
Home » Posts tagged 'GENENTECH'
Tag Archives: GENENTECH
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves third oncology drug Rozlytrek (entrectinib) that targets a key genetic driver of cancer, rather than a specific type of tumor
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
///////////////Rozlytrek, entrectinib, accelerated approval, priority Review, Breakthrough Therapy, Orphan Drug designation, fda 2019, Genentech, cancer
GNE-0877

GNE-0877
Maybe DNL-151 ?
CAS 1374828-69-9
Chemical Formula: C14H16F3N7
Molecular Weight: 339.31895
2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile
Denali Therapeutics Inc, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease
GNE-0877 is a highly potent and selective LRRK2 inhibitor. Leucine-rich repeat kinase 2 (LRRK2) has drawn significant interest in the neuroscience research community because it is one of the most compelling targets for a potential disease-modifying Parkinson’s disease therapy.
- Developer Denali Therapeutics Inc
- Class Antiparkinsonians; Small molecules
- Mechanism of Action LRRK2 protein inhibitors
- Phase I Parkinson’s disease
- 20 Dec 2017 Denali Therapeutics plans clinical studies for Parkinson’s disease
- 13 Nov 2017 Phase-I clinical trials in Parkinson’s disease (In volunteers) in Netherlands (unspecified route)
- 13 Nov 2017 Preclinical trials in Parkinson’s disease in USA (unspecified route) before November 2017
Denali Therapeutics is developing DNL-151 (phase 1, in July 2019), a lead from a program of small-molecule inhibitors of LRRK2 originally licensed from Genentech, for the treatment of Parkinson’s disease.
Leucine-rich repeat kinase 2 (LRRK2) is a complex signaling protein that is a key therapeutic target, particularly in Parkinson’s disease (PD). Combined genetic and biochemical evidence supports a hypothesis in which the LRRK2 kinase function is causally involved in the pathogenesis of sporadic and familial forms of PD, and therefore that LRRK2 kinase inhibitors could be useful for treatment (Christensen, K.V. (2017) Progress in medicinal chemistry 56:37-80). Inhibition of the kinase activity of LRRK2 is under investigation as a possible treatment for Parkinson’s disease (Fuji, R.N. et al (2015) Science Translational Medicine 7(273):ral5;
Taymans, J.M. et al (2016) Current Neuropharmacology 14(3):214-225). A group of LRRK2 kinase inhibitors have been studied (Estrada, A.A. et al (2015) Jour. Med. Chem. 58(17): 6733-6746; Estrada, A.A. et al (2013) Jour. Med. Chem. 57:921-936; Chen, H. et al (2012) Jour. Med. Chem. 55:5536-5545; Estrada, A.A. et al (2015) Jour. Med. Chem. 58:6733-6746; US 8354420; US 8569281; US8791130; US 8796296; US 8802674; US 8809331; US 8815882; US 9145402; US 9212173; US 9212186; WO 2011/151360; WO 2012/062783; and WO 2013/079493.
PATENT
WO2012062783 , assigned to Hoffmann-La Roche , but naming inventors specifically associated with both Genentech and BioFocus (which had an agreement with Genentech for drug discovery programs); the compound was also later identified in J.Med.Chem (57(3), 921-936, 2014) in an article from these two companies, with the lab code GNE-0877. So while this represents the first application in the name of Denali Therapeutics Inc that focuses on this compound, it is likely that it provides the structure of DNL-151 , a lead from a program of small-molecule inhibitors of leucine-rich repeat kinase 2 (LRRK2) originally licensed from Genentech, being developed for the oral treatment of Parkinson’s disease, and which had begun phase I trials by December 2017 (when this application was lodged).
PATENT
WO2019104086 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019104086
claiming novel crystalline and amorphous forms of pyrimidinylamino-pyrazole compound, useful for treating Alzheimer’s disease, breast tumor, type I diabetes mellitus and Crohn’s disease.
Novel crystalline and amorphous forms of 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-1H-pyrazol-1-yl)propanenitrile (which is substantially pure form) and their anhydrous and solvates such as cyclohexanol solvate (designated as Forms B-D), processes for their preparation and compositions comprising them are claimed. The compound is disclosed to be leucine rich serine threonine kinase 2 inhibitor, useful for treating Gaucher disease, Alzheimer’s disease, motor neurone disease, Parkinson’s disease, prostate tumor, Lewy body dementia, mild cognitive impairment, breast tumor, type I diabetes mellitus and Crohn’s disease.
The present disclosure relates to crystalline polymorph or amorphous forms of a pyrimidinylamino-pyrazole kinase inhibitor, referred to herein as the Formula I compound and having the structure:
FORMULA I COMPOUND
The present disclosure includes polymorphs and amorphous forms of Formula I compound, (CAS Registry Number 1374828-69-9), having the structure:
and named as: 2-methyl-2-(3-methyl-4-(4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-ylamino)-lH-pyrazol-l-yl)propanenitrile (WO 2012/062783; US 8815882; US 2012/0157427, each of which are incorporated by reference). As used herein, the Formula I compound includes tautomers, and pharmaceutically acceptable salts or cocrystals thereof. The Formula I compound is the API (Active Pharmaceutical Ingredient) in formulations for use in the treatment of neurodegenerative and other disorders, with pKa when protonated calculated at 6.7 and 2.1.
CRYSTALLIZATION
Initial polymorph screening experiments were performed using a variety of
crystallization or solid transition methods, including: anti-solvent addition, reverse anti-solvent addition, slow evaporation, slow cooling, slurry at room temperature (RT), slurry at 50 °C, solid vapor diffusion, liquid vapor diffusion, and polymer induced crystallization. By all these methods, the Form A crystal type was identified. Polarized light microscopy (PLM) images of Form A obtained from various polymorph screening methods were collected (Example 5).
Particles obtained via anti-solvent addition showed small size of about 20 to 50 microns (pm) diameter while slow evaporation, slow cooling (except for THF/isooctane), liquid vapor diffusion and polymer-induced crystallization resulted in particles with larger size. Adding isooctane into a dichloromethane (DCM) solution of the Formula I compound produced particles with the most uniform size. Crude Formula I compound crystallized from THF///-heptane and then was micronized. A crystallization procedure was developed to control particle size.
A total of four crystal forms (Forms A, B, C, and D) and an amorphous form E of Formula I compound were prepared, including 3 anhydrates (Form A, C, and D) and one solvate (Form B). Slurry competition experiments indicated that Form D was thermodynamically more stable when the water activity aw< 0.2 at RT, while Form C was more stable when aw> 0.5 at RT. The 24 hrs solubility evaluation showed the solubility of Form A, C and D in FLO at RT was 0.18, 0.14 and 0.11 mg/mL, respectively. DVS (dynamic vapor sorption) results indicated that Form A and D were non-hygroscopic as defined by less than 0.1% reversible water intake in DVS, while Form C was slightly hygroscopic. Certain characterization data and observations of the crystal forms are shown in Table 1.
Table 1 Characterization summary for crystal forms of Formula I compound
Differential Scanning Calorimetry (DSC) analysis of Forms A and C showed that Form C had higher melting point and higher heat of fusion (Table 1), suggesting that the two forms are monotropic with Form C being the more stable form. Competitive slurry experiments with 1 : 1 Form A and C in a variety of solvents always produced Form C confirming that Form C was
more stable than Form A. In accordance with this, Form C was produced even when the crystallization batch was seeded with seeds of Form A.
PATENT
WO-2019126383
Methods of making leucine-rich repeat kinase 2 (LRRK2)-inhibiting, pyrimidinyl-4-aminopyrazole compounds (eg 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol-1-yl)propanenitrile), useful for treating LRRK2 mediated diseases such as Parkinson’s disease.
Example 1 Preparation of 2-(4-amino-3 -methyl- liT-pyrazol-l -yl)-2-methylpropanamide 5a
4a 5a
To a 20-L reactor containing dimethyl formamide (4.5 L) was charged 5-methyl-4-nitro-lH-pyrazole la (1.5 kg, 1.0 equiv). The solution was cooled to 0 °C and charged with finely ground K2CO3 (2.45 kg, 1.5 equiv) in three portions over ~l h. Methyl 2-bromo-2-methylpropanoate (3.2 kg, 1.5 equiv) was added dropwise to the mixture and then was allowed to warm to ~25 °C. The reaction mixture was maintained for 16 h and then quenched with water (15 L) and product was extracted with ethyl acetate. The combined organic layer was washed with water, and then with a brine. The organic layer was dried over anhydrous Na2S04, filtered, and concentrated under reduced pressure to give a light yellow solid. The crude product was purified by crystallization with petroleum ether (15 L), filtered, and dried to give methyl 2-m ethyl -2-(3 -methyl -4-nitro- l//-pyrazol- l -yl)propanoate 3a (2.25 kg, >99% purity by HPLC, 84 % yield) as an off-white solid. ¾ NMR (400 MHz, CDCb) 8.28 (s, 1H), 3.74 (s, 3H), 2.53 (s, 3H), 1.85 (s, 6H).
Methanol (23 L) and 2-methyl-2-(3-methyl-4-nitro-lif-pyrazol-l-yl)propanoate 3a (2.25 kg, 1.0 equiv) were charged into a 50-L reactor and cooled to approximately -20 °C. Ammonia gas was purged over a period of 5 h and then the reaction mixture warmed to 25 °C. After 16 h, the reaction mixture was concentrated under reduced pressure (~50 °C) to give the crude product. Ethyl acetate (23 L) was charged and the solution agitated in the presence of charcoal (0.1 w/w) and Celite® (0.1 w/w) at 45 °C. The mixture was filtered and concentrated under reduced pressure, and then the solid was slurried in methyl tert-butyl ether (MTBE, 11.3 L) at RT for 2 h. Filtration and drying at ~45 °C gave 2-m ethyl -2-(3 -m ethyl -4-ni tro- 1 //-pyrazol – 1 -yl)propanamide 4a (1.94 kg, >99% purity by HPLC, 92% yield).
Methanol (5 L) and 2-m ethyl-2-(3 -methyl -4-nitro-lif-pyrazol-l-yl)propanamide 4a (0.5 kg) were charged into a 10-L autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C (50 g). Hydrogen was charged (8.0 kg pressure/l 13 psi) and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced
pressure and then slurried in MTBE (2.5 L) for 2 h at 25 °C. Filtration and drying under reduced pressure (45 °C) gave 2-(4-amino-3-methyl- l//-pyrazol- l -yl)-2-methyl propanamide 5a (0.43 kg, >99% purity by HPLC, 99% yield).
Example 2 Preparation of 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanamide 7a
DCM
Into a first reactor was charged /-BuOH (or alternatively 2-propanol) (15.5 vol) and 2-(4-amino-3 -methyl- li7-pyrazol-l-yl)-2-methylpropanamide 5a (15 kg), followed by zinc chloride (13.5 kg, 1.2 equiv) at room temperature and the suspension agitated ~2 h. Into a second reactor was charged dichloromethane (DCM, 26.6 vol) and 2,4-dichloro-5-trifluoromethyl pyrimidine 6a (19.6 kg, 1.1 equiv) and then cooled to 0 °C. The contents from first reactor were added portion-wise to the second reactor. After addition, the reaction mixture was agitated at 0 °C for ~l h and then Et3N (9.2 kg, 1.1 equiv) was slowly charged. After agitation for 1 h, the temperature was increased to 25 °C and monitored for consumption of starting material. The reaction mixture was quenched with 5% aqueous NaHCO, and then filtered over Celite®. The DCM layer was removed and the aqueous layer was back-extracted with DCM (3x). The combined organics were washed with water, dried (Na2S04), and concentrated. Methanol (2.5 vol) was charged and the solution was heated to reflux for 1 h, then cooled to 0 °C. After 1 h, the solids were filtered and dried under reduced pressure to give 2-(4-((4-chloro-5-(tri fluoromethyl)pyri mi din-2-yl)amino)-3 -methyl – l//-pyrazol- l -yl)-2-methyl propanamide 7a
(31.2 kg (wet weight)). 1H NMR (600 MHz, DMSO-de) 10.05 (br. s., 1H), 8.71 (d, J= 11 Hz, 1H), 7.95 (app. d, 1H), 7.18 (br. s., 1H), 6.78 (br. s., 1H), 2.14 (s, 3H), 1.67 (s, 6H).
Example 3 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanamide 8a
A reactor was charged with anhydrous tetrahydrofuran (THF, 10 vol) and 2-(4-((4-chloro-5-(trifl uoromethyl )pyrimi din-2-yl)amino)-3 -methyl – l //-pyrazol- l -yl)-2-methylpropanamide 7a (21 kg) at room temperature with agitation. A solution of 2M
methylamine in THF (3.6 vol) was slowly charged to the reactor at 25 °C and maintained for ~3 h. The reaction mixture was diluted with 0.5 w/w aqueous sodium bicarbonate solution (10 w/w), and extracted with ethyl acetate (EtOAc, 4.5 w/w). The aqueous layer was extracted with EtOAc (4x), the organics were combined and then washed with H20 (7 w/w). The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. «-Heptane (3 w/v) was added to the residue, agitated, filtered and dried under reduced pressure to give 2-m ethyl -2-(3 -methyl -4-((4-(methyl ami no)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol-1 -yl)propanamide 8a (19.15 kg, 93% yield). ¾ NMR (600 MHz, DMSO-d6) 8.85 (m, 1H), 8.10 (s, 1H), 8.00 (m, 1H), 7.16 (br. s., 1H), 6.94 (m, 1H), 6.61 (br. s., 1H), 2.90 (d, J = 4.3 Hz, 3H), 2.18 (br. s., 3H), 1.65 (s, 6H).
Example 4 Preparation of 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)- lH-pyrazol- 1 -yl)propanenitrile 9a
To a reactor was charged 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifl uoromethyl )pyri mi din-2-yl)amino)- l //-pyrazol- l -yl)propan amide 8a (15 kg, 1 equiv) at room temperature followed by EtOAc (2 vol) and 6.7 vol T3P (50% w/w in EtOAc). The reaction mixture was heated to 75 °C over 1 h and then agitated for 16 h until consumption of starting material. The reaction mixture was cooled between -10 to -15 °C then added drop-wise 5N aqueous NaOH (7 vol) resulting in pH 8-9. The layers were separated and the aqueous layer back-extracted with EtOAc (2 x 4 vol). The combined organic extracts were washed with 5 %
aqueous NaHCO, solution, and then distilled to azeotropically remove water. The organics were further concentrated, charged with «-heptane (2 vol) and agitated for 1 h at room temperature. The solids were filtered, rinsed with «-heptane (0.5 vol) and then dried under vacuum (<50 °C). The dried solids were dissolved in EtOAc (1.5 vol) at 55 °C, and then «-heptane (3 vol) was slowly added followed by 5-10% of 9a seeds. To the mixture was slowly added «-heptane (7 vol) at 55 °C, agitated for 1 h, cooled to room temperature and then maintained for 16 h. The suspension was further cooled between 0-5 °C, agitated for 1 hour, filtered, and then rinsed the filter with chilled 1 :6.5 EtOAc/«-heptane (1 vol). The product was dried under vacuum at 50 °C to give 2-methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1 //-pyrazol – 1 -yl )propaneni tri 1 e 9a (9.5 kg, first crop), 67% yield). ‘H NMR (600 MHz, DMSO-d6) 8.14 (s, 1H), 8.13 (br. s., 1H), 7.12 (br. s., 1H), 5.72 (br. s, 1H), 3.00 (d, J= 4.6 Hz, 3H),
2.23 (s, 3H), 1.96 (s, 3H).
Example 5 Preparation of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a
Following the procedure of Example 1, a mixture of methanol and methyl 2-methyl-2-(3-methyl-4-nitro-lH-pyrazol-l-yl)propanoate 3a (0.5 kg) was charged into an autoclave under nitrogen atmosphere, followed by slow addition of 10 % (50% wet) Pd/C. Hydrogen was charged under pressure and the reaction mixture agitated at 25 °C until complete. The mixture was filtered, concentrated under reduced pressure and then slurried in MTBE for 2 h at 25 °C. Filtration and drying under reduced pressure gave methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a (LC-MS, M+l=l98).
Example 6 Preparation of methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)-3 -methyl- lH-pyrazol- 1 -yl)-2-methylpropanoate 11a
Following the procedure of Example 2, a mixture of methyl 2-(4-amino-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 10a and DIPEA (1.2 equiv) in /-BuOH was warmed to 80 °C. Then a solution of 2,4-dichloro-5-trifluoromethyl pyrimidine 6a in /-BuOH was added slowly drop wise at 80 °C. After 15 minutes, LCMS showed the reaction was complete, including later eluting 59.9% of product ester 11a, earlier eluting 31.8% of undesired regioisomer (ester), and no starting material 10a. After completion of reaction, the mixture was cooled to room temperature and a solid was precipitated. The solid precipitate was filtered and dried to give methyl 2-(4-((4-chloro-5-(trifluoromethyl)pyrimi din-2 -yl)amino)-3-methyl-lH-pyrazol-l-yl)-2-methylpropanoate 11a (LC-MS, M+l=378).
PAPER
J.Med.Chem (57(3), 921-936, 2014
https://pubs.acs.org/doi/full/10.1021/jm401654j
2-Methyl-2-(3-methyl-4-((4-(methylamino)-5-(trifluoromethyl)pyrimidin-2-yl)amino)-1H-pyrazol-1-yl)propanenitrile (11)

