
Home » Posts tagged 'Ltd'
Tag Archives: Ltd
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
WO-2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
![]()

WO-2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
(WO2018001353) METHOD FOR PREPARING APREMILAST
ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
DU, Xiaoqiu; (CN).
ZHOU, Lianchao; (CN).
LIU, Jiegen; (CN)
EN)Method one: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L-leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an aprotic solvent to produce the compound of formula I; method two: (S) -1- (3-ethoxy-4-methoxyphenyl) -2- (methylsulfonyl) ethylamine N-acetyl-L- leucine salt of formula II is reacted with 3-acetylaminophthalic anhydride of formula III in an organic solvent in the presence of an organic alkaline or an alkali metal hydride to produce the compound of formula I. The method for preparing apremilast requires inexpensive raw materials and reagents , is suitable for industrialized production, and has great economic effects.
////////////WO 2018001353, APREMILAST, NEW PATENT, ZHEJIANG HUAHAI PHARMACEUTICAL CO., LTD
IMIGLIPTIN, NEW PATENT, WO 2017211293, XUANZHU PHARMA CO., LTD.
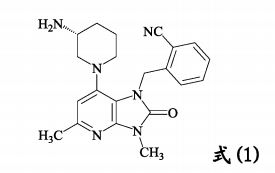
(WO2017211293) CRYSTALLINE FORM OF SUCCINATE USED AS DIPEPTIDYL PEPTIDASE-4 INHIBITOR
WO-2017211293,
XUANZHU PHARMA CO., LTD. [CN/CN]; 2518, Tianchen Street, National High-Tech Development Zone Jinan, Shandong 250101 (CN)
SHU, Chutian; (CN)
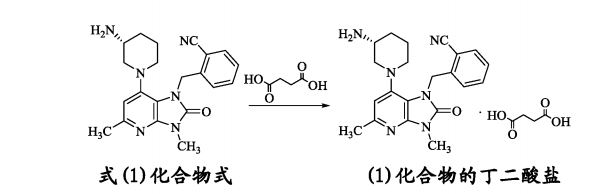
| The present invention relates to a crystalline form of a succinate used as a dipeptidyl peptidase-4 inhibitor, and a manufacturing method, pharmaceutical composition, and application thereof. The invention specifically relates to a dipeptidyl peptidase-4 inhibitor compound as represented by formula (1), a crystalline form of a succinate, wherein the succinate is an (R)-2-((7-(3-aminopiperidin-1-yl)-3,5-dimethyl-2-oxo-2,3-dihydro-1H-imidazo(4,5-b)pyridin-1-yl)methyl)benzonitrile, and a manufacturing method, pharmaceutical composition, and application thereof.
Example 1: Preparation of the succinate salt form I of the compound of formula (1) [0056]
[0057] The compound of formula (1) (44.6 g, 0.12 mol) was added to a 2 L round bottom flask and suspended in 1593 mL of acetonitrile. The mixture was heated to 80 ° C. and dissolved in free form. Immediately after the addition of 15.4 g A white solid precipitated, maintained at 80 ℃ for 1 hour and then cooled to room temperature, filtered and the filter cake was dried in vacuo at 40 ℃ for 10 hours, weighed 57.6g, yield 98.3%. The succinate salt Form I was tested by XRPD. [0058] Example 2: Preparation of the succinate salt form I of the compound of formula (1) II [0059] A quantity of succinate salt of the compound of formula (1) was weighed into glass vials in a total of 26 parts. A total of 26 vials of methanol, ethanol, isopropanol, isobutanol, 2-butanone, tetrahydrofuran, acetonitrile, methyl tert-butyl ether, acetone, water, toluene, Isopropyl acetate, n-propanol, isoamyl alcohol, butyl acetate, ethyl formate, 1,4-dioxane, n-butanol, pentane, heptane, cyclohexane, Ketone, xylene, isobutyl acetate, diethyl ether). After stirring, ultrasound and other means to make the sample fully dissolved. Subsequently, about 2 mL of liquid was removed from each bottle and filtered into 26 reagent tubes numbered 1-26. The resulting 26 filtrates were distributed in two 96-well plates. One or two of the above 1-13 solvents are sequentially added into the first 96-well plate, one or two of the above-mentioned 14-26 kinds of solvents are sequentially added into the second 96-well plate, Zha Kong sealing film sealed, placed in a fume hood, the natural environment to dry. Wherein Form I is obtained in the following mixed solvent, and Form I is also precipitated in the methyl isobutyl ketone in the remaining solution after plating. [0060] The solvent used to prepare succinate salt Form I was prepared [0061] [Table 0001]
[0062] Example 3: Preparation of the succinate salt form II of the compound of formula (1) [0063] Take 8 parts of the compound of formula (1), 200mg each, placed in a 10mL round bottom flask, add the solvent in the following table to each solvent, warmed until the solvent is refluxed, after dissolving it, add 69mg (1.1eq) succinic acid and cool to At room temperature, the solid precipitated and was filtered. The resulting solid was subjected to XRPD testing as succinate crystal form II. [0064] [Table 0002]
|
Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD

Sacubitril, WO 2016180275, New patent, SUZHOU PENGXU PHARMATECH CO., LTD
AHU-377 INTERMEDIATES AND METHOD FOR PREPARING AHU-377 AND AHU-377 INTERMEDIATES PATENT
WO2016180275, new patent, SUZHOU PENGXU PHARMATECH CO., LTD. [CN/CN]; 3rd Floor Building 7, 2358 Chang An Road, Wujiang Suzhou, Jiangsu 215200 (CN)
WANG, Peng; (CN).
LI, Pixu; (CN).
GU, Xiangyong; (CN)

