
Home » Posts tagged 'Hoffmann La Roche'
Tag Archives: Hoffmann La Roche
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CRD 1152, CURADEV PHARMA PRIVATE LTD
Several candidates….one is…….CRD1152
ONE OF THEM IS CRD 1152
Kynurenine pathway regulators (solid tumors)

Compound 2
CAS1638121-21-7
N3-(3-Chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine

COMPD 190
CAS 1638118-99-6

COMPD248
7-Chloro-N3- (3-chloro-4- fluorophenyl) furo[2,3- c]pyridine-2,3- diamine, 166
DMSO-d6: δ 7.87 (d, J = 5.1 Hz, 1H), 7.25 (s, 2H), 7.16-7.10 (m, 2H), 6.88 (d, J = 5.1 Hz, 1H), 6.59 (dd, J′ = 6.2 Hz, J″ = 2.6 Hz, 1H), 6.48 (dt, J′ = 8.8 Hz, J″ = 6.7 Hz, J′′′ = 3.4 Hz, 1H) M + H] 312
OR
N3-(3,4- difluorophenyl)- 7-(pyridin-4- yl)furo[2,3- c]pyridine-2,3- diamine, 184
CD3CN: δ 8.72 (s, 2H), 8.26 (s, 3H), 7.07-7.03 (m, 2H), 6.47-6.40 (m, 2H), 5.74 (s, 1H), 5.55 (s, 2H) M + H] 339
OR

COMPD73
CAS 1638117-85-7
Several candidates………..CRD1152
 67
67
 66
66
| Company | Curadev Pharma Pvt. Ltd. |
| Description | Small molecule dual indoleamine 2,3-dioxygenase 1 (IDO1) and tryptophan 2,3-dioxygenase (TDO1; IDO) inhibitor |
| Molecular Target | Indoleamine 2,3-dioxygenase (INDO) (IDO) ; Tryptophan 2,3-dioxygenase (TDO2) (TDO) |
| Mechanism of Action | Indoleamine 2,3-dioxygenase (INDO) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Preclinical |
| Standard Indication | Cancer (unspecified) |
| Indication Details | Treat cancer |
| Regulatory Designation | |
| Partner | Roche |
Hoffmann-La Roche partners with Curadev Pharma Ltd. for IDO1 and TDO inhibitors (April 20, 2015)
Curadev Pharma Pvt Ltd., founded in 2010 and headquartered in New Delhi, announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors to treat cancer. The agreement covers the development of CRD1152, the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2,3-dioxygenase-1) and TDO (tryptophan-2,3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response. Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments, as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev is also eligible for milestones and royalties on any additional products resulting from the research collaboration.




Curadev Announces Research Collaboration and Licensing Agreement to Develop Cancer Immunotherapeutic
Curadev’s dual IDO and TDO immune tolerance inhibitor – a novel approach in cancer immunotherapy
Apr 20, 2015, 06:30 ET from Curadev
NEW DELHI, India, April 20, 2015 /PRNewswire/ —
Curadev Pharma Private Ltd. today announced that it has entered into a research collaboration and exclusive license agreement with Roche for the development and commercialization of IDO1 and TDO inhibitors. The agreement covers the development of the lead preclinical immune tolerance inhibitor and a research collaboration with Roche’s research and early development organization to further explore the IDO and TDO pathways.
IDO1 (indoleamine-2, 3-dioxygenase-1) and TDO (tryptophan-2, 3-dioxygenase) are enzymes that mediate cancer-induced immune suppression. This mechanism is exploited by tumor cells as well as certain type of immune cells, limiting the anti-tumor immune response.
Dual inhibition of the IDO1 and TDO pathways promises to maintain the immune response, prevent local tumor immune escape and potentially avoid resistance to other immunotherapies when used in combination, and could lead to new treatment options for cancer patients. Curadev’s preclinical lead-compound, a small-molecule that shows potent inhibition of the two rate-limiting enzymes in the tryptophan – to kynurenine metabolic pathways, has the potential for mono therapy as well as combination with Roche’s broad oncology pipeline and portfolio.
“We are very excited to be working with the global leader in oncology with their unrivalled expertise in clinical development,” said Arjun Surya, PhD, Chief Scientific Officer, Curadev. “The collaboration acknowledges our focused research efforts on patient-critical drug targets that have yielded a drug candidate that could make a significant difference in the development of novel treatments for patients suffering from cancer.”
Under the terms of agreement, which includes a research collaboration with Roche’s research and early development organization to further extend Curadev’s findings, Curadev will receive an upfront payment of $25 million and will be eligible to receive up to $530 million in milestone payments based on achievement of certain predetermined events and sales levels as well as escalating royalties potentially reaching double digits for the first product from the collaboration developed and commercialized by Roche. Curadev would also be eligible for milestones and royalties on any additional products resulting from the research collaboration. Roche will fund future research, development, manufacturing and commercialization costs and will also provide additional research funding to Curadev for support of the research collaboration.
About Curadev
Headquartered in New Delhi, India, Curadev Pharma Private Limited was founded in 2010 by a team of professionals from the pharmaceutical and biotech sectors with the mission to improve human health and enhance the quality of human life by accelerating the discovery and delivery of new drugs. Curadev focuses on the creation and out-licensing of pre-IND assets and IND packages for drug development.
For further information:
Curadev Partnering
Manish Tandon – VP and Chief Financial Officer, manish@curadev.in
PATENT
US20160046596) INHIBITORS OF THE KYNURENINE PATHWAY
Monali Banerjee
Sandip Middya
Ritesh Shrivastava
Sushil Raina
Arjun Surya
Dharmendra B. Yadav
Veejendra K. Yadav
Kamal Kishore Kapoor
Aranapakam Venkatesan
Roger A. Smith
Scott K. Thompson
ONE ………….Example 2
Synthesis of N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine (Compound 2)
Step 1: 3-Methoxymethoxy-pyridine