aReagents and conditions: (a) NaH, methyl 2-bromo-2-methylpropanoate, DMF, 70%; (b) LiOH, THF-H2O, 90%; (c) (i) (COCl)2, CH2Cl2, (ii) R-NH2, THF; (d) Pd/C, H2, MeOH; (e) 26, Et3N, n-BuOH, 120 °C; (f) 26, TFA, 2-methoxyethanol, 70 °C; (g) POCl3, 90 °C, 42%.
GNE-9605
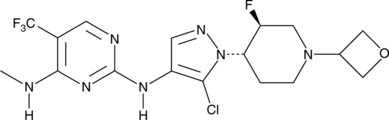
CAS № 1536200-31-3
GNE-9065 is an orally bioavailable and potent inhibitor of leucine-rich repeat kinase 2 (LRRK2; IC50 = 18.7 nM).1 It is selective for LRRK2 over 178 kinases, inhibiting only TAK1-TAB1 >50% at a concentration of 0.1 μM. GNE-9065 (10 and 50 mg/kg) inhibits LRRK2 Ser1292 autophosphorylation in BAC transgenic mice expressing human LRRK2 protein with the G2019S mutation found in families with autosomal Parkinson’s disease.
CNC1=C(C(F)(F)F)C=NC(NC2=C(Cl)N([C@H]3CCN(C4COC4)C[C@@H]3F)N=C2)=N1
N2-(5-Chloro-1-((trans)-3-fluoro-1-(oxetan-3-yl)piperidin-4-yl)-1H-pyrazol-4-yl)-N4-methyl-5-(trifluoromethyl)pyrimidine-2,4-diamine (20)

