
Home » Posts tagged 'Glucokinase Activator'
Tag Archives: Glucokinase Activator
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Astellas Pharma Inc. new Glucokinase Activator, ASP ? for Type 2 Diabetes
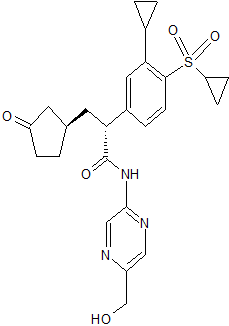
ASP ?
(2R)-2-(4-cyclopropanesulfonyl-3-cyclopropylphenyl)-N-[5-(hydroxymethyl)pyrazin-2-yl]-3-[(R)-3-oxocyclopentyl]propanamide
- Molecular Weight, 483.58
- [α]D20 −128.7 (c 1.00, MeOH);
- 1H NMR (DMSO-d6, 400 MHz) δ 11.07 (s, 1H), 9.20 (d, J = 1.4 Hz, 1H), 8.41 (d, J = 1.4 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.41 (dd, J = 8.2, 1.8 Hz, 1H), 7.15 (d, J = 1.8 Hz, 1H), 5.52 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 6.0 Hz, 2H), 4.04 (t, J = 7.6 Hz, 1H), 3.03–2.97 (m, 1H), 2.79 (tt, J = 8.4, 5.1 Hz, 1H), 2.25–1.81 (m, 8H), 1.53–1.47 (m, 1H), 1.17–1.12 (m, 2H), 1.08–1.02 (m, 4H), 0.89–0.84 (m, 2H);
- 13C NMR (DMSO-d6, 101 MHz) δ 218.5, 171.8, 152.1, 147.3, 145.7, 143.2, 140.3, 138.2, 134.8, 129.0, 125.3, 125.1, 62.5, 49.9, 44.4, 38.4, 38.2, 34.8, 32.1, 29.1, 12.4, 10.8, 10.7, 5.8;
- FTIR (ATR, cm–1) 3544, 3257, 1727, 1692, 1546, 1507, 1363, 1285, 1149, 719;
- HRMS (ESI) m/z [M + Na]+ calcd for C25H29N3O5S 506.1726, found 506.1747.
- Anal. Calcd for C25H29N3O5S: C, 62.09; H, 6.04; N, 8.69. Found: C, 61.79; H, 6.19; N, 8.62.
![]()
| Inventors | Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino |
| Applicant | Astellas Pharma Inc. |
Synthesis

contd…………………………..

PATENT
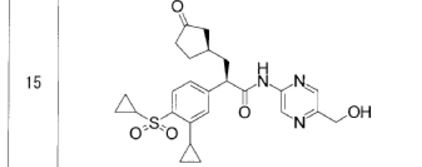
- PAPER
A Practical and Scalable Synthesis of a Glucokinase Activator via Diastereomeric Resolution and Palladium-Catalyzed C–N Coupling Reaction
 , Yasuhiro Morinaga†, Makoto Kasai†, Takao Hashimoto†, Yuji Takahama†, Atsushi Ohigashi†, Satoshi Yonishi‡, and Motohiro Akazome§
, Yasuhiro Morinaga†, Makoto Kasai†, Takao Hashimoto†, Yuji Takahama†, Atsushi Ohigashi†, Satoshi Yonishi‡, and Motohiro Akazome§
Here we describe the research and development of a process for the practical synthesis of glucokinase activator (R)-1 as a potential drug for treating type-2 diabetes. The key intermediate, chiral α-arylpropionic acid (R)-2, was synthesized in high diastereomeric excess through the diasteromeric resolution of 7 without the need for a chiral resolving agent. The counterpart 2-aminopyrazine derivative 3 was synthesized using a palladium-catalyzed C–N coupling reaction. This efficient process was demonstrated at the pilot scale and yielded 19.0 kg of (R)-1. Moreover, an epimerization process to obtain (R)-7 from the undesired (S)-7 was developed.
Hayakawa, M.; Kido, Y.; Nigawara, T.; Okumura, M.; Kanai, A.; Maki, K.; Amino, N. PCT Int. Appl. WO/2009/091014 A1 20090723,2009.
https://www.astellas.com/en/ir/library/pdf/3q2017_rd_en.pdf
///////////1174229-89-0, ASTELLAS, Glucokinase Activator, TYPE 2 DIABETES, PRECLINICAL, ASP ?, WO 2009091014, Masahiko Hayakawa, Yoshiyuki Kido, Takahiro Nigawara, Mitsuaki Okumura, Akira Kanai, Keisuke Maki, Nobuaki Amino, WO2009091014,
O=C(Nc1cnc(cn1)CO)[C@H](C[C@@H]2CC(=O)CC2)c3ccc(c(c3)C4CC4)S(=O)(=O)C5CC5
AM 2394
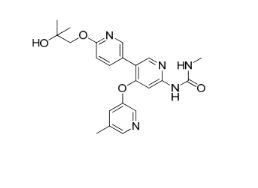
AM 2394
1-(6′-(2-hydroxy-2-methylpropoxy)-4-((5-methylpyridin-3-yl)oxy)-[3,3′-bipyridin]-6-yl)-3-methylurea
Urea, N-[6′-(2-hydroxy-2-methylpropoxy)-4-[(5-methyl-3-pyridinyl)oxy][3,3′-bipyridin]-6-yl]-N‘-methyl-
CAS 1442684-77-6
Chemical Formula: C22H25N5O4
Exact Mass: 423.1907
Array Biopharma Inc., Amgen Inc. INNOVATORS
AM-2394 is a potent and selective Glucokinase agonist (GKA), which catalyzes the phosphorylation of glucose to glucose-6-phosphate. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes
Type 2 diabetes mellitus (T2DM) is a disease characterized by elevated plasma glucose in the presence of insulin resistance and inadequate insulin secretion. Glucokinase (GK), a member of the hexokinase enzyme family, catalyzes the phosphorylation of glucose to glucose-6-phosphate in the presence of ATP.