Heart failure is a very high mortality syndrome, for patients with heart failure, so far no drug can significantly improve mortality and morbidity, and thus a new type of therapy is necessary. AHU-377 (CAS No. 149709-62-6) is an enkephalinase inhibitor, which is a prodrug ester groups can be lost through hydrolysis, converted to pharmaceutically active LBQ657, inhibit endorphin enzyme (NEP) the role of the main biological effects of NEP is to natriuretic peptides, bradykinin and other vasoactive peptide degradation failure. AHU-377 and angiotensin valsartan composition according to the molar ratio of 1 LCZ696. LCZ696 is an angiotensin receptor enkephalinase inhibitors, which can lower blood pressure, treat heart failure may become a new drug. Clinical data show, LCZ696 is more effective for the treatment of hypertension than valsartan alone.
Patents US 5,217,996 and US 5,354,892 reported the first synthesis of AHU-377, the synthetic route is as follows:
Reaction with unnatural D-tyrosine derivative as a substrate, more expensive, while the second step in the synthesis is necessary to use Pd-catalyzed Suzuki coupling reaction, whereby preparative route costs than the AHU-377 high.
Patent US 8,115,016 above routes also reported the departure from the pyroglutamate, through multi-step process for preparing a reaction AHU-377, which is more difficult methylation reaction, and the yield is not high. Patent US 8,580,974 also reported a carbonyl group of the a- introducing N, N- dimethyl enamine is converted to methyl, however, there are some problems in the route for constructing methyl chiral centers, are not suitable for scale-up synthesis route as follows:
About the latest AHU377 synthesis intermediates, Patent WO2014032627A1 reported using a Grignard reagent to react with epichlorohydrin, a quicker been important intermediates, synthetic route Compound AHU377 synthesized as follows:
However, the second step of the synthetic route use succinimide nitrogen atoms introduced by Mitsunobu reaction with hydrochloric acid hydrolysis to remove, then converted to Boc protected at the end of the synthesis process AHU377 Boc will have to take off protection, then any connection with succinic anhydride reaction product introduced into the structure of succinic acid portion, so that this method of atom economy and the economy of the steps are low.
Example 1
Synthesis of Compound 2
In inert atmosphere, a solution of three 500mL flask was added compound 1 (10g, 1eq), dissolved after 90mL THF, was added CuI (4.814g, 0.1eq), the system moves to the low temperature in the cooling bath to -20 ℃ when, biphenyl magnesium bromide dropwise addition, the internal temperature was controlled not higher than -10 ℃. Bi closed refrigeration drop, return to room temperature overnight. Completion of the reaction, the reaction solution was poured into saturated the NH 4 of Cl (10vol, 100 mL) was stirred at room temperature for 0.5h. Suction filtered, the filter cake was rinsed with a small amount of EA, and the filtrate was transferred to a separatory funnel carved, and the aqueous phase was extracted with EA (10vol × 2,100mL × 2) and the combined organic phases with saturated NaHC [theta] 3 , the NH 4 of Cl, each Brine 150mL (15vol) washed once, dried over anhydrous over MgSO 4 dried, suction filtered, and concentrated to give a white solid. Product obtained was purified by column 15.2g, yield 78%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.52 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.38-7.25 (m, 8H), 4.62-4.47 ( m, 2H), 4.09 (dd, J = 6.7,3.5Hz, 1H), 3.54 (dd, J = 9.5,3.5Hz, 1H), 3.43 (dd, J = 9.4 , 6.9Hz, 1H), 2.84 ( d, J = 6.6Hz, 2H), 2.38 (s, 1H).
Example 2
Synthesis of Compound 3
In an inert gas, at room temperature was added to the flask 500mL three Ph3P (18.54g, 2eq), 240mL DCM dissolution, butyryl diimide (of 6.44 g), compound 2 (15g), an ice-water bath cooling to 0 ℃ or so, was added dropwise DIAD (14mL) was complete, the reaction go to room temperature.Starting material the reaction was complete, the system was added to water (100 mL) quenched the reaction was stirred for 10min; liquid separation, the aqueous phase was extracted with DCM (100mL × 2), the combined organic phases with saturated Brine 100mL × 2), dried over anhydrous over MgSO 4 dried , filtration, spin dry to give a white solid; product was purified by column 15.4g, yield 82%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.56 (D, J = 7.4Hz, 2H), 7.49 (D, J = 8.0Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.37-7.30 (m, 3H), 7.27 ( d, J = 6.7Hz, 3H), 7.22 (d, J = 8.0Hz, 2H), 4.75 (s, 1H), 4.56 (d, J = 12.0Hz, 1H), 4.45 (d, J = 12.0Hz, 1H ), 4.06 (t, J = 9.6Hz, 1H), 3.70 (dd, J = 10.0,5.2Hz, 1H), 3.23 (dd, J = 13.8,10.3Hz, 1H) , 3.14-3.00 (m, 1H), 2.48 (d, J = 4.0Hz.4H).
Example 3
Synthesis of Compound 4
Protection of inert gas, at room temperature was added to the flask 1L three compound 3 (18.81g), 470mL EtOH was dissolved, was added Pd / C, replaced the H 2 three times, move heated on an oil bath at 60 ℃ reaction. Raw reaction was complete, the system was removed from the oil bath, the reaction solution was suction filtered through Celite and concentrated to give the crude product. It was purified by column pure 11.8g, a yield of 81.2%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.8Hz, 2H), 7.51 (D, J = 7.8Hz, 2H), 7.42 (T, J = 7.5Hz, 2H), 7.33 (T , J = 7.2Hz, 1H), 7.26 (d, J = 7.2Hz, 2H), 4.55 (d, J = 5.2Hz, 1H), 4.06-3.97 (m, 1H), 3.86 (dd, J = 12.0, 3.1Hz, 1H), 3.16 (dd , J = 8.1,2.9Hz, 2H), 2.58 (t, J = 7.0Hz, 4H), 1.26 (s, 2H).
Example 4
Synthesis of Compound 7
Protection of inert gas, at room temperature to a 25mL flask was added three Dess-Martin oxidant (767.7mg), 10mL DCM was dissolved, the system was cooled down to -10 deg.] C, was added 4 (500mg). Starting material the reaction was complete, to the system was added saturated NaHCO3 and Na2S2O3 each 5mL, quench the reaction stirred for 10min; aqueous phase was extracted with DCM (10mL × 3) and the combined organic phases with saturated NaHCO3, Brine 30mL each wash, dried over anhydrous MgSO4, filtration, spin dried to give the crude product used directly in the next reaction cast.
Example 5
Synthesis of Compound 8
Inert gas, at room temperature for three to 500mL flask 7 (497.5mg), 10mL DCM to dissolve an ice water bath to cool, added phosphorus ylide reagent (880.6mg), the system was removed from the ice water bath at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. Liquid separation, the aqueous phase was extracted with DCM (10mL × 2), organic phases were combined, washed with saturated Brine 20mL × 2, dried over anhydrous MgSO4, filtration, spin crude done. Product obtained was purified by column 563mg, 90% yield.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.60-7.53 (m, 2H), 7.51 (D, J = 8.1Hz, 2H), 7.42 (T, J = 7.6Hz, 2H), 7.33 (D, J = 7.3Hz, 1H), 7.23 (d , J = 8.1Hz, 2H), 7.13 (dd, J = 9.2,1.5Hz, 1H), 5.26 (td, J = 9.5,6.9Hz, 1H), 4.25-4.05 ( m, 2H), 3.40 (dd , J = 13.7,9.7Hz, 1H), 3.13 (dd, J = 13.8,6.7Hz, 1H), 2.53 (d, J = 2.2Hz, 4H), 1.85 (d, J = 1.4Hz, 3H), 1.30 ( t, J = 7.1Hz, 3H).
Example 6
Synthesis of Compound 9
Protection of inert gas, at room temperature to a 50mL flask was added three 8 (365mg, 1eq), 9mL of ethanol and stirred to dissolve, the system was replaced with hydrogen three times, was added Pd / C (25% w / w) at room temperature. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. The reaction mixture was suction filtered through Celite and concentrated to give the crude product. Product was purified by column, yield 80.2%, purity 97.2%.
Example 7
Synthesis of Compound 10
Equipped with Compound 9 (100mg) acetic acid A reaction flask (9mL), hydrochloric acid (1mL). The reaction was heated oil bath at 80 deg.] C. The reaction material completely stop the reaction, the system was added to water (5mL) to quench the reaction. After saturated NaHCO3 and extracted with EA and concentrated to give crude product. Product obtained was purified by column 90mg, yield 84%.
NMR data for the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.54 (m, 2H), 7.53-7.48 (m, 2H), 7.41 (dd, J = 10.5,4.9Hz, 2H), 7.31 (dd, J = 8.3 , 6.4Hz, 1H), 7.22 ( d, J = 8.2Hz, 2H), 5.93 (t, J = 9.7Hz, 1H), 4.34-4.00 (m, 3H), 2.91-2.71 (m, 2H), 2.68 -2.57 (m, 2H), 2.55 (ddd, J = 9.4,7.0,4.3Hz, 1H), 2.42 (dt, J = 13.3,6.8Hz, 2H), 1.97-1.74 (m, 1H), 1.64-1.46 (m, 1H), 1.23 ( td, J = 7.1,3.3Hz, 3H), 1.14 (dd, J = 7.1,3.9Hz, 3H)
Example 8
Synthesis of Compound 5
Example 8-1: The reaction flask was added compound 4 (1eq) was added water (2VOL), concentrated hydrochloric acid (2VOL), 110 ℃ reaction was heated in an oil bath overnight, complete conversion of starting material, the HPLC peak area 97%. 10% NaOH solution was added to adjust the pH to about 10, filtration products. Yield 85%.