Step 2: 3-Methoxymethoxy-pyridine-4-carbaldehyde

Step 3: 3-Hydroxy-pyridine-4-carbaldehyde

Step 4: 4-{[3-Chloro-4-fluoro-phenylimino]-methyl}-pyridin-3-ol

Step 5: N3-(3-Chloro-4-fluoro-phenyl)-furo[2,3-c]pyridine-2,3-diamine

Monali Banerjee – Director, R&D
Ms. Banerjee has more than 10 years of research experience, during which she has held positions of increasing responsibility. Her past organizations include TCG Lifesciences (Chembiotek) and Sphaera Pharma. Ms. Banerjee is a versatile scientist with a deep understanding of the fundamental issues that underlie various aspects of drug discovery. At Curadev, she has been responsible for target selection, patent analysis, pharmacophore design, assay development, ADME/PK and in vivo and in vitro pharmacology. Ms. Banerjee holds a Masters in Biochemistry and a Bachelors in Chemistry both from Kolkata University.
writeup
|
The essential amino acid Tryptophan (Trp) is catabolized through the kynurenine (KYN) pathway. The initial rate-limiting step in the kynurenine pathway is performed by heme-containing oxidoreductase enzymes, including tryptophan 2,3-dioxygenase (TDO), indoleamine 2,3-dioxygenase-1 (IDO1), and indoleamine 2,3-dioxygenase-2 (IDO2). IDO1 and IDO2 share very limited homology with TDO at the amino acid level and, despite having different molecular structures, each enzyme has the same biochemical activity in that they each catalyze tryptophan to form N-formylkynurenine. IDO1, IDO2, and/or TDO activity alter local tryptophan concentrations, and the build-up of kynurenine pathway metabolites due to the activity of these enzymes can lead to numerous conditions associated with immune suppression.
|
|
Kynurenine pathway dysregulation and IDO1 and/or TDO activity also correlate with cardiovascular risk factors, and kynurenines and IDO1 are markers for Atherosclerosis and other cardiovascular heart diseases such as coronary artery disease (Platten et al., Science, 2005, 310(5749):850-5, Wirlietner et al. Eur J Clin Invest. 2003 July; 33(7):550-4) in addition to kidney disease. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease (Pawlak et al., Atherosclerosis, 2009, (204)1:309-314). Studies show that kynurenine pathway metabolites are associated with endothelial dysfunction markers in the patients with chronic kidney disease (Pawlak et al., Advances in Medical Sciences, 2010, 55(2):196-203).
|
///////CRD1152, CRD-1152, CRD 1152, CURADEV PHARMA PRIVATE LTD, ROCHE, IDO1 and TDO inhibitors, COLLABORATION, CANCER, indoleamine-2,3-dioxygenase-1, Hoffmann-La Roche, kynurenine pathway regulators, solid tumors
RO-28-1675 for Type 2 Diabetes