Glucokioase i exok ase IV or D> is a glycolytic enssyiris that plays, an importaat. role irt blood sugar regulation .related to glucose utifeattoti a»d metabolism in the liver and pancreatic beta •cells. Serving as a glucose sessor, gtoeokiuase controls lasma glucose, levels. Glucokinaae plays a doal rob in .reducing plasma glucose levels; glucose-mediated activation of the en¾ymc in hepatocytes facilitates hepatic giocose npiafcc aad glycogen synthesis, while that la pancreatic beta ceils ultimately induces ins lin seeretio«. Both of these effects in turn reduce plasma glucose levels.
Clinical evidence has shown that, glueokitiase variants with, decreased, and increased activities are associated with mature easel, diabetes of the y ung { O0Y2) and persistent: hyperinsul nemic hypoglycemia &( infancy (PHHI), respectively. lso, aoo n.sulin dependent diabetes rneilitos (NIDDM) patients have been reported to have inappropriately lo giueokaiase activity; Ftirtherrnare. overexpressioa of glucokiuase it* dietary or gesetie animal models of diabetes either prevents, aoKiiorafes, or reverses the progress of pathological. symptoms in the disease. For these reasons, compounds that activate gfecokiaase have been sought by the pitasaaceatjeai liidustry.
International patent application, Publication No. WO 2 7/OS3345, which was published on May 10, 200?, discloses as giocokinase act ators certain 2-an«.aopyridiiie derivatives bearing at the 3 -position a meihyieneoxy-dkrked aromatic group a d on. the ammo group a heteroaryl ring, such as dna/oly! or i A4-lmadiazoiyl
it has .now been found that pyridyl ureas are useful as glneokirtase activators. Cettain of these •compounds have been, found to have an outstanding combination of properties that especially adapts them, for oral use to control plasma glucose levels.
![]()

Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00140

Glucokinase (GK) catalyzes the phosphorylation of glucose to glucose-6-phosphate. We present the structure–activity relationships leading to the discovery of AM-2394, a structurally distinct GKA. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes.

PATENT
WO 2013086397
http://www.google.com/patents/WO2013086397A1?cl=en
COPYING ERROR
Example. 1734 t¾^Jtiyi¾rea

Step A: In 100 mL of DMA were corafeiaed 1 ^545miSO- -ll«omp ridinr2-yl)-3-i«e hir8a- (17.5 g, 70,5 ii!-!to!). 5-o:ieS:t}yI yiidlii~3- ). (9,24 g, S4.7 ΪΗΪΪΪΟ!}, sad CO · (10.1 g, 77.6 mmo!) mid heated to 90 *C for 5 days. After that time, the reaction was om lete a d to it was added water arid DCM and stirred vigorously for 3 hr. The resulting solid was isolated via vacuum .filtratiott nd the cake was wasted mill rater and DCM. The DCM in tli aqueous rime was dried vdth a stream of aidogeji aad vigorous sbrriug. Use resulting solid was then collected via vacuum filtration aad these solids were
Stirred vig rousl in f 0% MeOH irt EtOAc arid die res dtipg solid was colleeied. via vactiiars fiirfati m.
Trie two batches wen i coiiibiaed to yield I-(5-bmmo-4 5^»ie†fey pyiidin-3-yl xy)p Tidin-2- d 3~ metbySurea (I S J g, 5 3.7 om»)i, 76% yield).
S e .8: In 2 niL ofc ioxane

yI) iyridMJ-2-yios:y)pf¾ps3i-2-oI (0,098 g, 0.33 «ΜΠΟΪ), “ -i5-bs¾tao-4-{5-a3fidiy I py f idia-3 – ylosy)f5yridia-2-yl)-3-raethyl«rea (0.075 g, 0.22 tn ol.. t, and.2M poiass.ua» carbonate (0.33 ml, 0.67 m oi} artd tfets was s parged wi h At .for 10 mia before PdC§4dppl)*DCM (0.01 g g, 0.022 msttol) was added and dre reae!io a was sparged for aaotber 5 ma-, ir efore a was sealed and heated to 100 oversight The react! art was then loaded directly onto s ilica gel (50% acetone to PCM w4i. }%
MH40H) to afford i – (6′-(2diydioxy-2i-H5eth:ylpropCis:y) -4-{ 5″i:t re th y Ipy r i d i rt -3- io s y ) -3 ,3 : -bipyr id i rt -6- yl)-3-aie5¾ylt)rea φ.? 42 , 0.096 m ol, 43 % yield). !1 1 HMR (400 Mife, CDCij) 3 ppm 9.06 is,. !H),
S.33 is, 1H>, 8,27 (rs 2H), 8. Π (s, I H): K. (s, IHU 82 (dd, j-S.fi, 5.9 H HI), 1.21 (S !H), 6,«8
(d, Hz, i i i ). 6. ,4 (s:. m>, 4.25 (s, 2H), 2,87 (dj =4,3 Hz„ 3H) 2,37 (s, 3H>. 1 .33 is, <SH). Mass speetram (apci) tar/, : – 423.9 (M÷H).
REFERENCES
Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
Paul J. Dransfield, Vatee Pattaropong, Sujen Lai, Zice Fu, Todd J. Kohn, Xiaohui Du, Alan Cheng, Yumei Xiong, Renee Komorowski, Lixia Jin, Marion Conn, Eric Tien, Walter E. DeWolf Jr., Ronald J. Hinklin, Thomas D. Aicher, Christopher F. Kraser, Steven A. Boyd, Walter C. Voegtli, Kevin R. Condroski, Murielle Veniant-Ellison, Julio C. Medina, Jonathan Houze, and Peter Coward
Publication Date (Web): May 23, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00140
/////////Glucokinase activator, GKA, AM-2394, 1442684-77-6, AM 2394, Amgen
O=C(NC)NC1=CC(OC2=CC(C)=CN=C2)=C(C3=CC=C(OCC(C)(O)C)N=C3)C=N1
Pfizer’s PF 04991532 a Hepatoselective Glucokinase Activator Clinical Candidate for Treating Type 2 Diabetes Mellitus

PF 04991532
GKA PF-04991532
(S)-6-{3-cyclopentyl-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propanamido}nicotinic acid
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
MW 396.36, MF C18 H19 F3 N4 O3
CAS 1215197-37-7
3-Pyridinecarboxylic acid, 6-[[(2S)-3-cyclopentyl-1-oxo-2-[4-(trifluoromethyl)-1H-imidazol-1-yl]propyl]amino]-
http://www.biochemj.org/content/441/3/881
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public epidemic affecting over 300 million people worldwide. This disease is characterized by elevated fasting plasma glucose (FPG), insulin resistance, abnormally elevated hepatic glucose production (HGP), and reduced glucose-stimulated insulin secretion (GSIS). Moreover, long-term lack of glycemic control increases risk of complications from neuropathic, microvascular, and macrovascular diseases.
The standard of care for T2DM is metformin followed by sulfonylureas, dipeptidyl peptidase-4 (DPP-IV) inhibitors, and thiazolidinediones (TZD) as second line oral therapies. As disease progression continues, patients typically require injectable agents such as glucagon-like peptide-1 (GLP-1) analogues and, ultimately, insulin to help maintain glycemic control. Despite these current therapies, many patients still remain unable to safely achieve and maintain tight glycemic control, placing them at risk of diabetic complications and highlighting the need for novel therapeutic options.