aReagents and conditions: (a) (±)-(cis)-tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate, PPh3, diisopropyl azodicarboxylate, THF; (b) TFA, DCM, 58% over two steps; (c) oxetan-3-one, DIPEA, NaBH(OAc)3, acetic acid, DCE, 85%; (d) LiHMDS then C2Cl6, THF, −78 °C, 65%; (e) iron dust, NH4Cl, EtOH, 90 °C; (f) 26, TFA, 2-methoxyethanol, 90 °C, 40%, two steps.
REFERENCES
1: Estrada AA, Chan BK, Baker-Glenn C, Beresford A, Burdick DJ, Chambers M, Chen H, Dominguez SL, Dotson J, Drummond J, Flagella M, Fuji R, Gill A, Halladay J, Harris SF, Heffron TP, Kleinheinz T, Lee DW, Pichon CE, Liu X, Lyssikatos JP, Medhurst AD, Moffat JG, Nash K, Scearce-Levie K, Sheng Z, Shore DG, Wong S, Zhang S, Zhang X, Zhu H, Sweeney ZK. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J Med Chem. 2014 Jan 15. [Epub ahead of print] PubMed PMID: 24354345.
/////////////DNL-151, DNL 151, DNL151, Alzheimer’s disease, breast tumor, type I diabetes mellitus, Crohn’s disease, phase 1, Parkinson’s disease, GNE0877, GNE 0877, GNE-0877, GNE-9605, GNE 9605, GNE9605, Genentech
CC(N1N=C(C)C(NC2=NC=C(C(F)(F)F)C(NC)=N2)=C1)(C)C#N
FDA approves new drug to treat multiple sclerosis Ocrevus (ocrelizumab)
On March 28, the U.S. Food and Drug Administration approved Ocrevus (ocrelizumab) to treat adult patients with relapsing forms of multiple sclerosis (MS) and primary progressive multiple sclerosis (PPMS). This is the first drug approved by the FDA for PPMS. Ocrevus is an intravenous infusion given by a health care professional.
“Multiple sclerosis can have a profound impact on a person’s life,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This therapy not only provides another treatment option for those with relapsing MS, but for the first time provides an approved therapy for those with primary progressive MS.”
MS is a chronic, inflammatory, autoimmune disease of the central nervous system that disrupts communication between the brain and other parts of the body. It is among the most common causes of neurological disability in young adults and occurs more frequently in women than men. For most people with MS, episodes of worsening function (relapses) are initially followed by recovery periods (remissions). Over time, recovery may be incomplete, leading to progressive decline in function and increased disability. Most people experience their first symptoms of MS between the ages of 20 and 40.
PPMS is characterized by steadily worsening function from the onset of symptoms, often without early relapses or remissions. The U.S. Centers for Disease Control and Prevention estimates that approximately 15 percent of patients with MS have PPMS.
The efficacy of Ocrevus for the treatment of relapsing forms of MS was shown in two clinical trials in 1,656 participants treated for 96 weeks. Both studies compared Ocrevus to another MS drug, Rebif (interferon beta-1a). In both studies, the patients receiving Ocrevus had reduced relapse rates and reduced worsening of disability compared to Rebif.
In a study of PPMS in 732 participants treated for at least 120 weeks, those receiving Ocrevus showed a longer time to the worsening of disability compared to placebo.
Ocrevus should not be used in patients with hepatitis B infection or a history of life-threatening infusion-related reactions to Ocrevus. Ocrevus must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks. Ocrevus can cause infusion-related reactions, which can be serious. These reactions include, but are not limited to, itchy skin, rash, hives, skin redness, flushing, low blood pressure, fever, tiredness, dizziness, headache, throat irritation, shortness of breath, swelling of the throat, nausea, and fast heartbeat. Additionally, Ocrevus may increase the risk for malignancies, particularly breast cancer. Delay Ocrevus treatment for patients with active infections. Vaccination with live or live attenuated vaccines is not recommended in patients receiving Ocrevus.
In addition to the infusion-related reactions, the most common side effect of Ocrevus seen in the clinical trials for relapsing forms of MS was upper respiratory tract infection. The most common side effects in the study of PPMS were upper respiratory tract infection, skin infection, and lower respiratory tract infection.
The FDA granted this application breakthrough therapy designation, fast track designation, and priority review.
The FDA granted approval of Ocrevus to Genentech, Inc.
GNE-272
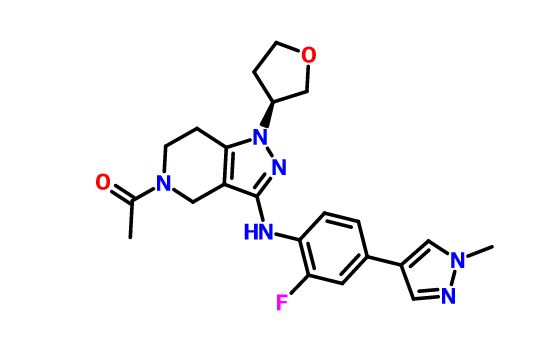

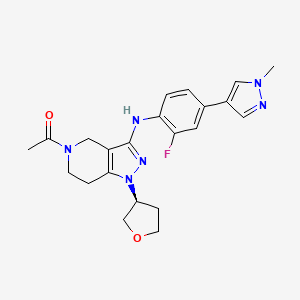
GNE-272
(S)-1-(3-((2-fluoro-4-(1-methyl-1H-pyrazol-4- yl)phenyl)amino)-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridin- 5(4H)-yl)ethanone
1-[3-[2-fluoro-4-(1-methylpyrazol-4-yl)anilino]-1-[(3S)-oxolan-3-yl]-6,7-dihydro-4H-pyrazolo[4,3-c]pyridin-5-yl]ethanone
CAS 1936428-93-1
| Molecular Formula: | C22H25FN6O2 |
|---|---|
| Molecular Weight: | 424.471303 g/mol |
GENENTECH, INC. [US/US]; 1 DNA Way South San Francisco, California 94080-4990 (US).
CONSTELLATION PHARMACEUTICALS, INC. [US/US]; 215 First Street Suite 200 Cambridge, Massachusetts 02142 (US)
ROMERO, F. Anthony; (US).
MAGNUSON, Steven; (US).
PASTOR, Richard; (US).
TSUI, Vickie Hsiao-Wei; (US).
MURRAY, Jeremy; (US).
CRAWFORD, Terry; (US).
ALBRECHT, Brian, K.; (US).
COTE, Alexandre; (US).
TAYLOR, Alexander, M.; (US).
LAI, Kwong Wah; (CN).
CHEN, Kevin, X.; (CN).
BRONNER, Sarah; (US).
ADLER, Marc; (US).
EGEN, Jackson; (US).
LIAO, Jiangpeng; (CN).
WANG, Fei; (CN).
CYR, Patrick; (US).
ZHU, Bing-Yan; (US).
KAUDER, Steven; (US)
Chromatin is a complex combination of DNA and protein that makes up chromosomes. It is found inside the nuclei of eukaryotic cells and is divided between heterochromatin (condensed) and euchromatin (extended) forms. The major components of chromatin are DNA and proteins. Histones are the chief protein components of chromatin, acting as spools around which DNA winds. The functions of chromatin are to package DNA into a smaller volume to fit in the cell, to strengthen the DNA to allow mitosis and meiosis, and to serve as a mechanism to control expression and DNA replication. The chromatin structure is controlled by a series of post-translational modifications to histone proteins, notably histones H3 and H4, and most commonly within the “histone tails” which extend beyond the core nucleosome structure. Histone tails tend to be free for protein-protein interaction and are also the portion of the histone most prone to post-translational modification. These modifications include acetylation, methylation, phosphorylation, ubiquitinylation, and SUMOylation. These epigenetic marks are written and erased by specific enzymes that place the tags on specific residues within the histone tail, thereby forming an epigenetic code, which is then interpreted by the cell to allow gene specific regulation of chromatin structure and thereby transcription.
Of all classes of proteins, histones are amongst the most susceptible to post-translational modification. Histone modifications are dynamic, as they can be added or removed in response to specific stimuli, and these modifications direct both structural changes to chromatin and alterations in gene transcription. Distinct classes of enzymes, namely histone acetyltransferases (HATs) and histone deacetylases (HDACs), acetylate or de-acetylate specific histone lysine residues (Struhl K., Genes Dev., 1989, 12, 5, 599-606).
Bromodomains, which are approximately 1 10 amino acids long, are found in a large number of chromatin-associated proteins and have been identified in approximately 70 human proteins, often adjacent to other protein motifs (Jeanmougin F., et al., Trends Biochem. Sc , 1997, 22, 5, 151-153; and Tamkun J.W., et al., Cell, 1992, 7, 3, 561-572).
Interactions between bromodomains and modified histones may be an important mechanism underlying chromatin structural changes and gene regulation. Bromodomain-containing proteins have been implicated in disease processes including cancer, inflammation and viral replication. See, e.g., Prinjha et al,, Trends Pharm. Sci., 33(3):146-153 (2012) and Muller et al , Expert Rev. , 13 (29): 1 -20 (September 201 1 ).
Cell-type specificity and proper tissue functionality requires the tight control of distinct transcriptional programs that are intimately influenced by their environment.
Alterations to this transcriptional homeostasis are directly associated with numerous disease states, most notably cancer, immuno-inflammation, neurological disorders, and metabolic diseases. Bromodomains reside within key chromatin modifying complexes that serve to control distinctive disease-associated transcriptional pathways. This is highlighted by the observation that mutations in bromodomain-containing proteins are linked to cancer, as well as immune and neurologic dysfunction. Hence, the selective inhibition of bromodomains across a specific family, such as the selective inhibition of a bromodomain of CBP/EP300, creates varied opportunities as novel therapeutic agents in human dysfunction.
There is a need for treatments for cancer, immunological disorders, and other
CBP/EP300 bromodomain related diseases.
PATENT

