
Home » Posts tagged 'IMMU 132'
Tag Archives: IMMU 132
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Sacituzumab govitecan-hziy

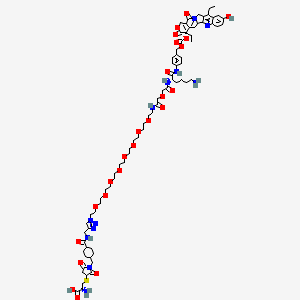

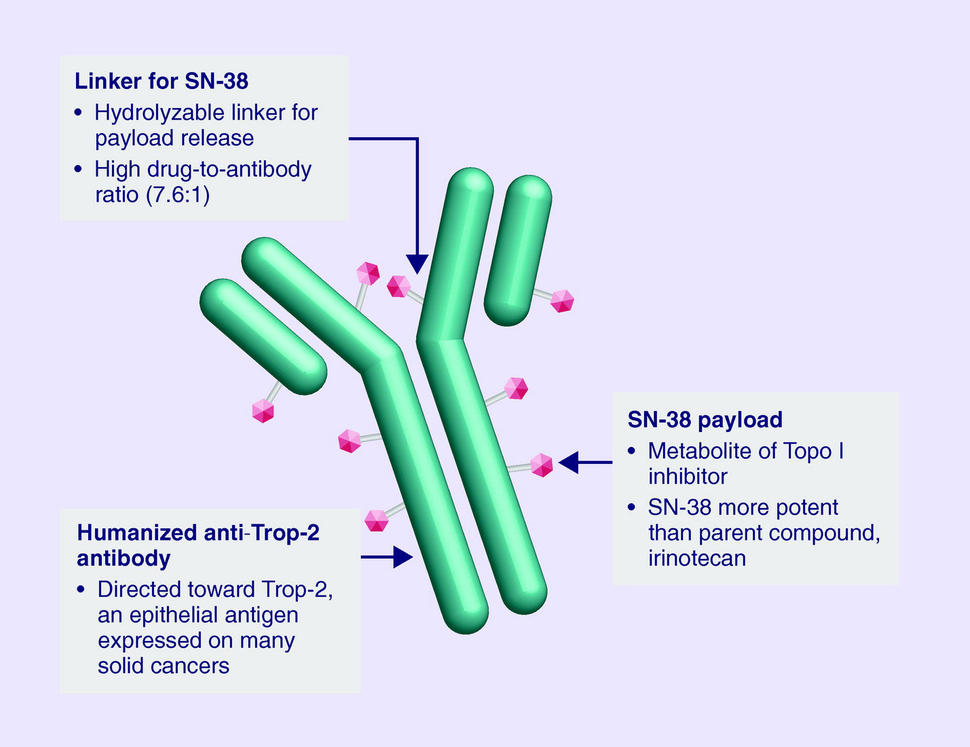
Sacituzumab govitecan-hziy
1601.8 g/mol
(2R)-2-amino-3-[1-[[4-[[1-[2-[2-[2-[2-[2-[2-[2-[2-[2-[[2-[2-[[(2S)-6-amino-1-[4-[[(19S)-10,19-diethyl-7-hydroxy-14,18-dioxo-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaen-19-yl]oxycarbonyloxymethyl]anilino]-1-oxohexan-2-yl]amino]-2-oxoethoxy]acetyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethyl]triazol-4-yl]methylcarbamoyl]cyclohexyl]methyl]-2,5-dioxopyrrolidin-3-yl]sulfanylpropanoic acid
Trodelvy
- hRS 7SN38
- hRS7-SN38
- IMMU 132
- IMMU-132
CAS: 1491917-83-9
UNII-DA64T2C2IO component ULRUOUDIQPERIJ-PQURJYPBSA-N
UNII-SZB83O1W42 component ULRUOUDIQPERIJ-PQURJYPBSA-N
| Efficacy | Antineoplastic, Topoisomerase I inhibitor |
|---|---|
| Disease | Breast cancer (triple negative) |
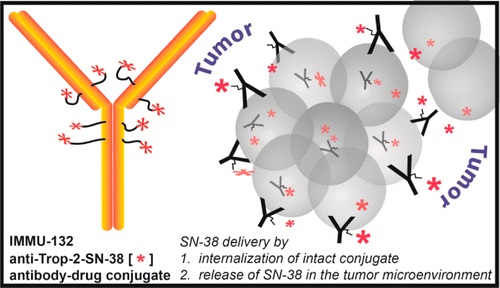

Sacituzumab Govitecan is an antibody drug conjugate containing the humanized monoclonal antibody, hRS7, against tumor-associated calcium signal transducer 2 (TACSTD2 or TROP2) and linked to the active metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38), with potential antineoplastic activity. The antibody moiety of sacituzumab govitecan selectively binds to TROP2. After internalization and proteolytic cleavage, SN-38 selectively stabilizes topoisomerase I-DNA covalent complexes, resulting in DNA breaks that inhibit DNA replication and trigger apoptosis. TROP2, also known as epithelial glycoprotein-1 (EGP-1), is a transmembrane calcium signal transducer that is overexpressed by a variety of human epithelial carcinomas; this antigen is involved in the regulation of cell-cell adhesion and its expression is associated with increased cancer growth, aggressiveness and metastasis.
