
Home » Posts tagged 'idiopathic pulmonary fibrosis'
Tag Archives: idiopathic pulmonary fibrosis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Admilparant
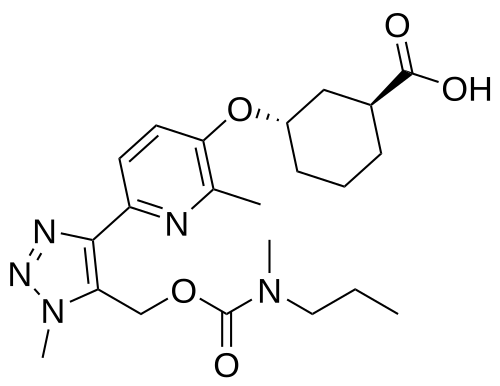
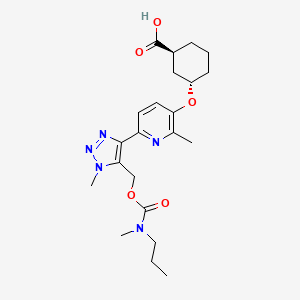
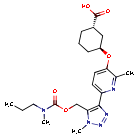
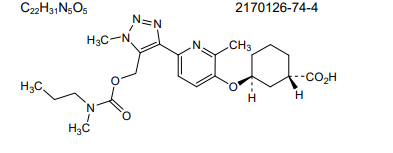
Admilparant, (BMS-986278)
CAS 2170126-74-4
MF C22H31N5O5 MW 445.5 g/mol
(1S,3S)-3-({2-methyl-6-[1-methyl-5-({[methyl(propyl)carbamoyl]oxy}methyl)-1H-1,2,3-triazol-4-l]pyridin-3-yl}oxy)cyclohexane-1-carboxylic acid
lysophosphatidic acid receptor 1 (LPA1) antagonist
- 4UN9AOU6G8
- BMS986278
- (1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic acid
Admilparant is an investigational new drug being developed by Bristol-Myers Squibb for the treatment of idiopathic pulmonary fibrosis (IPF) and progressive pulmonary fibrosis (PPF). It is a first-in-class lysophosphatidic acid receptor 1 (LPA1) antagonist.[1][2]
As of 2024, admilparant is in Phase III clinical trials for both IPF and PPF.[2][3]
SYN
Publication Name: Journal of Medicinal Chemistry, Publication Date: 2021-10-28, PMID: 34709814
DOI: 10.1021/acs.jmedchem.1c01256
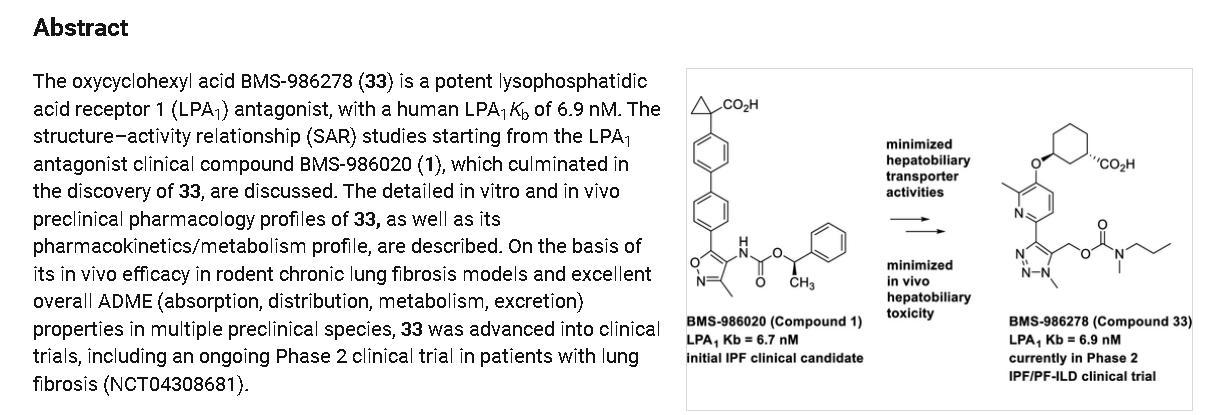
(1S,3S)-3-((2-Methyl-6-(1-methyl-5-(((methyl(propyl)carbamoyl)-oxy)methyl)-1H-1,2,3-triazol-4-yl)pyridin-3-yl)oxy)cyclohexane-1-carboxylic Acid (33). Compound 33 was prepared using the same
synthetic sequence as 25, except that intermediate 42 was reacted with
N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine. 1H NMR (500 MHz, DMSO-d6, 100 °C) δ 11.99−11.46 (m,1H), 7.82 (d, J = 8.3 Hz, 1H), 7.43 (d, J = 8.8 Hz, 1H), 5.65 (s, 2H),
4.89−4.62 (m, 1H), 4.10 (s, 3H), 3.12 (br t, J = 7.2 Hz, 2H), 2.79 (s,3H), 2.69 (tt, J = 9.4, 4.4 Hz, 1H), 2.44 (s, 3H), 2.03 (dt, J = 13.8, 4.5Hz, 1H), 1.92−1.86 (m, 1H), 1.86−1.79 (m, 2H), 1.74−1.68 (m, 1H),
1.68−1.58 (m, 2H), 1.58−1.51 (m, 1H), 1.43 (dq, J = 14.4, 7.1 Hz,2H), 0.76 (br t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, DMSO-d6, 100°C) δ 175.4, 154.7, 150.1, 147.7, 143.9, 141.4, 129.6, 120.0, 118.6, 71.8,
54.5, 49.5, 37.4, 34.4, 33.4, 31.6, 28.7, 27.2, 19.8, 19.4, 18.6, 10.1. m/z446 [M + H]+
. HPLC/UV purity: 99.9% using the following reverse phase chromatographic conditions: Agilent HPLC; Phenomenex Kinetex-C-18; 100 (L) × 4.6 mm2 (i.d.) column; 2.6 μm particle size; wavelength, 220−380 nm; flow rate, 1.0 mL/min; temperature, 35°C; injection volume, 4 μL of 0.25 mg/mL in 1:1 MeCN:H2O; mobilephase A, H2O−0.05% TFA; mobile phase B, MeCN−0.05% TFA; gradient elution, starting at 10−80% B over 10 min and ending at 95% Bafter an additional 4 min; retention time = 8.28 min. Stereoisomeric purity was >99.5% using the following chiral chromatographic conditions: UPC2 Analytical SFC, ChromegaChiral CC4; 250 (L) ×4.6 mm2 (i.d.); 5 μm column; flow rate, 3 mL/min; temperature, 40 °C;injection volume, 10 μL of 0.25 mg/mL in MeCN:MeOH (1:1);mobile phase, 30% MeOH and 70% CO2 at 120 bar retention time =6.05 min. Accurate mass, [M + H]+ at m/z = 446.2398 (−2.03 ppmfrom theoretical for C22H32N5O5). [α]20D = +28.24° (MeOH, c = 0.51).
Elem. Anal. (theoretical): C, 59.31; H, 7.01; N, 15.72. Found: C, 59.35;H, 6.78; N, 15.69. UV (MeOH) at 254 nm (ε = 17,856), 290 nm (ε =7,519), and 296 nm (ε = 8,288). Concentration: adjusted for purity,
0.05154840 g/L or 0.0001157047 mol/L. Melting point = 152−154°C. Accurate mass, [M + H]+ at m/z 466.2398 (−2.03 ppm fromtheoretical for C22H32N5O5).
synthetic sequence as 25, except that intermediate 42 was reacted with N-methylpropan-1-amine instead of 1-cyclobutyl-N-methylmethanamine
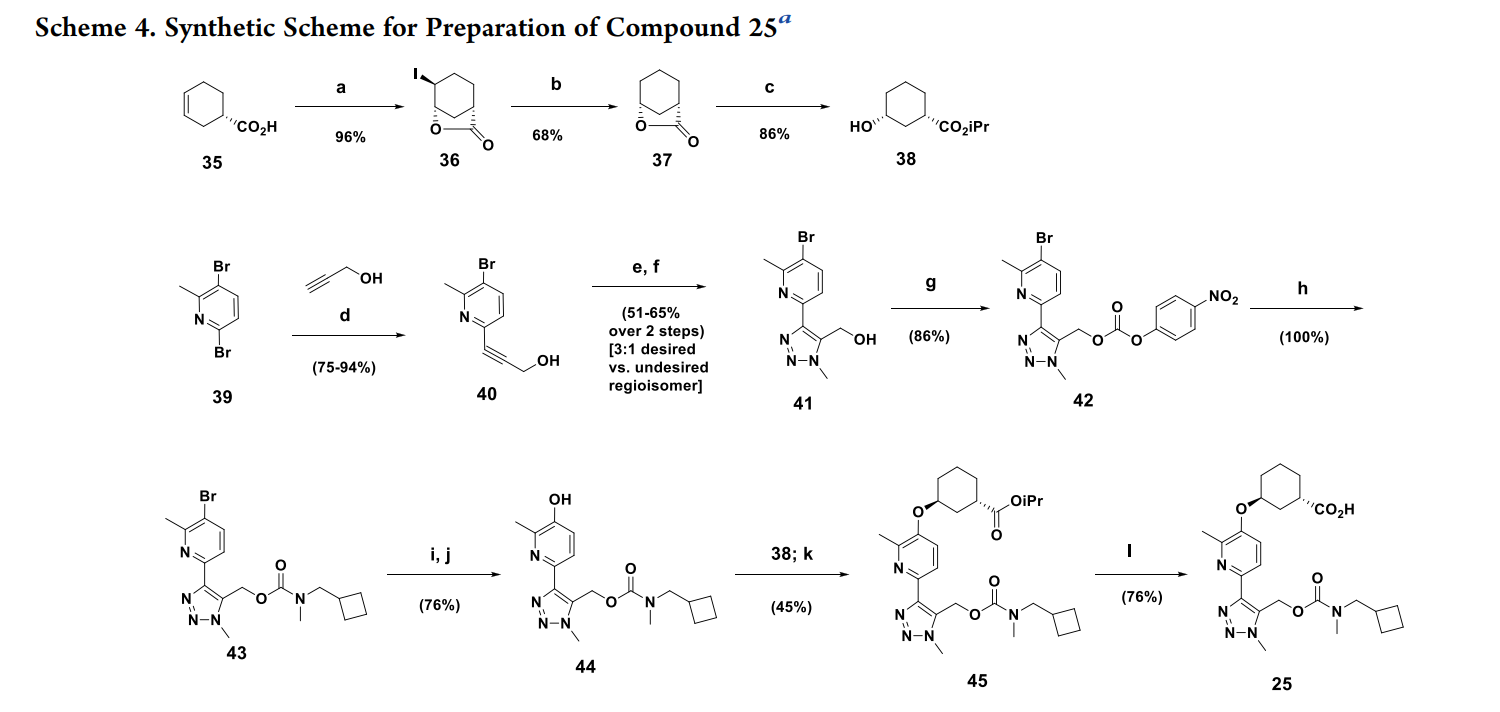
a
Reagents and conditions: (a) I2 (1.1 equiv)/KI (2.5 equiv)/NaHCO3 (3 equiv)/water (96%); (b) H2 (50 psi)/ Pd/C (cat)/Et3N (2 equiv)/EtOAc (68%); (c) CH3COCl (2.5 equiv)/iPrOH (87−95%); d) (Ph3P)2PdCl2 (5%)/ Et3N/CuI (5%)/RT (75−94%); (e) Ru(II)-(Ph3P)2(Me5Cyp)Cl (5%)/TMSCH2N3/dioxane 50 °C/15 h; (f) Bu4NF/0 °C to RT (51−65% over 2 steps; 3:1 desired:undesired regioisomer); (g) 4-nitrophenyl chloroformate/pyridine/CH2Cl2 (86%); (h) N-cyclobutyl N-methylamine/iPr2NEt/CH2Cl2 (100%); (i) B2(pin)2/KOAc/PdCl2(dppf)/THF/80 °C; (j) NaH2BO4/H2O/RT (76% over 2 steps); (k) 38; 1,1′-(azodicarbonyl)dipiperidine/Bu3P/toluene/50 °C (45%); (l)LiOH/H2O/MeOH (76%).
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US208146892&_cid=P20-MFS2PF-83792-1
PATENT
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2022249443-A1Priority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: US-RE49352-EPriority Date: 2016-06-21Grant Date: 2023-01-03
- Carbamoyloxymethyl triazole cyclohexyl acids as LPA antagonistsPublication Number: AU-2021209334-B2Priority Date: 2016-06-21Grant Date: 2023-06-01
- Carbamoyloxymethyltriazole cyclohexylates as LPA antagonistsPublication Number: JP-7312295-B2Priority Date: 2016-06-21Grant Date: 2023-07-20
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: US-2023390249-A1Priority Date: 2016-06-21
- Carbamoyloxymethyltriazolylcyclohexanes as LPA antagonistsPublication Number: CN-109963843-BPriority Date: 2016-06-21Grant Date: 2022-03-11
- Carbamoyloxymethyltriazole cyclohexyl acid as LPA antagonistPublication Number: CN-114601830-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acid as an LPA antagonistPublication Number: KR-102377340-B1Priority Date: 2016-06-21Grant Date: 2022-03-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-20220038537-APriority Date: 2016-06-21
- Carbamoyloxymethyl triazole cyclohexyl acids as lpa antagonistsPublication Number: KR-102463621-B1Priority Date: 2016-06-21Grant Date: 2022-11-03