RO-28-1675
- (2R)-3-Cyclopentyl-2-[4-(methanesulfonyl)phenyl]-N-(thiazol-2-yl)propionamide
- Ro 028-1675
- Ro 0281675
- Ro 28-1675
3-Cyclopentyl-2(R)-[4-(methylsulfonyl)phenyl]-N-(2-thiazolyl)propionamide
| MW | 378.51 | .-70.4 °
Conc 0.027 g/100mL; chloroform, 589 nm; 23 °C
|
|
|---|---|---|---|
| Formula | C18H22N2O3S2 | ||
| CAS No | 300353-13-3 |
Glucokinase Activators
Ro 28-1675 (Ro 0281675) is a potent allosteric GK activator with a SC1.5 value of 0.24± 0.0019 uM.
Roche (Innovator)
PHASE 1 Type 2 DIABETES,
IC50 value: 0.24± 0.0019 uM (SC1.5) [1]
Target: Glucokinase activator
The R stereoisomer Ro 28-1675 activated GK with a SC1.5 of 0.24 uM, while the S isomer did not activated GK up to 10 uM. Oral administration of Ro 28-1675 (50 mg/Kg) to male C57B1/6J mice caused a statistically significant reduction in fasting glucose levels and improvement in glucose tolerance relative to the vehicle treated animals [1].
Comparison of rat PK parameters indicated that Ro 28-1675 displayed lower clearance and higher oral bioavailability compared to 9a.
Following a single oral dose, Ro 28-1675 reduced fasting and postprandial glucose levels following an OGTT, was well tolerated, and displayed no adverse effects related to drug administration other than hypoglycemia at the maximum dose (400 mg).
 .
.
RO-28-1675 as glucokinase activator.
Joseph Grimsby et al., of Roche have recently discovered activators of glucokinase that increase kcat and decrease the S0.5 for glucose, and these may offer a treatment for type II diabetes. Glucokinase (GK) plays a key role in whole-body glucose homeostasis by catalyzing the phosphorylation of glucose in cells that express this enzyme, such as pancreatic β cells and hepatocytes.
By screening of a library of 120,000 structurally diverse synthetic compounds, they found one small molecule that increased the enzymatic activity of GK. Chemical optimization of this initial molecule led to the synthesis of RO-28-0450 as a lead GK activator which is a class of antidiabetic agents that act as nonessential, mixed-type GK activators (GKAs) that increase the glucose affinity and maximum velocity (Vmax) of GK. RO-28-0450 is a racemic compound.
Activation of GK was exquisitely sensitive to the chirality of the molecule: The R enantiomer, RO-28-1675, was found to be a potent GKA, whereas the S enantiomer, RO-28-1674, was inactive. RO-28-1675 also reversed the inhibitory action of the human glucokinase regulatory protein (GKRP). The activators binding in a glucokinase regulatory site originally was discovered in patients with persistent hyperinsulinemic hypoglycemi.
The result of RO-28-1675 as a potent small molecule GKA may shed light to the chemical biologists to devise strategy for developing activators. Thus for a success to this end we must focus on highly regulated enzymes, or cooperative enzymes such as glucokinase, where nature has provided binding sites that are designed to modulate catalysis.
.SYNTHESIS


Paper

Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl-propionamide (2.10 g, 74%) as a white foam.
[α] 23 589 = –70.4° (c=0.027, chloroform).
EI-HRMS m/e calcd for C18H22N2O3S2 (M+ ) 378.1072, found 378.1081.
1 H NMR (400 MHz, CHLOROFORM-d) δ ppm 10.48 (br. s., 1 H), 7.88 (d, J=8.6 Hz, 2 H), 7.53 (d, J=8.6 Hz, 2 H), 7.50 (d, J=3.5 Hz, 1 H), 7.06 (d, J=3.5 Hz, 1 H), 3.76 (t, J=7.7 Hz, 1 H), 3.03 (s, 3 H), 2.28 (dt, J=13.6, 7.7 Hz, 1 H), 1.88 – 1.98 (m, 1 H), 1.42 – 1.84 (m, 7 H), 1.07 – 1.19 (m, 2 H).
Anal. Calcd for C18H22N2O3S2: C, 56.94; H, 5.59; N, 7.28. Found: C, 57.12; H, 5.86; N, 7.40.
PATENT
WO 2000058293
http://www.google.com/patents/WO2000058293A2?cl=en
Example 3 (A) 3-CyclopentyI-2-(4-methanesulfonyl-phenyI)-N-thiazol-2-yI-propionamide

A solution of dπsopropylamine (3.3 mL, 23.5 mmol) in dry tetrahydrofuran (50 mL) and 1.3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdιnone (10 mL) was cooled to -78°C under nitrogen and then treated with a 10M solution of n-butyllithium m hexanes (2.35 mL, 23 5 mmol) The yellow reaction mixture was stiπed at -78°C for 30 mm and then treated dropwise with a solution of 4-methylsulfonylphenylacetιc acid (2.40 g, 11.2 mmol) in a small amount of dry tetrahydrofuran. After approximately one-half of the 4- methylsulfonylphenylacetic acid m dry tetrahydrofuran was added, a precipitate formed Upon further addition of the remaining 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture became thick in nature After complete addition of the 4-methylsulfonylphenylacetιc acid in dry tetrahydrofuran, the reaction mixture was very thick and became difficult to stir An additional amount of dry tetrahydrofuran (20 mL) was added to the thick reaction mixture, and the reaction mixture was stirred at –
78 C for 45 mm, at which time, a solution of lodomethylcyclopentane (2.35 g, 11.2 mmol) in a small amount of dry tetrahydrofuran was added dropwise The reaction mixture was allowed to warm to 25°C where it was stiπed for 15 h. The reaction mixture was quenched with water (100 mL), and the resulting yellow reaction mixture was concentrated in vacuo to remove tetrahydrofuran. The aqueous residue was acidified to pH = 2 using concentrated hydrochloπc acid The aqueous layer was extracted with ethyl acetate The organic phase was dπed over magnesium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 230-400 mesh, 1/3 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (1.80 g, 52%) as a white solid: mp 152-154°C; EI-HRMS m/e calcd for C15H20O4S (Nf) 296.1082, found 296.1080
A solution of 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)propιonιc acid (4.91 g, 16.56 mmol) and tnphenylphosphine (6.52 g, 24.85 mmol) m methylene chloπde (41 mL) was cooled to 0°C and then treated with N-bromosuccinimide (5.01 g, 28.16 mmol) m small portions The reaction mixture color changed from light yellow to a darker yellow then to brown After the complete addition of N-bromosuccinimide, the reaction mixture was allowed to warm to 25°C over 30 min. The brown reaction mixture was then treated with 2-aminothiazole (4.98 g, 49.69 mmol). The resulting reaction mixture was stiπed at 25°C for 19 h. The reaction mixture was then concentrated in vacuo to remove methylene chloride. The remaining black residue was diluted with a 10% aqueous hydrochloric acid solution (400 mL) and then extracted with ethyl acetate (3 x 200 mL). The combined organic layers were washed with a saturated aqueous sodium chloride solution (1 x 200 mL), dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate then 1/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4-methanesulfonyl-phenyl)-N-thiazol-2- yl-propionamide (4.49 g, 72%) as a white solid: mp 216-217°C; EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1071.
Example 13
(2R)-3-Cyclopentyl-2-(4-methanesuIfonylphenyl)-N-thiazol-2-yl-propionamide

