
Home » Posts tagged 'GENERICS'
Tag Archives: GENERICS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Indian pharma’s struggle to tighten standards paves way for M&A deals

MUMBAI – India’s smaller generic drugmakers, struggling to cope with a bruised reputation and tougher regulation in the United States, are under pressure to consider branching out to new, less-profitable markets or sell out to larger rivals.
Two years after its most high-profile regulatory setback to date in the United States – Ranbaxy’s $500 million U.S. fine for drug safety violations – India’s $15 billion a year generic drug industry is still rebuilding its image in its biggest market.
Many of its top firms are facing sanctions at some of their factories, as the U.S. Food and Drug Administration (FDA) tightens checks and its approvals process.
Combined with government-mandated price controls on drugs at home, that is piling pressure on smaller players.
“If they want to have a presence globally, they have to make investments. If they can’t, then they’ll have to focus on other markets or scale back their ambition outside of India, and that’s probably what will happen,” said Subhanu Saxena, CEO of Cipla , India’s fourth-largest drugmaker by revenue.
Ashok Anand, president of Hikal Ltd , a Mumbai-based drugmaker with a market value of $167 million, said some peers were putting themselves on the block.
“If they cannot deal with the stricter regulations, they might just prefer to sell out,” he said.
Pressure on U.S. sales has been felt across the Indian industry, with all drugmakers hit by delays in FDA approvals as the U.S. safety body overhauls its review process. Growth in U.S. revenue for drugmakers slowed to 14 percent in the year to March 2015, less than half what it was in the year to March 2012, according to brokerage Edelweiss.

But for larger players who want to plug gaps or, for the likes of Glenmark and Aurobindo who aim to grow in the United States, this pressure has lowered prices and could pave the way for attractive deals, bankers said.
“Now that some of the smaller companies are reeling under intensive regulatory scrutiny and want to cash out on their investments, valuations would be much more realistic,” said the head of India M&A at a large European bank in Mumbai.
SPENDING SPREE
Indian manufacturers say they have spent millions in high-end testing equipment, improved training and have hired larger teams in quality control since Ranbaxy was fined for manipulating clinical data.
Some consultants estimate spending on compliance has more than doubled to reach about 6 to 7 percent of sales for the larger companies.
But while the number of U.S. export bans issued to Indian companies fell to eight in 2014 from 21 in 2013, according to FDA data, the agency continues to find manufacturing violations at the plants of some of the biggest drugmakers in the country, an indication of the pervasiveness of the problem.
Sun Pharmaceutical Industries , Wockhardt , Dr Reddy’s Laboratories and Cadila Healthcarehave all faced FDA rebukes over the past year.
Smaller firms Ipca and Aarti Drugs faced FDA bans on their plants this year.
These failures – which executives blame on India’s “quick fix” culture and consultants blame on a failure to prioritize compliance – have clouded short-term growth prospects and added to pressure on smaller players, pushing some to look elsewhere.
“They can choose to be in lesser-regulated markets, such as Latin America, where there is a lot of demand. But they will have to live with much thinner margins,” said the finance director of a small Indian drugmaker, who did not want to be named. “It’s survival of the fittest.” REUTERS
http://m.todayonline.com/business/indian-pharmas-struggle-tighten-standards-paves-way-ma-deals
///////
Europe to boost cooperation with international partners on generics
DRUG REGULATORY AFFAIRS INTERNATIONAL
07/08/2014
Europe to boost cooperation with international partners on generics
European system to be used as model to facilitate assessment of medicines
The European Union’s decentralised procedure is being used as a model to accelerate the assessment of applications for generic medicines as part of theInternational Generic Drug Regulators Pilot![]() (IGDRP).
(IGDRP).
The European Union (EU) is leading an international pilot project through which, upon request from a generic pharmaceutical company, it will share the assessment reports generated as part of the decentralised procedure in real time with collaborating regulatory agencies outside the EU.
By offering to share its assessment reports, the EU aims to reinforce collaboration and information-sharing between regulatory authorities across the world, contributing to facilitating and strengthening the scientific assessment process for medicines. This should enable medicines to be authorised in different territories in a coordinated way at approximately the same time.
The first phase of the…
View original post 177 more words