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- “Admilparant (BMS-986278): Idiopathic Pulmonary Fibrosis Likelihood of Approval”. Pharmaceutical Technology. 25 December 2023. Retrieved 2024-11-23.
- Corte TJ, Behr J, Cottin V, Glassberg MK, Kreuter M, Martinez FJ, et al. (October 2024). “Efficacy and Safety of Admilparant, an LPA1 Antagonist in Pulmonary Fibrosis: A Phase 2 Randomized Clinical Trial”. American Journal of Respiratory and Critical Care Medicine. 211 (2): 230–238. doi:10.1164/rccm.202405-0977OC. PMID 39393084.
- Splete H (16 September 2024). “Admilparant Affects Biomarkers in Pulmonary Fibrosis”. Medscape. Retrieved 2024-11-23.
| Clinical data | |
|---|---|
| Other names | BMS-986278 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2170126-74-4 |
| PubChem CID | 132232205 |
| DrugBank | DB18011 |
| ChemSpider | 115009679 |
| UNII | 4UN9AOU6G8 |
| KEGG | D12657 |
| ChEMBL | ChEMBL5087506 |
| Chemical and physical data | |
| Formula | C22H31N5O5 |
| Molar mass | 445.520 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
/////////Admilparant, BMS 986278, PHASE 3, Bristol-Myers Squibb, idiopathic pulmonary fibrosis, 4UN9AOU6G8
AK 3280
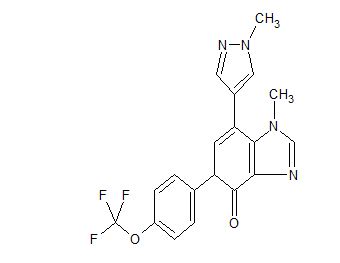
AK-3280
AK 3280; GDC3280; RG 6069
Ci8Hi4N502F3, mass 389.3 g/mol),
ROCHE,
Ark Biosciences , under license from Roche , is developing AK-3280, an antifibrotic agent, for the potential oral treatment of IPF. In July 2018, Ark intended to further clinical development of the drug, for IPF. In June 2019, a phase I trial was planned in Sweden.
- Originator Genentech
- Mechanism of Action Undefined mechanism
- Phase I Interstitial lung diseases
- 19 Jun 2019Ark Biosciences plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in Sweden (PO, Tablet), in August 2019 , (NCT03990688)
- 28 Sep 2018GDC 3280 is still in phase I trials for Interstitial lung diseases (Genentech pipeline, September 2018)
- 28 Jun 2018No recent reports of development identified for phase-I development in Fibrosis(In volunteers) in United Kingdom (PO)
Introduction
GDC 3280 (also known as RG 6069), an orally administered drug, is being developed by Genentech, for the treatment of interstitial lung diseases. Early stage clinical development is underway in the UK.
Company Agreements
In September 2018, Genentech licensed exclusive worldwide development and commercialisation rights of GDC 3280 to Ark Biosciences, for the treatment of idiopathic pulmonary fibrosis
Key Development Milestones
As at September 2018, GDC 3280 is still in phase I development for interstitial lung disease (Genentech pipeline, September 2018).
In December 2015, Genentech completed a phase I trial that evaluated the safety, pharmacokinetics and tolerability of GDC 3280 in healthy volunteers, compared with placebo (GB29751; EudraCT2015-000560-33; NCT02471859). The randomised, double-blind, single and multiple oral dose trial was initiated in June 2015 and enrolled eight volunteers in the UK .
PATENT
WO-2019152863
Novel crystalline salt forms of 1-methyl-7-(1-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-1,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (compound I; presumed to be AK-3280 ), processes for their preparation and compositions comprising them are claimed.
Compound I is an orally available small molecule having the structure:
[0004] Compound I has therapeutic value in several different indications that display fibrotic pathophysiology, including idiopathic pulmonary fibrosis (IPF).
[0005] Idiopathic pulmonary fibrosis is a disease of unknown etiology that occurs mainly in middle-aged and elderly patients, which is characterized by progressive fibrosis of the lung, leading to pulmonary insufficiency and death. Because fibrosis has long been considered to be a clinically irreversible process, treatments have traditionally been focused on managing the symptoms and complications, with little hope of significantly slowing progression of the condition. For many years, mainstay treatments have been typically anti inflammatory, immunosuppressive, and anti-oxidant agents. The effectiveness of these therapies in the treatment of IPF and other fibrotic conditions appears to be minimal and variable, and their side effects are often poorly tolerated by patients.
[0006] New treatment options have only recently become available. Both pirfenidone and nintedanib have been approved for use in the treatment of IPF. Current research efforts to develop new anti-fibrotic agents are targeting multiple mechanisms proposed to be linked to the underlying molecular pathogenic processes. This changing landscape has raised hopes and expectations for what might be achievable with new single agents or combination therapies targeting additional pathways.
Preparation of Compound I and its salts
[0045] A synthesis of Compound I and its tosylate salt is shown in the scheme below:
[0046] l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (5) was synthesized in 4 steps, including a copper-catalyzed coupling reaction e.g., a Goldberg-Ullmann coupling reaction. In another aspect of the invention, intermediate (5) is synthesized using any transition metal-catalyzed coupling reaction. The skilled chemist would know that intermediate (5) could be synthesized from intermediate (4) and compounds
LG
of the general formula: OCF3 , wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
[0047] Compound I was synthesized in 6 steps, using a transition metal cross-coupling reaction, e.g., a Suzuki reaction. In another aspect of the invention, Compound I is synthesized using any cross -coupling reaction. Compound I is synthesized from intermediate 6 containing any leaving group. For example, the skilled chemist would use compounds of
the general formula:
, wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
An alternative synthesis of Compound I and its salts is shown in the scheme below:
Example 13 – Synthesis of Compound I Tosylate Salt
[00183] A process for the formation of mono- and di-tosylate salts of Compound I was developed and a batch was performed to successfully produce the mono-tosylate salt.
Step 1 : Synthesis of2-chloro-N-methyl-3-nitropyridin-4-amine
[00184] A reactor was charged with 2,4-dichloro-3-nitropyridine and 3.0 volumes of DMF. The solution was stirred at 20-25 °C until a clear solution was obtained. The solution was then cooled to 0-5 °C, and 2.1 equivalents of 40% methylamine in water were slowly added over at least 2 hours at 0-5 °C. The reaction mixture was stirred for at least 2 hours at 0-5 °C until conversion to the product was 95% (as measured by HPLC). The reaction mixture was diluted by slowly adding 10 volumes of water over at least 30 minutes at 0-5 °C. The obtained suspension was stirred for at least 60 minutes at 0-5 °C. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 10 volumes of water at 0-5 °C. The damp filter cake was then dried in a flow of dry nitrogen to yield 2-chloro-A-methyl-3-nitropyridin-4-amine in 78% yield.
Step 2: Synthesis of 2-chloro-N4 -methylpyridine-3, 4-diamine
[00185] A reactor was charged with catalyst [2% Pt on charcoal, 59 %wt. water] (0.0004 equivalents Pt), damp 2-chloro-/V-methyl-3-nitropyridin-4-amine from step 1 and 9.4 volumes of THF. The solution was stirred, and then the suspension was transferred from the glass-reactor to an autoclave. The line was rinsed with 1.2 volumes of THF into the autoclave, and the autoclave was purged with nitrogen for 15 minutes at 50 rpm, followed by hydrogen for 15 minutes at 150 rpm. The autoclave was closed, and the hydrogen pressure was adjusted to 2 bar at 20-30 °C. The reaction mixture was stirred for 4-8 hours at 2 bar and 20-30 °C.
[00186] Next, the autoclave was released to atmospheric pressure and purged with nitrogen for at least 15 minutes. Conversion to the product was verified by HPLC, and then the catalyst was removed by filtration. The filtered catalyst was rinsed with 1.3 volumes of THF and the filtrates were combined. The combined filtrates were charged to a second reactor via a particle filter, and the line was rinsed with 0.5 volumes of THF. The solution was concentrated to a final volume of 2.5 volumes by distillation under reduced pressure at 40-45 °C.
[00187] The solution was then diluted with 10 volumes of THF in portions while concentrating the solution to a final volume of 2.5 volumes by distillation under reduced pressure at 45-50 °C. The reactor was purged with nitrogen to atmospheric pressure, and 5.0 volumes of heptane were added to the residue at 40-50 °C. The reaction mixture was cooled over 2 hours to 20-25 °C, and stirring was continued for 1 hour. The reaction mixture was then further cooled to 0-5 °C over 1 hour, and stirring was continued for 1 hour. The precipitated product was collected by filtration, rinsed via the reactor with 5.0 volumes of heptane, and the damp filter cake was dried in a vacuum drying oven at max. 40 °C until loss on drying was < 2 % weight, giving 2-chloro-/V4-methylpyridine-3, 4-diamine in 85% yield.
Step 3 : Synthesis of -inelhyl- 1 ,5-dihvdro-4H-iinidazoi4,5-c h yridin-4-one
[00188] A reactor was charged with 2-chloro-/V4-methylpyridine-3, 4-diamine and 4 volumes of formic acid. The reaction mixture was heated to smooth reflux within one hour, and reflux was maintained for 6 hours. The reaction mixture was then cooled to
approximately 60 °C, and conversion to the product was verified by HPLC.
[00189] The reaction mixture was then concentrated by distillation under reduced pressure at 60-80 °C to a final volume of 2 volumes. The temperature of the solution was adjusted to 60 °C, maintaining the temperature above 50 °C to avoid precipitation.
[00190] Next, a second reactor was charged with 10 volumes of acetone, and heated to gentle reflux. The product solution from the first reactor was slowly transferred to the acetone in the second reactor over 20 minutes, and the line was rinsed with approximately 0.05 volumes of formic acid. Reflux of the obtained suspension was maintained for 15 minutes. The slurry was cooled to 0 °C within 1 hour, and stirring was continued for 1 hour at that temperature. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 3.7 volumes of cold acetone at 0-10 °C. The filter cake was dried in a flow of dry nitrogen or in a vacuum drying oven at 50 °C until loss on drying was < 2% of weight, giving 1 -methyl- 1 ,5-dihydiO-4/7-imidazo[4,5-c]pyndin-4-onc in 95% yield.
Step 4: Synthesis of l-methyl-5-(4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one
[00191] A first reactor (Reactor A) was charged with 1 -methyl- 1 ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent), Cu(0Ac)2 H20 (0.1 mol equivalents), and K2C03 (1.1 mol equivalents). The reactor was closed and the atmosphere replaced with nitrogen.
[00192] Next, l-bromo-4-(trifluoromethoxy)benzene (1.5 mol equivalents) and N-methylpyrrolidinone (5.4 volume equivalents) were added, whereupon a suspension was formed. The suspension was stirred until the temperature had fallen again to approximately 20-25 °C and gas evolution had slowed. The reaction mixture was heated to approximately 130-150 °C at which time a blue/green color was observed, changing to dark brown after some time. The reaction was stirred at 130-150 °C for at least 40 hours. Stirring times of 40 hours up to 72 hours were required to reach an acceptable level of conversion. In general, higher reaction temperatures supported faster conversion.
[00193] Next, the reaction mixture was cooled to approximately 20-30 °C, and 25% aqueous NH3 (0.7 volume equivalents) was added, followed by water (3.5 volume equivalents). The resulting suspension was transferred into a second reactor (Reactor B). Additional water was added (18.1 volume equivalents) to the reaction mixture via Reactor A, followed by n-heptane (3.2 volume equivalents). The resulting suspension was cooled to approximately 0-5 °C, and stirred for approximately 2 hours.
[00194] The suspension was filtered, and the filter cake was washed with water (9.7 volume equivalents). The filter cake was then dissolved in dichloromethane (14.1 volume equivalents) and transferred back into reactor B. To this solution was added water (5.7 volume equivalents) via the filter, followed by 25% aq. NH3(1.6 volume equivalents). The mixture was stirred for approximately 1 hour at approximately 15-25 °C.