Scheme 1


Scheme 2

Scheme 3

Scheme 4

General procedure for Intermediates A & B

Intermediate A

Intermediate
General procedure for Intermediates F & G

Intermediate F

Intermediate G
Step 1:
(R)-tetrahydrofuran-3-yI methanesulfonate

To a solution of (^)-tetrahydrofuran-3-ol (25 g, 253.7 mmol) in DCM (250 mL) at 0 °C was added triethylamine (86 g, 851.2 mmol) and mesyl chloride (39 g, 340.48 mmol) dropwise. The mixture was stirred at room temperature for 12 h. The reaction was quenched with water (100 mL) and extracted with DCM (100 mL x 2). The combined organic layers were dried over anhydrous Na2S04, filtered and concentrated in vacuo to give the title compound (47 g, 99%) as a brown oil. Ή NMR (400 MHz, CDC13) δ 5.35 – 5.27 (m, 1H), 4.05 – 3.83 (m, 4H), 3.04 (s, 3 H), 2.28 – 2.20 (m, 2 H).
Step 2:
(S)-tert-butyl 3-bromo-l-(tetrahydrofuran-3-yI)-6,7-dihydro-li/-pyrazolo[43- c] pyridine-5(4H)-carboxylate

To a solution of tert-butyl 3-bromo-6,7-dihydro-lH-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate (Intermediate A, 24.8 g, 82 mmol) in DMF (200 mL) was added Cs2C03 (79 g, 246 mmol) and (/?)-tetrahydrofuran-3-yl methanesulfonate (17.4 g, 98 mmol). The mixture was heated to 80 °C for 12 h. After cooling the reaction to room temperature, the mixture was concentrated in vacuo. The crude residue was purified by silica gel chromatography
(petroleum ether / EtOAc = from 10 : 1 to 3 : 1) to give the title compound (Intermediate F, 50 g, 71 %) as a yellow oil. Ή NMR (400 MHz, DMSO-i ) δ 4.97 – 4.78 (m, 1H), 4.13 (s, 2H), 3.98 – 3.86 (m, 2H), 3.81 – 3.67 (m, 2H), 3.56 (t, J= 5.6 Hz, 2H), 2.68 (t, J= 5.6 Hz, 2H), 2.33 – 2.08 (m, 2H), 1.38 (s, 9H).
Step 3:
(5)-l-(3-bromo-l-(tetrahydrofuran-3-yl)-6,7-dihydro-lH-pyrazoIo[4,3-c]pyridin-5(4//)- yl)ethanone

To a solution of (S)-tert-buty\ 3-bromo- 1 -(tetrahydrofuran-3-yl)-6,7-dihydro-lH-pyrazolo [4,3 -c]pyridine-5(4H)-carboxy late (29 g, 78 mmol) in DCM (300 mL) was added trifluroacetic acid (70 mL) dropwise. The mixture was stirred at room temperature for 2 h. The solvent was concentrated in vacuo and the crude residue was re -dissolved in DMF (100 mL). The mixture was cooled to 0 °C before triethylamine (30 g, 156 mmol) and acetic anhydride (8.7 g, 86 mmol) were added dropwise. The mixture was stirred at room temperature for an additional 2 h. The reaction was quenched with water (200 mL) at 0 °C and extracted with EtOAc (150 mL x 3). The combined organic layers were dried over anhydrous Na2S0 , filtered and concentrated in vacuo. The crude residue was purified by silica gel chromatography (DCM / MeOH = 30 : 1) to give the title compound (Intermediate G, 21.3 g, 87%) as a white solid. lH NMR (400 MHz, CDC13) δ 4.78 – 4.67 (m, 1H), 4.45 -4.29 (m, 2H), 4.15 – 4.06 (m, 2H), 3.96 – 3.92 (m, 2H), 3.88 – 3.70 (m, 2H), 2.71 – 2.67 (m, 2H), 2.38 – 2.34 (m, 2H), 2.16 (s, 3H).
PATENT
| Example 300 | 1-[3-[2-fluoro-4-(1-methylpyrazol-4- yl)anilino]-1-[(3S)-tetrahydrofuran-3- yl]-6,7-dihydro-4H-pyrazolo[4,3- c]pyridin-5-yl]ethanone |
1H NMR (400 MHz, DMSO- d6) δ 8.03 (s, 1H), 7.83-7.68 (m, 3H), 7.36-7.33 (m, 1H), 7.32-7.21 (m, 1H), 4.88- 4.84 (m, 1H), 4.40-4.33 (m, 2H), 4.03-3.99 (m, 2H), 3.84- 3.67 (m, 7H), 2.79-2.64 (m, 2H), 2.26-2.21 (m, 2H), 2.08-2.05 (m, 3H) | 425 |
General Procedure for Intermediates F & G

Step 1
(R)-tetrahydrofuran-3-yl methanesulfonate

Step 2
(S)-tert-butyl 3-bromo-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridine-5(4H)-carboxylate

Step 3
(S)-1-(3-bromo-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridin-5(4H)-yl)ethanone

OTHER ISOMER

| Example 299 | 1-[3-[2-fluoro-4-(1-methylpyrazol-4- yl)anilino]-1-[(3R)-tetrahydrofuran-3- yl]-6,7-dihydro-4H-pyrazolo[4,3- c]pyridin-5-yl]ethanone |
1H NMR (400 MHz, DMSO- d6) δ 8.03 (s, 1H), 7.83-7.67 (m, 3H), 7.39-7.34 (m, 1H), 7.26-7.21 (m, 1H), 4.87- 4.77 (m, 1H), 4.41-4.34 (m, 2H), 4.02-3.97 (m, 2H), 3.83 (s, 3H), 3.81-3.67 (m, 4H), 2.77-2.66 (m, 2H), 2.26- 2.22 (m, 2H), 2.08-2.05 (m, 3H) | 425 |
PAPER