Glucokinase (hexokinase IV) continues to be a compelling target for the treatment of type 2 diabetes given the wealth of supporting human genetics data and numerous reports of robust clinical glucose lowering in patients treated with small molecule allosteric activators. Recent work has demonstrated the ability of hepatoselective activators to deliver glucose lowering efficacy with minimal risk of hypoglycemia.
While orally administered agents require a considerable degree of passive permeability to promote suitable exposures, there is no such restriction on intravenously delivered drugs. Therefore, minimization of membrane diffusion in the context of an intravenously agent should ensure optimal hepatic targeting and therapeutic index.
Diabetes is a major public health concern because of its increasing prevalence and associated health risks. The disease is characterized by metabolic defects in the production and utilization of carbohydrates which result in the failure to maintain appropriate blood glucose levels. Two major forms of diabetes are recognized. Type I diabetes, or insulin-dependent diabetes mellitus (IDDM), is the result of an absolute deficiency of insulin. Type II diabetes, or non-insulin dependent diabetes mellitus (NIDDM), often occurs with normal, or even elevated levels of insulin and appears to be the result of the inability of tissues and cells to respond appropriately to insulin. Aggressive control of NIDDM with medication is essential; otherwise it can progress into IDDM.
As blood glucose increases, it is transported into pancreatic beta cells via a glucose transporter. Intracellular mammalian glucokinase (GK) senses the rise in glucose and activates cellular glycolysis, i.e. the conversion of glucose to glucose-6-phosphate, and subsequent insulin release. Glucokinase is found principally in pancreatic β-cells and liver parenchymal cells. Because transfer of glucose from the blood into muscle and fatty tissue is insulin dependent, diabetics lack the ability to utilize glucose adequately which leads to undesired accumulation of blood glucose (hyperglycemia). Chronic hyperglycemia leads to decreases in insulin secretion and contributes to increased insulin resistance. Glucokinase also acts as a sensor in hepatic parenchymal cells which induces glycogen synthesis, thus preventing the release of glucose into the blood. The GK processes are thus critical for the maintenance of whole body glucose homeostasis.
It is expected that an agent that activates cellular GK will facilitate glucose-dependent secretion from pancreatic beta cells, correct postprandial hyperglycemia, increase hepatic glucose utilization and potentially inhibit hepatic glucose release. Consequently, a GK activator may provide therapeutic treatment for NIDDM and associated complications, inter alia, hyperglycemia, dyslipidemia, insulin resistance syndrome, hyperinsulinemia, hypertension, and obesity.
Several drugs in five major categories, each acting by different mechanisms, are available for treating hyperglycemia and subsequently, NIDDM (Moller, D. E., “New drug targets for Type II diabetes and the metabolic syndrome” Nature 414; 821-827, (2001)): (A) Insulin secretogogues, including sulphonyl-ureas (e.g., glipizide, glimepiride, glyburide) and meglitinides (e.g., nateglidine and repaglinide) enhance secretion of insulin by acting on the pancreatic beta-cells. While this therapy can decrease blood glucose level, it has limited efficacy and tolerability, causes weight gain and often induces hypoglycemia. (B) Biguanides (e.g., metformin) are thought to act primarily by decreasing hepatic glucose production. Biguanides often cause gastrointestinal disturbances and lactic acidosis, further limiting their use. (C) Inhibitors of alpha-glucosidase (e.g., acarbose) decrease intestinal glucose absorption. These agents often cause gastrointestinal disturbances. (D) Thiazolidinediones (e.g., pioglitazone, rosiglitazone) act on a specific receptor (peroxisome proliferator-activated receptor-gamma) in the liver, muscle and fat tissues. They regulate lipid metabolism subsequently enhancing the response of these tissues to the actions of insulin. Frequent use of these drugs may lead to weight gain and may induce edema and anemia. (E) Insulin is used in more severe cases, either alone or in combination with the above agents.
Ideally, an effective new treatment for NIDDM would meet the following criteria: (a) it would not have significant side effects including induction of hypoglycemia; (b) it would not cause weight gain; (c) it would at least partially replace insulin by acting via mechanism(s) that are independent from the actions of insulin; (d) it would desirably be metabolically stable to allow less frequent usage; and (e) it would be usable in combination with tolerable amounts of any of the categories of drugs listed herein.
Substituted heteroaryls, particularly pyridones, have been implicated in mediating GK and may play a significant role in the treatment of NIDDM. For example, U.S. Patent publication No. 2006/0058353 and PCT publication Nos. WO2007/043638, WO2007/043638, and WO2007/117995 recite certain heterocyclic derivatives with utility for the treatment of diabetes. Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for diabetes, particularly NIDDM.