[00195] Next, the layers were separated, and dichloromethane was added (3.6 volume equivalents) to the aqueous layer. The biphasic mixture was stirred at approximately 15-25 °C for approximately 20-30 minutes. The layers were separated over a period of at least 1 hour, and to the combined organic layers was added a solution of NH4Cl (2.5 mol equivalents) in water (7.0 volume equivalents). The biphasic mixture was stirred at approximately 15-25 °C for about 20-30 minutes, then the layers were separated over the course of 1 hour.
[00196] The lower organic layer was filtered through a particle filter and diluted with toluene (7.1 volume equivalents) via the filter. The organic layer was concentrated under ambient pressure at approximately 80 °C, until no further liquid was seen to evaporate and a precipitate began to form. Toluene was added (16.6 volume equivalents), then concentrated in vacuo, followed by addition of more toluene (7.1 volume equivalents) and again concentrated in vacuo. The suspension was cooled to approximately 0-5 °C, stirred for approximately 2 hours, and filtered. The filter cake was washed with toluene (2.9 volume equivalents), and dried in vacuo at approximately 50 °C until the loss on drying was 0.5% of the weight to give l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a beige-colored solid in 83.1% yield.
Step 5 : Synthesis of 7-bromo- 1 -methyl-5-(4-( trifluoromethoxy Iphenyl )- l,5- 4H-
imidaz.o[4,5-clpyridin-4-one
[00197] A first reactor (Reactor A) was charged with water (1.8 volume equivalents) and cooled to approximately 0-5 °C, to which was slowly added 96% sulfuric acid (14 mol. equivalents) at approximately 0-20 °C. The temperature of the solution was adjusted to approximately 0-5 °C, and l -mcthyl-5-(4-(tnfluoromcthoxy)phcnyl)-l ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent) was added in 3-4 portions at approximately 0-5 °C. The temperature of the mixture was adjusted to approximately 0-5 °C, and N-bromosuccinimide (1.0 mol equivalents) was slowly added in 3-4 portions, while maintaining the temperature at approximately 0-5 °C.
[00198] The reaction mixture was stirred for about 1 hour at approximately 0-5 °C, and then for an additional 4-16 hours at approximately 0-22 °C. Conversion to the product was confirmed by HPLC, then the reaction mixture was cooled to approximately 0-5 °C.
[00199] A second reactor (Reactor B) was charged with water (42.7 volume equivalents) and cooled to approximately 0-5 °C. The reaction mixture from Reactor A was transferred into the pre-cooled water in Reactor B at a temperature below 30 °C over 2 hours. The reaction was rinsed with water (1.6 volume equivalents), and 50% aqueous sodium hydroxide (25 mol. equivalents) was carefully added at approximately 0-30 °C over about 2 hours until the pH reached 2-5.
[00200] Next, MTBE (6.5 volume equivalents) was added at approximately 0-20 °C, and the mixture was stirred for about 5 minutes. Additional 50% aqueous sodium hydroxide (2 mol. equivalents) was added at approximately 0-30 °C until the pH of the solution was in the range of 10-14. The reaction was stirred for at least 1.5 hours at approximately 15-25 °C, and then the layers were allowed to separate over a period of at least 1 hour. The suspension was filtered, taking care to capture the product, which accumulated at the interface of the aqueous and organic layers. The filter cake was washed with MTBE (1.7 volume equivalents), water (3.0 volume equivalents), and then MTBE again (3.0 volume equivalents). The product was dried in vacuo at below 50 °C until the loss on drying was < 1% of the weight, giving 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a pale beige-colored solid in 97.6% yield.
Step 6: Synthesis of 1 -methyl-7 -( 1 -methyl-lH-pyraz.ol-4-yl )-5-(4-( trifluoromethoxy )pheml )-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00201] A reactor was charged with 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalents), ( 1 -methyl- 1 //-pyrazol-4-yl)boronic acid pinacol ester (l-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l//-pyrazole, 1.6 mol equivalents), Pd[Ph3]4 (0.025 mol equivalents, and K2C03 (2.0 mol equivalents), to which were added acetonitrile (10.0 volume equivalents) and water (3.0 volume equivalents). The reaction mixture was stirred for approximately 10-20 minutes at about 20-25 °C to form a suspension.
[00202] The mixture was heated to slight reflux, whereupon a biphasic, yellow solution formed. The mixture was stirred at slight reflux for at least 10 hours. The reaction mixture was cooled to between 30-50 °C, then passed through a particle filter. The filter was washed with acetonitrile (2.6 volume equivalents), the filtrates were combined, and the solution was concentrated to a final volume of approximately 120 mL (4.8 volume equivalents) under reduced pressure at below 60 °C.
[00203] To the resulting suspension was added water (1.9 volume equivalents), methanol (26 mL, 1.0 volume equivalents), and dichloromethane (14.8 volume equivalents). The mixture was warmed to about 30-35 °C and stirred until two clear layers were observed. The layers were allowed to separate without stirring at about 30-35 °C, and additional dichloromethane (3.7 volume equivalents) was added to the aqueous layer. The mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes, and then the layers were allowed to separate at approximately 30-35 °C.
[00204] To the combined organic layers was added water (1.9 volume equivalents), and the mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes. The layers were separated at approximately 30-35 °C. Charcoal was added to the combined organic layers and stirred for 30-60 minutes at approximately 30-35 °C. The charcoal was removed by filtration, and the filter was washed with dichloromethane (39 mL, 1.6 volume equivalents).
[00205] The solution was concentrated to approximately 4.0 volume equivalents at ambient pressure and at below 50 °C, then diluted with methanol (5.0 volume equivalents). The solution was again concentrated to approximately 4.0 volume equivalents at ambient pressure and below 60 °C, diluted with methanol (5.0 volume equivalents), and concentrated to a final volume of approximately 3.0 volume equivalents under reduced pressure below 60 °C.
[00206] To the resulting suspension was added methanol (2.9 volume equivalents), and the suspension was warmed to approximately 45-55 °C and stirred for about 1 hour. The suspension was cooled to approximately 0-5 °C within approximately 1 hour, stirred for 1 hour at approximately 0-5 °C, and then filtered. The filter cake was washed with cold methanol (pre-cooled to approximately 0-10 °C, 2.9 volume equivalents), and the product was dried under a stream of nitrogen and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I (l-methyl-7-(l-methyl-l -pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one) as a white solid in 88.5% yield.
Step 7: Recrystallization of 1 -methyl-7 -(1 -methyl- lH-pyraz.ol-4-yl)-5-( 4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00207] A reactor was charged with crude l-methyl-7-(l -methyl- l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one from step 6, and to this was added glacial acetic acid (1.5 volume equivalents). The suspension was warmed to approximately 50-60 °C and stirred until a clear solution was obtained, approximately 10-20 minutes. The warm solution was passed through a particle filter into a second reactor.
[00208] To this solution was added ethanol (10.0 volume equivalents) at approximately 45-55 °C over 2 hours. The suspension was stirred for approximately 30 minutes at approximately 45-55 °C, then cooled to approximately 0-5 °C over about 4 hours. The suspension was then stirred for approximately 4-16 hours at about 0-5 °C.
[00209] Next, the suspension was filtered and the filter cake was washed with cold isopropanol (4.2 volume equivalents) at approximately 0-20 °C. The product was dried under a nitrogen stream and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I ( 1 – mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(tnfluoromcthoxy)phcnyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one) as a white solid in 93.0% yield.
Step 8 : Synthesis of 1 -methyl-7 -( 1 -methyl- 1 H-pyrazol-4-yl )-5-(4-( trifluoromethoxy )phenyl )- 1 ,5-dihvdro-4H-imidaz.oi 4,5-clpyridin-4-one, mono – mono -tosylate
salt)
[00210] A reactor was charged with Compound I ( 1 -mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one, 1.00 mol equivalent), para-toluenesulfonic acid monohydrate (1.05 mol equivalents), acetone (6.75 volume equivalents), and water (0.75 volume equivalents). The mixture was stirred at 15-25 °C until a clear solution formed, and then this solution was filtered through a particle filter into a second reactor.
[00211] The filter was washed with acetone (2.5 volume equivalents), and to the combined filtrates was added MTBE (7.5 volume equivalents) at 15-25 °C and Compound I mono-tosylate seeding crystals (0.001 mol equivalents).
[00212] The resulting suspension was stirred at 15-25 °C for approximately 30-60 minutes, and MTBE was added (22.5 volume equivalents) at 15-25 °C during a period of
approximately 30 minutes. Stirring was continued at 15-25 °C for approximately 30-60 minutes, and then the suspension was filtered. The filter was washed with MTBE (2.5 volume equivalents), and the material was dried in vacuo at below 55 °C to give Compound I mono-tosylate salt (l-methyl-7-(l-methyl-l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one, mono-tosylate salt) as a white, crystalline solid in 93% yield.
PATENT
WO2018102323 ,
claiming use of a specific compound, orally administered, in combination with food (eg low, medium or high fat meal) for treating fibrotic, inflammatory or autoimmune disorders eg idiopathic pulmonary fibrosis IPF, assigned to Genentech Inc ,
References
-
Roche licenses IPF candidate to Ark Biosciences. Internet-Doc 2019;.
Available from: URL: https://scrip.pharmaintelligence.informa.com/deals/201820364
-
Roche Q3 2018. Internet-Doc 2018;.
Available from: URL: https://www.roche.com/dam/jcr:f9cad8fc-8655-4692-9a85-efbe1cf7a59b/en/irp181017.pdf
-
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Ascending, Single- and Multiple-Oral-Dose, Safety, Tolerability, and Pharmacokinetic Study of GDC-3280 in Healthy Subjects
// AK-3280, AK 3280, AK3280, GDC 3280, RG 6069, PHASE 1, Idiopathic pulmonary fibrosis
Tanzisertib
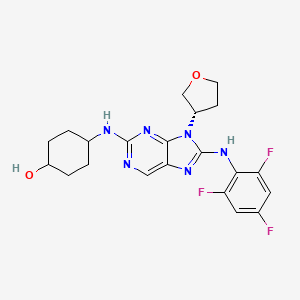
Tanzisertib
CAS 899805-25-5
trans-4-((9-((3S)-Tetrahydrofuran-3-yl)-8-((2,4,6-trifluorophenyl)amino)-9H-purin-2-yl)amino)cyclohexanol
4-[[9-[(3S)-oxolan-3-yl]-8-(2,4,6-trifluoroanilino)purin-2-yl]amino]cyclohexan-1-ol
C21-H23-F3-N6-O2, 448.4467
- CC 930
- CC-930
- Tanzisertib
- UNII-M5O06306UO
- A c-Jun amino-terminal kinase inhibitor.UNII, M5O06306UO
Treatment of Idiopathic Pulmonary Fibrosis (IPF)
- Originator Celgene Corporation
- Class Antifibrotics; Small molecules
- Mechanism of ActionJ NK mitogen-activated protein kinase inhibitors
- Orphan Drug Status Yes – Idiopathic pulmonary fibrosis
- Discontinued Discoid lupus erythematosus; Idiopathic pulmonary fibrosis
- 16 Jul 2012 Celgene Corporation terminates a phase II trial in Discoid lupus erythematosus in USA (NCT01466725)
- 23 Feb 2012 Celgene initiates enrolment in a phase II trial for Discoid lupus erythematosus in the USA (NCT01466725)
- 08 Nov 2011The Committee for Orphan Medicinal Products (COMP) recommends orphan drug designation for tanzisertib in European Union for Idiopathic pulmonary fibrosis
Tanzisertib has been granted orphan drug status by the FDA for the treatment of idiopathic pulmonary fibrosis. A positive opinion has been received from the EU Committee for Orphan Medicinal Products (COMP
Tanzisertib has been used in trials studying the treatment of Fibrosis, Discoid Lupus, Pulmonary Fibrosis, Interstitial Lung Disease, and Lung Diseases, Interstitial, among others.
PATENT
https://patents.google.com/patent/US20090048275A1/de