The single bromodomain of the closely related transcriptional regulators CBP/EP300 is a target of much recent interest in cancer and immune system regulation. A co-crystal structure of a ligand-efficient screening hit and the CBP bromodomain guided initial design targeting the LPF shelf, ZA loop, and acetylated lysine binding regions. Structure–activity relationship studies allowed us to identify a more potent analogue. Optimization of permeability and microsomal stability and subsequent improvement of mouse hepatocyte stability afforded 59 (GNE-272, TR-FRET IC50 = 0.02 μM, BRET IC50 = 0.41 μM, BRD4(1) IC50 = 13 μM) that retained the best balance of cell potency, selectivity, and in vivo PK. Compound 59 showed a marked antiproliferative effect in hematologic cancer cell lines and modulates MYC expression in vivo that corresponds with antitumor activity in an AML tumor model.
Discovery of a Potent and Selective in Vivo Probe (GNE-272) for the Bromodomains of CBP/EP300
UNDESIRED R ISOMER
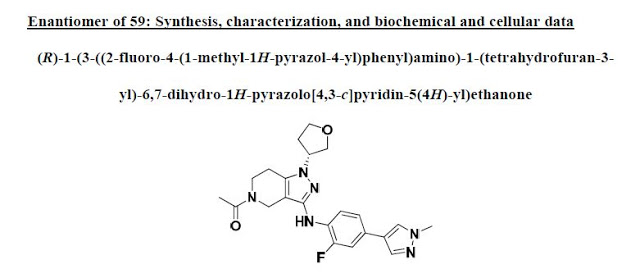
In a similar procedure to59, the title compound was prepared from (S)-tetrahydrofuran-3-yl
methanesulfonate and purified by Prep-TLC (DCM / MeOH = 15 : 1) to give the title
compound as a light yellow solid.
1H NMR (400 MHz, CDCl3) δ 7.76–7. 42 (m,1H), 7.68 (s, 1H), 7.53 (s, 1H), 7.20–7.12 (m, 2H), 5.86–5.77 (m, 1H), 4.79–4.69 (m, 1H),4.47–4.29 (m, 2H), 4.25–4.08 (m, 2H), 4.06–3.72 (m, 4H), 3.99 (s, 3H), 2.76–2.65 (m, 2H),
2.49–2.28 (m, 2H), 2.25–2.12 (m, 3H).
13C NMR (100 MHz, CDCl3) δ 169.81, 169.36,151.71 (d, J = 238.9 Hz), 145.51, 144.64, 137.83, 136.32, 135.89, 126.35, 121.41, 116.44 (d,J = 26.0 Hz), 111.88, 103.09 (d, J = 24.0 Hz), 71.94, 68.10, 57.65, 43.24, 42.24, 39.02, 37.83,32.49, 22.01.
LCMS M/Z (M+H) 425.
[α]27D +8.8 (c 0.78, CHCl3, 99% ee).
DESIRED S ISOMER
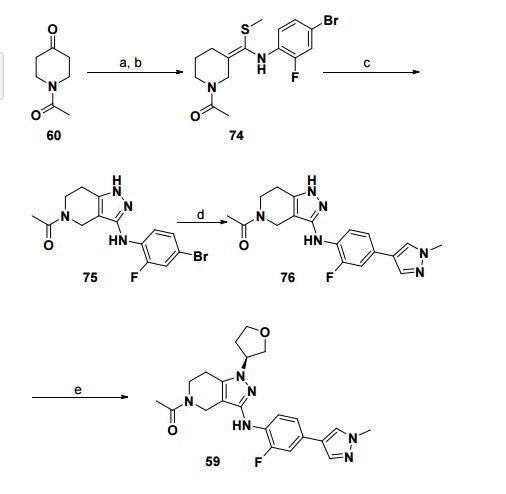
(S)-1-(3-((2-fluoro-4-(1-methyl-1H-pyrazol-4- yl)phenyl)amino)-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1H-pyrazolo[4,3-c]pyridin- 5(4H)-yl)ethanone
aReagents and conditions: (a) 4-bromo-2-fluoro-1-isothiocyanato-benzene, KOtBu, THF, rt (b) CH3I, 40 °C, 51%; (c) hydrazine monohydrate, EtOH, 85 °C; 96%; (d) 1-methyl-4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole, dioxane / water, Na2CO3, Pd(dppf)Cl2, 100 °C, 63%; (e) (R)-tetrahydrofuran-3-yl methanesulfonate, Cs2CO3, DMF, 90 oC, 42%.
The crude residue was purified by silica gel chromatography (DCM / MeOH = 100:1) to give (S)-1-(3-((2-fluoro-4-(1- methyl-1H-pyrazol-4-yl)phenyl)amino)-1-(tetrahydrofuran-3-yl)-6,7-dihydro-1Hpyrazolo[4,3-c]pyridin-5(4H)-yl)ethanone as a light yellow solid.
1H NMR (400 MHz, CDCl3) δ 7.76–7.72 (m, 1H), 7.68 (s, 1H), 7.53 (s, 1H), 7.20–7.12 (m, 2H), 5.86–5.77 (m, 1H), 4.79–4.69 (m, 1H), 4.47–4.29 (m, 2H), 4.25–4.08 (m, 2H), 4.06– 3.72 (m, 4H), 3.99 (s, 3H), 2.76–2.65 (m, 2H), 2.49–2.28 (m, 2H), 2.25–2.12 (m, 3H).
13C NMR (100 MHz, CDCl3) δ 169.8, 169.4, 151.7 (d, J = 238.9 Hz), 145.5, 144.64, 137.83, 136.3, 135.9, 126.4, 121.4, 116.4 (d, J = 26.0 Hz), 111.9, 103.1 (d, J = 24.0 Hz), 71.9, 68.1, 57.7, 43.2, 42.2, 39.0, 37.8, 32.5, 22.0.
LCMS m/z (M+H) 425.
[α]27 D -11.0 (c 1.0, CHCl3, 99% ee).
HRMS m/z 425.2093 (M + H+ , C22H25FN6O2, requires 425.2057).
//////////GNE-272, Genentech, CBP, EP300, cancer, immune system regulation, 1936428-93-1
[H][C@@]1(CCOC1)N1N=C(NC2=C(F)C=C(C=C2)C2=CN(C)N=C2)C2=C1CCN(C2)C(C)=O
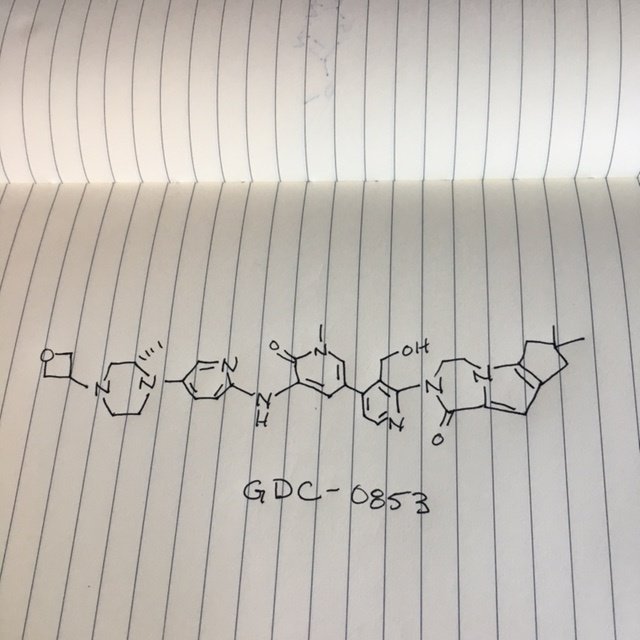
GDC 0853, Fenebrutinib
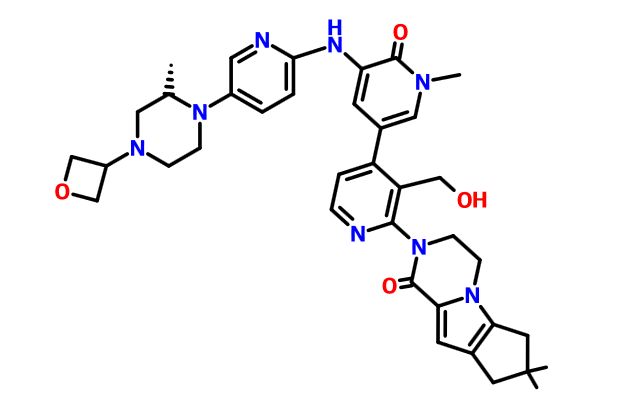
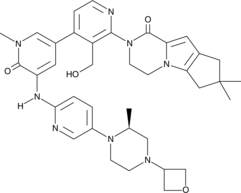
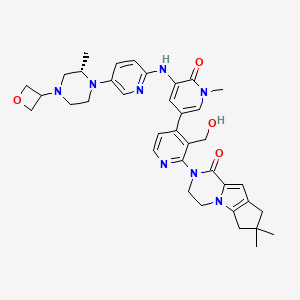
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
 , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† 
Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
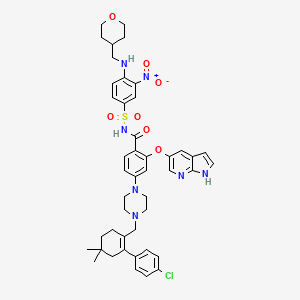
AbbVie’S Investigational Oncology Compound ABT-199/GDC-0199, Venetoclax

ABT 199, RG 7601, GDC 0199
Venetoclax
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
SYNTHESIS UPDATED BELOW …………..

CAS 1257044-40-8 [RN]
2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)-4-(4-((2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enyl)methyl)piperazin-1-yl)-N-(3-nitro-4-((tetrahydro-2H-pyran-4-yl)methylamino)phenylsulfonyl)benzamide
4-[4-[[2-(4-chlorophenyl)-4,4-dimethylcyclohexen-1-yl]methyl]piperazin-1-yl]-N-[3-nitro-4-(oxan-4-ylmethylamino)phenyl]sulfonyl-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
ABT 199
- Molecular Formula: C45H50ClN7O7S
- Average mass: 868.439209 Da
- Monoisotopic mass: 867.318115 Da
-
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
NORTH CHICAGO, Ill., May 31, 2014/NEWS.GNOM.ES/ — AbbVie (NYSE: ABBV) released interim results from a Phase Ib clinical trial of ABT-199/GDC-0199, an investigational B-cell lymphoma 2 (BCL-2) selective inhibitor, in combination with rituximab (Abstract 7013). Results showed anoverall response rate (ORR) of 84 percent, in patients with relapsed/refractory chronic lymphocytic leukemia(CLL), the most common leukemia in the UnitedStates. These results were presented at the 50thAnnual Meeting of the American Society of ClinicalOncology (ASCO), May 30 – June 3 in Chicago.
ABT-199 is a so-called BH3-mimetic drug, which is designed to block the function of the protein Bcl 2. In 1988, it was discovered that Bcl-2 allowed leukaemia cells to become long-lived, a discovery made at the Walter and Eliza Hall Institute by Professors David Vaux, Suzanne Cory and Jerry Adams. Subsequent research led by them and other institute scientists, including Professors Andreas Strasser, David Huang, Peter Colman and Keith Watson, has explained much about how Bcl-2 and related molecules function to determine if a cell lives or dies. These discoveries have contributed to the development of a new class of drugs called BH3-mimetics that kill, and thereby rapidly remove, leukaemic cells by blocking Bcl-2. (source:http://www.wehi.edu.au).
|
Highlights of recent research using this agent |
GDC-0199 (RG7601) is a novel small molecule Bcl-2 selective inhibitor designed to restore apoptosis, also known as programmed cell death, by blocking the function of a pro-survival Bcl-2 family protein. The Bcl-2 family proteins, which are expressed at high levels in many tumors, play a central role in regulating apoptosis and, consequently, are thought to impact tumor formation, tumor growth and resistance.
Venetoclax (previously: GDC-0199, ABT-199, RG7601 )[1] is a small molecule oral drug being investigated to treat chronic lymphocytic leukemia (CLL).[2][3]
In 2015, the FDA granted Breakthrough Therapy Designation to venetoclax for CLL in previously treated (relapsed/refractory) patients with the 17p deletion genetic mutation.[3]
Mechanism of action
Venetoclax (a BH3-mimetic[4]) acts as a Bcl-2 inhibitor, ie. it blocks the anti-apoptotic B-cell lymphoma-2 (BCL2) protein, leading toprogrammed cell death in CLL cells.[2]
Clinical trials
A phase 1 trial established a dose of 400mg/day.[2]
A trial of venetoclax in combination with rituximab had an encouraging complete response rate.[5]
A phase 2 trial met its primary endpoint which was overall response rate.[3] Interim results from a Phase 2b trial are encouraging, especially in patients with the 17p deletion.[2]
A phase 3 trial (NCT02005471)[1] has started.[3]
NOW IN PHASE 3 UPDATED…………
4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (hereafter, “Compound 1”) is a potent and selective Bcl-2 inhibitor having, inter alia, antitumor activity as an apoptosis-inducing agent. Compound 1 has the formula:

Compound 1 is currently the subject of ongoing clinical trials for the treatment of chronic lymphocytic leukemia. U.S. Patent Publication No. 2010/0305122 describes Compound 1, and other compounds which exhibit potent binding to a Bcl-2 family protein, and pharmaceutically acceptable salts thereof. U.S. Patent Publication Nos. 2012/0108590 and 2012/0277210 describe pharmaceutical compositions comprising such compounds, and methods for the treatment of neoplastic, immune or autoimmune diseases comprising these compounds. U.S. Patent Publication No. 2012/0157470 describes pharmaceutically acceptable salts and crystalline forms of Compound 1. The disclosures of U.S. 2010/0305122; 2012/0108590; 2012/0157470 and 2012/0277210 are hereby incorporated by reference in their entireties.



PATENT
US 2015183783
http://www.google.com/patents/US20150183783
PATENT
CN 104370905
http://www.google.com/patents/CN104370905A?cl=en
ABT-199 is developed AbbVie Bel-2 inhibitors, I trial (NCT01328626) enrolled 84 patients with relapsed type / refractory CLL / SLL patients and 44 cases of relapsing / refractory non-Hodgkin lymphoma patients. ABT-199 treatment response CLL / SLL rate of 79% (complete response rate of 22%), median duration of response time was 20.5 months; ABT-199 treatment of non-Hodgkin’s lymphoma response rate of 48% (complete response rate was 7.5%). The efficacy of ABT-199 is capable of obinutuzumab, idelalisib, ibrutinib rival, is expected to become the first listed Bcl_2 inhibitors, ABT-199 is currently ongoing Phase III clinical study.
ABT-199 compound CAS number 1257044-40-8, the compound is structured as follows:

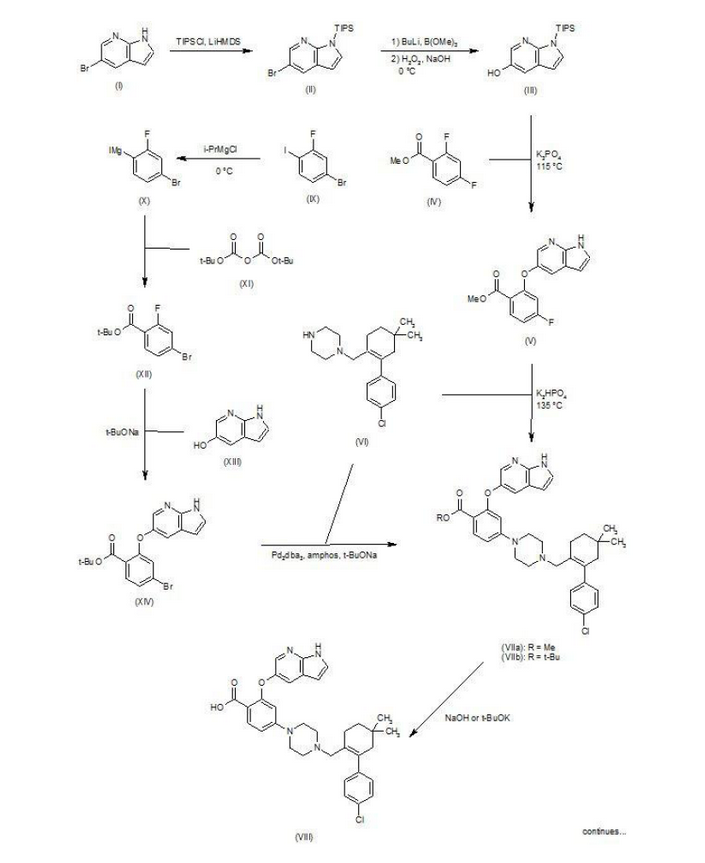
Patent W02012058392, W02012071336, W02010138588 et al. Discloses the preparation of ABT-199 in order to -IH- 5-bromo-pyrrolo [2, 3-b] pyridine as raw material to protect hydroxylation, after replacing the compound 5, and reaction of compound 6, hydrolysis to give compound 9, compound 10 and compound 9 obtained by condensation of ABT-199, a specific line as follows:

use of 2-fluoro-4-nitrobenzoate (A) as a raw material, and substituted 5-hydroxy-7-aza-indole (B), reduction to produce compound ( D), the compound (D) with the compound by cyclization after (H) substitution, hydrolysis to yield compound (J), and then with the compound (K) to afford ABT-199.

Preparation of a compound of Example (F) of the
Example

First step: Synthesis of Compound (C)
2-fluoro-4-nitrobenzoate in IL three-necked flask 50. 0g, dissolved with dimethylformamide N’N- 250ml, was added successively 5-hydroxy-7-aza-indole indole 33. 6g, potassium carbonate 34. 7g, the reaction was heated to 50 degrees under nitrogen gas protection for 2 hours, poured into 2L of ice water was added and extracted three times with ethyl acetate, the organic phase was dried with saturated sodium chloride spin dry to give Compound (C) crude 82. 0g, crude without purification in the next reaction direct investment.
Step two: Synthesis of Compound (D)
The compound of the previous step (C) of the crude product was dissolved in methanol 400ml, was added 10% palladium on carbon 4. 0g, through the reaction of hydrogen at atmospheric pressure, after the end of the reaction by TLC spin solvent to give compound (D) The crude product 73. 2g, crude without purification in the next reaction direct investment.
The third step: Synthesis of compound (F)
Take the previous step the compound (D) crude 20. 0g, t-butanol were added 150ml, compound (E) 10. g, potassium carbonate 9. 7g, completion of the addition the reaction was refluxed for 48 hours the reaction solution was cooled, added acetic acid ethyl ester was diluted, washed with water three times, the combined aqueous phases extracted once with ethyl acetate, the combined ethyl acetate phases twice, dried over anhydrous sodium sulfate and the solvent was spin, the crude product obtained was purified by silica gel column chromatography to give 13. 9g, three-step overall yield of 57.4%.
Preparation Example II Compound (H),

[0029] Take compound (G) (prepared according to W02012058392 method) 5. 0g, dissolved with 50ml of dichloromethane, was added triethylamine 5. 6ml, the reaction solution was cooled to 0-5 ° with stirring, was added dropwise methanesulfonyl chloride 2. 7g, the addition was complete the reaction was warmed to room temperature overnight, after the end of the reaction by TLC the reaction was quenched with water, the organic phase was dried over anhydrous sodium sulfate and the solvent was spin, purified by silica gel column chromatography to give compound (H) 6. 5g , a yield of 99%.
Three ABT-199 Preparation of Example

First step: Synthesis of Compound (I)
In IOOml three-necked flask were added the compound (F) 2. 5g, compound ⑶2. 3g, potassium carbonate I. 9g, Ν ‘was added and reacted at 50 degrees N- dimethylformamide 15ml, nitrogen atmosphere, TLC detection After the reaction, the reaction solution was poured into ice-water, extracted with ethyl acetate twice added ethyl acetate phase was dried over anhydrous sodium sulfate spin, and purified by silica gel column chromatography to give compound (I) 3. 6g, yield 88 %.
Step two: Synthesis of Compound (J)
In IOml single jar Compound (I) I. 0g, followed by adding water 5ml, ethanol 5ml, tetrahydrofuran 5ml, sodium hydroxide 136mg, the reaction was stirred at room temperature the reaction, ethyl acetate was added after dilution of the reaction by TLC, adjusted with IN hydrochloric acid PH4-5, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate and spin dried to give compound (J) 907mg, 93% yield.
Step two: Synthesis of ABT-199
In a 25ml single neck flask was added the compound (J) 100mg, EDCI67mg, dichloromethane 10ml, the reaction was stirred for 30 minutes, was added the compound (K) (prepared in accordance with W02012058392) 55mg, finally added a catalytic amount of DMAP, the force After opening the reaction was stirred overnight, after the end of the reaction by TLC the solvent was spin, HPLC purified preparation obtained by pure ABT-199 ^ 9811, 65% yield.
PATENT
WO 2014165044
http://www.google.com/patents/WO2014165044A1?cl=en
PATENT
US 2014275540
http://www.google.com/patents/US20140275540