FDA Guidance on Polymorphic Compounds in Generic Drugs

The guidance issued by the US Food and Drug Administration advises companies on how to treat polymorphic drug compounds—those that exhibit multiple structural forms—in filing abbreviated new drug applications (ANDAs). The bottom line, according to the guidance, is that generic drug products containing the polymorphs be the “same” as the reference listed drug (RLD) in active ingredients, bioavailability, and bioequivalence.
The guidance pertains to orally available drugs that are either solid- or suspension-dosage products.
Polymorphisms arise when compounds are identical chemically, but not structurally. This can happen when two solids take on different crystalline forms—such as graphite and diamond; when molecules are disordered and fail to produce a repeatable crystal lattice, as is the case for the molecules in glass; or when solvent is trapped inside the crystal structure—as in hydrates, where water molecules are found within crystals.
The guidance notes that different polymorphisms may alter physical properties of compounds and affect their solubility, which in turn can alter their bioavailability or bioequivalence. In addition, polymorphic forms of a compound may alter the way the compound behaves during production, which again, may alter the finished drug’s biological activities.
On this latter point, the guidance specifically states, “Since an ANDA applicant should demonstrate that the generic drug product can be manufactured reliably using a validated process, we recommend that you pay close attention to polymorphism as it relates to pharmaceutical processing.”
The guidance also emphasizes the effect polymorphisms may have on drug stability, which again, may alter the drug’s biological activity. But the guidance goes on to say that “it is the stability of the drug product and not stability of the drug substance polymorphic form that should be the most relevant measure of drug equality.” Otherwise, a generic drug can be considered the “same” as the active ingredient in an RLD if the generic compound conforms to the standards set out in a United States Pharmacopeia (USP) monograph, if one exists for that particular drug substance.
These standards generally include the chemical name, empirical formula, and molecular structure of the compound. However, the “FDA may prescribe additional standards that are material to the sameness of a drug substance.” But as concerns polymorphisms, the guidance goes on to say “…differences in drug substance polymorphic forms do not render drug substances different active ingredients for the purposes of ANDA approvals….”
Finally, the guidance reminds ANDA applicants that the biological performance characteristics of a drug are also dependent on the drug’s formulation and advises applicants to consider the properties of both the drug substance and formulation excipients, when assessing “sameness.”
A sponsor of an Abbreviated New Drug Application (ANDA) must have information to show that the proposed generic product and the innovator product are both pharmaceutically equivalent and bioequivalent, and therefore, therapeutically equivalent.
Many pharmaceutical solids exist in several crystalline forms and thus exhibit polymorphism. Polymorphism may result in differences in the physico-chemical properties of the active ingredient and variations in these properties may render a generic drug product to be bioinequivalent to the innovator brand. For this reason, in ANDAs, careful attention is paid to the effect of polymorphism in the context of generic drug product equivalency.
This review ..Adv Drug Deliv Rev. 2004 Feb 23;56(3):397-414……discusses the impact of polymorphism on drug product manufacturability, quality, and performance. Conclusions from this analysis demonstrate that pharmaceutical solid polymorphism has no relevance to the determination of drug substance “sameness” in ANDAs.
Three decision trees for solid oral dosage forms or liquid suspensions are provided for evaluating when and how polymorphs of drug substances should be monitored and controlled in ANDA submissions. Case studies from ANDAs are provided which demonstrate the irrelevance of polymorphism to the determination of drug substance “sameness”. These case studies also illustrate the conceptual framework from these decision trees and illustrate how their general principles are sufficient to assure both the quality and the therapeutic equivalence of marketed generic drug products.