A solution of ^-( ethanesulfonyl)phenyl acetic acid (43 63 g, 0.204 mol) in methanol (509 mL) was treated slowly with concentrated sulfunc acid (2 mL) The resulting reaction mixture was heated under reflux for 19 h The reaction mixture was allowed to cool to 25°C and then concentrated in vacuo to remove methanol The residue was diluted with ethyl acetate (800 mL) The organic phase was washed with a saturated aqueous sodium bicarbonate solution (1 x 200 mL), washed with a saturated aqueous sodium chlonde solution (1 x 200 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 1/1 hexanes/ethyl acetate) afforded 4-(methanesulfonyl)phenyl acetic acid methyl ester (45.42 g, 98%) as a yellow oil which solidified to a cream colored solid upon sitting over time at 25°C mp 78-80°C, EI-HRMS m/e calcd for Cι0H12O4S (M+) 228 0456, found 228 0451.
A mechanical stiπer was used for this reaction A solution of dnsopropylamme (29.2 mL, 0.21 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro- 2(lH)-pyπmιdιnone (62 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (83 4 mL, 0.21 mol) The yellow-orange reaction mixture was stiπed at -78°C for 35 min and then slowly treated with a solution of 4- (methanesulfonyl)phenyl acetic acid methyl ester (45.35 g, 0.20 mol) in dry tetrahydrofuran (186 mL) and l,3-dιmethyl-3,4,5,6-tetrahydro-2(lH)-pyπmιdmone (62 mL) The reaction mixture turned dark in color. The reaction mixture was then stiπed at -78°C for 50 mm, at which time, a solution of lodomethylcyclopentane (50.08 g, 0.24 mol) in a small amount of dry tetrahydrofuran was added slowly. The reaction mixture was then stiπed at -78°C for 50 mm, and then allowed to warm to 25°C, where it was stirred for 36 h. The reaction mixture was quenched with water (100 mL), and the resulting reaction mixture was concentrated in vacuo to remove tetrahydrofuran The remaining residue was diluted with ethyl acetate (1.5 L). The organic phase was washed with a saturated aqueous sodium chloπde solution (1 x 500 mL), dned over sodium sulfate, filtered, and concentrated in vacuo Flash chromatography (Merck Silica gel 60, 70-230 mesh, 3/1 hexanes/ethyl acetate) afforded 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid methyl ester (41.79 g, 68%) as a yellow viscous oil EI-HRMS m/e calcd for Cι6H22O4S (M+) 310.1239. found 310.1230.
A solution of 3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid methyl ester (50 96 g, 0.16 mol) in methanol (410 mL) was treated with a IN aqueous sodium hydroxide solution (345 mL, 0.35 mol). The reaction mixture was stirred at 25°C for 24 h. The reaction mixture was concentrated in vacuo to remove methanol. The resulting aqueous residue was acidified to pH = 2 with concentrated hydrochlonc acid and then extracted with ethyl acetate (5 x 200 mL) The combined organic layers were dned over sodium sulfate, filtered, and concentrated in vacuo to afford pure 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (43 61 g, 90%) as a white solid which was used without further puπfication. mp 152-154°C, EI-HRMS m e calcd for C15H20O4S (M+) 296.1082, found 296.1080.
Two separate reactions were setup in parallel: (1) A solution of (R)-(+)-4-benzyl-2- oxazohdmone (3.67 g, 20.73 mmol) m dry tetrahydrofuran (35 mL) was cooled to -78°C and then treated with a 2.5M solution of n-butylhthium in hexanes (7.9 mL, 19.86 mmol). The resulting reaction mixture was stiπed at -78°C for 30 mm and then allowed to warm to 25°C, where it was stirred for 1.5 h (2) A solution of racemic 3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonιc acid (5.12 g, 17.27 mmol) in dry tetrahydrofuran (35 mL) was cooled to 0°C and then treated with tnethylamme (2.8 mL, 19.86 mmol). The reaction mixture was stiπed at 0°C for 10 nun and then treated dropwise with tπmethylacetyl chlonde (2.6 mL, 20.73 mmol). The resulting reaction mixture was stiπed at 0°C for 2 h and then cooled to -78°C for the addition of the freshly prepared chiral oxazolidmone. The reaction mixture containing the oxazolidmone was then added to the cooled (-78°C) mixed anhydπde solution The resulting reaction mixture was stiπed as -78°C for 1 h and allowed to gradually warm to 25°C. The reaction mixture was then stiπed at 25°C for 3 d. The resulting reaction mixture was quenched with water (100 mL) and then concentrated in vacuo to remove tetrahydrofuran. The resulting aqueous residue was diluted with ethyl acetate (600 mL). The organic layer was washed with a saturated aqueous sodium chloπde solution (1 x 300 mL), dπed over sodium sulfate, filtered, and concentrated in vacuo Thin layer chromatography using 13/7 hexanes/ethyl acetate as the developing solvent indicated the presence of two products The higher moving product had a Rf =0.32 and the lower moving product had