-

-
An exemplary reaction according to Scheme 2 is shown below.
 Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302.
Scheme 3 below. Compound (E) is commercially available or may be prepared by techniques known in the art, e.g., as shown in U.S. Pat. No. 3,813,443 and Proceedings of the Chemical Society, London, 1907, 22, 302. -
 Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218.
Scheme 4 below. Compound (M) is commercially available or may be prepared by techniques known in the art, e.g., as shown in GB 585940 and J. Am. Chem. Soc., 1950, 72, 1215-1218. -

-
In another embodiment, the compound of formula (1) is prepared from compound (D) and compound (I) as shown in Scheme 5 below. Compound (J) may be prepared by techniques known in the art, e.g., as shown in WO 2009/117626 and Organometallics, 2008, 27 (21), 5605-5611.
-

 Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H).
Example 1 Synthesis of tert-butyl 4-bromo-2-fluorobenzoate (Compound (C))To a 100 ml jacketed reactor equipped with a mechanical stirrer was charged 4-bromo-2-fluoro1-iodobenzene, “Compound (A)” (5 g, 1.0 eq) and THF (25 ml). The solution was cooled to −5° C. 2 M isopropyl magnesium chloride in THF (10.8 ml, 1.3 eq) was slowly added maintaining the internal temperature below 0° C. The mixture was stirred at 0° C. for 1 h. Di-tert-butyl dicarbonate (5.44 g, 1.5 eq) in THF (10 ml) was added. After 1 h, the solution was quenched with 10% citric acid (10 ml), and then diluted with 25% NaCl (10 ml). The layers were separated and the organic layer was concentrated to near dryness and chased with THF (3×10 ml). The crude oil was diluted with THF (5 ml), filtered to remove inorganics, and concentrated to dryness. The crude oil (6.1 g, potency=67%, potency adjusted yield=88%) was taken to the next step without further purification. 1H NMR (DMSO-d6): δ 1.53 (s, 9H), 7.50-7.56 (m, 1H), 7.68 (dd, J=10.5, 1.9 Hz, 1H), 7.74 (t, J=8.2 Hz, 1H). -
Example 2 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-bromobenzoate (Compound (D))
-
To a 3 L three-neck Morton flask were charged 1H-pyrrolo[2,3-b]pyridin-5-ol (80.0 g, 1.00 eq.), tert-butyl 4-bromo-2-fluorobenzoate (193 g, 1.15 eq.), and anhydrous DMF (800 mL). The mixture was stirred at 20° C. for 15 min. The resulting solution was cooled to about zero to 5° C. A solution of sodium tert-butoxide (62.0 g) in DMF (420 mL) was added slowly over 30 min while maintaining the internal temperature at NMT 10° C., and rinsed with DMF (30 mL). The reaction mixture was stirred at 10° C. for 1 hour (an off-white slurry) and adjusted the internal temperature to ˜45° C. over 30 min. The reaction mixture was stirred at 45-50° C. for 7 hr and the reaction progress monitored by HPLC (IP samples: 92% conversion % by HPLC). The solution was cooled to ˜20° C. The solution was stirred at 20° C. overnight.
-
Water (1200 mL) was added slowly to the reaction mixture at <30° C. over 1 hour (slightly exothermic). The product slurry was adjusted to ˜20° C., and mixed for NLT 2 hours. The crude product was collected by filtration, and washed with water (400 mL). The wet-cake was washed with heptane (400 mL) and dried under vacuum at 50° C. overnight to give the crude product (236.7 g).
-
Re-crystallization or Re-slurry: 230.7 g of the crude product, (potency adjusted: 200.7 g) was charged back to a 3 L three-neck Morton flask. Ethyl acetate (700 mL) was added, and the slurry heated slowly to refluxing temperature over 1 hr (small amount of solids left). Heptane (1400 mL) was added slowly, and the mixture adjusted to refluxing temperature (78° C.). The slurry was mixed at refluxing temperature for 30 min., and cooled down slowly to down to ˜−10° C. at a rate of approximate 10° C./hour), and mixed for 2 hr. The product was collected by filtration, and rinsed with heptane (200 ml).
-
The solid was dried under vacuum at ˜50° C. overnight to give 194.8 g, 86% isolated yield of the product as an off-white solid. MS-ESI 389.0 (M+1); mp: 190-191° C. (uncorrected). 1H NMR (DMSO-d6): δ 1.40 (s, 9H), 6.41 (dd, J=3.4, 1.7 Hz, 1H), 7.06 (d, J=1.8 Hz, 1H), 7.40 (dd, J=8.3, 1.8 Hz, 1H), 7.51 (t, J=3.4 Hz, 1H), 7.58 (d, J=2.6 Hz, 1H), 7.66 (d, J=8.3 Hz, 1H), 8.03 (d, J=2.7 Hz, 1H), 11.72 (s, 1H, NH).
-
Example 3 Synthesis of 2-chloro-4,4-dimethylcyclohexanecarbaldehyde (Compound (F))
-
To a 500 mL RB flask were charged anhydrous DMF (33.4 g, 0.456 mol) and CH2Cl2 (80 mL). The solution was cooled down <−5° C., and POCl3 (64.7 g, 0.422 mol) added slowly over 20 min @<20° C. (exothermic), rinsed with CH2Cl2 (6 mL). The slightly brown solution was adjusted to 20° C. over 30 min, and mixed at 20° C. for 1 hour. The solution was cooled back to <5° C. 3,3-Dimethylcyclohexanone (41.0 g, 90%, ˜0.292 mol) was added, and rinsed with in CH2Cl2 (10 mL) (slightly exothermic) at <20° C. The solution was heated to refluxing temperature, and mixed overnight (21 hours).
-
To a 1000 mL three neck RB flask provided with a mechanical stirrer were charged 130 g of 13.6 wt % sodium acetate trihydrate aqueous solution, 130 g of 12% brine, and 130 mL of CH2Cl2. The mixture was stirred and cooled down to <5° C. The above reaction mixture (clear and brown) was transferred, quenched into it slowly while maintaining the internal temperature <10° C. The reaction vessel was rinsed with CH2Cl2 (10 mL). The quenched reaction mixture was stirred at <10° C. for 15 min. and allowed to rise to 20° C. The mixture was stirred 20° C. for 15 min and allowed to settle for 30 min. (some emulsion). The lower organic phase was separated. The upper aq. phase was back extracted with CH2Cl2 (50 mL). The combined organic was washed with a mixture of 12% brine (150 g)-20% K3PO4 aq. solution (40 g). The organic was dried over MgSO4, filtered and rinsed with CH2Cl2 (30 ml). The filtrate was concentrated to dryness under vacuum to give a brown oil (57.0 g, potency=90.9 wt % by qNMR, ˜100%). 1H NMR (CDCl3): δ 0.98 (s, 6H), 1.43 (t, J=6.4 Hz, 2H), 2.31 (tt, J=6.4, 2.2 Hz, 2H), 2.36 (t, J=2.2 Hz, 2H), 10.19 (s, 1H).
-
Example 4 Synthesis of 2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-enecarbaldehyde (Compound (G))
-
To a 250 mL pressure bottle were charged 2-chloro-4,4-dimethylcyclohex-1-enecarbaldehyde (10.00 g), tetrabutylammonium bromide (18.67 g), and acetonitrile (10 mL). The mixture was stirred at 20° C. for 5 min. 21.0 wt % K2CO3 aq. solution (76.0 g) was added. The mixture was stirred at room temperature (rt) for NLT 5 min. followed by addition of 4-chlorophenylboronic acid (9.53 g) all at once. The mixture was evacuated and purged with N2 for three times. Palladium acetate (66 mg, 0.5 mol %) was added all at once under N2. The reaction mixture was evacuated and purged with N2 for three times (an orange colored mixture). The bottle was back filled with N2 and heated to ˜35° C. in an oil bath (bath temp ˜35° C.). The mixture was stirred at 30° C. overnight (15 hours). The reaction mixture was cooled to RT, and pulled IP sample from the upper organic phase for reaction completion, typically starting material <2% (orange colored mixture). Toluene (100 mL) and 5% NaHCO3-2% L-Cysteine aq. solution (100 mL) were added. The mixture was stirred at 20° C. for 60 min. The mixture was filtered through a pad of Celite to remove black solid, rinsing the flask and pad with toluene (10 mL). The upper organic phase was washed with 5% NaHCO3 aq. solution-2% L-Cysteine (100 mL) once more. The upper organic phase was washed with 25% brine (100 mL). The organic layer (105.0 g) was assayed (118.8 mg/g, 12.47 g product assayed, 87% assayed yield), and concentrated to ˜1/3 volume (˜35 mL). The product solution was directly used in the next step without isolation. However, an analytical sample was obtained by removal of solvent to give a brown oil. 1HNMR (CDCl3): δ 1.00 (s, 6H), 1.49 (t, J=6.6 Hz, 2H), 2.28 (t, J=2.1 Hz, 2H), 2.38 (m, 2H), 7.13 (m, 2H), 7.34 (m, 2H), 9.47 (s, 1H).
-
Example 5 Synthesis of tert-butyl 4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine-1-carboxylate (Compound (H))
-