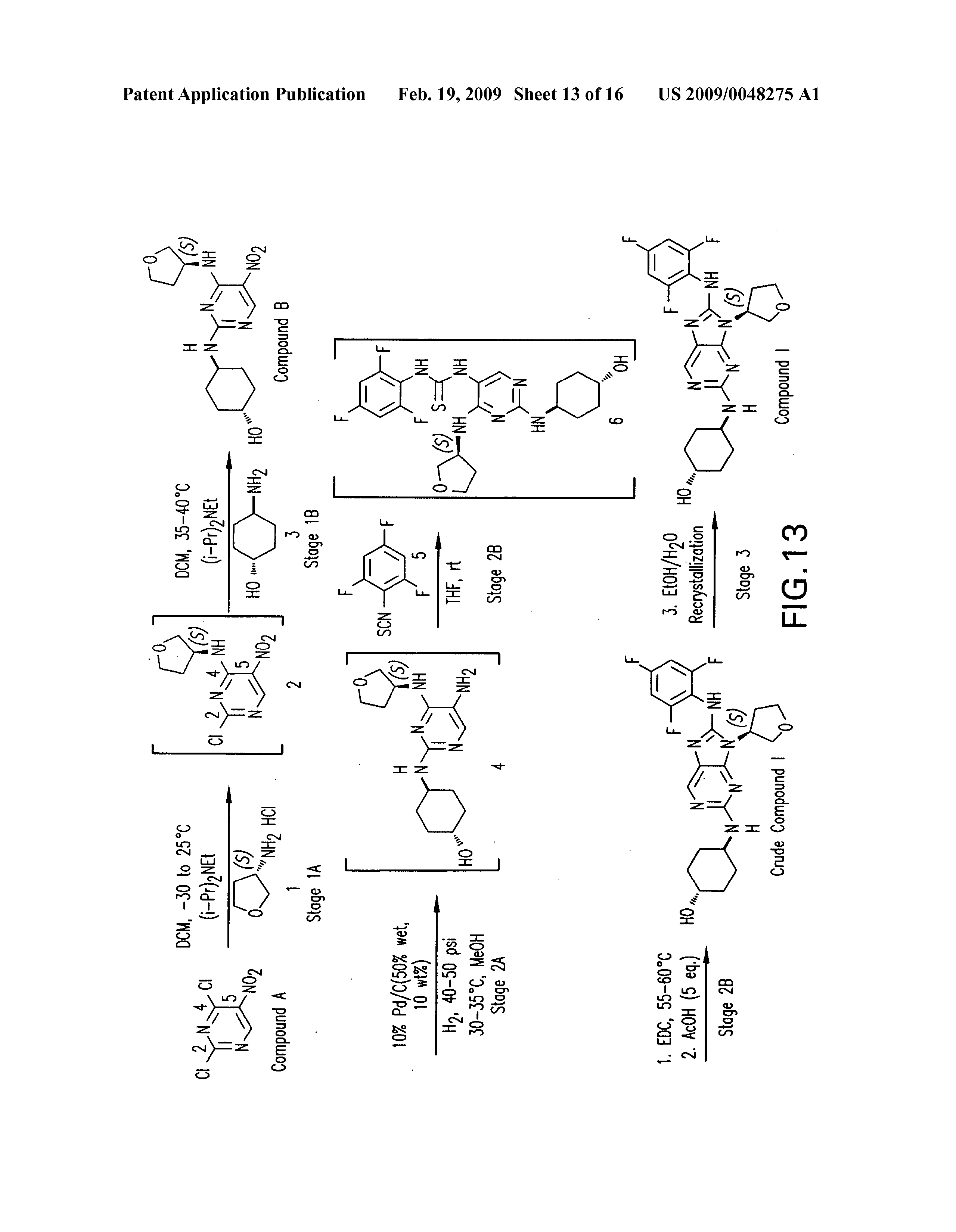

PATENT
WO 2006076595
US 20070060598
WO 2008057252
US 20080021048
US 20140094456
WO 2014055548
PATENT
WO 2015153683
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015153683
/////////Tanzisertib, CC 930, Idiopathic Pulmonary Fibrosis, Orphan Drug, phase II, CELGENE
c1c(c(c(cc1F)F)Nc2n(c3nc(ncc3n2)N[C@H]4CC[C@@H](CC4)O)[C@@H]5COCC5)F
HEC-68498
HEC-68498, CT-365
CAS 1621718-37-3
N-[5-(3-Cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxy-3-pyridinyl]-2,4-difluorobenzenesulfonamide
HEC Pharm , Calitor Sciences Llc; Sunshine Lake Pharma Co Ltd
PHASE 1, idiopathic pulmonary fibrosis and solid tumors
Phosphoinositide 3-kinase inhibitor; mTOR inhibitor

- Originator HEC Pharm
- Developer HEC Pharm; Sunshine Lake Pharma
- Class Anti-inflammatories; Antifibrotics; Isoenzymes
- Mechanism of Action 1 Phosphatidylinositol 3 kinase inhibitors; MTOR protein inhibitors
- Phase I Idiopathic pulmonary fibrosis
- 22 May 2018 Phase-I clinical trials in Idiopathic pulmonary fibrosis in USA (PO) (NCT03502902)
- 24 Apr 2018 Sunshine Lake Pharma in collaboration with Covance plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in China , (NCT03502902)
- 19 Apr 2018 Preclinical trials in Idiopathic pulmonary fibrosis in China (PO)
- US 20140234254
- CN 103965199
CN 103965199

CN 103965199

Sunshine Lake Pharma , a subsidiary of HEC Pharm is developing an oral capsule formulation of HEC-68498 (phase 1, in July 2019) sodium salt, a dual inhibitor of phosphoinositide-3 kinase and the mTOR pathway, for the treatment of idiopathic pulmonary fibrosis and solid tumors
HEC 68498 is an oral inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin in clinical development at HEC Pharm for the treatment of idiopathic pulmonary fibrosis. A phase I trial is under way in healthy volunteers.
The phosphoinositide 3-kinases (PI3 kinases or PI3Ks), a family of lipid kinases, have been found to play key regulatory roles in many cellular processes including cell survival, proliferation and differentiation. The PI3K enzymes consist of three classes with variable primary structure, function and substrate specificity. Class I PI3Ks consist of heterodimers of regulatory and catalytic subunits, and are subdivided into 1A and 1B based on their mode of activation. Class 1A PI3Ks are activated by various cell surface tyrosine kinases, and consist of the catalytic pl lO and regulatory p85 subunits. The three known isoforms of Class 1A pl lO are pl lOot, rΐ ΐqb, and rΐ ΐqd, which all contain an amino terminal regulatory interacting region (which interfaces with p85), a Ras binding domain, and a carboxy terminal catalytic domain. Class IB PI3Ks consist of the catalytic (pl lOy) and regulatory (p 101 ) subunits and are activated by G-protein coupled receptors. (“Small-molecule inhibitors of the PI3K signaling network” Future Med. Chem ., 2011, 3, 5, 549-565).
[0004] As major effectors downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs), PI3Ks transduce signals from various growth factors and cytokines into intracellular massages by generating phospholipids, which activate the serine-threonine protein kinase ART (also known as protein kinase B (PKB)) and other downstream effector pathways. The tumor suppressor or PTEN (phosphatase and tensin
homologue) is the most important negative regulator of the PI3K signaling pathway. (“Status of PBK/Akt/mTOR Pathway Inhibitors in Lymphoma.” Clin Lymphoma, Myeloma Leuk , 2014, 14(5), 335-342.)
[0005] The signaling network defined by phosphoinositide 3-kinases (PI3Ks), AKT and mammalian target of rapamycin (mTOR) controls most hallmarks of cancer, including cell cycle, survival, metabolism, motility and genomic instability. The pathway also contributes to cancer promoting aspects of the tumor environment, such as angiogenesis and inflammatory cell recruitment. The lipid second messenger produced by PI3K enzymes, phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3; also known as PIP3), is constitutively elevated in most cancer cells and recruits cytoplasmic proteins to membrane-localized‘onco’ signal osomes.
[0006] Cancer genetic studies suggest that the PI3K pathway is the most frequently altered pathway in human tumors: the PIK3CA gene (encoding the PI3K catalytic isoform pl lOa) is the second most frequently mutated oncogene, and PTEN (encoding phosphatase and tensin homolog, the major PtdIns(3,4,5)P3 phosphatase) is among the most frequently mutated tumor suppressor genes. In accord, a recent genomic study of head and neck cancer found the PI3K pathway to be the most frequently mutated. Indeed, even in cancer cells expressing normal PI3K and PTEN genes, other lesions are present that activate the PI3K signaling network (that is, activated tyrosine kinases, RAS and AKT, etc ). As a net result of these anomalies, the PI3K pathway is activated, mutated or amplified in many malignancies, including in ovarian cancer (Campbell et al., Cancer Res., 2004, 64, 7678-7681; Levine et al., Clin. Cancer Res., 2005, 11, 2875-2878; Wang et al., Hum. Mutat., 2005, 25, 322; Lee et al., Gynecol. Oncol. ,2005, 97, 26-34), cervical cancer, breast cancer (Bachman et al.,· Cancer Biol., Ther, 2004, 3, 772-775; Levine et al., supra; Li et al., Breast Cancer Res. Treat., 2006, 96, 91-95; Saal et al., Cancer Res., 2005, 65, 2554-2559; Samuels and Velculescu, Cell Cycle, 2004, 3, 1221-1224), colorectal cancer (Samuels et al., Science, 2004, 304, 554; Velho et al., Eur. J. Cancer, 2005, 41, 1649-1654), endometrial cancer (Oda et al ., Cancer Res., 2005, 65, 10669-10673), gastric carcinomas (Byun et al., M. J. Cancer, 2003 , 104, 318-327; Li et al., supra; Velho et al., supra; Lee et al., Oncogene, 2005 , 24, 1477-1480), hepatocellular carcinoma (Lee et al., id), small and non-small cell lung cancer (Tang et al., Lung Cancer 2006, 11, 181-191; Massion et al , Am. J. Respir. Crit. Care Med., 2004, 170, 1088-1094), thyroid carcinoma (Wu et al., J. Clin. Endocrinol. Metab., 2005, 90, 4688-4693),
acute myelogenous leukemia (AML) (Sujobert et al., Blood, 1997, 106, 1063-1066), chronic myelogenous leukemia (CML) (Hickey et al., J. Biol. Chem ., 2006, 281, 2441-2450), glioblastomas (Hartmann et al. Jlcta Neuropathol (Bert ), 2005, 109, 639-642; Samuels et al., supra), Hodgkin and non-Hodgkin lymphomas (“PI3K and cancer: lessons, challenges and opportunities”, Nature Reviews Drug Discovery., 2014, 13, 140).
[0007] The PI3K pathway is hyperactivated in most cancers, yet the capacity of PI3K inhibitors to induce tumor cell death is limited. The efficacy of PI3K inhibition can also derive from interference with the cancer cells’ ability to respond to stromal signals, as illustrated by the approved PI3K5 inhibitor idelalisib in B-cell malignancies. Inhibition of the leukocyte-enriched PI3K5 or RI3Kg may unleash antitumor T-cell responses by inhibiting regulatory T cells and immune-suppressive myeloid cells. Moreover, tumor angiogenesis may be targeted by PI3K inhibitors to enhance cancer therapy. (“Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy”, Cancer Discov., 2016, 6(10), 1090-1105.)
[0008] mTOR is a highly conserved serine-threonine kinase with lipid kinase activity and participitates as an effector in the PI3K/AKT pathway. mTOR exists in two distinct complexes, mTORCl and mTORC2, and plays an important role in cell proliferation by monitoring nutrient avaliability and cellular energy levels. The downstream targets of mTORCl are ribosomal protein S6 kinase 1 and eukaryotic translation initiation factor 4E-binding protein 1, both of which are crucial to the regulation of protein synthesis. (“Present and future of PI3K pathway inhibition in cancer: perspectives and limitations”, Current Med. Chem., 2011, 18, 2647-2685).
[0009] Knowledge about consequences of dysregulated mTOR signaling for tumorigenesis comes mostly from studies of pharmacologically disruption of mTOR by repamycin and its analogues such as temsirolimus (CCI-779) and everolimus (RADOOl).Rapamycin was found to inhibit mTOR and thereby induce G1 arrest and apoptosis. The mechanism of rapamycin growth inhibition was found to be related to formation of complexes of rapamycin with FK-binding protein 12 (FKBP-12). These complexes then bound with high affinity to mTOR, preventing activation and resulting in inhibition of protein translation and cell growth. Cellular effects of mTOR inhibition are even more pronounced in cells that have concomitant inactivation of PTEN. Antitumor activity of rapamycin was subsequently identified, and a number of rapamycin analogues such as temsirolimus and everolimus have been approved by the US Food and Drug
Administration for the treatment certain types of cancer.
[0010] Fibrosis is the formation of excess fibrous connective tissue in an organ or tissue in a reparative or reactive process. Examples of fibrosis include, but are not limited to pulmonary fibrosis, liver fibrosis, dermal fibrosis, and renal fibrosis. Pulmonary fibrosis, also called idiopathic pulmonary fibrosis (IPF), interstitial diffuse pulmonary fibrosis, inflammatory pulmonary fibrosis, or fibrosing alveolitis, is a lung disorder and a heterogeneous group of conditions characterized by abnormal formation of fibrous tissue between alveoli caused by alveolitis comprising cellular infiltration into the alveolar septae with resulting fibrosis. The effects of IPF are chronic, progressive, and often fatal.
[0011] The clinical course of IPF is variable and largely unpredictable. IPF is ultimately fatal, with historical data suggesting a median survival time of 2-3 years from diagnosis. A decline in forced vital capacity (FVC) is indicative of disease progression in patients with IPF and change in FVC is the most commonly used endpoint in clinical trials. A decline in FVC of 5% or 10% of the predicted value over 6-12 months has been associated with increased mortality in patients with IPF.
[0012] Our understanding of the pathogenesis of IPF has evolved from that of a predominantly inflammatory disease to one driven by a complex interplay of repeated epithelial cell damage and aberrant wound healing, involving fibroblast recruitment, proliferation and differentiation, and culminating in excess deposition of extracellular matrix. This shift in knowledge prompted a change in the type of compounds being investigated as potential therapies, with those targeted at specific pathways in the development and progression of fibrosis becoming the focus.
[0013] In patients with IPF, the mechanisms whereby PI3K/mTOR inhibitors act may involve inhibition of kinases such as PI3Ks and mTOR. This results in inactivation of cellular receptors for mediators involved in the development of pulmonary fibrosis. As a result, fibroblast proliferation is inhibited and extracellular matrix deposition is reduced. (“Update on diagnosis and treatment of idiopathic pulmonary fibrosis”, J Bras Pneumol. 2015, 41(5), 454-466.)
[0014] Accordingly, small-molecule compounds that specially inhibit, regulate and/or modulate the signal transduction of kinases, particularly including PI3K and mTOR as described above, are desirable as a means to prevent, manage, or treat proliferative disorders and fibrosis, particular idiopathic pulmonary fibrosis in a patient. One such small-molecule is A-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfon-amide, which has the chemical structure as shown in the following:
[0015] WO 2014130375A1 described the synthesis of N-(5 -(‘3 -cyanopyrazol o [l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (Example 3) and also disclosed the therapeutic activity of this molecule in inhibiting, regulating and modulating the signal transduction of protein kinases.
[0016] Different salts and solid state forms of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for example, if they serve to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
Different salts and solid state forms of /V-(5-(3-cyanopyrazolo[l,5- ]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide are described herein.
PATENT
WO2014130375 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014130375
claiming new pyrazolo[1,5-a]pyridine derivatives are PI3K and mTOR inhibitors, useful for treating proliferative diseases
Example 3 N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
Step 1) 5-bromopyrazolo[1,5-a]pyridine
[196] A solution of ethyl 5-bromopyrazolo[1,5-a]pyridine-3-carboxylate (240
mmol) in 40% H2SO4 (12 mL) was stirred at 100 °C for 4 hours, then cooled to rt, and neutralized to pH=7 with aq. NaOH (6 M) in ice bath. The resulted mixture was extracted with DCM (25 mL x 2). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (175 mg, 99.5%).
MS (ESI, pos. ion) m/z: 196.9 [M+H]+.
Step 2) 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde
[197] To a solution of 5-bromopyrazolo[1,5-a]pyridine (175 mg, 0.89 mmol) in DCM (6 mL) was added (chloromethylene)dimethyliminium chloride (632 mg, 3.56 mmol). The reaction was stirred at 44 °C overnight, and concentrated in vacuo. The residue was dissolved in saturated NaHCO3 aqueous solution (25 mL) and the resulted mixture was then extracted with EtOAc (25 mL x 3). The combined organic phases were dried over anhydrous Na2SO4 and concentrated in vacuo to give the title compound as a light yellow solid (225 mg, 100%).
MS (ESI, pos. ion) m/z: 225.0 [M+H]+.
Step 3) (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime
[198] To a suspension of 5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde (225 mg, 1 mmol) in EtOH (10 mL) and H2O (5 mL) was added hydroxylamine hydrochloride (104 mg, 1.5 mmol). The reaction was stirred at 85 °C for 2 hours, then cooled to rt, and concentrated in vacuo. The residue was adjusted to pH=7 with saturated NaHCO3 aqueous solution. The resulted mixture was then filtered and the filter cake was dried in vacuo to give title compound as a yellow solid (240 mg, 99%).
MS (ESI, pos. ion) m/z: 240.0 [M+H]+.
Step 4) 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile
[199] A solution of (E)-5-bromopyrazolo[1,5-a]pyridine-3-carbaldehyde oxime (240 mg,
1 mmol) in Ac2O (6 mL) was stirred at 140 °C for 18 hours, then cooled to rt, and concentrated in vacuo. The residue was washed with Et2O (1 mL) to give the title compound as a yellow solid (44 mg, 22.5%).
MS (ESI, pos. ion) m/z: 222.0 [M+H]+.
Step 5) N-(5-(3-cyanopyrazolo[1,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide
[200] 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (612 mg, 1.5 mmol), 5-bromopyrazolo[1,5-a]pyridine-3-carbonitrile (222 mg, 1 mmol), Pd(dppf)Cl2·CH2Cl2 (16 mg, 0.02 mmol) and Na2CO3 (85 mg, 0.8 mmol) were placed into a two-neck flask, then degassed with N2 for 3 times, and followed by adding 1,4-dioxane (5 mL) and water (1 mL). The resulted mixture was degassed with N2 for 3 times, then heated to 90 °C and stirred further for 5 hours. The mixture was cooled to rt and filtered. The filtrate was concentrated in vacuo and the residue was purified by a silica gel column chromatography (PE/EtOAc (v/v) = 1/2) to give the title compound as a light yellow solid (400 mg, 81.6%).
MS (ESI, pos. ion) m/z: 442.0 [M+H]+;
1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.37 (s, 1H), 9.02 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.60 (d, J = 2.2 Hz, 1H), 8.26-8.16 (m, 2H), 7.82-7.72 (m, 1H), 7.57 (dd, J = 13.2, 5.8 Hz, 2H), 7.21 (t, J= 8.5 Hz, 1H), 3.67 (s, 3H).
PATENT
WO-2019125967
The invention relates to salts of pyrazolo[l,5-a]pyridine derivatives and use thereof, specifically relates to salt of /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl) -2,4-difluorobenzenesulfonamide (compound of formula (I)) and use thereof, further relates to composition containing said salts above. The salts or the composition can be used to inhibit/modulate protein kinases, further prevent, manage or treat proliferative disorders or pulmonary fibrosis in a patient.
Amorphous form of mono-sodium salt of HEC-68498 , useful for treating a proliferative disorder or pulmonary fibrosis.
The invention is further illustrated by the following examples, which are not be construed as limiting the invention in scope.
[00108] /V-(5-(3-cyanopyrazolo[l,5-a]pyridin-5-yl)-2-methoxypyridin-3-yl)-2,4-difluoroben zenesulfonamide can be prepared according to the synthetic method of example 3 disclosed in WO2014130375 Al.
//////////////HEC-68498, HEC 68498, HEC68498, HEC Pharm , Calitor Sciences, Sunshine Lake Pharma, PHASE 1, proliferative disorder, pulmonary fibrosis, idiopathic pulmonary fibrosis, solid tumors, CT-365 , CT 365 , CT365
Fc1ccc(c(F)c1)S(=O)(=O)Nc2cc(cnc2OC)c3ccn4ncc(C#N)c4c3
GSK 3008348
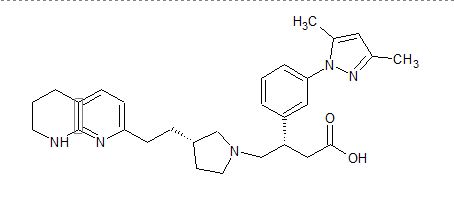
GSK 3008348
(3S)-3-[3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl]-4-{(3S)-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-1-pyrrolidinyl}butanoic acid
cas 1629249-33-7
1-Pyrrolidinebutanoic acid, β-[3-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl]-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-, (βS,3R)-
(S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8- naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid
- (βS,3R)-β-[3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl]-3-[2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl]-1-pyrrolidinebutanoic acid
- Molecular Formula C29H37N5O2
- Average mass 487.636 Da
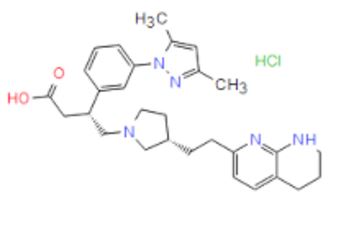
| CAS Number: | 1629249-40-6 |
| Molecular Weight: | 524.1 |
| Molecular Formula: | C29H38ClN5O2 |
- Originator GlaxoSmithKline
- Mechanism of Action Integrin alphaV antagonists
- Phase I Idiopathic pulmonary fibrosis
- 06 Mar 2017 GlaxoSmithKline plans a phase I trial for Idiopathic pulmonary fibrosis (NCT03069989)
- 01 Jun 2016 GlaxoSmithKline completes a first-in-human phase I trial for Idiopathic pulmonary fibrosis in United Kingdom (Inhalation) (NCT02612051)
- 01 Dec 2015 Phase-I clinical trials in Idiopathic pulmonary fibrosis in United Kingdom (Inhalation) (NCT02612051)
Inventors Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard
Applicant Glaxosmithkline Intellectual Property Development Limited