NEW DRUG APPROVALS
one time
$10.00
CLIP
FDA Approves Trodelvy®, the First Treatment for Metastatic Triple-Negative Breast Cancer Shown to Improve Progression-Free Survival and Overall Survival
– Trodelvy Significantly Reduced the Risk of Death by 49% Compared with Single-Agent Chemotherapy in the Phase 3 ASCENT Study –
– Trodelvy is Under Regulatory Review in the EU and in the United Kingdom, Canada, Switzerland and Australia as Part of Project Orbis –April 07, 2021 07:53 PM Eastern Daylight Time
FOSTER CITY, Calif.–(BUSINESS WIRE)–Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted full approval to Trodelvy® (sacituzumab govitecan-hziy) for adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. The approval is supported by data from the Phase 3 ASCENT study, in which Trodelvy demonstrated a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)), extending median PFS to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also extended median overall survival (OS) to 11.8 months vs. 6.9 months (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death.
Trodelvy is directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including TNBC, where high expression is associated with poor survival and relapse. Prior to the FDA approval of Trodelvy, patients with previously treated metastatic TNBC had few treatment options in this high unmet-need setting. The FDA granted accelerated approval to Trodelvy in April 2020 based on objective response rate and duration of response results in a Phase 1/2 study. Today’s approval expands the previous Trodelvy indication to include treatment in adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“Women with triple-negative breast cancer have historically had very few effective treatment options and faced a poor prognosis,” said Aditya Bardia, MD, MPH, Director of Breast Cancer Research Program, Mass General Cancer Center and Assistant Professor of Medicine at Harvard Medical School, and global principal investigator of the ASCENT study. “Today’s FDA approval reflects the statistically significant survival benefit seen in the landmark ASCENT study and positions sacituzumab govitecan-hziy as a potential standard of care for pre-treated TNBC.”
“A metastatic TNBC diagnosis is frightening. As an aggressive and difficult-to-treat disease, it’s a significant advance to have an FDA-approved treatment option with a proven survival benefit for patients with metastatic disease that continues to progress,” said Ricki Fairley, Founder and CEO of Touch, the Black Breast Cancer Alliance. “For far too long, people with metastatic TNBC had very few treatment options. Today’s news continues the progress of bringing more options to treat this devastating disease.”
Among all patients evaluable for safety in the ASCENT study (n=482), Trodelvy had a safety profile consistent with the previously approved FDA label. The most frequent Grade ≥3 adverse reactions for Trodelvy compared to single-agent chemotherapy were neutropenia (52% vs. 34%), diarrhea (11% vs. 1%), leukopenia (11% vs. 6%) and anemia (9% vs. 6%). Adverse reactions leading to treatment discontinuation occurred in 5% of patients receiving Trodelvy.
“Today’s approval is the culmination of a multi-year development program and validates the clinical benefit of this important treatment in metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Building upon this milestone, we are committed to advancing Trodelvy with worldwide regulatory authorities so that, pending their decision, Trodelvy may become available to many more people around the world who are facing this difficult-to-treat cancer.”