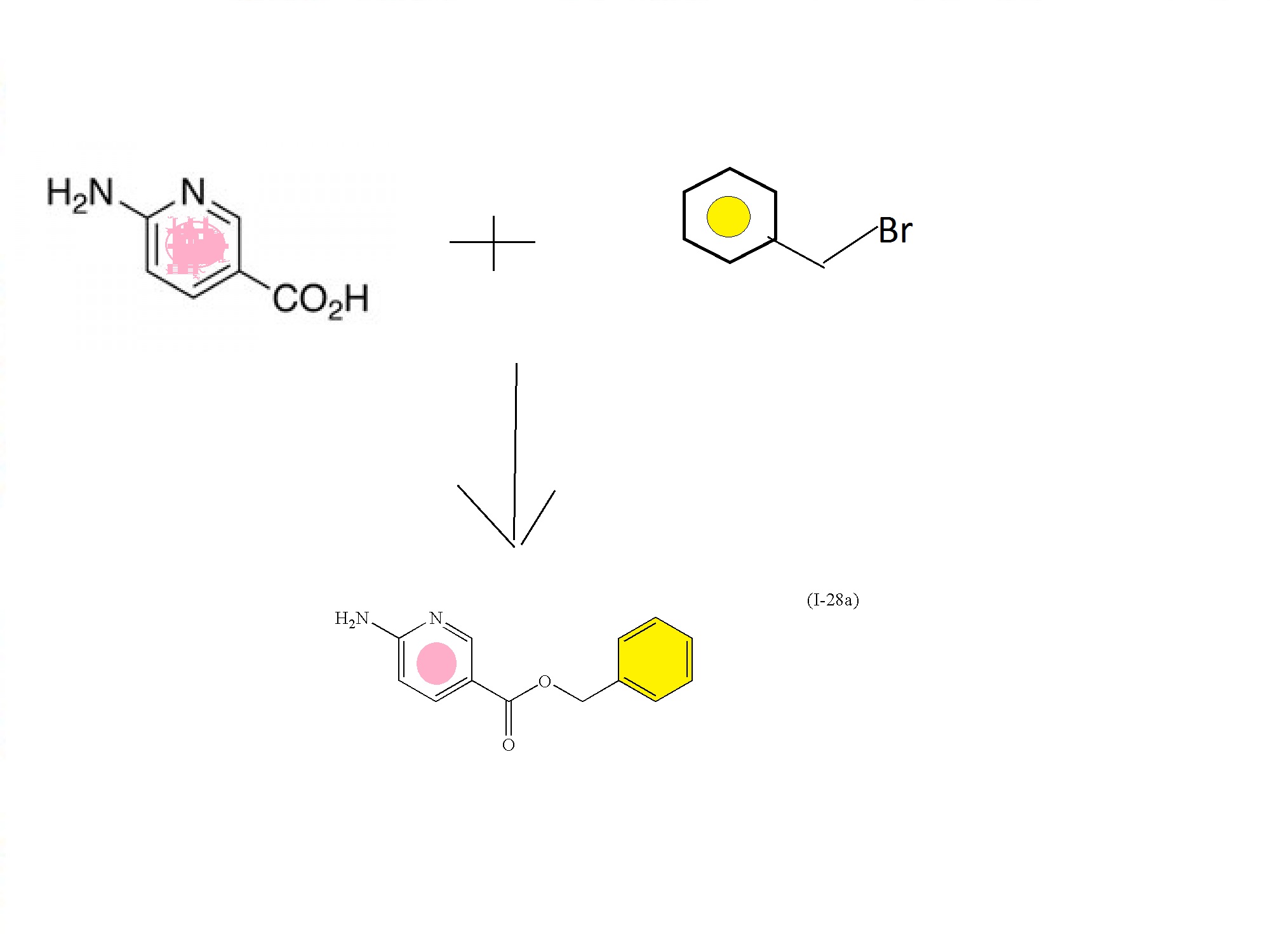
PATENT
US 20100063063
http://www.google.com/patents/US20100063063
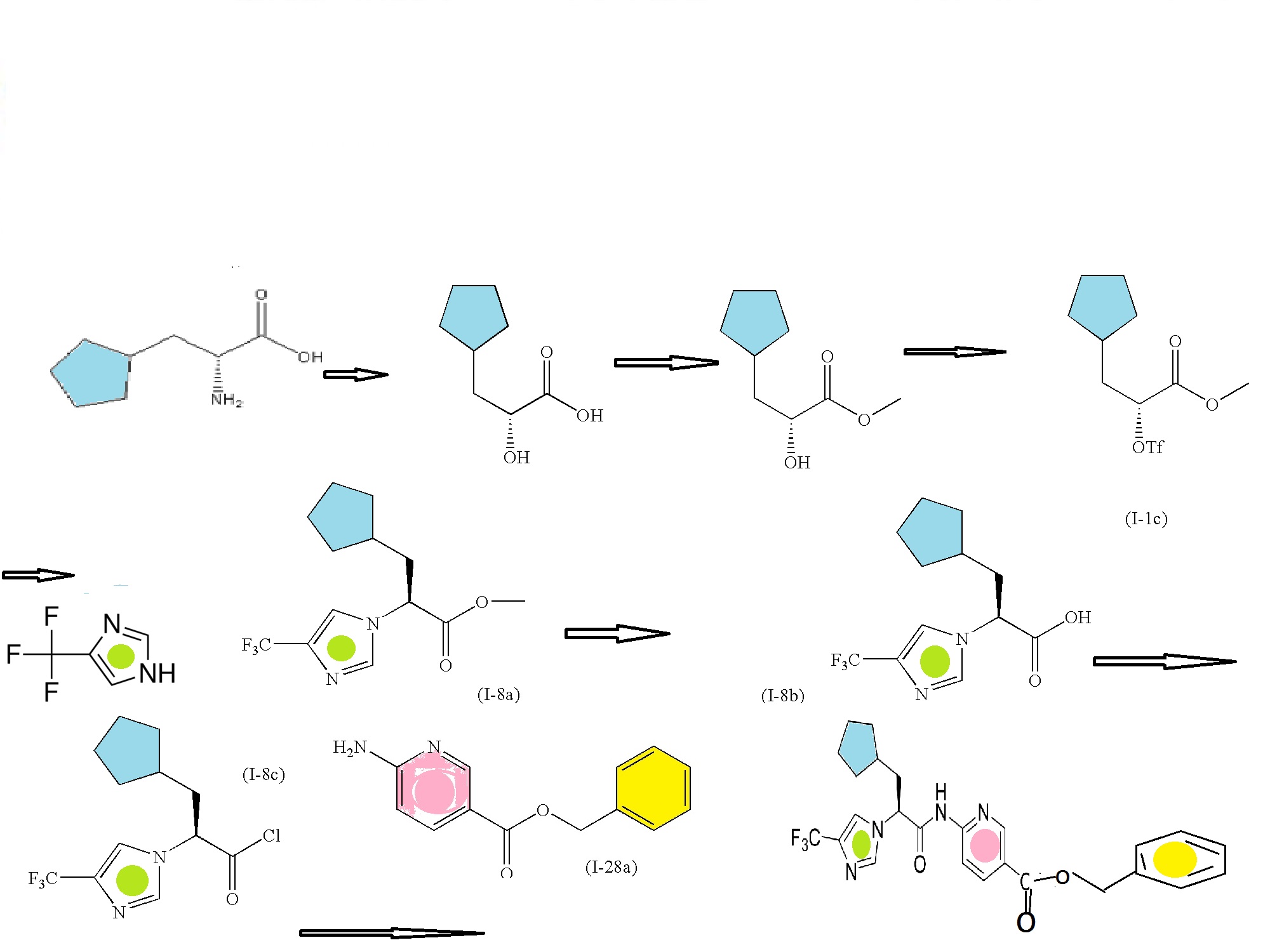
SYNTHESIS CONSTRUCTION