Example 8-2: The reaction flask was added compound 4 (1eq) was added ethanol (5 vol), water (5 vol), potassium hydroxide (8 eq), was heated in an oil bath overnight at 110 ℃ reaction, complete conversion of the starting material, the HPLC peak area 99%. Water was added (5Vol), filtered to obtain the product. Yield 95%. Product was dissolved in toluene, was added ethanolic hydrochloric acid, the precipitated hydrochloride Compound 5.
NMR data for the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.31 (S, 3H), 7.70-7.61 (m, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.31 (m, 3H), 4.09 (the dq- , J = 42.6,7.1Hz, 1H), 3.62-3.51 (m, 1H), 3.50-3.41 (m, 1H), 3.11-3.00 (m, 1H), 2.95-2.84 (m, 1H), 1.30-1.10 (m, 1H).
EXAMPLE 9
Synthesis of Compound 6
To the reactor was added compound 5, was added absolute ethanol (3vol). Temperature of the outer set 30 ℃ heating, stirring was continued after the temperature reached 25 ℃ 20min. Was added 30% NaOH aqueous solution (1.1eq). External temperature 65 ℃ heating provided, after the internal temperature reached 60 deg.] C was slowly added (of Boc) 2 O (1.1 eq). Stirring 0.5h, reaction monitoring. After completion of the reaction, water was added slowly dropwise (8vol), turn off the heating and natural cooling. The system temperature was lowered to 25 deg.] C and continue stirring for 2h. Filter cake at 50 ℃ blast oven drying to obtain the product.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Synthesis of Intermediate Compound 6 to Compound 10, i.e., the AHU-377, a synthetic route in the background of the present invention, the cited patent application WO2014032627A1 loaded in detail, not in this repeat.
Example 10
Synthesis of Compound 2
Benzyl glycidyl ether preparation (50g) in THF (200mL) was added. Under inert gas protection, the biphenyl magnesium bromide (365mmol) was added to THF (1020mL) was added the reaction flask is placed in a low temperature bath -40 ℃ cooling. Cuprous iodide (O.leq) when the internal temperature dropped to -9 ℃. Continued to decrease the temperature of -23 ℃ dropwise addition of benzyl glycidyl ether in THF was added dropwise to control the internal temperature process of not higher than -15 deg.] C, 47 min when used, the addition was completed the cooling off the reaction was stirred overnight. The cooling system to -20 ℃ quenched with 1N HCl aqueous solution, <10 ℃ Go stirred 30min at room temperature. Liquid separation, the aqueous phase was extracted with THF, the combined THF phases. Respectively saturated ammonium chloride (250mL), saturated brine (250mL) washed. Rotary evaporation to remove THF, and water (200 mL) Continue rotary evaporation 1h, cool to precipitate a solid. Suction crude. Crude n-heptane was added 2Vol beating, suction filtration to obtain the product in a yield of 90 ~ 95%, HPLC peak area 94%. In another column purification was pure, columned yield 88.6%, HPLC 99.1%.
Example 11
Synthesis of Compound 3
Preparation Example 9, said compound taking the embodiment 2 (5g) added to the reaction flask, the reaction flask was added toluene (80mL), phthalimide (2.55 g of) and triphenylphosphine (5.35g of), the nitrogen was replaced protection. An ice-salt bath cooling to -5 deg.] C, was added dropwise DIAD (4.12g), dropwise addition was exothermic, the temperature was raised to 5 ℃. The reaction was continued 1h sampling HPLC test material substantially complete reaction. Join 12g silica spin column done to collect the product (including DIEA derivative).
Example 12
Synthesis of Compound 11
Compound 3 (3g) was added to the reaction flask embodiment taken in Preparation Example 10, was added ethanol (30 mL), with stirring. Was added hydrazine hydrate (2g) was heated in an oil bath reflux 1h, when supplemented with 20mL ethanol was stirred difficulties, the reaction was continued to 2.5h, HPLC showed the starting material the reaction was complete. Add EA / H2O 100mL each liquid separation, the EA phase was washed with water (100mL) and the combined organic phases were washed with water (100mL) and saturated brine (100mL) washed. Anhydrous magnesium sulfate and filtered spin column was done product 1.88g, yield 88%, HPLC 94%.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 7.64 (D, J = 7.2Hz, 2H), 7.57 (D, J = 8.1Hz, 2H), 7.45 (T, J = 7.6Hz, 2H), 7.39-7.32 ( m, 5H), 7.29 (d , J = 8.1Hz, 3H), 4.55-4.43 (m, 2H), 3.38-3.23 (m, 3H), 3.18-3.10 (m, 1H), 2.82-2.74 (m, 1H), 2.61-2.52 (m, 1H ).
Example 13
Synthesis of Compound 11
To the toluene solution of the compound 2 was added phthalimide (1.1 eq), triphenylphosphine (1.3 eq) with stirring. External bath set -10 ℃, to cool the system, the internal temperature dropped to 0 ~ 5 ℃, start dropping DIAD (1.3eq), control the internal temperature -5 ~ 5 ℃. Completion of the dropwise addition, the cooling bath was turned off outside the reaction was stirred at room temperature. The reaction was stirred for 1 to 4 hours. The reaction solution to give compound 3, administered directly in the next reaction. To the above reaction mixture was added hydrazine hydrate (6 eq), heated to 70 ~ 80 ℃, to complete the reaction, filtered hot, the filtrate. Aqueous sodium hydroxide solution (20vol 10%) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. Water was added (20vol) was stirred for 0.5h, allowed to stand for liquid separation from toluene phase. The toluene phase was added hydrochloric acid (20vol, 3N), stirred for 0.5h, to form a solid precipitate. Filtration and drying to obtain a product, i.e. compound 11, the hydrochloride salt, yield 60% in two steps.
NMR data of the product are as follows:
1 the H NMR (400MHz, of DMSO) [delta] 8.46 (S, 3H), 7.63 (dd, J = 16.4,7.7Hz, 4H), 7.47 (T, J = 7.6Hz, 2H), 7.42-7.22 (m, 8H ), 4.56 (d, J = 12.1Hz, 1H), 4.48 (d, J = 12.1Hz, 1H), 3.58 (d, J = 7.9Hz, 2H), 3.47 (dd, J = 10.9,6.3Hz, 1H ), 3.11 (dd, J = 13.5,4.9Hz, 1H), 2.92 (dd, J = 13.4,9.1Hz, 1H).
Example 14
Synthesis of Compound 12
Weigh Compound 11 (1.38g) was added to the reaction flask. To the reaction flask plus DCM (14ml) and Et3N (462mg, 0.73ml). Weighed (of Boc) 2O (1.23 g of) was added to DCM (5ml) was dissolved. Room temperature (8 ℃), a solution (of Boc) 2 DCM solution O was added dropwise to the reaction, (2ml) rinsed with DCM. The reaction mixture was stirred at room temperature, detected by HPLC, the reaction ends 4h. Reaction mixture was washed (15ml) 3 times with Brine (15ml) The reaction solution was washed 1 times. Inorganic sulfate, concentrated and purified by column PE:EA = 15:1 give product 560mg, yield 30.8%, HPLC 99.92%.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) [delta] 7.57 (D, J = 7.6Hz, 2H), 7.49 (D, J = 7.4Hz, 2H), 7.43 (T, J = 7.3Hz, 2H), 7.39-7.28 (m, 5H), 7.24 ( d, J = 9.0Hz, 3H), 5.00-4.80 (br, 1H), 4.51 (q, J = 11.8Hz, 2H), 4.08-3.85 (br, 1H), 3.43 ( d, J = 2.9Hz, 2H) , 3.02-2.77 (m, 2H), 1.42 (s, 9H).
Example 15
Synthesis of Compound 6
Weigh Compound 12 (250mg) and methanol (9ml) was added to the reaction flask. Added Pd / C (138mg, 1 / 4w / w, water content 55%). The H 2replaced 3 times, 50 ℃ stirred and heated. HPLC detection reaction, the reaction end 30h. Filtered off Pd / C, 40 ℃ concentrated under reduced pressure to remove methanol. PE:EA = 3:1 florisil column to give the product 196mg, 100% yield, 99.34% purity.
NMR data of the product are as follows:
1 the H NMR (400MHz, CDCl 3 ) δ7.61-7.50 (m, 4H), 7.61-7.50 (m, 4H), 7.46-7.39 (m, 2H), 7.48-7.38 (m, 2H), 7.38-7.23 (m, 3H), 7.37-7.26 ( m, 3H), 4.82 (d, J = 7.9Hz, 1H), 4.82 (d, J = 7.9Hz, 1H), 3.91 (s, 1H), 3.70 (d, J = 11.0Hz, 1H), 3.77-3.54 (m, 2H), 3.65-3.47 (m, 1H), 2.88 (d, J = 7.0Hz, 2H), 2.88 (d, J = 7.0Hz, 2H), 2.51 (s, 1H), 2.51 (s, 1H), 1.42 (s, 9H), 1.42 (s, 9H).
Method for preparing the AHU-377, characterized by comprising the steps of: (a) Compound (1) S- benzyl glycidyl ether and biphenyl Grignard reagent produced by the reaction of the compound (2) in an organic solvent; ( b) compound (2) with a succinimide or phthalimide Mitsunobu reaction occurs in an organic solvent to form a compound (3); (C) compound (3) in an organic solvent in the role of a catalyst under removal debenzylation protected form compound (4); (D) compound (4) with an oxidizing agent oxidation reaction occurs in an organic solvent to form a compound (7); (E) compound (7) with a phosphorus ylide reagent in an organic solvent to give the compound (8); (F.) compound (8) in an organic solvent in the selective catalytic hydrogenation of the compound (9); and (g) of the compound (9) in an organic solvent in the hydrolysis reaction of the amide compound occurs in the presence of an acid ( 10), i.e., AHU-377;
HAO 472
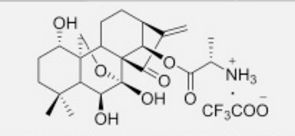
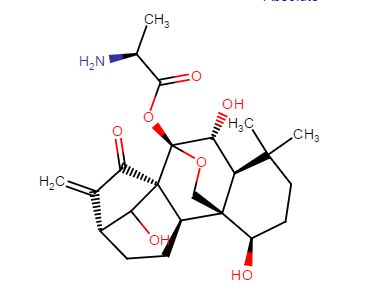 .CF3COOH
.CF3COOH
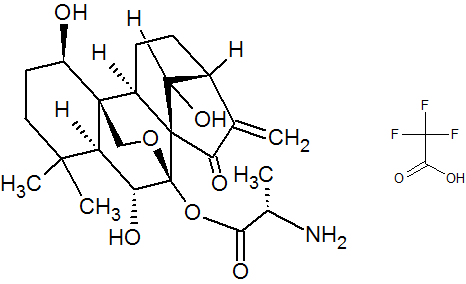
HAO 472
PHASE 1 CHINA