read
ANDAs: Pharmaceutical Solid Polymorphism – Food and Drug … click here
also
Issues of Polymorphism and Abbreviated New Drug Applications click here
and
POLYMORPHISM OF DRUGS – Seventh Street Development Group click here
An Overview of Solid Form Screening During Drug … – ICDD..http://www.icdd.com/ppxrd/10/presentations/PPXRD-10_Ann_Newman.pdf
http://www.ivtnetwork.com/sites/default/files/Polymorphism_01.pdf
Although polymorph/salt screening should ideally be performed to select the optimum solid form upon selection of the lead compound prior to animal pharmacokinetic (PK) studies, these screening study can be costly and time consuming. But the consequences of late discovery of a thermodynamic form are grave, so there must be a strategy to minimize the risk without spending a large amount of resources.
We find this right strategy based on early BCS classification of new compounds. We tailor the upfront polymorph/salt studies based on the risk in bioavailability, stability and manufacture-ability. Since regulatory agencies worldwide require the use of the same salt across preclinical and clinical studies, for insoluble or unstable compounds, salt screening is done early to enable further compound development.
Once salt is selected, the polymorph screening of the selected salt if soluble may be done a little later after animal study. However it is paramount to confirm 1) the polymorph in use is stable in the toxicological vehicle, 2) no changes of solid forms during shipping and storage, 3) no significant degradation upon storage.
Should there be polymorphic changes such as formation of a hydrate in the animal vehicle resulting in lowered solubility and precipitation of the hydrate, or formation of a hydrate when exposed to humidity during shipping and storage, early discovery of the stable forms will enable consistent animal exposure and avoid study repeats and delays in timelines.
Therefore, although most companies do not perform comprehensive polymorph screening until late in the development cycle, we recommend identification of a thermodynamic stable form within the confine of not only the API manufacture processes but also in the designated animal and human formulations.
For instance, for a drug product manufactured by direct compression, the solidstate properties of the active ingredient will likely be critical to the manufacture of the drug product, particularly when it constitutes the bulk of the tablet mass.
On the other hand, for a drug product manufactured by wet granulation, the solidstate properties of the active ingredient may no longer be important but the potential for polymorphic conversion is high in the presence of high moisture contents. In the context of the effect of polymorphism on pharmaceutical processing, what is most relevant is the ability to consistently manufacture a drug product that conforms to applicable in-process controls and release specifications.
This upfront work is especially critical to insoluble compounds prone to varied oral bioavailability in animal and human.
How Long Is A Drug Patent Good For?
 Patents are good for 20 years after the invention of a drug–not after the drug comes to market. It can easily take eight years for the pharmaceutical companies to gather enough data to get approval for their new invention from the U.S. Food and Drug Administration. Meanwhile the FDA can send the drug company back for more clinical studies (experiments using humans as subjects to test the drugs’ efficacy and side effects) and more data, and all the while the patent clock is ticking.
Patents are good for 20 years after the invention of a drug–not after the drug comes to market. It can easily take eight years for the pharmaceutical companies to gather enough data to get approval for their new invention from the U.S. Food and Drug Administration. Meanwhile the FDA can send the drug company back for more clinical studies (experiments using humans as subjects to test the drugs’ efficacy and side effects) and more data, and all the while the patent clock is ticking.
That’s why the name of the game for pharmaceutical companies is working to extend those patents for a top-selling drug
read all at
How Long Is A Drug Patent Good For? – Drugsdb.com http://www.drugsdb.com/blog/how-long-is-a-drug-patent-good-for.html#ixzz2evb9L5rn
Research Perspective in Academia and Generic Pharmaceutical Industry