6-aminonicotinic acid

BENZYL BROMIDE

FIRST KEY INTERMEDIATE
SECOND SERIES FOR NEXT INTERMEDIATE

(R)-2-amino-3-cyclopentylpropanoic acid

(R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)

(R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
Trifluoromethanesulfonic acid anhydride

(R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
CONDENSED WITH

4-Trifluoromethyl-1H-imidazole
TO GIVE PRODUCT SHOWN BELOW

(S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)

(S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)

CONVERTED TO ACID CHLORIDE, (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
AND CONDENSED WITH
WILL GIVE BENZYL DERIVATIVE AS BELOW

THEN DEBENZYLATION TO FINAL PRODUCT
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoic acid (I-1a)
To a stirred solution of (R)-2-amino-3-cyclopentylpropanoic acid (5.0 grams; Chem-Impex International, Inc., Wood Dale, Ill.) and 1 M H2SO4 (45.1 mL) at 0° C., was added a solution of NaNO2 (3.12 g) in H2O (15.6 mL) drop wise over 10 minutes. The reaction mixture was stirred for 3 hours at 0° C., then for 2 hours at room temperature. The solution was then extracted (3 times) with diethyl ether. The combined organic extracts were dried over MgSO4, filtered, and the filtrate concentrated to afford 2.36 g of (I-1a). 1H NMR (400 MHz, CDCl3) δ 4.26-4.28 (1H), 1.99-2.07 (1H), 1.76-1.81 (4H), 1.60-1.62 (4H), 1.12-1.16 (2H); LCMS for C8H14O3 m/z 157.1 (M−H)−.
Intermediate: (R)-methyl 3-cyclopentyl-2-hydroxypropanoate (I-1b)
To a stirred solution of 2.36 g of (I-1a) in anhydrous methanol (15 mL) at room temperature was added SOCl2(1.64 mL). The resulting mixture was heated at reflux for 2 hours. It was then cooled and concentrated under reduced pressure. The residue was partitioned between ethyl acetate and aqueous saturated NaHCO3 solution. The biphasic mixture was separated and the aqueous portion was extracted with ethyl acetate. The combined extracts were dried over MgSO4, filtered, and the filtrate concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel, heptanes/ethyl acetate) to afford 1.5 g of (I-1b) as a clear oil.1H NMR (400 MHz, CDCl3) δ 4.15-4.20 (1H), 3.77 (3H), 2.62-2.63 (1H), 1.97-2.05 (1H), 1.49-1.86 (8H), 1.06-1.17 (2H); LCMS for C9H16O3 m/z 171.6 (M)+. Intermediate (I-1b) can alternatively be prepared by the method described below.
A 0.2M solution of Li2CuCl4 was prepared as follows: Anhydrous CUCl2 (26.9 g, 200 mol) and anhydrous LiCl (17.0 g, 400 mmol) were dissolved in THF (1000 mL). The mixture required gentle heating to completely dissolve the solids. After cooling the solution is ready for use.
A solution of Li2CuCl4 (0.2 M in THF, 125 mL, 25.0 mmol) was added slowly to a suspension of cyclopentylmagnesium bromide (2 M in diethyl ether, 135 mL, 270 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) and THF (500 mL) at −50° C. over 2-3 mins. The pale grey/brown suspension was then allowed to warm slowly to −10° C. over 30 mins, by which time the color had developed to a dark grey. The mixture was re-cooled to −78° C. and (R)-methyl oxirane-2-carboxylate (25.0 g, 245 mmol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) was added neat via syringe over 90 seconds. The reaction was then stirred at −78° C. for 20 mins, before removing the ice-bath and allowing to warm to approximately −50° C. over 30 mins. Saturated NH4Cl (aq, 700 mL) was then added and the mixture stirred for 30 mins. The organic layer was collected and the aqueous layer extracted with diethyl ether (2×250 mL). The combined organics were washed with saturated NH4Cl (aq, 350 mL), dried over MgSO4, and evaporated. Distillation of the crude residue (68-70° C. at 0.8 mbar) yielded 65-70% of (I-1b) as a pale yellow oil. A small amount of less volatile material remained in the still pot. 1H NMR (400 MHz; CDCl3): δ 4.17(1H), 3.76 (3H), 2.67 (1H), 2.01 (1H), 1.48-1.88 (8H), 1.11 (2H).
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c)
Intermediate: (R)-methyl 3-cyclopentyl-2-(trifluoromethylsulfonyloxy)propanoate (I-1c
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H).
Intermediate (I-1b) (6.37 g, 37.0 mmol) was dissolved in dry dichloromethane (260 mL) and stirred under nitrogen in an ice bath. 2,6-Lutidine (9.0 mL, 77 mmol) was added. Trifluoromethanesulfonic acid anhydride (11 mL, 65 mmol) in dry dichloromethane (75 mL) was added dropwise. The reaction was stirred in the ice bath for 60 minutes, concentrated under reduced pressure, and taken up in 1N HCl and methyl t-butyl ether. The aqueous layer was separated, and the organic layer was washed with additional 1N HCl to insure the removal of all the lutidine. The combined organic layer was then washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-1c) (11.3 g, 37 mmol, 100%), which was used immediately without further purification; 1H NMR (400 MHz, CDCl3) δ 5.10-5.14 (1H), 3.82 (3H), 2.02-2.12 (1H), 1.79-1.98 (4H), 1.51-1.66 (4H), 1.08-1.18 (2H)
Intermediate: (S)-methyl 3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoate (I-8a)
4-Trifluoromethyl-1H-imidazole (5.0 g, 37.0 mmol; Apollo Scientific Ltd., Bredbury, Cheshire, UK) was stirred in dry THF (180 mL) under nitrogen at room temperature. Lithium hexamethyldisilazide (1M in THF, 33.4 mL, 33.4 mmol) was added dropwise via addition funnel. The mixture was stirred at room temperature for 50 minutes and then chilled in an ice bath. A solution of (I-1c) (11.3 g, 37 mmol) in dry THF (45 mL), which had been chilled in an ice bath, was added in one portion. The reaction was allowed to warm to room temperature, stirred for 2 hours, quenched with saturated aqueous ammonium chloride solution (20 mL) and allowed to stir overnight. The aqueous layer was separated, and the organic layer was concentrated and then diluted with water and ethyl acetate. The organic layer was washed in series with dilute aqueous phosphoric acid, aqueous 10% potassium carbonate, and brine. The organic layer was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a brown oil. The crude material, containing the undesired regioisomer as a small impurity, was purified by chromatography on a 330 g pre-packed silica gel column, eluting with 10% ethyl acetate/heptane, linear gradient to 70% ethyl acetate/heptane. The product fractions were located by spotting on a silica TLC plate and visualizing with KMnO4 stain. TLC (1:1 ethyl acetate/heptane, developed in potassium permanganate) located the pure and mixed fractions. The clean product fractions were combined, evaporated, and dried under high vacuum to afford (I-8a) as a clear oil (6.61 g, 22.4 mmol, 67%). 1H NMR (400 MHz, CDCl3) δ 7.57 (1H), 7.38 (1H), 4.71-4.74 (1H), 3.76 (3H), 2.01-2.14 (2H), 1.45-1.79 (7H), 1.03-1.18 (2H); m/z 291.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoyl chloride (I-8c)
To a suspension of intermediate (I-8b) (0.25 g, 0.9 mmol) in dichloromethane (5 mL) was added oxalyl chloride (0.35 g, 2.7 mmol) and N,N-dimethylformamide (1 drop) at room temperature. The mixture was stirred for 2 hours at room temperature. The reaction mixture was concentrated in vacuo, and the residue was chased with dichloromethane two times and concentrated in vacuo to afford (I-8c) (0.27 g, 100%) as an oil, which was used in the next step directly.
Intermediate: (S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido) nicotinoyl chloride (I-21a)
-
 Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.
Thionyl chloride (225 mg, 1.89 mmol) was added to a solution of the compound of Example 48 (150 mg, 0.387 mmol) in dichloromethane (1.5 mL) and the reaction stirred at room temperature for 1 hour. LCMS of an aliquot in methanol showed ˜67% methyl ester. To the reaction mixture was added another 25 uL of thionyl chloride and this was stirred at room temp for another 30 minutes. Solvents were evaporated to afford 157 mg (100%) of (I-21a) as a grayish-white solid. LCMS in methanol to generate the methyl ester gave m/z 395.9 (M+H)+.