PRoject Name: HAO472 treatment Phase I clinical trial in relapsed / refractory AML, M2b type of AML
The main purpose: to determine HAO472 treatment of relapsed / refractory C the maximum tolerated dose (MTD). Secondary objectives: 1) evaluation of drug safety and tolerability; 2) study HAO472 in pharmacokinetic characteristics of the human body; 3) the effectiveness of HAO472 treatment of relapsed / refractory M2b type of AML.
Introduction Test
Acute myelogenous leukemia
HAO472
Phase I
Test Number: CTR20150246
Sponsor Name:
Jiangsu Hengrui Medicine Co., Ltd. 1/
2 Ruijin Hospital, Shanghai Jiaotong University School of Medicine /
3 Jiangsu Hengrui Medicine Co., Ltd. /
4 Shanghai Hengrui Medicine Co., Ltd. /

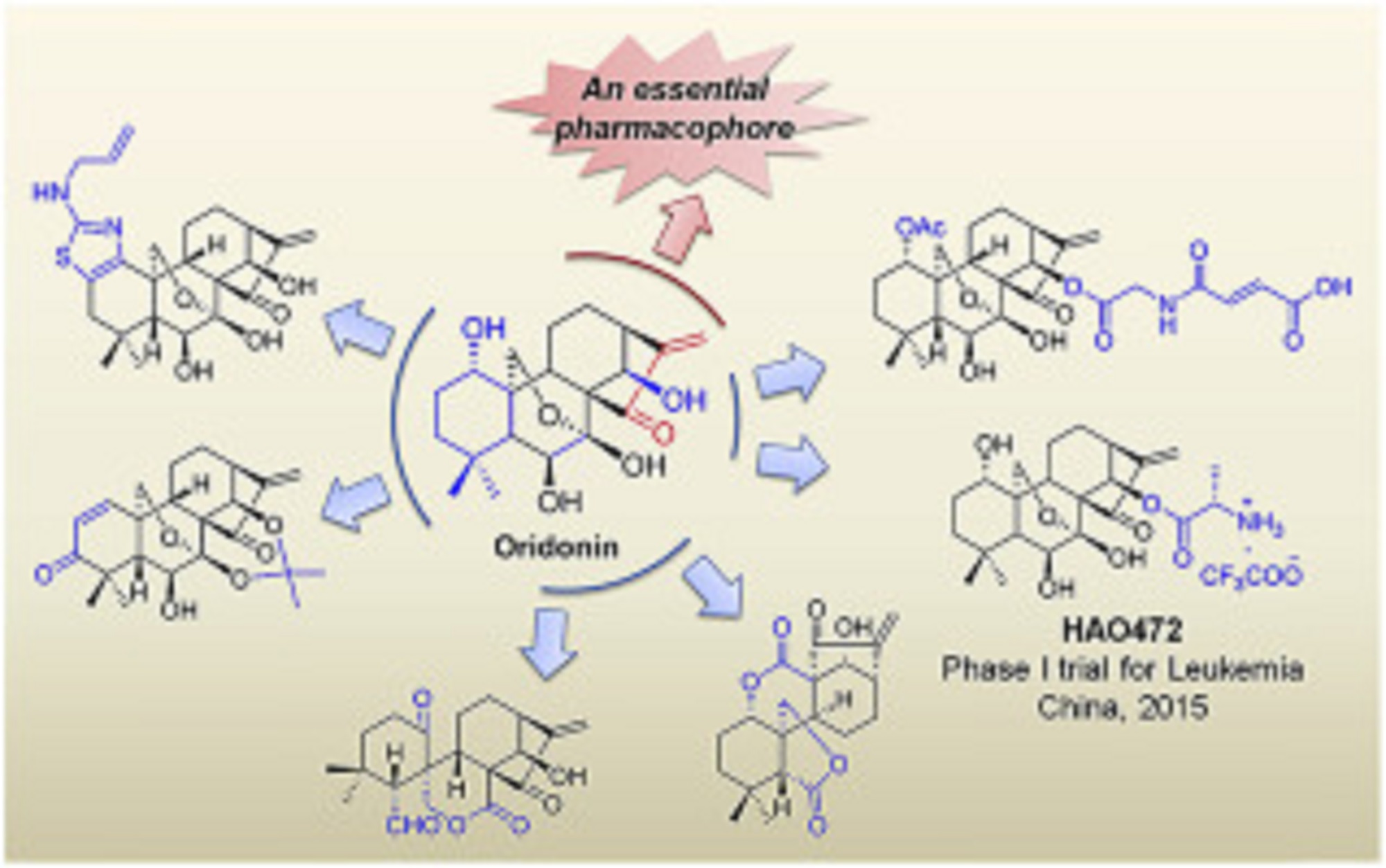
Natural products have historically been, and continue to be, an invaluable source for the discovery of various therapeutic agents. Oridonin, a natural diterpenoid widely applied in traditional Chinese medicines, exhibits a broad range of biological effects including anticancer and anti-inflammatory activities. To further improve its potency, aqueous solubility and bioavailability, the oridonin template serves as an exciting platform for drug discovery to yield better candidates with unique targets and enhanced drug properties. A number of oridonin derivatives (e.g. HAO472) have been designed and synthesized, and have contributed to substantial progress in the identification of new agents and relevant molecular mechanistic studies toward the treatment of human cancers and other diseases. This review summarizes the recent advances in medicinal chemistry on the explorations of novel oridonin analogues as potential anticancer therapeutics, and provides a detailed discussion of future directions for the development and progression of this class of molecules into the clinic.
Highlights
Oridonin displays significant anticancer activities via multi-signaling pathways.
Recent advances in medicinal chemistry of oridonin-like compounds are presented.
The article summarizes the SAR and mechanism studies of relevant drug candidates.
The milestones and future direction of oridonin-based drug discovery are discussed.
Volume 122, 21 October 2016, Pages 102–117

Discovery and development of natural product oridonin-inspired anticancer agents
- a Chemical Biology Program, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, TX, 77555, United States
- b Department of Clinical Cancer Prevention, Division of Cancer Prevention and Population Sciences, The University of Texas MD Anderson Cancer Center, Houston, TX, 77030, United States