a Rf = 0.19. Flash chromatography (Merck Silica gel 60, 230-400 mesh, 9/1 then 13/7 hexanes/ethyl acetate) afforded two products: (1) The higher Rf product (4R, 2’S)-4-benzyl-3-[3- cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazohdm-2-one (2.12 g, 54%) as a white foam- mp 62-64°C; [c.]23 589 = +6.3° (c=0.24, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M+) 455.1766, found 455.1757. (2) The lower Rf product (4R, 2R)-4- benzyl-3-[3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.88 g, 99%) as a white foam: mp 59-61°C; [α]23 589 = -98.3° (c=0.35, chloroform); EI-HRMS m/e calcd for C25H29NO5S (M +) 455.1766, found 455.1753. The combined mass recovery from the two products was 6.00 g, providing a 76% conversion yield for the reaction
An aqueous solution of lithium hydroperoxide was freshly prepared from mixing a solution of anhydrous lithium hydroxide powder (707.3 mg, 16.86 mmol) m 5.27 mL of water with a 30% aqueous hydrogen peroxide solution (3.44 mL, 33.71 mmol). This freshly prepared aqueous lithium hydroperoxide solution was cooled to 0°C and then slowly added to a cooled (0°C) solution of (4R, 2’R)-4-benzyl-3-[3-cyclopentyl-2-(4- methanesulfonylphenyl)propιonyl]-oxazolιdm-2-one (3.84 g, 8.43 mmol) in tetrahydrofuran (33 mL) and water (11 mL). The reaction mixture was stiπed 0°C for 1.5 h The reaction mixture was then quenched with a 1.5N aqueous sodium sulfite solution (25 mL) The reaction mixture was further diluted with water (300 mL) The resulting aqueous layer was continuously extracted with diethyl ether until thm layer chromatography indicated the absence of the recovered chiral oxazolidmone in the aqueous layer The aqueous layer was then acidified to pH = 2 with a 10% aqueous hydrochlonc acid solution and extracted with ethyl acetate (300 mL) The organic extract was dned over sodium sulfate, filtered, and concentrated in vacuo to afford (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white solid (2.23 g, 89%) which was used without further puπfication Flash chromatography (Merck Silica gel 60, 70-230 mesh, 30/1 methylene chlonde/methanol then 10/1 methylene chlonde/methanol) was used to obtain a punfied sample for analytical data and afforded pure (2R)-3- cyclopentyl-2-(4-methanesulfonylphenyl)propιomc acid as a white foam- mp 62-64°C (foam to gel), [α]23 589 = -50.0° (c=0.02, chloroform), EI-HRMS m/e calcd for C15H20O4S (M+) 296 1082, found 296 1080
A solution of tnphenylphosphme (3.35 g, 12.79 mmol) m methylene chloπde (19 mL) was cooled to 0°C and then slowly treated with N-bromosuccmimide (2.28 g, 12.79 mmol) in small portions. The reaction mixture was stiπed at 0°C for 30 mm, and dunng this time penod, the color of the reaction mixture changed from light yellow to a darker yellow then to a purple color. The cooled purple reaction mixture was then treated with the (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)propιonιc acid (2.23 g, 7.52 mmol) The resulting reaction mixture was then allowed to warm to 25°C over 45 mm, at which time, the reaction mixture was then treated with 2-amιnothιazole (1.88 g, 18.81 mmol) The resulting reaction mixture was stiπed at 25°C for 12 h. The reaction mixture was then concentrated in vacuo to remove methylene chloπde The remaining black residue was diluted with ethyl acetate (300 mL) and then washed well with a 10% aqueous hydrochlonc acid solution (2 x 100 mL), a 5% aqueous sodium bicarbonate solution (3 x 100 mL), and a saturated aqueous sodium chloride solution (1 x 200 mL). The organic layer was then dried over sodium sulfate, filtered, and concentrated in vacuo. Flash chromatography (Merck Silica gel 60, 70-230 mesh, 9/1, 3/1, and then 11/9 hexanes/ethyl acetate) afforded (2R)-3-cyclopentyl-2-(4-methanesulfonylphenyl)-N-thiazol-2-yl- propionamide (2.10 g, 74%) as a white foam: mp 78-80°C (foam to gel); [α]23 589 = -70.4° (c=0.027, chloroform); EI-HRMS m/e calcd for C18H22N2O3S2 (M+) 378.1072, found 378.1081.
REFERENCES
Glucokinase (GK) is a glucose sensor that couples glucose metabolism to insulin release. The important role of GK in maintaining glucose homeostasis is illustrated in patients with GK mutations. In this publication, identification of the hit molecule 1 and its SAR development, which led to the discovery of potent allosteric GK activators 9a and 21a, is described. Compound 21a (RO0281675) was used to validate the clinical relevance of targeting GK to treat type 2 diabetes.
http://www.nature.com/nrd/journal/v8/n5/fig_tab/nrd2850_T2.html
NMR…..http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-NMR-HY-10595-13569-2014.pdf
http://www.medchemexpress.com/product_pdf/HY-10595/Ro%2028-1675-Lcms_Ms-HY-10595-13569-2014.pdf
///////////RO-28-1675, Ro 0281675
O=C(Nc1nccs1)[C@H](CC2CCCC2)c3ccc(cc3)S(C)(=O)=O