(I-8b
Intermediate: (S)-3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanoic acid (I-8b)
6N HCl (140 mL) was added to (I-8a) (6.61 g, 22.4 mmol) and the mixture was warmed to 95° C. for 16 hours and then allowed to cool. Solid potassium carbonate (58 g) was added in portions to bring the pH to about 4. A precipitate crashed out. Ethyl acetate was added, and the mixture was stirred until everything dissolved. The aqueous layer was extracted once with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered, concentrated under reduced pressure, and dried under high vacuum to afford (I-8b) as a clear glass (6.15 g, 21.9 mmol, 98%). 1H NMR (400 MHz, CDCl3) δ 7.73 (1H), 7.34 (1H), 6.85-7.15 (1H), 4.66-4.70 (1H), 1.98-2.17 (2H), 1.41-1.75 (7H), 1.01-1.19 (2H); m/z 277.4 (M+H)+.
(I-28a
Intermediate: benzyl 6-aminonicotinate (I-28a)
To a stirred suspension of 6-aminonicotinic acid (100 g, 0.72 mol; Aldrich Chemical Company, Inc., Milwaukee, Wis.) in N,N-dimethylformamide (700 mL) with brisk mechanical stirring was added potassium carbonate (150 g, 1.08 mol) and the reaction was stirred for 10 min before the portionwise addition of benzyl bromide (95 mL, 0.80 mol). The reaction was stirred at room temperature overnight, then the solids were filtered off and washed thoroughly with ethyl acetate, and the solvent was removed under vacuum. The filter cake was dissolved in water and extracted with ethyl acetate. The residue after evaporation of N,N-dimethylformamide was combined with the ethyl acetate extracts (total volume 2 L of ethyl acetate) and the combined organic extracts washed with brine (5×500 mL), dried (MgSO4) and the solvent removed under reduced pressure. The crude product was refluxed with 1:1 diethyl ether:hexane for 30 min then the solids filtered off (warm), washed with diethyl ether:hexane (1:1), and dried. This solid was precipitated from hot toluene (hot filtration required to remove dibenzylated material) and dried to afford (I-28a) (107.2 g, 65%) as an off-white solid; 1H NMR (DMSO-d6): δ 8.50 (1H), 7.82 (1H), 7.34-7.29 (5H), 6.84 (2H), 6.43 (1H), 5.23 (2H); m/z 229.4 (M+H)+.
Example 47
(S)-benzyl 6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinate
Formula (1A-4) wherein R4 is
To Intermediate (I-8b) (16.28 g, 59.8 mmol) stirring in dry dichloromethane (400 mL) at room temperature under nitrogen was added 2 drops of DMF. Oxalyl chloride (11 mL, 130 mmol) was added dropwise. After the bubbling subsided the reaction was left stirring for 90 minutes and then concentrated under reduced pressure. Two successive portions of 1,2-dichloroethane were added and evaporated to remove all excess oxalyl chloride. The crude acid chloride was taken up in dichloromethane (150 mL) and stirred at room temperature. Intermediate (I-28a) (14.3 g, 62.5 mmol) and pyridine (10 mL, 130 mmol) were stirred in 400 mL dry dichloromethane. This was added to the acid chloride solution, using another 50 mL dry dichloromethane to complete the transfer. The mixture was left stirring at room temperature under nitrogen for 18 hours. The reaction was diluted with dichloromethane and water, and 1M aqueous phosphoric acid was added. The organic layer was separated and washed sequentially with dilute aqueous potassium carbonate, and brine. This was then dried over sodium sulfate, filtered, and concentrated under reduced pressure to a glass, which was taken up in hot ethyl acetate and stirred at room temperature. A precipitate appeared at about 30 minutes. The mixture was stirred for 16 hours and then filtered. The precipitate was washed with ethyl acetate and then diethyl ether and dried under high vacuum at 60° C. to afford the title compound as a white solid (17.8 g, 36.6 mmol, 61%). The mother liquor was evaporated and purified by silica gel chromatography on a 120 g pre-packed column, eluting with 40% ethyl acetate/heptane. The product fractions were combined, concentrated under reduced pressure, dried under high vacuum to a glass, and converted as previously described to additional product (3.5 g, 7.2 mmol, 12%, total yield 73%). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (1H), 8.87-8.88 (1H), 8.29-8.32 (1H), 8.12-8.14 (1H), 7.93-7.94 (2H), 7.39-7.46 (2H), 7.30-7.37 (3H), 5.32 (2H), 5.21-5.25 (1H), 2.06-2.19 (2H), 1.26-1.63 (8H), 1.01-1.06 (1H); m/z 487.5 (M+H)+.
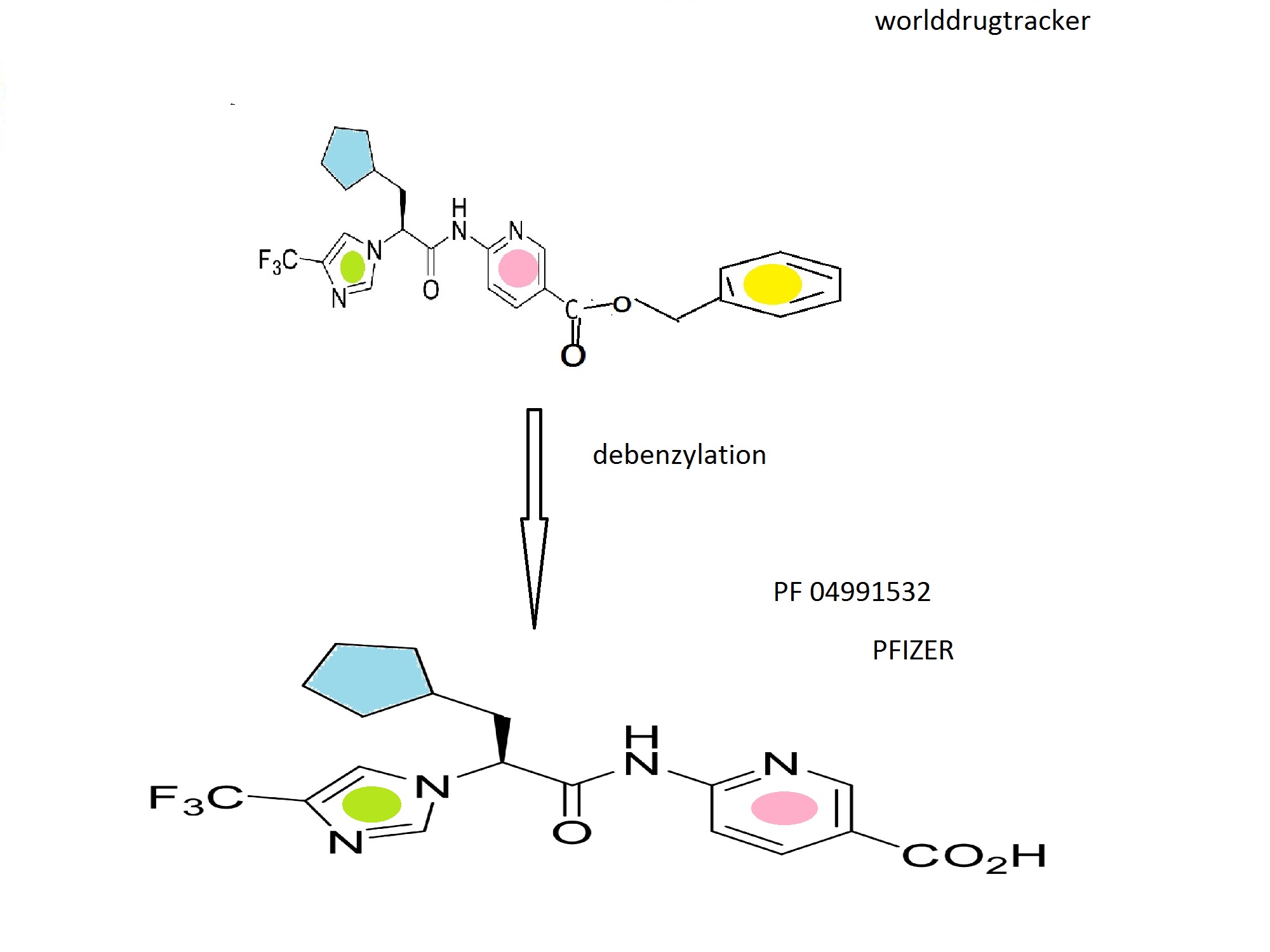
Example 48
(S)-6-(3-cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic acid
Formula (1A-4) wherein R4 is