Rakeshwar Bandichhor
Director at Dr. Reddy’s Laboratories
Research Perspective in Academia and Generic Pharmaceutical Industry
Research per say is an exploratory endeavor that allows us to discover or invent not only medicine and material but how to make them in sustainable manner. In particular, research primarily involves identification of the target and its significance in terms of applicability to meet the societal needs ranging from education to commercial production of the goods.
| Research Perspective in Academia and Generic Pharmaceutical Industry | ||
| Rakeshwar Bandichhor* | ||
|

Research Perspective in Academia and Generic Pharmaceutical Industry
Research Perspective in Academia and Generic Pharmaceutical Industry
| Research Perspective in Academia and Generic Pharmaceutical Industry | ||
| Rakeshwar Bandichhor* | ||
| Associate Director, API, R&D, Dr. Reddy′s Laboratories, Hyderabad 500072, AP, India | ||
|
Dr. Reddy’s Announces the Launch of Decitabine for Injection
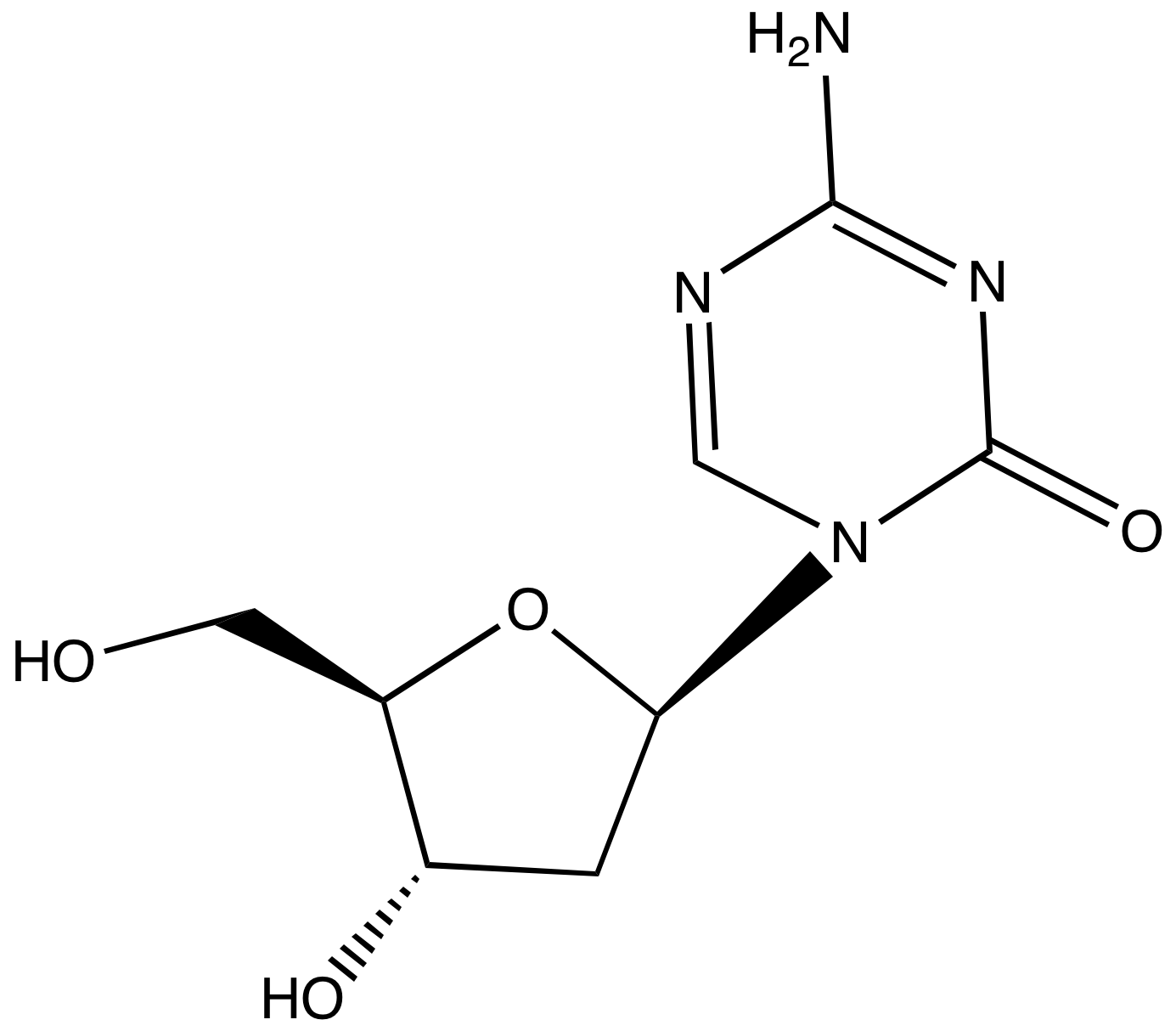
Decitabine
Hyderabad, India, July 12, 2013 — Dr. Reddy’s Laboratories announced today that it has launched Decitabine for Injection (50mg) a therapeutic equivalent generic version of Dacogen (Decitabine for Injection) in the US market on July 11, 2013, following the approval by the United States Food & Drug Administration (USFDA) of Dr. Reddy’s ANDA for Decitabine for Injection.
The Dacogen brand has U.S. sales of approximately $260 Million MAT for the most recent twelve months ending in July 2013 according to IMS Health*.
Dr. Reddy’s Decitabine for Injection 50 mg is available as a single dose vial.
About Dr. Reddy’s