Chemical structures of Roche’s glucokinase activators (GKAs) RO-28-1675 and piragliatin, as well as the related GKA 1.
RG-1577, EVT 302, Sembragiline, RO-4602522
RG-1577, EVT 302, Sembragiline, RO-4602522
CAS 676479-06-4, MW 342.36
- C19 H19 F N2 O3
- Acetamide, N-[(3S)-1-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-3-pyrrolidinyl]-
UNII-K3W9672PNJ
RG-1577, a selective and reversible monoamine oxidase B inhibitor, for treating AD (phase 2 clinical, as of May 2015).
Family members of the product case for RG-1577 (WO2004026825) hold protection in EU until 2023 and expire in US in 2024 with US154 extension. Follows on from WO2006097197, claiming a process for preparing RG-1577.
Alzheimer‘s Disease is a brain disease that slowly destroys memory and thinking skills, up to loss of the ability to carry out the simplest tasks. It is the most common cause of dementia among older people. Mild Alzheimer‘s Disease manifests itself in memory loss and small changes in other cognitive abilities, e.g getting lost, trouble handling money and managing daily tasks, having some mood and personality changes, etc.
In the stage of Moderate Alzheimer‘s Disease, the control of language, reasoning, sensory processing, and conscious thought are impacted. Memory loss and con usion grow worse, e.g patients have problems recognizing family and friends and become unable to learn new things, etc. hallucinations, delusions, and paranoia may occur. .Severe Alzheimer‘s Disease is the final stage. Patients cannot communicate anymore and are completely dependent.
N-[(3S)-l-[4-[(3-fluorophenyl)methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide has previously been described in the art. 1 WO 2006/097197 2 and WO 2006/0972703 relate to methods for preparing enantiomerically pure 4-pyrrolidinophenylbenzyl ether derivatives.
![]()
The processes of the prior art hamper from several drawbacks (e.g. long reaction sequence, low overall yield also due to loss of half of the product in the classical resolution step, the need for a chromatographic purification to remove by-products formed in the Mitsunobu reaction) and are therefore less suitable for the preparation of N-[(3S)-l-[4-[(3-fluorophenyl) methoxy]phenyl]-5-oxo-pyrrolidin-3-yl]acetamide on large scale.
Most Recent Events
- 01 Aug 2014Roche completes a phase I trial in volunteers in USA (NCT02104648)
- 14 May 2014Roche completes enrolment in the MAyflOwer RoAD trial for Alzheimer’s disease (combination therapy, adjunctive treatment) in Australia, Canada, Czech Republic, France, Germany, Italy, Poland, South Korea, Spain, Sweden the United Kingdom and the USA (NCT01677754)
- 01 Apr 2014Roche initiates enrolment in a phase I trial in healthy volunteers in USA (NCT02104648)
http://www.evotec.com/uploads/media_library/10/2012-09_Evotec_Company_presentation_September_e.pdf