////////Natural product, Oridonin, Diterpenoids, Anticancer agents, Drug discovery, Chemical biology, AML, HAO 472, relapsed / refractory AML. Jiangsu Hengrui Medicine Co., Ltd, PHASE1, LEUKEMIA
C[C@H](N)C(=O)O[C@]15OC[C@@]2([C@H](O)CCC(C)(C)[C@@H]2[C@H]1O)[C@H]3CC[C@@H]4C(=C)C(=O)[C@@]35C4O
FDA approves new diagnostic imaging agent FLUCICLOVINE F-18 to detect recurrent prostate cancer

FLUCICLOVINE F-18
Cyclobutanecarboxylic acid, 1-amino-3-(fluoro-18F)-, trans- [
- Molecular FormulaC5H818FNO2
- Average mass132.124 Da
May 27, 2016
Release
The U.S. Food and Drug Administration today approved Axumin, a radioactive diagnostic agent for injection. Axumin is indicated for positron emission tomography (PET) imaging in men with suspected prostate cancer recurrence based on elevated prostate specific antigen (PSA) levels following prior treatment.
Prostate cancer is the second leading cause of death from cancer in U.S. men. In patients with suspected cancer recurrence after primary treatment, accurate staging is an important objective in improving management and outcomes.
“Imaging tests are not able to determine the location of the recurrent prostate cancer when the PSA is at very low levels,” said Libero Marzella, M.D., Ph.D., director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research. “Axumin is shown to provide another accurate imaging approach for these patients.”
Two studies evaluated the safety and efficacy of Axumin for imaging prostate cancer in patients with recurrent disease. The first compared 105 Axumin scans in men with suspected recurrence of prostate cancer to the histopathology (the study of tissue changes caused by disease) obtained by prostate biopsy and by biopsies of suspicious imaged lesions. Radiologists onsite read the scans initially; subsequently, three independent radiologists read the same scans in a blinded study.
The second study evaluated the agreement between 96 Axumin and C11 choline (an approved PET scan imaging test) scans in patients with median PSA values of 1.44 ng/mL. Radiologists on-site read the scans, and the same three independent radiologists who read the scans in the first study read the Axumin scans in this second blinded study. The results of the independent scan readings were generally consistent with one another, and confirmed the results of the onsite scan readings. Both studies supported the safety and efficacy of Axumin for imaging prostate cancer in men with elevated PSA levels following prior treatment.
Axumin is a radioactive drug and should be handled with appropriate safety measures to minimize radiation exposure to patients and healthcare providers during administration. Image interpretation errors can occur with Axumin PET imaging. A negative image does not rule out the presence of recurrent prostate cancer and a positive image does not confirm the presence of recurrent prostate cancer. Clinical correlation, which may include histopathological evaluation of the suspected recurrence site, is recommended.
The most commonly reported adverse reactions in patients are injection site pain, redness, and a metallic taste in the mouth.
Axumin is marketed by Blue Earth Diagnostics, Ltd., Oxford, United Kingdom
Patent
http://www.google.com/patents/WO2014023775A1?cl=en
The non-natural amino acid [ F]-l-amino-3-fluorocyclobutane-l-carboxylic acid
([18F]-FACBC, also known as [18F]-Fluciclovine) is taken up specifically by amino acid transporters and has shown promise for tumour imaging with positron emission tomography (PET).
A known synthesis of [18F]-FACBC begins with the provision of the protected precursor compound 1 -(N-(t-butoxycarbonyl)amino)-3 –
[((trifluoromethyl)sulfonyl)oxy]-cyclobutane-l-carboxylic acid ethyl ester. This precursor compound is first labelled with [18F]-fluoride:

II before removal of the two protecting groups:

IT III
EP2017258 (Al) teaches removal of the ethyl protecting group by trapping the [18F]- labelled precursor compound (II) onto a solid phase extraction (SPE) cartridge and incubating with 0.8 mL of a 4 mol/L solution of sodium hydroxide (NaOH). After 3 minutes incubation the NaOH solution was collected in a vial and a further 0.8 mL 4 mol/L NaOH added to the SPE cartridge to repeat the procedure. Thereafter the SPE cartridge was washed with 3 mL water and the wash solution combined with the collected NaOH solution. Then 2.2 mL of 6 mol/L HCl was then added with heating to 60°C for 5 minutes to remove the Boc protecting group. The resulting solution was purified by passing through (i) an ion retardation column to remove Na+ from excess NaOH and Cl~ from extra HCl needed to neutralise excess of NaOH to get a highly acidic solution before the acidic hydrolysis step, (ii) an alumina column, and (iii) a reverse-phase column. There is scope for the deprotection step(s) and/or the
purification step in the production of [18F]-FACBC to be simplified.
Example 1: Synthesis of f FIFACBC
No-carrier- added [18F]fluoride was produced via the 180(p,n)18F nuclear reaction on a GE PETtrace 6 cyclotron (Norwegian Cyclotron Centre, Oslo). Irradiations were performed using a dual-beam, 30μΑ current on two equal Ag targets with HAVAR foils using 16.5 MeV protons. Each target contained 1.6 ml of > 96% [180]water (Marshall Isotopes). Subsequent to irradiation and delivery to a hotcell, each target was washed with 1.6 ml of [160]water (Merck, water for GR analysis), giving approximately 2-5 Gbq in 3.2 ml of [160]water. All radiochemistry was performed on a commercially available GE FASTlab™ with single-use cassettes. Each cassette is built around a one-piece-moulded manifold with 25 three-way stopcocks, all made of polypropylene. Briefly, the cassette includes a 5 ml reactor (cyclic olefin copolymer), one 1 ml syringe and two 5 ml syringes, spikes for connection with five prefilled vials, one water bag (100 ml) as well as various SPE cartridges and filters. Fluid paths are controlled with nitrogen purging, vacuum and the three syringes. The fully automated system is designed for single-step fluorinations with cyclotron-produced [18F]fluoride. The FASTlab was programmed by the software package in a step-by-step time-dependent sequence of events such as moving the syringes, nitrogen purging, vacuum, and temperature regulation. Synthesis of
[18F]FACBC followed the three general steps: (a) [18F]fluorination, (b) hydrolysis of protection groups and (c) SPE purification.
Vial A contained K222 (58.8 mg, 156 μπιοΐ), K2C03 (8.1 mg, 60.8 μπιοΐ) in 79.5% (v/v)
MeCN(aq) (1105 μΐ). Vial B contained 4M HC1 (2.0 ml). Vial C contained MeCN
(4.1ml). Vial D contained the precursor (48.4 mg, 123.5 μιηοΐ) in its dry form (stored at -20 °C until cassette assembly). Vial E contained 2 M NaOH (4.1 ml). The 30 ml product collection glass vial was filled with 200 mM trisodium citrate (10 ml). Aqueous
[18F]fluoride (1-1.5 ml, 100-200 Mbq) was passed through the QMA and into the 180-
H20 recovery vial. The QMA was then flushed with MeCN and sent to waste. The trapped [18F]fluoride was eluted into the reactor using eluent from vial A (730 μΐ) and then concentrated to dryness by azeotropic distillation with acetonitrile (80 μΐ, vial C). Approximately 1.7 ml of MeCN was mixed with precursor in vial D from which 1.0 ml of the dissolved precursor (corresponds to 28.5 mg, 72.7 mmol precursor) was added to the reactor and heated for 3 min at 85°C. The reaction mixture was diluted with water and sent through the tC18 cartridge. Reactor was washed with water and sent through the tC18 cartridge. The labelled intermediate, fixed on the tC18 cartridge was washed with water, and then incubated with 2M NaOH (2.0 ml) for 5 min after which the 2M NaOH was sent to waste. The labelled intermediate (without the ester group) was then eluted off the tC18 cartridge into the reactor using water. The BOC group was hydrolysed by adding 4M HC1 (1.4 ml) and heating the reactor for 5 min at 60 °C. The reactor content with the crude [18F]FACBC was sent through the HLB and Alumina cartridges and into the 30 ml product vial. The HLB and Alumina cartridges were washed with water (9.1 ml total) and collected in the product vial. Finally, 2M NaOH (0.9 ml) and water (2.1 ml) was added to the product vial, giving a purified formulation of [18F]FACBC with a total volume of 26 ml. Radiochemical purity was measured by radio-TLC using a mixture of MeCN:MeOH:H20:CH3COOH (20:5:5: 1) as the mobile phase. The radiochemical yield (RCY) was expressed as the amount of radioactivity in the [18F]FACBC fraction divided by the total used [18F]fluoride activity (decay corrected). Total synthesis time was 43 min.
The RCY of [18F]FACBC was 62.5% ± 1.93 (SD), n=4.
/////FDA, diagnostic imaging agent, recurrent prostate cancer, fda 2016, Axumin, marketed, Blue Earth Diagnostics, Ltd., Oxford, United Kingdom, fluciclovine F 18
C1[C@@](C[C@H]1[18F])(N)C(=O)O
UPDATE
![]()
SEE EMA
| Axumin : EPAR – Summary for the public | EN = English | 06/07/2017 |
The active substance fluciclovine (18F) is prepared from the precursor AH113487 by nucleophilic substitution
of a triflate group by 18F-fluoride, followed by two deprotection steps. Due to the short half-life of the 18Ffluorine
radioisotope, each batch is prepared on the day of clinical use.
The active substance is prepared in a proprietary automated synthesiser unit. The synthesiser module is
computer-controlled. A fluid path for synthesis is provided in the form of a single use cassette (FASTlab). The
cassette contains 3 reagent vials and 3 solid phase cartridges. Two other reagent vials are supplied
separately as they have a recommended storage temperature of 2-8°C. These 2 vials are inserted into the
cassette on the day of production.
Assessment report
EMA/237809/2017 Page 13/90
Fluciclovine (18F) is produced in a continuous operation from the precursor AH113487. Due to the radioactive
nature of the process, and the short half-life of [18F] fluorine, intermediates are not isolated and there is no
opportunity for operator intervention or in-process testing. Control of the synthesis of fluciclovine (18F) from
the precursor is achieved through the automated synthesis platform, which is pre-programmed with
synthesis parameters optimised for the process. On-board detectors record transfers of radioactivity through
the fluid path at critical points and monitor temperature and pressure as appropriate so that the operator
may track the progress of the synthesis.
The active substance fluciclovine (18F) progressses immediately to purification, formulation and dispensing as
the finished product within a single, continuous operation. Validation of the manufacturing process for
fluciclovine (18F) is therefore described as part of finished product validation.
The characterisation of the active substance is in accordance with the EU guideline on chemistry of new
active substances.
As mentioned, the manufacture of the active substance and finished product takes place in a single,
continuous process. The active substance is not isolated at any point. Therefore, relevant information about
impurities is given only for the finished product.
For the same reason, information for the container closure system is provided only for the finished product.http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004197/WC500230836.pdf
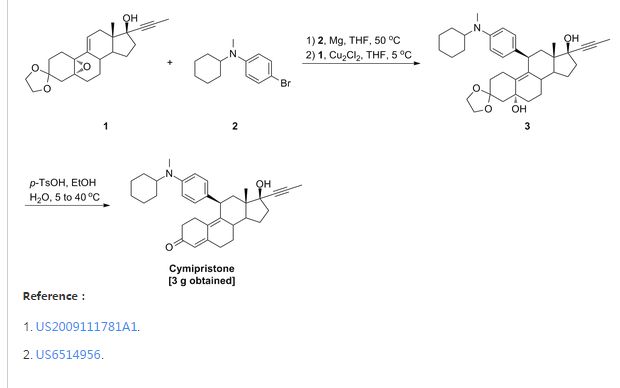
Cymipristone
Cymipristone
- Estra-4,9-dien-3-one, 11-[4-(cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propynyl)-, (11β,17β)- (9CI)
- (11β,17β)-11-[4-(Cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propyn-1-yl)estra-4,9-dien-3-one
- Saimisitong
NDA Filed china
Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
A progesterone receptor antagonist potentially for termination of intrauterine pregnancy.
![]()
CAS No.329971-40-6
- Molecular FormulaC34H43NO2
- Average mass497.711 Da
- Steroid Compounds, a Method for Preparation thereof, Pharmaceutical Compositions Containing the Same and Use thereof
-
This invention relates to steroid compounds and pharmaceutical acceptable salts thereof, a method for preparation thereof, pharmaceutical compositions containing the same as active component, and their use in the preparation of medicines for treating diseases associated with progestogen dependence and for fertility control, abortion or contraception and for anticancer use.
-
Mifepristone (11β-[4-(N,N-dimethylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in French Patent No. 2,497,807 to Rousell-Uclaf, published May 31, 1983. It is the first progesterone receptor antagonist put into clinical application and is a new type of anti-progestin. It binds to progesterone receptor and glucocorticoid receptor, having an affinity with progesterone receptor in rabbit endometrium five-fold higher than that of progesterone and thereby having strong anti-progesterone effect. It causes degeneration of pregnant villus tissue and decidual tissue, endogenous prostaglandin (PG) release, luteinizing hormone decrease, corpus luteum dissolution, and necrosis of embryo sac whose development depends on corpus luteum, leading to abortion. Therefore, it can be used as a non-surgical medicine for stopping early pregnancy. It can also be used, inter alia, in contraception and as an antineoplastic. (The Antiprogestin Steroid Ru486 and Human Fertility Control, 1985, New York: Plenum Press) .
-
Onapristone (11β-[4-(N,N-diemthylamino)phenyl]-17α-hydroxy-17β-(3-hydroxypropyl)-13α-4,9-estradiene-3-one), is a steroid compound which is disclosed in German Patent No. 3,321,826 to Schering AG, published Dec. 20, 1984. It has a strong antiprogestin activity and can be used in abortion (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), anticancer (Breast Cancer Research and Treatment, 1989, 14:275-288), etc. It was reported that onapristone had toxicity to human liver (European Journal of Cancer, 1999, 35(2):214-218).
-
Lilopristone (11β-[4-(N,N-dimethylamino) phenyl]-17α-[3-hydroxy-1(Z)-propenyl]-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in German Patent No. 3,347,126 to Schering AG, published July 11, 1985. It has a strong antiprogestin activity and can be used in abortion, contraception (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), etc. It was reported that the clinical effect of lilopristone in stopping early pregnancy was only equivalent to that of mifepristone (Human Reproduction, 1994, 9(1):57-63).
-
ZK112993 (11β-(4-acetylphenyl)-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is as steroid compound which is disclosed in German Patent No. 3,504,421 to Schering AG, published Aug. 7, 1986. It has a potent antiprogestin activity and can be used in, inter alia, anticancer (Anticancer Res., 1990, 10:683-688).
-
In European Patent No. 321,010 to Akzo NV, The Netherland published June 21, 1989 are disclosed “11-arylsteroid compounds” having a strong antiprogestin activity.
PATENT
WO 2001018026
http://www.google.com/patents/EP1219632A1?cl=en

The preparation method of the present invention includes the following single- or multi-step procedures:
1. Method for the preparation of 11β-[4-(N-methyl-N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (IV) which includes the following steps:
(1) Preparation of Grignard reagent (III)

4-bromo-N-methyl-N-cyclohexylaniline (II) is reacted with magnesium in tetrahydrofuran (THF) to obtain Grignard reagent of formula (III).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (III) prepared in step (1) are brought to an additive reaction to obtain compound of formula (V).
(3) Hydrolytic reaction

The compound of formula (V) prepared in step (2) is subjected to a hydrolytic reaction to obtain compound of form (VI).
2. Method for preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI) which includes the following steps:
(1) Preparation of Grignard reagent of formula (IX)

4-bromo-N-cyclohexylaniline (VII) is first protected by trimethylchlorosilane, then reacted with magnesium in THF to obtain Grignard reagent of formula (IX).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (IX) prepared in step (1) are brought to an additive reaction to obtain compound of formula (X).
(3) Hydrolytic reaction

The compound of formula (X) prepared in step (2) is subjects to a hydrolytic reaction to obtain compound of formula (XI).
Example 2:
-

-
9g 4-bromo-N-cyclohexylaniline (VII) (CA registration number [113388-04-8], see Synthetic Communications, 1986, 16(13): 1641-1645 for its preparation) was placed into a four-necked flask and 15 ml (1.5 mol/L) n-BuLi solution in n-hexane. The mixture was stirred for 30 min at room temperature. Then 8 g trimethylsilyl chloride (Me3SiCl) was added and the mixture was stirred for 1 hour. Solvent and excessive Me3SiCl was evaporated under reduced pressure to yield 4-bromo-(N-cyclohexyl-N-trimethylsilylaniline) (VIII) which was formulated into a solution with 7.5 ml anhydrous tetrahydrofuran for further use.
-
1.3 g magnesium was placed into a four-necked flask and a small amount of the above solution was added dropwise and slowly at 40°C. After completion of addition, the temperature was kept for 1 hour to yield a solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenylmagnesium bromide (IX) in tetrahydrofuran for further use.
- Preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI)(1) Preparation of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX)

(2) Preparation of 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cylohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).