Dr. Reddy’s Laboratories Ltd. (NYSE: RDY) is an integrated global pharmaceutical company, committed to providing affordable and innovative medicines for healthier lives. Through its three businesses – Pharmaceutical Services and Active Ingredients, Global Generics and Proprietary Products – Dr. Reddy’s offers a portfolio of products and services including APIs, custom pharmaceutical services, generics, biosimilars, differentiated formulations and NCEs. Therapeutic focus is on gastro-intestinal, cardiovascular, diabetology, oncology, pain management, anti-infective and pediatrics. Major markets include India, USA, Russia and CIS, Germany, UK, Venezuela, S. Africa, Romania, and New Zealand. For more information, log on to: http://www.drreddys.com.
Dacogen® is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals, Inc
Decitabine (trade name Dacogen), or 5-aza-2′-deoxycytidine, is a drug for the treatment of myelodysplastic syndromes, a class of conditions where certain blood cells are dysfunctional, and for acute myeloid leukemia (AML).[1] Chemically, it is a cytidine analog.
Decitabine is a hypomethylating agent.[2][3] It hypomethylates DNA by inhibiting DNA methyltransferase.
It functions in a similar manner to azacitidine, although decitabine can only be incorporated into DNA strands while azacitidine can be incorporated into both DNA and RNA chains.

Clinical uses
Decitabine is indicated for the treatment of myelodysplastic syndromes (MDS) including previously treated and untreated, de novo and secondary MDS of all French-American-British subtypes (refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia) and Intermediate-1, Intermediate-2, and High-Risk International Prognostic Scoring System groups. In patients with renal insufficiency, Batty and colleagues reported the first case series on the feasibility of therapy with hypomethylating agents in patients with renal insufficiency.[4]
Chemical synthesis
Decitabine can be synthesized from a benzoyl-protected chlorosugar:[5] 
- “EC Approves Marketing Authorization Of DACOGEN For Acute Myeloid Leukemia”. 2012-09-28. Retrieved 28 September 2012.
- Kantarjian H, Issa JP, Rosenfeld CS, et al. (April 2006). “Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study”. Cancer 106 (8): 1794–803. doi:10.1002/cncr.21792. PMID 16532500.
- Kantarjian HM, O’Brien S, Cortes J, et al. (August 2003). “Results of decitabine (5-aza-2’deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia”. Cancer 98 (3): 522–8. doi:10.1002/cncr.11543. PMID 12879469.
- Ravandi, F.; Cortés, J. E.; O’Brien, S.; Pierce, S.; Garcia-Manero, G.; McCue, D.; Santos, F. P. S.; Jabbour, E. et al. (2010). “Feasibility of Therapy with Hypomethylating Agents in Patients with Renal Insufficiency”. Clinical Lymphoma, Myeloma & Leukemia 10 (3): 205–210. doi:10.3816/CLML.2010.n.032. PMID 20511166.
|displayauthors=suggested (help) edit - Piml, J.; Sorm, F. (1964). Coll. Czech. Chem. Commun. 29: 2576.

NDA FDA-Nuvo reports FDA response to PENNSAID 2% , diclofenac sodium topical solution, 2% w/w

DICLOFENAC

PENNSAID 2%
7 MAR 2013
The US Food and Drug Administration (FDA) has issued a Complete Response Letter (CRL) to Nuvo Research’s US licensing partner, Mallinckrodt, following the review of Mallinckrodt’s New Drug Application (NDA) for diclofenac sodium topical solution, 2% w/w (PENNSAID 2%).
FDA in the letter mentioned that it requires Mallinckrodt’s complete pharmacokinetic study comparing PENNSAID 2% to original PENNSAID 1.5%.
FDA denied to review the similar pharmacokinetic studies submitted by Mallinckrodt with the NDA, as the reserve samples were not retained at the clinical site.
Pharmacokinetic studies are standard studies conducted during a drug development program to identify the total exposure or the amount of drug that reaches the blood stream after a patient receives both single and multiple doses of the product.
Mallinckrodt has suggested Nuvo that it expects to complete the study and submit the results to the FDA in the third quarter of 2013, and that it anticipates the FDA will provide a formal response to the filing within 6 months thereafter.
Nuvo’s Pain Group president Dr. Bradley Galer said with the new FDA’s letter the firm was disappointed that PENNSAID 2% will not be approved in this review cycle.
“We are pleased that the FDA has outlined a clear pathway to approval that we believe can be completed in a relatively short time frame,” Galer added.
“Upon approval, PENNSAID 2% will be the first and only topical NSAID in the U.S. featuring twice per day dosing and a metered dose pump bottle.”