……………………..
WO2004026825
http://www.google.com/patents/WO2004026825A1?cl=en
………………….
WO2006097197
http://www.google.com/patents/WO2006097197A1?cl=en
……………………………………………..
PATENT
WO 2015063001
Novel, crystalline polymorphic forms A and B of a pyrrolidone derivative ie RG-1577, useful for treating Alzheimer’s disease (AD). Roche and its Japanese subsidiary Chugai, under license from Evotec, which previously licensed the drug from Roche, are developing RG 1577
formula 1 via the following routes

In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route A

1
In a certain embodiment, present invention relates to a synthesis of a compound of formula he following route B

In a certain embodiment, present invention relates to a crystalline polymorph of a compound of formula 1.

synthesize a compound of formula 1 from a compound of formula 7

compound of formula 6 to a compound of formula 7

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 6 via the intermediate 6a to a compound of formula 7

further comprising reacting a compound of formula 3 with a compound of formula 5 to a compound of formula 6

comprising reacting a compound of formula 2 to a compound of formula 3

2 3
In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 10 to a compound of formula 6

eacting a compound of formula 9 with a compound of formula 5 to a compound of formula 10

In a certain embodiment, present invention relates to a process to synthesize a compound of formula 1 as described herein, further comprising reacting a compound of formula 8 to a compound of formula 9

(lS’)-N-[l-[4-(3-fluoro-benzyloxy)-phenyl]-5-oxo-pyrrolidin-3-yl-]acetamide (1)
To a suspension of chloride (7) (37.9 g, 100 mmol) in 2-methyltetrahydrofurane (600 ml) was added under vigorous stirring at 0°C 1.65 M potassium ie/t-butoxide in THF (75.5 ml, 125 mmol, ACROS) over 2.5 h. After additional stirring at 0°C for 1 h, the cold suspension was hydrolyzed with 0.1 M HCl (600 ml) and the reaction mixture was stirred at 30°C for 0.5 h. The organic layer was washed with water (300 ml), dried (Na2S04) and filtered. Removal of the solvent by rotary evaporation (50°C/>10 mbar) afforded 32.1 g crystalline residue, which was dissolved in 2-butanone (400 ml) at ca. 95°C and hot filtered. Crystallization, which was induced by seeding and cooling to room temperature and 0°C (4 h) afforded 25.4 g (74.2%) of the titled compound (1) as an off-white, crystalline powder,
Mp. 162-164°C (polymorph B).
Ee >99.8%, [cc]D20 = – 17.8 (DMF; c = 1).
1H NMR (400 MHz, DMSO- 6) δ ppm 1.82 (s, 3H), 2.34 (dd, J1=n. l, J2=3.9, 1H), 2.84 (dd, J/=17.1, J2=8.2, 1H), 3.55 (dd, J/=10.2, J2=3.2, 1H), 4.07 (dd, J/=10.2, J2=6.7, 1H), 4.32-4.41 (m, 1H), 5.13 (s, 2H), 7.02 & 7.55 (d, J=9.1, each 1H), 7.11-7.19 (m, 1H), 7.24-7.31 (m, 1H), 7.40-7.47 (m, 1H), 8.40 (d, J=6.4, 1H).
ESI-MS (m/z) 343 [M+H]+, 365 [M+Na]\. Anal.Calcd for Ci9H19FN203 (342.37): Calcd. C, 66.66; H, 5.59; N, 8.18; F, 5.02; O, 14.02. Found C, 66.76; H, 5.48; N, 8.13; F, 5.03; O, 13.99.
Crystallized (1) form previous step (9.5 g, 0.028 mol) was dissolved in 2-butanone (290 mL) upon heating. The hot solution was filtered over charcoal. The solution was concentrated by removal of 2-butanone (200 mL) by distillation prior to seeded cooling crystallization. Filtration, washing with chilled 2-butanone and drying at 50°C/25 mbar/16h afforded 9.18 g (93.9% corrected yield) of the title compound (1) as a crystalline powder of polymorphic form B with an assay of 100.4 %(w/w) and a purity of 99.97 %(area) (by HPLC).
Alternatively, to a stirred suspension of hydroxyamide (6) (30.0 g, 0.083 mol) in toluene (500 ml) was added at 50°C within 45 minutes thionyl chloride (10.40 g, 0.087 mol) and the resulting mixture was stirred for 3h at 50°C. The mixture was then heated up to 92°C and subsequently stirred at this temperature for 15 h. The Suspension was then cooled to 50°C and toluene was removed by distillation under reduced pressure. The distillation residue was cooled to ambient temperature and treated with N-methylpyrrolidone (210 ml) to obtain an almost clear solution. This solution was then cooled to -10°C and subsequently treated at this temperature within 2h with a solution of potassium iert-butoxide (12.40 g, 0.111 mol) in THF (60 g). The resulting mixture was stirred for another 60 minutes at -10°C, then warmed up to room temperature within 60 minutes and subsequently stirred at room temperature for 6 h. The reaction mixture was quenched with water (150 g) and the pH was adjusted with acetic acid (approx. 1.8 g) to pH 7-8. The mixture was then heated to 30-45°C and THF and toluene were distilled off under reduced pressure (<200 mbar) to obtain a clear NMP/water mixture (400 ml). This mixture was heated to 45°C and 260 mg of seed crystals were added. Water (320 ml) was then added within 3 h whereby the product crystallized. The resulting suspension was cooled to room temperature within 3 h and subsequently stirred at this temperature for 2 h. Filtration and washing of the filter cake with a mixture of water (100 ml) and N-methylpyrrolidone (20 ml) and subsequently only with water (150 ml) afforded after drying (70°C/10 mbar/20 h) 26.2 g (92%) of the title compound (1) as a crystalline powder with an assay of 99.6 %(w/w) and a purity of 99.7 %(area) (by HPLC).