Niall Anderson
![]()
GSK-3008348, an integrin alpha(v)beta6 antagonist, is being developed at GlaxoSmithKline in early clinical studies for the treatment of idiopathic pulmonary fibrosis (IPF).
Idiopathic pulmonary fibrosis (IPF) is a chronic disease characterised by a progressive decline in lung function, due to excessive deposition of extracellular matrix (collagen) within the pulmonary interstitium. It affects approximately 500,000 people in the USA and Europe and is poorly treated. IPF inexorably leads to respiratory failure due to obliteration of functional alveolar units. Patients’ mean life-expectancy is less than 3 years following diagnosis.
IPF therefore represents a major unmet medical need for which novel therapeutic approaches are urgently required.1 Pirfenidone (EsbrietTM from Roche), a non-selective kinase inhibitor, is approved for mild and moderate IPF patients in Japan, Europe, Canada and China and for all IPF patients in USA . Furthermore, nintedanib (OfevTM formerly BIBF-1120 from Boehringer-Ingelheim), a multiple tyrosine-kinase inhibitor targeting vascular endothelial factor receptor, fibroblast growth factor and platelet derived growth factor receptor is approved for all patients with IPF in USA and Europe. Both compounds are administered orally twice or three times per day at high total doses (pirfenidone at 2.4 g/day and nintedanib at 300 mg/day).

Patient compliance is limited by tolerability due to gastro-intenstinal and phototoxicity issues, which require dose titration. (S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8- naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid hydrochloride is a first in class compound (descovered by GlaxoSmithKline) undergoing currently Phase I clinical trials for the treatment of IPF. It is a non-peptidic αvβ6 integrin inhibitor and in cell adhesion assays has high affinity for the human receptor with a pIC50 of 8.4, and lower affinity for other integrins, such as αvβ3 6.0, αvβ5 5.9 and αvβ8 7.7. Inhibition of integrin αvβ6 is thought to prevent pulmonary fibrosis without exacerbating inflammation.
Integrin superfamily proteins are heterodimeric cell surface receptors, composed of an alpha and beta subunit. 18 alpha and 8 beta subunits have been reported, which have been demonstrated to form 24 distinct alpha/beta heterodimers. Each chain comprises a large extracellular domain (>640 amino acids for the beta subunit, >940 amino acids for the alpha subunit), with a transmembrane spanning region of around 20 amino acids per chain, and generally a short cytoplasmic tail of 30-50 amino acids per chain. Different integrins have been shown to participate in a plethora of cellular biologies, including cell adhesion to the extracellular matrix, cell-cell interactions, and effects on cell migration, proliferation, differentiation and survival (Barczyk et al, Cell and Tissue Research, 2010, 339, 269).
Integrin receptors interact with binding proteins via short protein-protein binding interfaces with ligands and the integrin family can be grouped into sub-families that share similar binding recognition motifs in such ligands. A major subfamily is the RGD-integrins, which recognise ligands that contain an RGD (Arginine-glycine-aspartic acid) motif within their protein sequence. There are 8 integrins in this sub-family, namely ανβι, ανβ3, νβ5ι νβ6ι ανβδ, αι¾β3, α5βι, α8βι, where nomenclature demonstrates that ανβι, ανβ3, νβ5ι νβ6ι & ανβδ share a common V subunit with a divergent β subunit, and ανβι, α5βι & α8βι share a common β!subunit with a divergent a subunit. The βι subunit has been shown to pair with 11 different a subunits, of which only the 3 listed above commonly recognise the RGD peptide motif. (Humphries et al, Journal of Cell Science, 2006, 119, 3901).
Within the 8 RGD-binding integrins are different binding affinities and specificities for different RGD-containing ligands. Ligands include proteins such as fibronectin, vitronectin, osteopontin, and the latency associated peptides (LAPs) of Transforming growth factor βι and β3 (ΤΰΡβι and ΤΰΡβ3). The binding to the LAPs of ΤΰΡβι and ΤΰΡβ3 has been shown in several systems to enable activation of the ΤΰΡβι and ΤΰΡβ3 biological activities, and subsequent ΤΰΡβ- driven biologies (Worthington et al, Trends in Biochemical Sciences, 2011, 36, 47). The specific binding of RGD integrins to such ligands depends on a number of factors, depending on the cell phenotype. The diversity of such ligands, coupled with expression patterns of RGD-binding integrins, generates multiple opportunities for disease intervention. Such diseases include fibrotic diseases (Margadant et al, EMBO reports, 2010, 11, 97), inflammatory disorders, cancer (Desgrosellier et al, Nature Reviews Cancer, 2010, 10, 9), restenosis, and other diseases with an angiogenic component (Weis et al, Cold Spring. Harb. Perspect Med.2011, 1, a006478).
A significant number of av integrin antagonists (Goodman et al, Trends in Pharmacological Sciences, 2012, 33, 405) have been disclosed in the literature including antagonist antibodies, small peptides and compounds. For antibodies these include the pan-av antagonist Intetumumab, the selective ανβ3 antagonist Etaracizumab, and the selective a 6 antagonist STX-100. Cilengitide is a cyclic peptide antagonist that inhibits both ανβ3 and ανβ5, and SB-267268 is an example of a compound (Wilkinson-Berka et al, Invest. Ophthalmol. Vis. Sci, 2006, 47, 1600), which inhibits both ανβ3 and ανβ5. Invention of compounds to act as antagonists of differing combinations of av integrins enables novel agents to be generated and tailored for specific disease indications.
Pulmonary fibrosis represents the end stage of several interstitial lung diseases, including the idiopathic interstitial pneumonias, and is characterised by the excessive deposition of extracellular matrix within the pulmonary interstitium. Among the idiopathic interstitial pneumonias, idiopathic pulmonary fibrosis (IPF) represents the commonest and most fatal condition with a median survival of 3 to 5 years following diagnosis. Fibrosis in IPF is generally progressive, refractory to current pharmacological intervention and inexorably leads to respiratory failure due to obliteration of functional alveolar units. IPF affects approximately 500,000 people in the USA and Europe. This condition therefore represents a major unmet medical need for which novel therapeutic approaches are urgently required (Datta A et al, Novel therapeutic approaches for pulmonary fibrosis, British Journal of ‘Pharmacology’2011163: 141-172).
There are strong in vitro, experimental animal and IPF patient immunohistochemistry data to support a key role for the epithelial-restricted integrin, α 6ι in the activation of TGF-βΙ. Expression of this integrin is low in normal epithelial tissues and is significantly up-regulated in injured and inflamed epithelia including the activated epithelium in IPF. Targeting this integrin therefore reduces the theoretical possibility of interfering with wider TGF-β homeostatic roles. Partial inhibition of the a 6 integrin by antibody blockade has been shown to prevent pulmonary fibrosis without exacerbating inflammation (Horan GS etal Partial inhibition of integrin a 6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med2008177: 56-65)
The ανβ3 integrin is expressed on a number of cell types including vascular endothelium where it has been characterised as a regulator of barrier resistance. Data in animal models of acute lung injury and sepsis have demonstrated a significant role for this integrin in vascular leak since knockout mice show markedly enhanced vessel leak leading to pulmonary oedema or death. Furthermore antibodies capable of inhibiting ανβ3 function caused dramatic increases in monolayer permeability in human pulmonary artery and umbilical vein endothelial cells in response to multiple growth factors. These data suggest a protective role for ανβ3 in the maintenance of vascular endothelial integrity following vessel stimulation and that inhibition of this function could drive pathogenic responses in a chronic disease setting (Su et al Absence of integrin ανβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation Am J Respir Crit Care Med 2012 185: 58-66). Thus, selectivity for cl over α 3 may provide a safety advantage.
It is an object of the invention to provide ανβ6 antagonists.
PATENT
| Inventors | Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard |
| Applicant | Glaxosmithkline Intellectual Property Development Limited |
Scheme 1