Regulatory submissions for Trodelvy in metastatic TNBC have been filed in the United Kingdom, Canada, Switzerland and Australia as part of Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE) that provides a framework for concurrent submission and review of oncology products among international partners, as well as in Singapore through our partner Everest Medicines.The European Medicines Agency has also validated a Marketing Authorization Application for Trodelvy in the European Union. All filings are based on data from the Phase 3 ASCENT study.
Trodelvy Boxed Warning
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein frequently expressed in multiple types of epithelial tumors, including metastatic triple-negative breast cancer (TNBC), where high expression is associated with poor survival and relapse.
Trodelvy is also being developed as an investigational treatment for metastatic urothelial cancer, hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER 2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
About Triple-Negative Breast Cancer (TNBC)
TNBC is an aggressive type of breast cancer, accounting for approximately 15% of all breast cancers. The disease is diagnosed more frequently in younger and premenopausal women and is more prevalent in African American and Hispanic women. TNBC cells do not have estrogen and progesterone receptors and have limited HER 2. Medicines targeting these receptors therefore are not typically effective in treating TNBC.
About the ASCENT Study
The Phase 3 ASCENT study, an open-label, active-controlled, randomized confirmatory trial, enrolled more than 500 patients with relapsed/refractory metastatic triple-negative breast cancer (TNBC) who had received two or more prior systemic therapies (including a taxane), at least one of them for metastatic disease. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary efficacy outcome was progression-free survival (PFS) in patients without brain metastases at baseline, as measured by a blinded, independent, centralized review using RECIST v1.1 criteria. Additional efficacy measures included PFS for the full population (all patients with and without brain metastases) and overall survival (OS). More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Important Safety Information for Trodelvy
BOXED WARNING: NEUTROPENIA AND DIARRHEA
- Severe, life-threatening, or fatal neutropenia may occur. Withhold TRODELVY for absolute neutrophil count below 1500/mm3 or neutropenic fever. Monitor blood cell counts periodically during treatment. Consider G-CSF for secondary prophylaxis. Initiate anti-infective treatment in patient with febrile neutropenia without delay.
- Severe diarrhea may occur. Monitor patients with diarrhea and give fluid and electrolytes as needed. Administer atropine, if not contraindicated, for early diarrhea of any severity. At the onset of late diarrhea, evaluate for infectious causes and, if negative, promptly initiate loperamide. If severe diarrhea occurs, withhold TRODELVY until resolved to ≤ Grade 1 and reduce subsequent doses.
CONTRAINDICATIONS
- Severe hypersensitivity to TRODELVY
WARNINGS AND PRECAUTIONS
Neutropenia: Dose modifications may be required due to neutropenia. Neutropenia occurred in 62% of patients treated with TRODELVY, leading to permanent discontinuation in 0.5% of patients. Grade 3-4 neutropenia occurred in 47% of patients. Febrile neutropenia occurred in 6%.
Diarrhea: Diarrhea occurred in 64% of all patients treated with TRODELVY. Grade 3 diarrhea occurred in 12% of patients. Neutropenic colitis occurred in 0.5% of patients. Withhold TRODELVY for Grade 3-4 diarrhea and resume when resolved to ≤ Grade 1. At onset, evaluate for infectious causes and if negative, promptly initiate loperamide, 4 mg initially followed by 2 mg with every episode of diarrhea for a maximum of 16 mg daily. Discontinue loperamide 12 hours after diarrhea resolves. Additional supportive measures (e.g., fluid and electrolyte substitution) may also be employed as clinically indicated. Patients who exhibit an excessive cholinergic response to treatment can receive appropriate premedication (e.g., atropine) for subsequent treatments.
Hypersensitivity and Infusion-Related Reactions: TRODELVY can cause severe and life-threatening hypersensitivity and infusion-related reactions, including anaphylactic reactions. Hypersensitivity reactions within 24 hours of dosing occurred in 37% of patients. Grade 3-4 hypersensitivity occurred in 1% of patients. The incidence of hypersensitivity reactions leading to permanent discontinuation of TRODELVY was 0.4%. Pre-infusion medication is recommended. Observe patients closely for hypersensitivity and infusion-related reactions during each infusion and for at least 30 minutes after completion of each infusion. Medication to treat such reactions, as well as emergency equipment, should be available for immediate use.