HPLC
Purity (HPLC): Column: XSelect Phenyl Hexyl x2, 150 x 4.6mm, 3.5um. Starting
Pressure: 226 bar; temp.: 50°C. Inj. vol.: 2.0 μΐ^ + wash. Flow: 1.0 ml/min. Det: 204 nm. A: Water + 5% ACN, 77-2% in 7 min., hold for 1 min.; B: 0.1% HCOOH, 18% isocratic; C: MeOH, 5-80% in 7 min., hold for 1 min. Sample prep.: 2 mg/ml ACN. Retention times: β-acid 5.93 min., diacid 6.18 min., cc-acid 6.89 min., diester 6.96 min.
ee determination(HPLC): Column: Chiralpak IA-3 100 x 4.6mm, 3um; 91 bar, 2ml/min; temp.: 30°C. Inj. vol.: 10.0 μL· Det.: 206 nm. A: n-heptane, 80%; B: EtOH, 20%. Sample prep.: 4 mg/ml EtOH. Retention times: D-enantiomer 2.21 min., L-enantiomer 2.71 min
………………….
US 20050065204
EXAMPLE 11
Preparation of (S)-1-(4-Hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic Acid
8.00 g Polyethyleneglycol 6000 was dissolved in 150 mL (100 mM) magnesium acetate buffer pH 6.0 under stirring, and the solution added to a stirred suspension of 10.00 g (42.51 mmol) (RS)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid methyl ester (99.7%) in 40 mL methylcyclohexane. The mixture was heated to 28° C. and the pH readjusted to 6.0 with 2 M NaOH. The reaction was started by adding 33.2 mg Candida cylindraceae cholesterase (16.88 kU/g), and the pH was maintained at 6.0 by the controlled addition of 1.0 M NaOH solution under stirring. After a total consumption of 20.35 mL (20.35 mmol) 1.0 M sodium hydroxide solution (after 17.1 h; 47.9% conversion) the reaction mixture was passed through a sintered glass filter. The filtrate spontaneously separated into an aqueous and an organic phase.The aqueous phase was washed with 2×200 mL ethyl acetate to remove uncleaved ester. The aqueous phase was set to pH 4.0 with 25% sulfuric acid and concentrated in vacuo to a volume of ca. 80 mL (bath 60° C.). The solution was cooled to 1° C. (formation of white precipitate/crystals) and the pH set to 1.5 with 25% sulfuric acid. The precipitate/crystals were stirred overnight at 1° C., filtered off on a sintered glass filter (washed with a minimum amount of water) and dried overnight on high vacuum (RT, 6×10−2 mbar) to give 4.32 g (19.53 mmol; 45.9%) (S)-1-(4-hydroxyphenyl)-5-oxo-pyrrolidine-3-carboxylic acid. Analysis: HPLC (area A226nm): 99.3%, 0.7% ester. 98.9%ee. The product contains 5.3% water (according to Karl Fischer determination) and 2.1% (w/w) PEG (according to NMR).
| Company | Evotec AG |
| Description | Small molecule monoamine oxidase B (MAO-B) inhibitor |
| Molecular Target | Monoamine oxidase B (MAO-B) |
| Mechanism of Action | Monoamine oxidase B (MAO-B) inhibitor |
| Therapeutic Modality | Small molecule |
| Latest Stage of Development | Phase II |
| Standard Indication | Alzheimer’s disease (AD) |
| Indication Details | Treat Alzheimer’s disease (AD) |
| Regulatory Designation | |
| Partner |
//////////
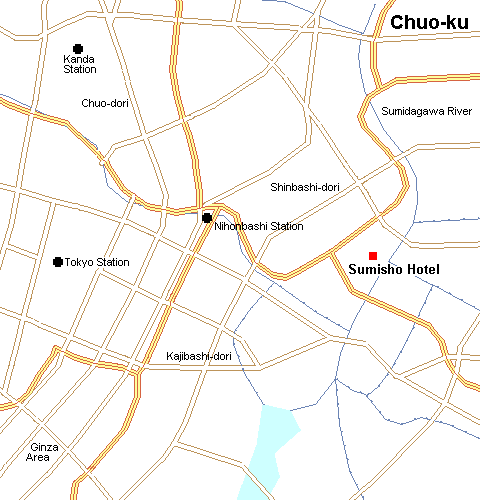
Chūō, japan




A Chūō Line (Rapid) E233 series (right) and A Chūō-Sōbu Line E231 series (June 2007)
 Chuo Dori street on a weekend afternoon
Chuo Dori street on a weekend afternoon