Reagents and conditions: (a) iodine, imidazole, triphenylphosphine, DCM, 0°C; (b) 2- methyl-[l,8]-naphthyridine, LiN(TMS)2, THF, 0°C; (c) 4M HQ in dioxane.
Scheme 2

Reagents and conditions: (a) isobutylene, cone. H2S04, diethyl ether, 24 h; (b) potassium acetate, acetonitrile, 60 °C, 4 h.


Scheme 3. Reagents and Conditions: (a) LiAIH4, THF; (b) H2, 5% Rh/C, EtOH


Intermediate 42
iate 39


Scheme 6. Reagents and Conditions: (a) EDC, HOBT, NMM, DCM; (b) H2, 5% Rh/C, EtOH; (c) TFA, DCM; (d) BH3.THF; (e) UAIH4, THF, 60°C
Example 1: 3-f3-f3,5-Dimethyl-l pyrazol-l-vnphenvn-4-ff/?)-3-f2-f5,6,7,8- tetrahvdro-l,8-naphthyridin- -vnethvnpyrrolidin-l-vnbutanoic acid

A solution of te/f-butyl 3-(3-(3,5-dimethyl-l pyrazol-l-yl)phenyl)-4-((>?)-3-(2-(5,6,7,8- tetrahydro-l,8-naphthyridin-2-yl)ethyl)pyrrolidin-l-yl)butanoate (Intermediate 14) (100 mg, 0.184 mmol) in 2-methylTHF (0.5 mL) was treated with cone. HCI (12M, 0.077 mL, 0.92 mmol) and stirred at 40 °C for 2 h. The solvent was evaporated in vacuo and the residual oil was dissolved in ethanol (2 mL) and applied to a SCX-2 ion-exchange cartridge (5 g), eluting with ethanol (2 CV) and then 2M ammonia in MeOH (2 CV). The ammoniacal fractions were combined and evaporated in vacuo to give the title compound (79 mg, 88%) as an off-white solid: LCMS (System A) RT= 0.86 min, 100%, ES+ve /77/Z488 (M+H)+; H NMR δ (CDCI3; 600 MHz): 7.42 – 7.37 (m, 1H), 7.31 (d, 7=1.5 Hz, 1H), 7.29 (d, 7=0.9 Hz, 1H), 7.23 (d, 7=7.7 Hz, 1H), 7.21 (d, 7=7.3 Hz, 1H), 6.31 (d, 7=7.3 Hz, 1H), 5.99 (s, 1H), 3.55 (br. s., 1H), 3.60 – 3.52 (m, 1H), 3.45 (t, 7=5.4 Hz, 2H), 3.27 (t, 7=10.6 Hz, 1H), 3.09 (br. S.,1H), 2.93 – 2.86 (m, 1H), 2.82 (d, 7=10.1 Hz, 1H), 2.86 – 2.75 (m, 2H), 2.72 (t, 7=6.2 Hz, 1H), 2.74 – 2.67 (m, 2H), 2.75 (d, 7=9.0 Hz, 1H), 2.61 – 2.50 (m, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 2.33 – 2.26 (m, 1H), 2.24 – 2.11 (m, 1H), 1.94 – 1.86 (m, 2H), 1.94 – 1.84 (m, 1H), 1.78 – 1.66 (m, 1H), 1.65 – 1.51 (m, 1H).
Example 1 was identified by a method described hereinafter as (^-S-iS-iS^-dimethyl-l pyrazol-l-yl)phenyl)-4-((>?)-3-(2-(5,6,7,8-tetrahydro-l,8-naphthyridin-2-yl)ethyl)pyrrolidin-l- yl)butanoic acid.

PAPER
Organic & Biomolecular Chemistry (2016), 14(25), 5992-6009
http://pubs.rsc.org/en/content/articlelanding/2016/ob/c6ob00496b#!divAbstract
Synthesis and determination of absolute configuration of a non-peptidic αvβ6 integrin antagonist for the treatment of idiopathic pulmonary fibrosis
Abstract
A diastereoselective synthesis of (S)-3-(3-(3,5-dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid (1), a potential therapeutic agent for the treatment of Idiopathic Pulmonary Fibrosis, which is currently undergoing Phase I clinical trials is reported. The key steps in the synthesis involved alkylation of 2-methylnaphthyridine with (R)-N-Boc-3-(iodomethyl)-pyrrolidine, and an asymmetric Rh-catalysed addition of an arylboronic acid to a 4-(N-pyrrolidinyl)crotonate ester. The overall yield of the seven linear step synthesis was 8% and the product was obtained in >99.5% ee proceeding with 80% de. The absolute configuration of 1 was established by an alternative asymmetric synthesis involving alkylation of an arylacetic acid using Evans oxazolidinone chemistry, acylation using the resulting 2-arylsuccinic acid, and reduction. The absolute configuration of the benzylic asymmetric centre was established as (S).

naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic acid (1a) FREE FORM
isopropylamine), flow-rate = 1 mL/min, detecting at 235 nm, RT=12.5 min, 100% (other
diastereoisomer not present RT=9.6 min);
J=5.4 Hz, 2H), 3.27 (t, J=10.6 Hz, 1H), 3.09 (br. s.,1H), 2.93 – 2.86 (m, 1H), 2.82 (d, J=10.1Hz, 1H), 2.86 – 2.75 (m, 2H), 2.72 (t, J=6.2 Hz, 1H), 2.74 – 2.67 (m, 2H), 2.75 (d, J=9.0 Hz,1H), 2.61 – 2.50 (m, 1H), 2.31 (s, 3H), 2.29 (s, 3H), 2.33 – 2.26 (m, 1H), 2.24 – 2.11 (m, 1H),1.94 – 1.86 (m, 2H), 1.94 – 1.84 (m, 1H), 1.78 – 1.66 (m, 1H), 1.65 – 1.51 (m, 1H);
126.2, 123.7, 123.2, 117.4, 109.7, 107.0, 63.3, 56.7 , 54.5, 44.1, 40.9, 40.0, 36.9, 35.5, 32.8,
30.3, 25.8, 19.9, 13.5, 12.5;





REFERENCES
| MacDonald, S.; Pritchard, J.; Anderson, N. Discovery of a small molecule alphavbeta6 inhibitor for idiopathic pulmonary fibrosis 253rd Am Chem Soc (ACS) Natl Meet (April 2-6, San Francisco) 2017, Abst MEDI 362 |
///////////////GSK 3008348, phase 1, idiopathic pulmonary fibrosis, GSK, Niall Andrew ANDERSON, Brendan John FALLON, John Martin Pritchard, Integrin alphaV antagonists
Next talk in #MEDI 1st time disclosures is Simon MacDonald of @GSK on a treatment for idiopathic pulmonary fibrosis
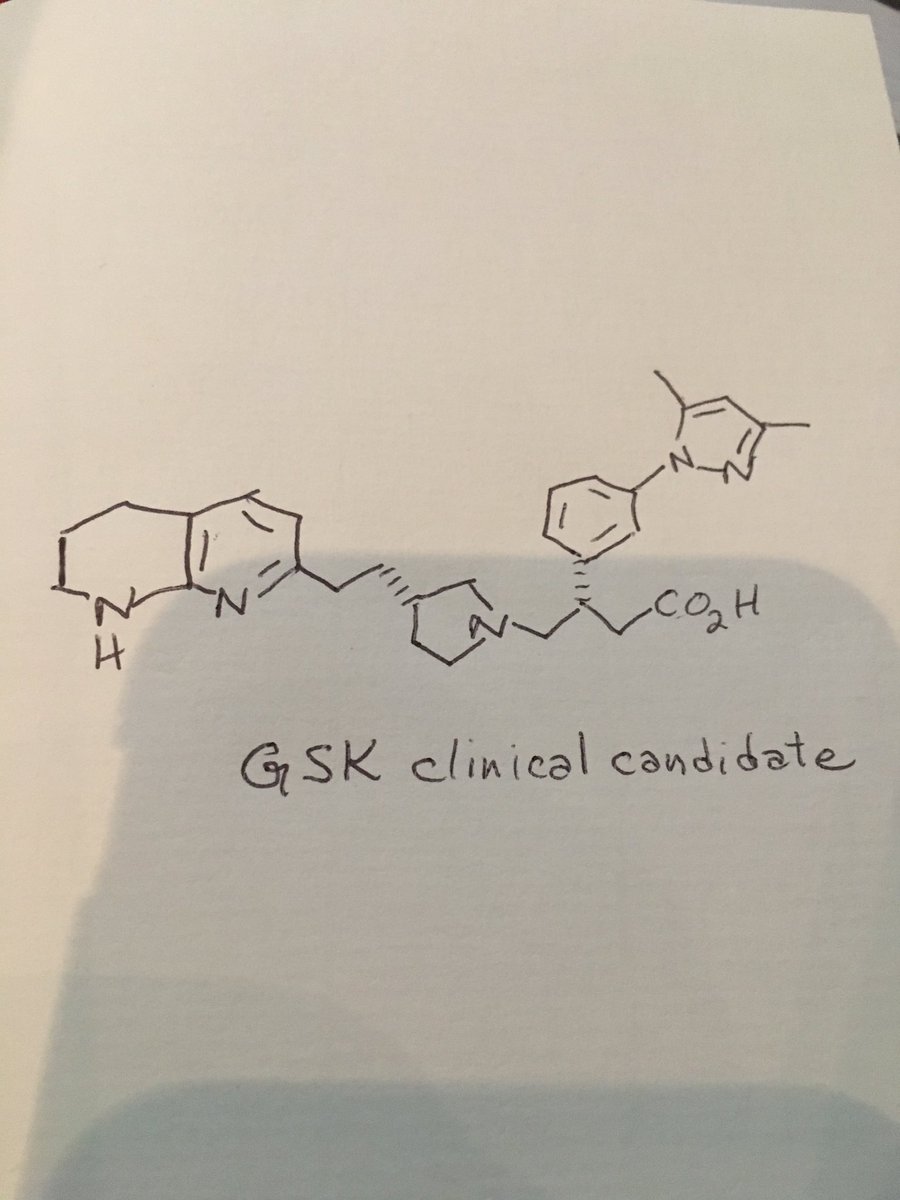
GLPG 1690
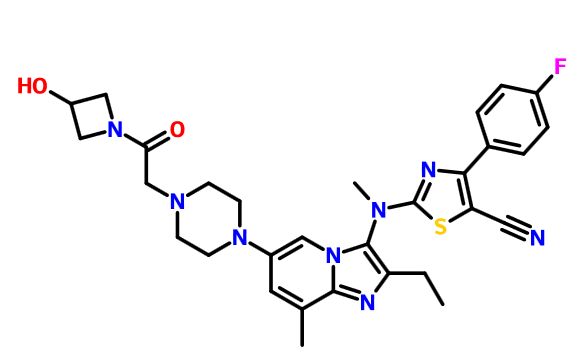