Nausea and Vomiting: Nausea occurred in 67% of all patients treated with TRODELVY. Grade 3-4 nausea occurred in 5% of patients. Vomiting occurred in 40% of patients and Grade 3-4 vomiting occurred in 3% of these patients. Premedicate with a two or three drug combination regimen (e.g., dexamethasone with either a 5-HT3 receptor antagonist or an NK-1 receptor antagonist as well as other drugs as indicated) for prevention of chemotherapy-induced nausea and vomiting (CINV). Withhold TRODELVY doses for Grade 3 nausea or Grade 3-4 vomiting and resume with additional supportive measures when resolved to Grade ≤ 1. Additional antiemetics and other supportive measures may also be employed as clinically indicated. All patients should be given take-home medications with clear instructions for prevention and treatment of nausea and vomiting.
Increased Risk of Adverse Reactions in Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1)*28 allele are at increased risk for neutropenia, febrile neutropenia, and anemia and may be at increased risk for other adverse reactions with TRODELVY. The incidence of Grade 3-4 neutropenia in genotyped patients was 69% in patients homozygous for the UGT1A1*28, 48% in patients heterozygous for the UGT1A1*28 allele and 46% in patients homozygous for the wild-type allele. The incidence of Grade 3-4 anemia in genotyped patients was 24% in patients homozygous for the UGT1A1*28 allele, 8% in patients heterozygous for the UGT1A1*28 allele, and 10% in patients homozygous for the wild-type allele. Closely monitor patients with known reduced UGT1A1 activity for adverse reactions. Withhold or permanently discontinue TRODELVY based on severity of the observed adverse reactions in patients with evidence of acute early-onset or unusually severe adverse reactions, which may indicate reduced UGT1A1 function.
Embryo-Fetal Toxicity: Based on its mechanism of action, TRODELVY can cause teratogenicity and/or embryo-fetal lethality when administered to a pregnant woman. TRODELVY contains a genotoxic component, SN-38, and targets rapidly dividing cells. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TRODELVY and for 6 months after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TRODELVY and for 3 months after the last dose.
ADVERSE REACTIONS
In the ASCENT study (IMMU-132-05), the most common adverse reactions (incidence ≥25%) were nausea, neutropenia, diarrhea, fatigue, alopecia, anemia, vomiting, constipation, rash, decreased appetite, and abdominal pain. The most frequent serious adverse reactions (SAR) (>1%) were neutropenia (7%), diarrhea (4%), and pneumonia (3%). SAR were reported in 27% of patients, and 5% discontinued therapy due to adverse reactions. The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the ASCENT study were reduced hemoglobin, lymphocytes, leukocytes, and neutrophils.
DRUG INTERACTIONS
UGT1A1 Inhibitors: Concomitant administration of TRODELVY with inhibitors of UGT1A1 may increase the incidence of adverse reactions due to potential increase in systemic exposure to SN-38. Avoid administering UGT1A1 inhibitors with TRODELVY.
UGT1A1 Inducers: Exposure to SN-38 may be substantially reduced in patients concomitantly receiving UGT1A1 enzyme inducers. Avoid administering UGT1A1 inducers with TRODELVY
Please see full Prescribing Information, including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Sacituzumab govitecan, sold under the brand name Trodelvy, is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate indicated for the treatment of metastatic triple-negative breast cancer (mTNBC) in adult patients that have received at least two prior therapies.[1][2]
The most common side effects are nausea, neutropenia, diarrhea, fatigue, anemia, vomiting, alopecia (hair loss), constipation, decreased appetite, rash and abdominal pain.[1][2] Sacituzumab govitecan has a boxed warning about the risk of severe neutropenia (abnormally low levels of white blood cells) and severe diarrhea.[1][2] Sacituzumab govitecan may cause harm to a developing fetus or newborn baby.[1] Women are advised not to breastfeed while on sacituzumab govitecan and 1 month after the last dose is administered.[3]
The U.S. Food and Drug Administration (FDA) considers it to be a first-in-class medication.[4]
Mechanism
Sacituzumab govitecan is a conjugate of the humanized anti-Trop-2 monoclonal antibody linked with SN-38, the active metabolite of irinotecan.[5] Each antibody having on average 7.6 molecules of SN-38 attached.[6] SN-38 is too toxic to administer directly to patients, but linkage to an antibody allows the drug to specifically target cells containing Trop-2.