- 5g 3,3-ethylenedioxy-5,10-epoxy-17α-(1-propinyl)-17β-hydroxy-9(11)-estrene (IV) was placed into a four-necked flask and 10 ml anhydrous tetrahydrofuran and a catalytic amount of cuprous chloride (Cu2Cl2) added. Then solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX) in tetrahydrofuran was added dropwise and slowly while controlling the temperature below 5°C. After completion of addition, the mixture was allowed to react for 2 hours at room temperature and to stand overnight. Saturated ammonium chloride aqueous solution was added and the tetrahydrofuran layer separated which was washed with saturated ammonium chloride solution. The solution in tetrahydrofuran was washed with saturated saline and dried over anhydrous sodium sulfate. Evaporation of tetrahydrofuran under reduced pressure yielded a residual which was chromatographed on silica gel column using cyclohexane: acetone (5:1) as developing agent to yield 3 g 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).
-
IR (KBr) cm-1: 3420 (C5, C17-OH), 1610, 1510 (benzene backbone), 840, 808 (ArH).
1H NMR (CDCl3) δ ppm: 0.52(3H, S, C13-CH3), 2.72(3H, S, N-CH3), 3.92(4H, m, -O-CH2CH2-O-), 4.24(1H, m, C11-H), 6.65-7.00 (4H, ArH).
(3) Preparation of 11β- [4- (N-cyclohexylamino)phenyl] -17α- (1-propinyl) -17β-hydroxy-4,9-estradiene-3-one (XI).

- 1.5g 3,3-ethylenedioxy-5,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene (X) and 0.75 g para-toluenesulfonic acid (PTS) were dissolved in 15 ml 90 % ethanol (v/v). The mixture was stirred for 2 hours while controlling the temperature at 40°C-50°C. After completion of the reaction, the reactant was poured into diluted sodium hydroxide aqueous solution, extracted with dichloroethane, washed with water to neutrality, and dried over anhydrous sodium sulfate. Evaporation of the solvent and chromatography on silica gel column using cyclohexane: ethyl acetate (5:1) as developing agent yielded 0.9 g 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI).
-
IR (KBr) cm-1: 3400 (C17-OH), 1658 (unsaturated ketone), 1613, 1514 (benzene backbone), 865, 810 (ArH).
1H NMR (CDCl3) δ ppm: 0.50 (3H, S, C13-CH3), 1.76 (3H, S, C≡C-CH3), 4.32(1H, S, C11-H), 5.75(1H, S, C4-H), 6.9-7.10 (4H, ArH).
PATENT
PATENT
Example 1
Race meters mifepristone synthetic routes:

Epoxy adduct match rice mifepristone
(N- hexylamino methylcyclohexyl) phenyl magnesium bromide (1) 4-
In the four-necked flask, 1.4 g of magnesium into pieces (Mg) and 10 ml of anhydrous tetrahydrofuran (THF), no iodine or add a little change, at about 50 ° C, a solution of 10.86 g of 4-bromo-methyl -N- cyclohexyl aniline (dissolved in 24 ml of anhydrous tetrahydrofuran) dropwise Bi, incubation was continued for 1 hour with stirring to give 4- (N- methyl-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran solution (to be used in the next step an addition reaction ).
(2) 3,3-ethylenedioxy -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propyl block-yl) -9 (10) – Preparation of estra-ene (adduct) of
In the four-necked flask, into 5 g of 3,3-ethylenedioxy-5,10-epoxy -17 α – (1- propynyl) – 17 (3 – hydroxy – 9 (11) – estra-ene (epoxy), 29.1 ml anhydrous tetrahydrofuran (THF) and 0.1 g cuprous chloride (of Cu 2 of Cl 2 ), a solution of 4- (N- methyl -N-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran
Nan solution, temperature control 5. C, the drop was completed, the incubation was continued for 5 hours, the reaction was completed, the reaction solution was poured into saturated aqueous ammonium chloride solution, points to the water layer, the organic layer was washed with saturated ammonium chloride solution, the aqueous layer extracted with ethyl acetate number times, the organic layers combined, washed with saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, and concentrated under reduced pressure, a silica gel column, eluent cyclohexane: acetone = (5: 1) to give 3,3-ethylene dioxo -5 α, 17 β – dihydroxy -11 β – [4- (- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – female steroidal women (adduct) solid 6 grams.
IR. ‘KBi cm- ^ SlS OI ^ ^ -OH lS jSlS benzene backbone), 819 (aromatic hydrogen). NMR Ή: (CDC1 3 ) ppm by [delta]: 0.47 (3H, the S, the C IR CH 3 ), 1.88 (3H, the S, the C ≡ the C-CH 3 ), 2.72 (3H, the S, the N-CH 3 ), 6.65- 7.03 (4H, ArH) O
(3) 11 β – [4- (N- methyl -N- cyclohexylamino) phenyl] -17 α – (1- propynyl) -17 β – hydroxy-estra-4,9-diene – Preparation of 3-one (match rice mifepristone) of
‘2.5 g of p-toluenesulfonic acid (PTS) and 5 grams of 3,3-ethylenedioxythiophene -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – estra-ene (adduct) was dissolved in 50 ml of ethanol 90% (V / V), and at 5 ° C – 40 ° C the reaction was stirred 3 hours, the reaction solution was poured into dilute aqueous sodium hydroxide solution, the precipitated solid was suction filtered, washed with water until neutral, the filter cake was dissolved in 50 ml of ethyl acetate, then with saturated aqueous sodium chloride solution to the water layer was evaporated part of the solvent, the precipitated solid was suction filtered, and dried to give a pale yellow solid 11 β – [4- (Ν- -N- methyl-cyclohexylamino)] -17 α – (1- propynyl) -17 β – hydroxy estra-4,9-dien-3-one (match rice mifepristone) 3 grams.
^ Cm & lt IRCKB 1 : 3447 (the C . 17 -OH), among 1655 (unsaturated ketone), 1607,1513 (benzene backbone), 865,819 (aromatic hydrogen).
NMR ¾: (CDC1 3 ) ppm by [delta]: 0.56 (3H, the S 5 the C 13 -CH 3 ), 1.89 (3H, the S 5 -C ≡ the C-the CH3), 2.74 (3H, the S, the N-the CH3), 4.34 ( lH, the S, the C N -H), 5.75 (lH, the S, the C 4 -H), 6.68-6.99 (4H, ArH).
PATENT
PATENT
PAPER
Volume 878, Issues 7–8, 1 March 2010, Pages 719–723
Determination of cymipristone in human plasma by liquid chromatography–electrospray ionization-tandem mass spectrometry
doi:10.1016/j.jchromb.2010.01.027
Abstract
A rapid, specific and sensitive liquid chromatography–electrospray ionization-tandem mass spectrometry method was developed and validated for determination of cymipristone in human plasma. Mifepristone was used as the internal standard (IS). Plasma samples were deproteinized using methanol. The compounds were separated on a ZORBAX SB C18 column (50 mm × 2.1 mm i.d., dp 1.8 μm) with gradient elution at a flow-rate of 0.3 ml/min. The mobile phase consisted of 10 mM ammonium acetate and acetonitrile. The detection was performed on a triple-quadruple tandem mass spectrometer by selective reaction monitoring (SRM) mode via electrospray ionization. Target ions were monitored at [M+H]+m/z 498 → 416 and 430 → 372 in positive electrospray ionization (ESI) mode for cymipristone and IS, respectively. Linearity was established for the range of concentrations 0.5–100 ng/ml with a coefficient correlation (r) of 0.9996. The lower limit of quantification (LLOQ) was identifiable and reproducible at 0.5 ng/ml. The validated method was successfully applied to study the pharmacokinetics of cymipristone in healthy Chinese female subjects.
CHEMICAL ABSTRACTS, vol. 115, no. 25, 23 December 1991 (1991-12-23) Columbus, Ohio, US; abstract no. 270851g, X. ZHAO ET AL.: “Synthesis and terminating early pregnancy effect of mifepristone derivatives” page 117; XP002219009 & ZHONGGUO YAOKE DAXUE XUEBAO, vol. 22, no. 3, 1991, pages 133-136,
//////////Cymipristone, Saimisitong, NDA Filed , china, Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
WO 2016018024, DAPAGLIFLOZIN, HANMI FINE CHEMICAL CO., LTD, NEW PATENT
![]()