Picture credit….Bethany Halford
GLPG 1690
2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]-methylamino]-4-(4-fluorophenyl)-1,3-thiazole-5-carbonitrile
5- Thiazolecarbonitrile, 2-[[2-ethyl-6-[4-[2-(3-hydroxy-1-azetidinyl)-2-oxoethyl]- 1-piperazinyl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)-
CAS 1628260-79-6
![]()
$GLPG compound for treating idiopathic pulmonary fibrosis
| Molecular Formula: | C30H33FN8O2S |
|---|---|
| Molecular Weight: | 588.698823 g/mol |
| Galapagos Nv |
http://files.glpg.com/docs/website_1/Poster_ERS_2015_final.pdf
http://www.glpg.com/docs/view/56b360a81f6b2-en
Phase I Idiopathic pulmonary fibrosis
| Description | Selective autotaxin (ENPP2; ATX) inhibitor |
| Molecular Target | Autotaxin (ENPP2) (ATX) |
- Originator Galapagos NV
- Class Anti-inflammatories; Small molecules
- Mechanism of Action ENPP2 protein inhibitors
- 23 Sep 2015 Pharmacodynamics data from a preclinical trial in Indiopathic pulmonary fibrosis released by Galapagos
- 22 Sep 2015 Pharmacokinetics data from a phase I trial in healthy volunteers released by Galapagos
- 22 Sep 2015 Updated adverse events data from a phase I trial in healthy volunteers released by Galapagos
GLPG1690
GLPG1690 is a selective autotaxin inhibitor discovered by Galapagos, with potential application in idiopathic pulmonary disease (IPF). In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015. GLPG1690 is fully proprietary to Galapagos.
| Source: Galapagos NV
- Fully owned and proprietary clinical asset for pulmonary fibrosis
- GLPG1690 acts on autotaxin target
- Novel mode of action, originating from Galapagos target discovery engine
- Filing for Phase 2 clinical trial in 2015
MECHELEN, Belgium, March 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced that Janssen Pharmaceutica NV and Galapagos have mutually agreed to terminate the inflammation alliance and option agreements between the companies. Galapagos views the molecules emerging from the alliance as strong additions to its growing proprietary pipeline. Among others, all rights to candidate drug GLPG1690, a selective autotaxin inhibitor, return to Galapagos. Galapagos has successfully completed a First-in-Human Phase 1 trial for GLPG1690 and is preparing a Phase 2 clinical trial in idiopathic pulmonary fibrosis (IPF).
“We are pleased to regain the rights to GLPG1690 to pursue the most suitable clinical application of autotaxin inhibition. There is a large unmet medical need in IPF, and our pre-clinical data with GLPG1690 supports its potential as a competitive and novel approach in this disease area,” said Dr Piet Wigerinck, Chief Scientific Officer of Galapagos. “The alliance with Janssen has been underway since October 2007 and has generated three clinical molecules, two of which are now proprietary Phase 2 assets of Galapagos: GLPG1205 and GLPG1690. This program is a valuable component of our development portfolio, and regaining the rights is a next step in our transformation into a mature biotech company with a proprietary product pipeline.”
Galapagos identified autotaxin as playing a key role in inflammation, using an inflammation assay in its unique target discovery platform. Pharmacology and translational studies published by other parties in the literature since then suggest autotaxin may play a key role in metabolic disease, arthritic pain, oncology, and lung disease.
GLPG1690 is a potent and selective inhibitor of autotaxin. In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015.
About IPF
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal disease characterized by a progressive decline in lung function. Pulmonary fibrosis involves scarring of lung tissue and is the cause of shortness of breath. Fibrosis is usually associated with a poor prognosis. The term “idiopathic” is used because the cause of pulmonary fibrosis is still unknown. Estimated incidence of IPF is up to 16.3 per 100,000 persons in the US and 7.4 per 100,000 persons in Europe, with approximately 30,000-35,000 new patients diagnosed with IPF worldwide each year. The goals of treatment in IPF are essentially to reduce the symptoms, slow down disease progression, reduce acute exacerbations, and prolong survival. Approved treatments thus far have improved the overall survival of IPF patients, but unwanted side effects with these treatments are common, presenting an unmet need for effective treatments with safer side effect profiles.
| Source: Galapagos NV
MECHELEN, Belgium, Sept. 22, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext & NASDAQ: GLPG) presents pre-clinical and Phase 1 results for autotaxin inhibitor GLPG1690 at the European Respiratory Society Annual Meeting in Amsterdam, Netherlands. Galapagos expects to file an exploratory Phase 2 study in idiopathic pulmonary fibrosis before year end. GLPG1690 has potential application in other pulmonary diseases such as chronic obstructive pulmonary disease (COPD), as supported by the presentation on pre-clinical findings at ERS this year:
“Pharmacological profile and efficacy of GLPG1690, a novel ATX inhibitor for COPD treatment,” poster PA2129 in Poster Discussion Session: “New targets and modalities for the treatment of asthma and COPD” (September 28, 2015; Room D201-202, 10:45 AM – 12:45 PM)
Galapagos is the first to show efficacy of an autotaxin inhibitor in pre-clinical models for COPD and IPF, pointing to novel therapeutic areas for autotaxin inhibition. The poster shows how GLPG1690 acts as a potent inhibitor of mouse and human autotaxin (IC50: 100 -500 nM range). Furthermore, GLPG1690 reduces inflammation in a mouse steroid-resistant tobacco smoke model to a similar extent as a standard therapy for COPD.
Galapagos also presents the topline results with GLPG1690 in Phase 1 in healthy human volunteers: “Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in COPD,” oral presentation OA484 in session “Advances in the future treatment of COPD” (September 27, 2015; Room 2.1, 10:45 AM – 12:45 PM)
GLPG1690 was safe and well tolerated up to a single oral dose of 1500 mg and up to 1000 mg twice daily for 14 days, with no significant adverse effects on ECGs, vital signs or laboratory parameters. The compound also showed good oral bioavailability with a half-life of 5 hours and a dose-proportional increase in exposure. GLPG1690 showed concentration-dependent reduction of a relevant biomarker (plasma LPA18:2 levels) with a maximum of approximately 90%. At steady state, continuous reduction of this biomarker levels of >60% was observed from 0 to 24 hours. The presentation will also include relevant pre-clinical model data for COPD and IPF with GLPG1690.
Both the presentation and the posters will be made available on the Galapagos website after the conference.
About Galapagos
Galapagos (Euronext & NASDAQ: GLPG) is a clinical-stage biotechnology company specialized in the discovery and development of small molecule medicines with novel modes of action, with a pipeline comprising three Phase 2 programs, two Phase 1 trials, five pre-clinical studies, and 20 discovery small-molecule and antibody programs in cystic fibrosis, inflammation, and other indications. In the field of inflammation, AbbVie and Galapagos signed a collaboration agreement for the development and commercialization of filgotinib. Filgotinib is an orally-available, selective inhibitor of JAK1 for the treatment of rheumatoid arthritis and potentially other inflammatory diseases, currently in Phase 2B studies in RA and in Phase 2 in Crohn’s disease. Galapagos reported good activity and a favorable safety profile in both the DARWIN 1 and 2 trials in RA. AbbVie and Galapagos also signed a collaboration agreement in cystic fibrosis to develop and commercialize molecules that address mutations in the CFTR gene. Potentiator GLPG1837 is currently in a Phase 1 trial, and corrector GLPG2222 is at the pre-clinical candidate stage. GLPG1205, a first-in-class inhibitor of GPR84 and fully-owned by Galapagos, is currently being tested in a Phase 2 proof-of-concept trial in ulcerative colitis patients. GLPG1690, a fully proprietary, first-in-class inhibitor of autotaxin, has shown favorable safety in a Phase 1 trial and is expected to enter Phase 2 in idiopathic pulmonary fibrosis. The Galapagos Group, including fee-for-service subsidiary Fidelta, has approximately 400 employees, operating from its Mechelen, Belgium headquarters and facilities in The Netherlands, France, and Croatia. More info at www.glpg.com
CONTACT
Galapagos NV
Elizabeth Goodwin, Head of Corporate Communications & IR
Tel: +31 6 2291 6240
ir@glpg.com
MECHELEN, Belgium, Feb. 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced today that GLPG1690, a first-in-class molecule for pulmonary disease, has demonstrated target engagement, a good safety profile, and favorable drug properties in a Phase 1 study. Galapagos is developing GLPG1690 within its alliance with Janssen Pharmaceutica NV.
The aim of the Phase 1 study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of oral single and multiple ascending doses of GLPG1690. The randomized, double-blind, placebo-controlled, single center study was conducted in 40 healthy volunteers in Belgium. In the first part of the study, single ascending doses were evaluated. In the second part, the new compound was administered daily for 14 days.
GLPG1690 proved to be safe and well-tolerated over a wide dose range in healthy volunteers. Engagement of the thus far undisclosed novel target was confirmed using a relevant biomarker. GLPG1690 displayed a favorable pharmacokinetic and pharmacodynamic profile. The data shown in Phase 1 encourage Galapagos to explore a Phase 2 study design in pulmonary disease.
“GLPG1690 is the first molecule against this target ever to be evaluated clinically, and we are pleased with the outcome of the Phase 1 study,” said Dr Piet Wigerinck, CSO of Galapagos. “Galapagos continues to deliver novel therapeutics from its unique target and drug discovery engine.”
In 2007, Galapagos announced an alliance agreement with Janssen Pharmaceutica NV providing the option to worldwide, commercial licenses to certain Galapagos internal inflammatory disease programs. These programs are based on novel targets for inflammatory disorders that were identified and validated by Galapagos using its proprietary target discovery engine. Subsequent Galapagos research led to the discovery of GLPG1690, a first-in-class molecule that entered the clinic for inflammatory disorders. Galapagos is responsible for execution of Phase 1 and Phase 2A studies with GLPG1690.
SYNTHESIS
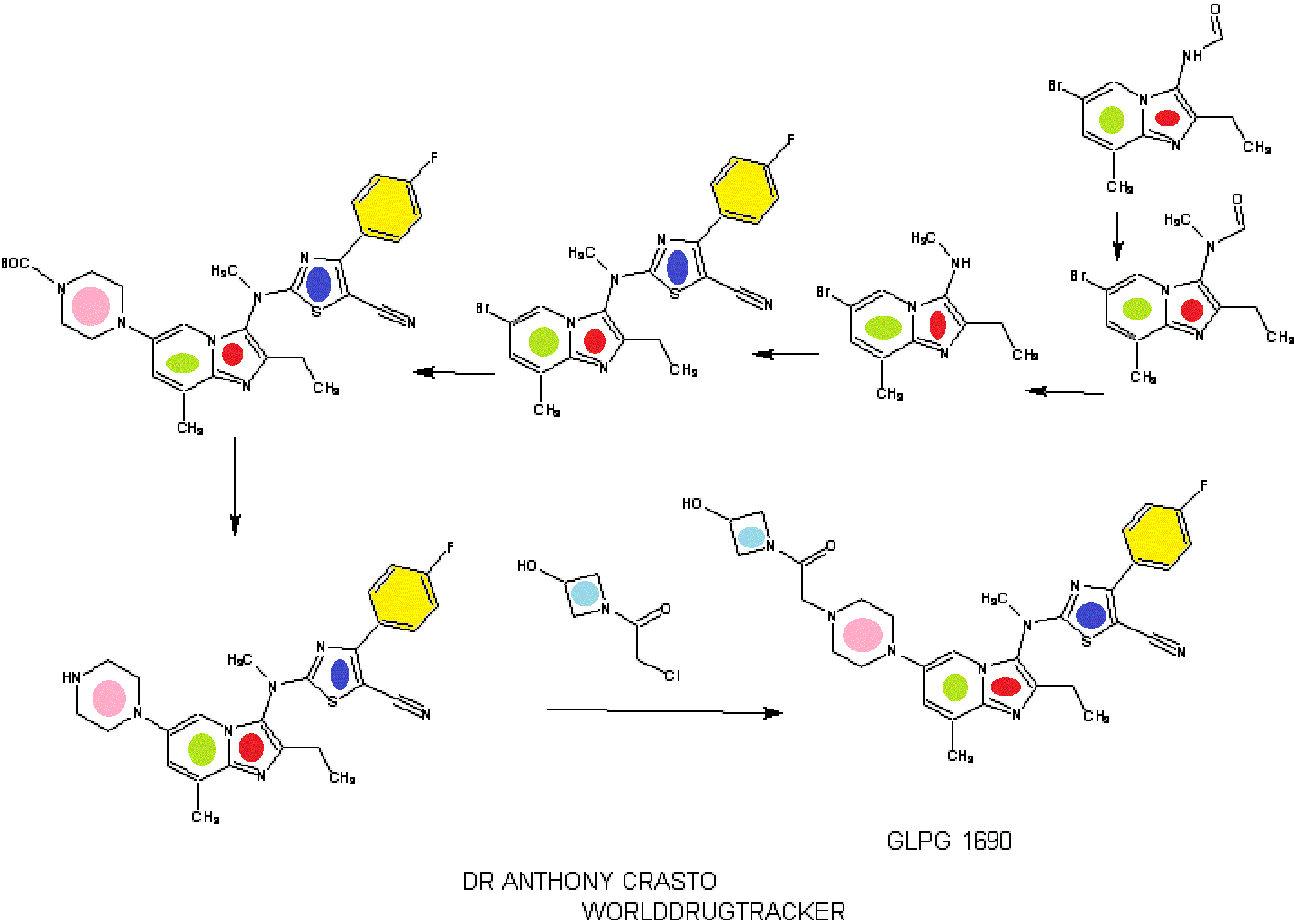
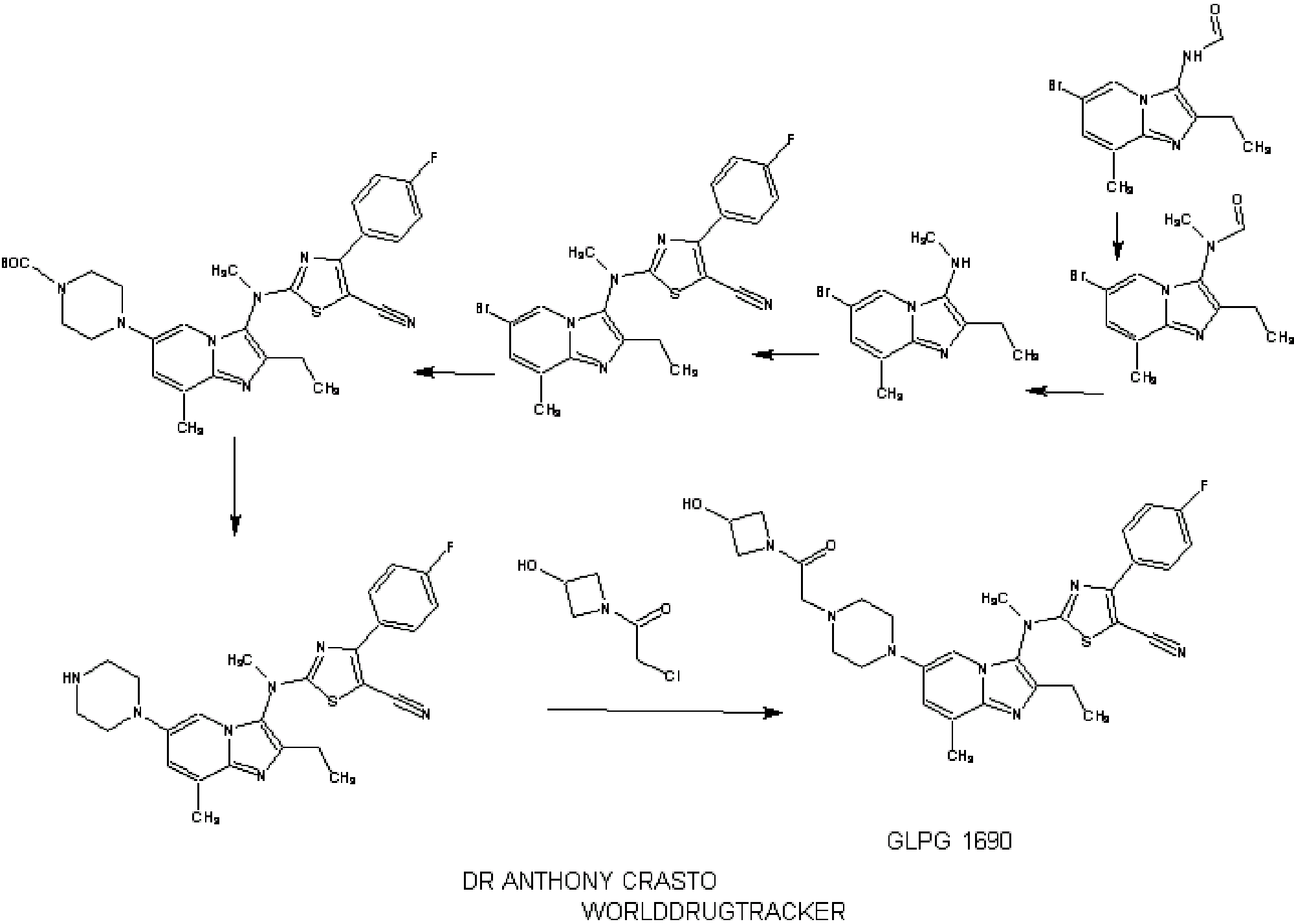
INTRODUCTION
relates to compounds that are inhibitors of autotaxin, also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2), that is involved in fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases. The present invention also provides methods for the production of a compound of the invention, pharmaceutical compositions comprising a compound of the invention, methods for the prophylaxis and/or treatment of diseases involving fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases by administering a compound
STAGE 1