Sacituzumab govitecan is a Trop-2-directed antibody and topoisomerase inhibitor drug conjugate, meaning that the drug targets the Trop-2 receptor that helps the cancer grow, divide and spread, and is linked to topoisomerase inhibitor, which is a chemical compound that is toxic to cancer cells.[1] Approximately two of every ten breast cancer diagnoses worldwide are triple-negative.[1] Triple-negative breast cancer is a type of breast cancer that tests negative for estrogen receptors, progesterone receptors and human epidermal growth factor receptor 2 (HER2) protein.[1] Therefore, triple-negative breast cancer does not respond to hormonal therapy medicines or medicines that target HER2.[1]
Development
Immunomedics announced in 2013, that it had received fast track designation from the US Food and Drug Administration (FDA) for the compound as a potential treatment for non-small cell lung cancer, small cell lung cancer, and metastatic triple-negative breast cancer. Orphan drug status was granted for small cell lung cancer and pancreatic cancer.[7][8] In February 2016, Immunomedics announced that sacituzumab govitecan had received an FDA breakthrough therapy designation (a classification designed to expedite the development and review of drugs that are intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition) for the treatment of patients with triple-negative breast cancer who have failed at least two other prior therapies for metastatic disease.[9][10]
History
Sacituzumab govitecan was added to the proposed INN list in 2015,[11] and to the recommended list in 2016.[12]
Sacituzumab govitecan-hziy was approved for use in the United States in April 2020.[1][13][14][2]
Sacituzumab govitecan-hziy was approved based on the results of IMMU-132-01, a multicenter, single-arm clinical trial (NCT01631552) of 108 subjects with metastatic triple-negative breast cancer who had received at least two prior treatments for metastatic disease.[1][14][2] Of the 108 patients involved within the study, 107 were female and 1 was male.[15] Subjects received sacituzumab govitecan-hziy at a dose of 10 milligrams per kilogram of body weight intravenously on days one and eight every 21 days.[14][15] Treatment with sacituzumab govitecan-hziy was continued until disease progression or unacceptable toxicity.[15] Tumor imaging was obtained every eight weeks.[14][2] The efficacy of sacituzumab govitecan-hziy was based on the overall response rate (ORR) – which reflects the percentage of subjects that had a certain amount of tumor shrinkage.[1][14] The ORR was 33.3% (95% confidence interval [CI], 24.6 to 43.1). [1][14][15] Additionally, with the 33.3% of study participants who achieved a response, 2.8% of patients experienced complete responses.[15] The median time to response in patients was 2.0 months (range, 1.6 to 13.5), the median duration of response was 7.7 months (95% confidence interval [CI], 4.9 to 10.8), the median progression free survival was 5.5 months, and the median overall survival was 13.0 months.[15] Of the subjects that achieved an objective response to sacituzumab govitecan-hziy, 55.6% maintained their response for six or more months and 16.7% maintained their response for twelve or more months.[1][14]
Sacituzumab govitecan-hziy was granted accelerated approval along with priority review, breakthrough therapy, and fast track designations.[1][14] The U.S. Food and Drug Administration (FDA) granted approval of Trodelvy to Immunomedics, Inc.[1]
References
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA Approves New Therapy for Triple Negative Breast Cancer That Has Spread, Not Responded to Other Treatments”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 22 April 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f “Drug Trial Snapshot: Trodelvy”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 29 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ (PDF)https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf. Missing or empty
|title=(help) - ^ “New Drug Therapy Approvals 2020”. U.S. Food and Drug Administration (FDA). 31 December 2020. Retrieved 17 January2021. This article incorporates text from this source, which is in the public domain.