The compound of Example 47 (4.07 g, 8.35 mmol) was added to a 500 mL Parr bottle, followed by ethyl acetate (50 mL) and ethanol (100 mL). The mixture was warmed until all of the solid dissolved, and then cooled to room temperature. 10% Pd/C (450 mg) was added, and the mixture was shaken under 50 psi hydrogen for 90 minutes. The reaction was filtered through a microfiber filter. The filtrate was concentrated under reduced pressure and dried under high vacuum at 50° C. to afford product as a glassy solid (3.0 g, 7.75 mmol, 90.6%). The glassy solid was stirred overnight in diethyl ether. The white solid precipitate was filtered, washed with diethyl ether, suction dried, and dried under high vacuum at 50° C. to afford the title compound as a white solid.
1H NMR (400 MHz, DMSO-d6) δ 13.10-13.25 (1H), 11.44 (1H), 8.83 (1H), 8.23-8.26 (1H), 8.09-8.12 (1H), 7.94-7.95 (2H), 5.22-5.26 (1H), 2.06-2.17 (2H), 1.29-1.64 (8H), 1.04-1.07 (1H);
m/z 397.3 (M+H)+.
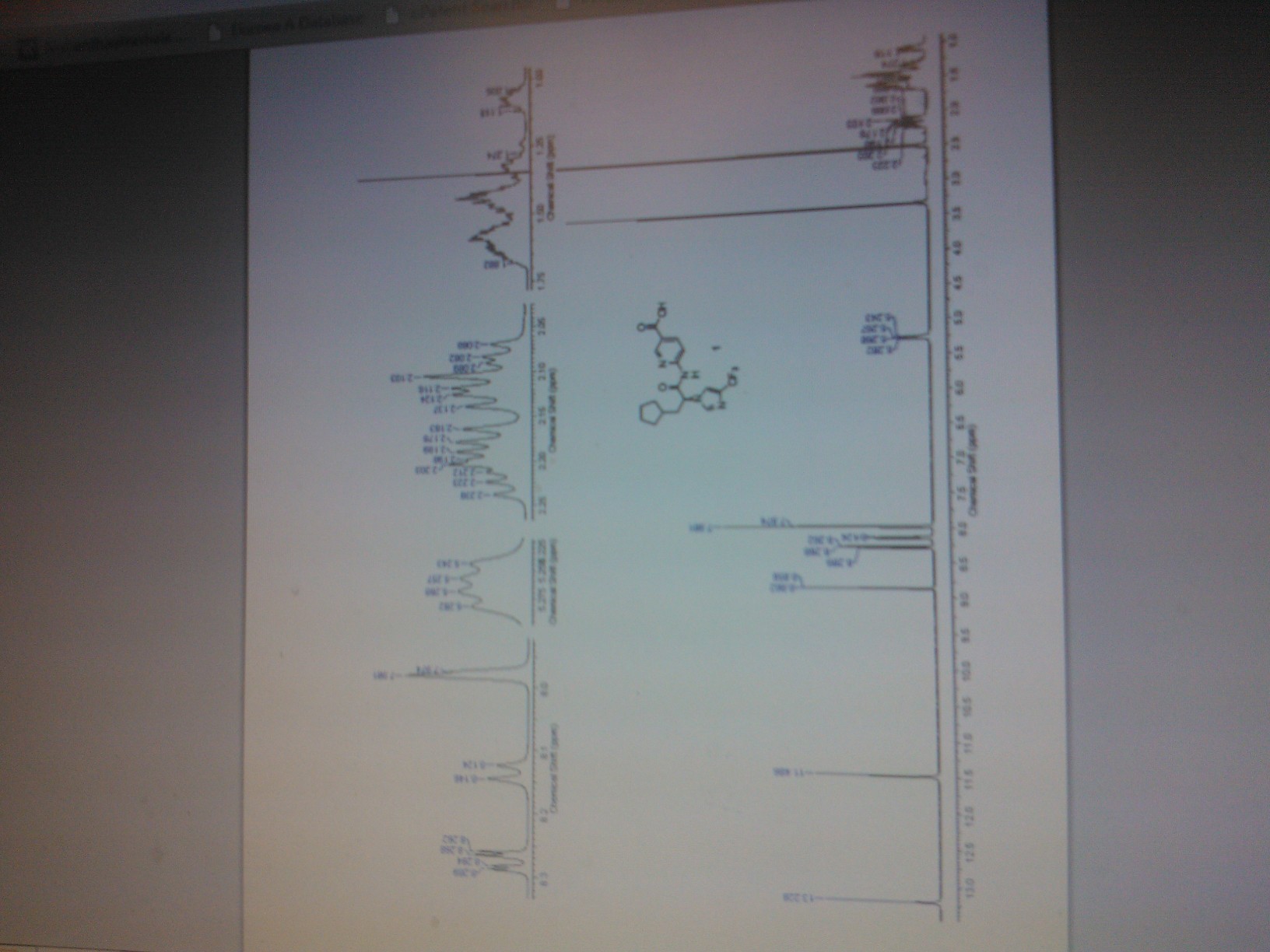
THIS NMR IS FROM SUPPORTING INFO OF A JOURNAL
PAPER
Organic Process Research & Development (2012), 16(10), 1635-1645
http://pubs.acs.org/doi/abs/10.1021/op300194c