STAGE2

STAGE 3

STAGE4

STAGE 5

FINAL

PATENT
US2014303140
http://www.google.com/patents/US20140303140
1.2.4.4. Illustrative Synthesis of Intermediate Gen-3-e: N-(6-bromo-2-ethyl-8-methylimidazo[1,2-a]pyridin-3-yl)-N-methylformamide
-

-
To a suspension of formamide Gen-2-d (720 g, 2.55 mol, 1 eq.) in 5 L of acetone were added potassium carbonate (1 kg, 7.66 mol, 3 eq.) and methyl iodide (700 g, 4.93 mol, 1.9 eq.). The reaction mixture was heated to 40° C. overnight. Additional methyl iodide (25 g, 0.18 mol, 0.07 eq.) was then introduced and stirring continued for 1 h at 40° C. The reaction mixture was filtered and washed with acetone (2×300 mL) and DCM (2×300 mL). The filtrate was concentrated in vacuo and the residue was partitioned between DCM (3 L) and water (1 L). The aqueous layer was further extracted with DCM. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with Et2O (1 L) at r.t. for 1 h, filtered off and dried to afford Intermediate Gen-3-e.
-
Rotamer A (Major): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.19 (1H, s), 7.78 (1H, s), 7.15 (1H, s), 3.24 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
Rotamer B (Minor): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.49 (1H, s), 7.65 (1H, s), 7.08 (1H, s), 3.36 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
LC-MS: MW (calcd): 295 (79Br), 297 (81Br); m/z MW (obsd): 296 (79Br M+1), 298 (81Br M+1)
1.2.5.2. Illustrative Synthesis of Intermediate Gen-4-d: (6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine
-

-
Intermediate Gen-3-e (80 g, 270 mmol, 1 eq.) was dissolved in a 1.25 M HCl solution in MeOH (540 mL, 2.5 eq.) and the resulting mixture was refluxed overnight. 270 mL of 1.25 M HCl solution in MeOH were added and heating continued overnight. After 48 h, additional 70 mL of the 1.25 M HCl solution in MeOH were introduced in the reaction mixture. Heating was maintained overnight until conversion was complete. The crude mixture was then concentrated in vacuo and the residue was partitioned between EtOAc (300 mL) and water (700 mL). A saturated NaHCO3 solution was added until pH reached 8-9. The aqueous layer was extracted twice with EtOAc (2×300 mL). The combined organic layers were then washed with brine (200 mL), dried over Na2SO4, filtered and concentrated in vacuo to give Intermediate Gen-4-d (6-bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine) as a free base.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.05 (1H, s), 7.04 (1H, s), 2.84-2.78 (5H, m), 2.60 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 267 (79Br), 269 (81Br); m/z MW (obsd): 268 (79Br M+1), 270 (81Br M+1)
1.2.6.4. Illustrative Synthesis of Intermediate Gen-5-t: 2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-
-
To a solution of amine Gen-4-d (4.4 g, 16.6 mmol, 1 eq.) in THF (44 mL) under argon was slowly added NaH (60% in oil suspension, 2.0 g, 50.0 mmol, 3 eq.). The reaction mixture was heated at 90° C. for 30 min then cooled to 40° C. before adding the chlorothiazole Gen-12-a (4.74 g, 19.9 mmol, 1.2 eq.). The reaction mixture was stirred at 90° C. overnight. After cooling to r.t. the mixture was slowly quenched by addition of water and then diluted with EtOAc. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic layers were then washed with water and brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was triturated in Et2O, filtered and washed with Et2O and MeCN. Recrystallization was performed in MeCN (180 mL) to afford Intermediate Gen-5-t (2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile).
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.15 (2H, dd), 7.80 (1H, s), 7.22-7.14 (3H, m), 3.62 (3H, s), 2.77 (2H, q), 2.64 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 469 (79Br), 471 (81Br); m/z MW (obsd): 470 (79Br M+1), 472 (81Br M+1)

1.2.7.1.4. Illustrative Synthesis of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of Intermediate Gen-5-t (24.2 g, 51.5 mmol, 1 eq.) in toluene under argon were successively added N-Boc piperazine (14.4 g, 77.3 mmol, 1.5 eq.), sodium tert-butoxide (9.9 g, 103 mmol, 2 eq.), JohnPhos (1.54 g, 5.15 mmol, 0.1 eq.) and Pd2(dba)3 (2.36 g, 2.58 mmol, 0.05 eq.). The reaction mixture was heated at 115° C. for 1 h. After cooling to r.t., the crude product was filtered on Celpure® P65 and the residue dissolved in EtOAc and washed with water. The organic layer was further washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by chromatography on silica gel (elution with heptane/EtOAc:90/10 to 20/80) to afford the expected product.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.16 (2H, dd), 7.17 (2H, app t), 6.99 (2H, bs), 3.62-3.53 (4H, m), 3.60 (3H, s), 3.04-2.93 (4H, m), 2.74 (2H, q), 2.62 (3H, s), 1.47 (9H, s), 1.33 (3H, t).
-
LC-MS: MW (calcd): 575; m/z MW (obsd): 576 (M+1)
1.2.7.8.4. Illustrative Synthesis of Compound 1: 2-[(2-Ethyl-8-methyl-6-piperazin-1-yl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-

-
4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester was prepared from intermediate Gen-5-t using Boc-piperazine and method Flb.
-
To a solution of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester (24.4 g, 42 mmol, 1 eq.) in MeOH (100 mL) was added a 2 M HCl solution in Et2O (127 mL, 254 mmol, 6 eq.). The reaction mixture was stirred at r.t. for 3.5 h then concentrated in vacuo. The residue was partitioned between EtOAc and water. The aqueous layer was extracted twice with EtOAc. A 2 M NaOH solution was added to the aqueous layer until pH reached 8-9 and further extraction with EtOAc was performed. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with heptane (100 mL) at r.t. overnight, filtered off, washed with heptane and Et2O, and dried to afford the expected compound.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.17 (2H, dd), 7.18 (2H, app t), 6.99 (2H, bs), 3.61 (3H, s), 3.09-2.98 (8H, m), 2.75 (2H, q), 2.61 (3H, s), 1.34 (3H, t).
-
LC-MS: MW (calcd): 475; m/z MW (obsd): 476 (M+1)
1.2.7.14. Illustrative Synthesis of Compound 2: 2-((2-ethyl-6-(4-(2-(3-hydroxyazetidin-1-yl)-2-oxoethyl)piperazin-1-yl)-8-methylimidazo[1,2-a]pyridin-3-yl)(methyl)amino)-4-(4-fluorophenyl)thiazole-5-carbonitrile
-

-
To a solution of amine compound 1 (12.6 g, 27 mmol, 1 eq.) in 100 mL of MeCN were added potassium carbonate (7.3 g, 53 mmol, 2 eq.) and Gen13-a (5.2 g, 34 mmol, 1.3 eq.). The reaction mixture was refluxed for 5.5 h then cooled to r.t. and stirred for 40 h. The crude product was filtered and washed with MeCN. The collected precipitate was then suspended in 300 mL of water, stirred for 1 h, filtered, and finally washed with water and MeCN. The solid obtained was dried in vacuo for 48 h to afford Compound 2.
-
1H NMR (400 MHz, CDCl3) δ ppm 8.20-8.12 (2H, m), 7.22-7.13 (2H, m), 6.99 (2H, s), 4.68 (1H, m), 4.43 (1H, dd), 4.26 (1H, dd), 4.14-4.05 (1H, m), 3.88 (1H, dd), 3.61 (3H, s), 3.58-3.52 (1H, m), 3.14-3.02 (6H, m), 2.74 (2H, q), 2.70-2.62 (4H, m), 2.59 (3H, s), 1.33 (3H, t)
-
LC-MS: MW (calcd): 588; m/z MW (obsd): 589 (M+1)
| US9249141 | Dec 17, 2014 | Feb 2, 2016 | Galapagos Nv | Compounds and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015111872 | 2015-04-23 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
| US2014303140 | 2014-10-09 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
////////////GLPG 1690, idiopathic pulmonary fibrosis, PHASE 1, GALAPAGOS, 1628260-79-6
n12c(c(nc1c(cc(c2)N3CCN(CC3)CC(=O)N4CC(C4)O)C)CC)N(C)c5nc(c(s5)C#N)c6ccc(cc6)F
CCC1=C(N2C=C(C=C(C2=N1)C)N3CCN(CC3)CC(=O)N4CC(C4)O)N(C)C5=NC(=C(S5)C#N)C6=CC=C(C=C6)F
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
The U.S. Food and Drug Administration approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
 Nintedanib
Nintedanib
FDA approves Ofev to treat idiopathic pulmonary fibrosis
10/15/2014
The U.S. Food and Drug Administration today approved Ofev (nintedanib) for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
see synthesis
https://newdrugapprovals.org/2014/05/21/in-battle-of-ipf-drugs-bis-nintedanib-impresses/

FDA approves Esbriet (pirfenidone ピルフェニドン 吡非尼酮) to treat idiopathic pulmonary fibrosis

The U.S. Food and Drug Administration today approved Esbriet (pirfenidone)
ピルフェニドン 吡非尼酮
for the treatment of idiopathic pulmonary fibrosis (IPF).
read at
SYNTHESIS
Click for synthesis



//////////

{kind=link}