield 151 g (52%). Ratio of A and B was found to be 1.7% and 96.6%, respectively.
ii) The mixture of A and B obtained from Step (e)(i) (151 g) was dissolved in
EtOAc (755 mL, 5 times) by heating at 50-60 0C. After cooled to 25-30 0C, cyclohexane (1510 mL) was added and stirred the reaction mixture for 14-15 h at 20-35 0C. Solid separated out was frltered, washed with cyclohexane (3000 mL) and dried under reduced pressure (1-2 mm) for 4-5 hours. Yield 140 g (92%). Ratio ofA and B was found to be 0.2% and 98.1%, respectively.
Off white solid, mp 191-193 0C. 1H NMR (CDCI3): δ 2.13 (s, 3H), 2.60 (br s, 4H), 3.13 (br s, 4H), 3.41 (s, 2H), 6.24 (s, 1H), 6.977.29 (m, 9H), 7.53 (d, J = 5.6 Hz, 2H), 8.50 (S, 1H), 8.70 (d, J = 5.6 Hz, 2H). 13C NMR (CDCI3): δ 165.93, 151.77, 150.86, 139.74, 133.02, 131.99, 131.43, 129.92, 129.01, 127.79, 127.49, 121.74, 119.09, 116.18, 115.05, 112.48, 109.51, 54.87, 52.99, 48.93, 9.77. MS: m/z (%) 520 (100) [M+U Anal. Calcd for C29H28F3N5O (519.56): C, 67.04; H, 5.43; N, 13.48. Found: C, 67.36; H, 5.71; N, 13.69.
The free base Λ/-{2-methyl-5-phenyl-3-[4-(3-trifluoromethyl-phenyl)-piperazin-1- ylmethyl]-pyrrol-1-yl}-isonicotinamide is obtained in a crystalline form having characteristic powder X-ray diffraction pattern given in Figure 1 with 2Θ values 4.85, 5.99, 6.83, 7.34, 9.15, 9.78, 10.93, 11.98, 13.17, 13.98, 14.33, 14.75, 15.73, 16.42, 17.11. 17.72, 17.95, 18.32, 19.11, 19.75, 20.32, 21.36, 22.04, 23.19, 25.17
Step (f): Preparation of /V-{2-methyl-5-phenyl-3-[4-(3-trifluoromethyl-phenyl)- piperazin-1-ylmethyl]-pyrrol-1-yl}-isonicotinamide hydrochloride
To a stirred solution of 6% w/v HCI-EtOAc solution (821.8 mL, 1.351 mol, 7.0 equivalent) in EtOAc (2000 mL) was added a solution of Λ/-{2-methyl-5-phenyl-3- [4-(3-trifluoromethyl-phenyl)-piperazin-1-ylmethyl]-pyrrol-1-yl}-isonicotinamide (100 g, 0.193 mol) in EtOAc (2000 mL) through dropping funnel at 15-20 0C. When the addition was completed (~60 min), the reaction mixture was stirred at 10-150C for 1 h and then nitrogen gas was passed through reaction mass for 1 h until all the excess HCI fumes were removed. Solid so obtained was filtered through suction in an inert atmosphere, washed with ethyl acetate (2×500 mL), diisopropyl ether (2×500 mL) and dried in vacuum oven under reduced pressure (1-2 mm) at 35-40 0C for 15-20 h. Yield 115 g (99%).
Yellow solid, mp 177-179 0C. 1H NMR (DMSO-d6): δ 2.21 (s, 3H), 3.11-3.42 (m, 6H), 3.93-4.23 (m, 4H), 6.62 (s, 1H), 7.09-7.51 (m, 9H), 8.19-8.21 (d, 2H, J = 4.6 Hz), 8.95-8.97 (d, 2H1 J = 4.6 Hz), 11.30 (br s, 1H), 12.86 (s, 1H). MS: m/z (%) 520 (100) [M+1]. Anal. Calcd for C29H28F3N5O.2HCI.3H2O (646.53): C, 53.87; H, 5.61; N, 10.83. Found: C, 53.67; H, 5.59; N, 10.86.
The product obtained was amorphous in nature having the characteristic X-ray powder diffraction pattern given in Figure 2.
| Cited Patent |
Filing date |
Publication date |
Applicant |
Title |
| WO2004026828A1 * |
Sep 20, 2002 |
Apr 1, 2004 |
Lupin Limited |
Pyrrole derivatives as antimycobacterial compounds |
| WO2005107809A2 * |
Aug 27, 2004 |
Nov 17, 2005 |
Lupin Limited |
Antimycobacterial pharmaceutical composition comprising an antitubercular drug |
| US3168532 * |
Jun 12, 1963 |
Feb 2, 1965 |
Parke Davis & Co |
1, 5-diarylpyrrole-2-propionic acid compounds |
| Reference |
| 1 |
* |
BIAVA M ET AL: “SYNTHESIS AND MICROBIOLOGICAL ACTIVITIES OF PYRROLE ANALOGS OF BM 212, A POTENT ANTITUBERCULAR AGENT” MEDICINAL CHEMISTRY RESEARCH, BIRKHAEUSER, BOSTON, US, vol. 9, no. 1, 1999, pages 19-34, XP008016949 ISSN: 1054-2523 |
| 2 |
* |
BIAVA, MARIANGELA ET AL: “Antimycobacterial compounds. New pyrrole derivatives of BM212” BIOORGANIC & MEDICINAL CHEMISTRY , 12(6), 1453-1458 CODEN: BMECEP; ISSN: 0968-0896, 2004, XP002390961 |
| 3 |
* |
PARLOW J.J.: “synthesis of tetrahydonaphthaenes. part II” TETRAHEDRON, vol. 50, no. 11, 1994, pages 3297-3314, XP002391102 |
| 4 |
* |
R. RIPS , CH. DERAPPE AND N. BII-HOÏ: “1,2,5-trisubstituted pyrroles of pharmacologic interest” JOURNAL OF ORGANIC CHEMISTRY, vol. 25, 1960, pages 390-392, XP002390960 cited in the application |
///////////////LL 3858, SUDOTERB, TB, LUPIN
CC1=C(C=C(N1NC(=O)C2=CC=NC=C2)C3=CC=CC=C3)CN4CCN(CC4)C5=CC=CC(=C5)C(F)(F)F

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



































 PITAVASTATIN
PITAVASTATIN















 .
.


 Paço Municipal de Juiz de Fora
Paço Municipal de Juiz de Fora 








%200.5%20mL407.GIF)