This work describes the process development and manufacture of early-stage clinical supplies of a hepatoselective glucokinase activator, a potential therapy for type 2 diabetes mellitus. Critical issues centered on challenges associated with the synthesis of intermediates and API bearing a particularly racemization-prone α-aryl carboxylate functionality. In particular, a T3P-mediated amidation process was optimized for the coupling of a racemization-prone acid substrate and a relatively non-nucleophilic amine. Furthermore, an unusually hydrolytically-labile amide in the API also complicated the synthesis and isolation of drug substance. The evolution of the process over multiple campaigns is presented, resulting in the preparation of over 110 kg of glucokinase activator.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (1)
Pressure Hydrogenation
PAPER
Journal of Medicinal Chemistry (2012), 55(3), 1318-1333
http://pubs.acs.org/doi/abs/10.1021/jm2014887

Glucokinase is a key regulator of glucose homeostasis, and small molecule allosteric activators of this enzyme represent a promising opportunity for the treatment of type 2 diabetes. Systemically acting glucokinase activators (liver and pancreas) have been reported to be efficacious but in many cases present hypoglycaemia risk due to activation of the enzyme at low glucose levels in the pancreas, leading to inappropriately excessive insulin secretion. It was therefore postulated that a liver selective activator may offer effective glycemic control with reduced hypoglycemia risk. Herein, we report structure–activity studies on a carboxylic acid containing series of glucokinase activators with preferential activity in hepatocytes versus pancreatic β-cells. These activators were designed to have low passive permeability thereby minimizing distribution into extrahepatic tissues; concurrently, they were also optimized as substrates for active liver uptake via members of the organic anion transporting polypeptide (OATP) family. These studies lead to the identification of 19 as a potent glucokinase activator with a greater than 50-fold liver-to-pancreas ratio of tissue distribution in rodent and non-rodent species. In preclinical diabetic animals, 19 was found to robustly lower fasting and postprandial glucose with no hypoglycemia, leading to its selection as a clinical development candidate for treating type 2 diabetes.
(S)-6-(3-Cyclopentyl-2-(4-(trifluoromethyl)-1H-imidazol-1-yl)propanamido)nicotinic Acid (19)

PAPER
Bioorganic & Medicinal Chemistry Letters (2013), 23(24), 6588-6592
http://www.sciencedirect.com/science/article/pii/S0960894X13012638


Figure 1.
Structure of Hepatoselective GKA PF-04991532 (1).
References
Drug Metabolism & Disposition (2015), 43(2), 190-198
PLoS One (2014), 9(5), e97139/1-e97139/9,
Journal of Biological Chemistry (2012), 287(17), 13598-13610
Drug Discovery Today (2012), 17(9-10), 528-529
Biochemical Journal (2012), 441(3), 881-887.
///////////

Figure 1. Representative structures of glucokinase activators.