(S) – propylene glycol and water, 1: 1 crystalline complex
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
KIM, Ki Lim; (KR).
PARK, Chulhyun; (KR).
LEE, Jaeheon; (KR).
CHANG, Young-kil; (KR)

The present invention relates to a crystalline composite comprising dapagliflozin and a method for preparing the same. More specifically, the present invention provides a novel crystalline composite comprising dapagliflozin, which is an SGLT2 inhibitor, and a preparing method capable of economically preparing the novel crystalline composite at high purity.

Best Mode for Carrying out the Invention
Mode for the Invention
TABLE 1
| column | Ascentis Express RP-Amide 4.6mm × 150mm (diameter × height), 2.7㎛ (Aldrich) |
| The mobile phase | A: Formic acid 1mL/1000mL in H 2 OB: Formic acid 1mL/1000mL in Acetonitrile (ACN) |
| Test Solution | Acetonitrile Test specimen 5mg / 10mL in 50% (ACN) |
| Column temperature | 25 ℃ |
| Wavelength detector | UV, 220nm |
| Dose | 3 ㎕ |
| Flow rate | 0.7 mL / min |
| Operating hours | 40 min |
Table 2
| Gradient systems | ||
| Time (min) | Mobile phase A (%) | Mobile phase B (%) |
| 0 | 75 | 25 |
| 0-25 | 35 | 65 |
| 25-26 | 30 | 70 |
| 26-29 | 30 | 70 |
| 29-35 | 75 | 25 |
| 35-40 | 75 | 25 |
Claims
According to claim 1, wherein said crystalline complex is in the X- ray diffraction pattern of 9.7, 11.1, 13.7, 17.3, 18.7, 20.0, 20.4, 21.4, 27.5, 33.9, 36.2, 40.4, and the characteristic peaks at 2θ of 43.9 ± 0.2 ° containing crystalline complexes.
According to claim 4, wherein the mixing ratio by the spirit and mannitol dapa glyph is 1: 0.5 to 2 mole ratio, the method of producing a crystalline complex.
FIGURES
 CEO, YOUNG KIL CHANG
CEO, YOUNG KIL CHANG
/////////WO 2016018024, DAPAGLIFLOZIN, HANMI FINE CHEMICAL CO., LTD, New patent
β-GLUCAN . Food Ingredient Sourced From Yeast, Shanghai Genon Biotech Co.,Ltd

Diagram showing orientation and location of different beta-glucan linkages.

Examples of various β-glucan glycosidic linkages.
beta 1, 3- glucan and beta 1, 6- glucan.
Food Ingredient Sourced From Yeast

The shiitake mushroom contains beta-glucans.
β-Glucans (beta-glucans) are polysaccharides of D-glucose monomers linked by β-glycosidic bonds. β-glucans are a diverse group of molecules that can vary with respect to molecular mass, solubility, viscosity, and three-dimensional configuration. They occur most commonly as cellulose in plants, the bran of cereal grains, the cell wall of baker’s yeast, certain fungi, mushrooms and bacteria. Some forms of beta glucans are useful in human nutrition as texturing agents and as soluble fibersupplements, but can be problematic in the process of brewing.
Oat is a rich source of the water-soluble fibre (1,3/1,4) β-glucan, and its effects on health have been extensively studied the last 30 years. Oat β-glucans are the only dietary fiber currently recognized by the European Food Safety Authority (EFSA) to be able to reduce a disease risk. Oat β-glucans can be highly concentrated in different types of oat brans.
“Barley has more beta glucan fiber than any other grain” claims a report on DiabetesHealth website ; 11 sources are listed.
Yeast and medicinal mushroom derived β-glucans are notable for their ability to modulate the immune system. One study has shown that insoluble (1,3/1,6) β-glucan, has greater biological activity than that of its soluble (1,3/1,4) β-glucan counterparts.The differences between β-glucan linkages and chemical structure are significant in regards to solubility, mode of action, and overall biological activity.
β-GLUCAN
‘Gecono’ β-Glucan (GNP80), derived from fresh food grade brewer’s yeast or baker’s yeast, is akind of ‘new resource food material’ developed by unique innovative biotechnologies. Its maincomponent is yeast sourced immunocompetent polysaccharide which has two isomers structured asbeta 1, 3- glucan and beta 1, 6- glucan. The former one which can greatly enhance human immunity isproved to have the anti-tumor, anti-radiation, anti-aging and free radical scavenging activities. It is animportant biological effect response agent.
Appearance: White or light yellow powder.
Features:
●A good Immune activator.
●A powerful free radical scavenger.
●Activate macrophages or neutrophil leukocyte to scavenge cell debris caused by radiation.
●Help the macrophages recognize and destroy the mutated cells.
●Help speed up the recovery of damaged tissue to produce cell factor( IL-1) .
●Enhance the activities of the other substances like antibiotics, antifungal and antiparasitic.
●Reduce the low-density lipoprotein( LDL) level and increase the high-density lipoprotein(HDL)
level in the blood to reduce the hyperlipidemiaoccurrence.
Applications:
●Health food supplements.
●Capsules and tablet health food.
●Beverages & functional oral liquid.
●Pharmaceutical & cosmetic ingredients.
●Other anti-aging,anti-radiation functional foods.
Package:25kg / bag, double-ply composite bag.
Shelf Life:24 months
Storage:Please store in dry condition and avoid explosion in open air. No shipment with noxious
chemicals.
Shanghai Genon Biotech Co.,Ltd
Address:No.88 Cailun Road Zhangjiang Hi-Tech Park, Shanghai, China Post Code:201210
Contact:Amy Tel:0086-21-5138 0613 Mobile: 15201937160 Fax:0086-21-58951012
Email: guyimei@hotmail.com ; amygoo@cngenon.com
Website:http://www.breweryeast.cn
cut paste of mail
Dear Sirs,
We got your info on line, and we are professional manufacturer of Beta 1,3,1,6-D-Glucan from 100% natural Brewer’s Yeast in Shanghai, China.
Beta-glucan can strengthen the immune system and in turn fend off cold,flu and even cancer. Additonally,beta-glucan increases the body`s denfense against the harmful effects of stress.
In additon to being available in food, beta-glucan supplements can help with following health problems:
·allergies ·asthma ·cancer
·Crohn`s disease ·chronic fatigue syndrome
·diabetes ·fibromyalgia
·high cholesterol ·reheumatoid arthritis
·ulcerative colitis
Beta glucan 70%min / 80%min, the price is very competitive now.
We have 4 factories in China to produce brewer yeast series products: yeast powder, yeast extract (seasoning, fermentation), yeast cellwall, selenium yeast, etc.
Feel free to contact me if you are interested.
Best regards
Amy
Micro protein: Yeast extract, Beta-glucan, Yeast cell wall, Organic selenium and Brewer yeast etc.
Animal protein: Hemoglobin Plasma and Nutritional Peptide etc.
With Fine Quality and Competitive Price.
Skype: amy007387
Mobile: +86-15201937160
Tel:+86-21-51380613
E-mail: guyimei@hotmail.com
Fax:86-21-58951012
Shanghai Genon Biotech Co.,Ltd.