- ^ Sacituzumab Govitecan (IMMU-132), an Anti-Trop-2/SN-38 Antibody-Drug Conjugate: Characterization and Efficacy in Pancreatic, Gastric, and Other Cancers. 2015
- ^ “Novel Agents are Targeting Drivers of TNBC”. http://www.medpagetoday.com. 28 June 2016.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “Sacituzumab govitecan Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 22 April 2020.
- ^ “New Therapy Shows Early Promise, Continues to Progress in Triple-Negative Breast Cancer”. Cure Today.
- ^ “U.S. Food and Drug Administration (FDA) Grants Breakthrough Therapy Designation to Immunomedics for Sacituzumab Govitecan for the Treatment of Patients With Triple-Negative Breast Cancer”(Press release). Immunomedics. 5 February 2016. Retrieved 25 April 2020 – via GlobeNewswire.
- ^ World Health Organization (2015). “International nonproprietary names for pharmaceutical substances (INN): proposed INN: list 113”. WHO Drug Information. 29 (2): 260–1. hdl:10665/331080.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 151–3. hdl:10665/331046.
- ^ “Trodelvy: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 April 2020.
- ^ Jump up to:a b c d e f g h “FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer”. U.S. Food and Drug Administration (FDA). 22 April 2020. Retrieved 23 April 2020. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. The New England Journal of Medicine.
Further reading
- Bardia A, Mayer IA, Vahdat LT, et al. (February 2019). “Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer”. N. Engl. J. Med. 380 (8): 741–751. doi:10.1056/NEJMoa1814213. PMID 30786188.
- Weiss J, Glode A, Messersmith WA, et al. (August 2019). “Sacituzumab govitecan: breakthrough targeted therapy for triple-negative breast cancer”. Expert Rev Anticancer Ther. 19 (8): 673–679. doi:10.1080/14737140.2019.1654378. PMID 31398063. S2CID 199518147.
External links
- “Sacituzumab govitecan”. Drug Information Portal. U.S. National Library of Medicine.
- “Sacituzumab govitecan”. ADC Review.
- “Sacituzumab govitecan”. National Cancer Institute.
- Clinical trial number NCT01631552 for “Phase I/II Study of IMMU-132 in Patients With Epithelial Cancers” at ClinicalTrials.gov
- Sacituzumab govitecan at the US National Library of Medicine Medical Subject Headings (MeSH)
| Monoclonal antibody | |
|---|---|
| Type | ? |
| Source | Humanized (from mouse) |
| Target | Trop-2 |
| Clinical data | |
| Trade names | Trodelvy |
| Other names | IMMU-132, hRS7-SN-38, sacituzumab govitecan-hziy |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a620034 |
| License data | US DailyMed: Sacituzumab_govitecan |
| Pregnancy category | Contraindicated |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only |
| Identifiers | |
| CAS Number | 1491917-83-9 |
| PubChem CID | 91668186 |
| DrugBank | DB12893 |
| ChemSpider | none |
| UNII | M9BYU8XDQ6 |
| KEGG | D10985 |
| Chemical and physical data | |
| Formula | C76H104N12O24S |
| Molar mass | 1601.79 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| show |
//////////sacituzumab govitecan-hziy, fda 2021, approvals 2021, Trodelvy , hRS 7SN38, hRS7-SN38, IMMU 132, IMMU-132, MONOCLONAL ANTIBODY, Sacituzumab govitecan, sacituzumab govitecan-hziy, CANCER, MONOCLONAL ANTIBODIES
#sacituzumab govitecan-hziy, #fda 2021, #approvals 2021, #Trodelvy , #hRS 7SN38, #hRS7-SN38, #IMMU 132, #IMMU-132, #MONOCLONAL ANTIBODY, #Sacituzumab govitecan, #sacituzumab govitecan-hziy, #CANCER, #MONOCLONAL ANTIBODIES
CCC1=C2CN3C(=CC4=C(C3=O)COC(=O)C4(CC)OC(=O)OCC5=CC=C(C=C5)NC(=O)C(CCCCN)NC(=O)COCC(=O)NCCOCCOCCOCCOCCOCCOCCOCCOCCN6C=C(N=N6)CNC(=O)C7CCC(CC7)CN8C(=O)CC(C8=O)SCC(C(=O)O)N)C2=NC9=C1C=C(C=C9)O
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}