To a 2 L three neck RB flask provided with a mechanical stirrer were charged a solution of 4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-carbaldehyde (50.0 g) in toluene (250 mL), BOC-piperazine (48.2 g) and anhydrous THF (250 mL). The yellow solution was stirred at 20° C. for 5 min. Sodium triacetoxyborohydride (52.7 g) was added in portion (note: the internal temperature rose to ˜29.5° C. in 15 min cooling may be needed). The yellow mixture was stirred at ˜25° C. for NLT 4 hrs. A conversion of starting material to product of 99.5% was observed by HPLC after a 3 hour reaction time.
-
12.5 wt % brine (500 g) was added slowly to quench the reaction. The mixture was stirred at 20° C. for NLT 30 min and allowed to settle for NLT 15 min. The lower aq. phase (˜560 mL) was separated (note: leave any emulsion in the upper organic phase). The organic phase was washed with 10% citric acid solution (500 g×2). 500 g of 5% NaHCO3 aq. solution was charged slowly into the flask. The mixture was stirred at 20° C. for NLT 30 min., and allowed to settle for NLT 15 min. The upper organic phase was separated. 500 g of 25% brine aq. solution was charged. The mixture was stirred at 20° C. for NLT 15 min., and allowed to settle for NLT 15 min. The upper organic phase was concentrated to ˜200 mL volume under vacuum. The solution was adjusted to −30° C., and filtered off the inorganic salt. Toluene (50 mL) was used as a rinse. The combined filtrate was concentrated to ˜100 mL volume. Acetonitrile (400 mL) was added, and the mixture heated to ˜80° C. to achieve a clear solution. The solution was cooled down slowly to 20° C. slowly at rate 10° C./hour, and mixed at 20° C. overnight (the product is crystallized out at ˜45-50° C., if needed, seed material may be added at 50° C.). The slurry was continued to cool down slowly to ˜−10° C. at rate of 10° C./hours. The slurry was mixed at ˜−10° C. for NLT 6 hours. The product was collected by filtration, and rinsed with pre-cooled acetonitrile (100 mL). The solid was dried under vacuum at 50° C. overnight (72.0 g, 85%). MS-ESI: 419 (M+1); mp: 109-110° C. (uncorrected); 1H NMR (CDCl3): δ 1.00 (s, 6H), 1.46 (s, 9H), 1.48 (t, J=6.5 Hz, 2H), 2.07 (s, br, 2H), 2.18 (m, 4H), 2.24 (t, J=6.4 Hz, 2H), 2.80 (s, 2H), 3.38 (m, 4H), 6.98 (m, 2H), 7.29 (m, 2H).
-
Example 6 Synthesis of 1-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazine dihydrochloride (Compound (I))
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the Boc reductive amination product (Compound (H), 72.0 g) and IPA (720 mL). The mixture was stirred at rt for 5 min, and 59.3 g of concentrated hydrochloride aq. solution added to the slurry. The reaction mixture was adjusted to an internal temperature of ˜65° C. (a clear and colorless solution achieved). The reaction mixture was agitated at ˜65° C. for NLT 12 hours.
-
The product slurry was cooled down to −5° C. slowly (10° C./hour). The product slurry was mixed at ˜−5° C. for NLT 2 hours, collected by filtration. The wet cake was washed with IPA (72 mL) and dried at 50° C. under vacuum overnight to give 73.8 g (95%) of the desired product as a bis-hydrochloride IPA solvate (purity >99.5% peak area at 210 nm). MS-ESI: 319 (M+1); 1HNMR (CDCl3): δ 0.86 (s, 6H), 1.05 (d, J=6.0 Hz, 6H, IPA), 1.42 (t, J=6.1 Hz, 2H), 2.02 (s, br, 2H), 2.12 (m, 2H), 3.23 (m, 4H), 3.4 (s, br, 4H), 3.68 (s, 2H), 3.89 (septet, J=6.0 Hz, 1H, IPA), 7.11 (d, J=8.1 Hz, 2H), 7.41 (d, J=8.1 Hz, 2H).
-
Example 7 Synthesis of 3-nitro-4-(((tetrahydro-2H-pyran-4-yl)methyl)amino)-benzenesulfonamide (Compound (N))
-
To a 500 mL three-neck RB flask equipped with a mechanical stirrer were charged the 4-chloro-3-nitrobenzenesulfonamide, Compound M (10.0 g), diisopropylethylamine (17.5 g), (tetrahydro-2H-pyran-4-yl)methanamine (7.0 g) and acetonitrile (150 mL). The reaction mixture was adjusted to an internal temperature of 80° C. and agitated for no less than 12 hours.
-
The product solution was cooled down to 40° C. and agitated for no less than 1 hour until precipitation observed. The product slurry was further cooled to 20° C. Water (75 mL) was slowly charged over no less than 1 hour, and the mixture cooled to 10° C. and agitated for no less than 2 hours before collected by filtration. The wet cake was washed with 1:1 mix of acetonitrile:water (40 mL). The wet cake was then reslurried in water (80 mL) at 40° C. for no less than 1 hour before collected by filtration. The wet cake was rinsed with water (20 mL), and dried at 75° C. under vacuum to give 12.7 g of the desired product in 99.9% purity and in 91% weight-adjusted yield. 1H NMR (DMSO-d6): δ 1.25 (m, 2H), 1.60 (m, 2H), 1.89 (m, 1H), 3.25 (m, 2H), 3.33 (m, 2H), 3.83 (m, 2H), 7.27 (d, J=9.3 Hz, 1H), 7.32 (s, NH2, 2H), 7.81 (dd, J=9.1, 2.3 Hz, 1H), 8.45 (d, J=2.2 Hz, 1H), 8.54 (t, J=5.9 Hz, 1H, NH).
-
Example 8 Synthesis of tert-butyl 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (Compound (K))
-
General Considerations:
-
this chemistry is considered air and moisture sensitive. While the catalyst precursors in their solid, dry form can be handled and stored in air without special precautions, contact with even small amounts of solvent may render them susceptible to decomposition. As a result, traces of oxygen or other competent oxidants (e.g., solvent peroxides) must be removed prior to combination of the catalyst precursors with solvent and care must be used to prevent ingress of oxygen during the reaction. Also, care must be taken to use dry equipment, solvents, and reagents to prevent formation of undesirable byproducts. The sodium t-butoxide used in this reaction is hygroscopic and it should be properly handled and stored prior to or during use.
-
To a 2.0 L three-neck RB flask equipped with a mechanical stirrer were charged the bis-hydrochloride salt (Compound (I), 42.5 g) and toluene (285 ml). 20% K3PO4 (285 ml) was added and the biphasic mixture was stirred for 30 min. The layers were separated and the organic layer was washed with 25% NaCl (145 ml). The organic layer concentrated to 120 g and used in the coupling reaction without further purification.
-
NaOtBu (45.2 g) and Compound (I) in toluene solution (120 g solution −30 g potency adjusted) were combined in THF (180 ml) in a suitable reactor and sparged with nitrogen for NLT 45 min. Pd2dba3 (0.646 g), Compound (J) (0.399 g), and Compound (D) (40.3 g) were combined in a second suitable reactor and purged with nitrogen until oxygen level was NMT 40 ppm. Using nitrogen pressure, the solution containing Compound (I) and NaOtBu in toluene/THF was added through a 0.45 μm inline filter to the second reactor (catalyst, Compound (J) and Compound (D)) and rinsed with nitrogen sparged THF (30 ml).
-
The resulting mixture was heated to 55° C. with stirring for NLT 16 h, then cooled to 22° C. The mixture was diluted with 12% NaCl (300 g) followed by THF (300 ml). The layers were separated.
-
The organic layer was stirred with a freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated.
-
The organic layer was stirred with a second freshly prepared solution of L-cysteine (15 g), NaHCO3 (23 g), and water (262 ml). After 1 h the layers were separated. The organic layer was washed with 12% NaCl (300 g), then filtered through a 0.45 μm inline filter. The filtered solution was concentrated in vacuo to ˜300 mL, and chased three times with heptane (600 mL each) to remove THF.
-
The crude mixture was concentrated to 6 volumes and diluted with cyclohexane (720 ml). The mixture was heated to 75° C., held for 15 min, and then cooled to 65° C. over NLT 15 min. Seed material was charged and the mixture was held at 65° C. for 4 hours. The suspension was cooled to 25° C. over NLT 8 h, then held at 25° C. for 4 hours. The solids were filtered and washed with cyclohexane (90 ml) and dried at 50° C. under vacuum.
-
Isolated 52.5 g (88.9% yield) as a white solid. Melting point (uncorrected) 154-155° C. 1H NMR (DMSO-d6): δ 0.93 (s, 6H), 1.27 (s, 9H), 1.38 (t, J=6.4 Hz, 2H), 1.94 (s, 2H), 2.08-2.28 (m, 6H), 2.74 (s, 2H), 3.02-3.19 (m, 4H), 6.33 (dd, J=3.4, 1.9 Hz, 1H), 6.38 (d, J=2.4 Hz, 1H), 6.72 (dd, J=9.0, 2.4 Hz, 1H), 6.99-7.06 (m, 2H), 7.29 (d, J=2.7 Hz, 1H), 7.30-7.36 (m, 2H), 7.41-7.44 (m, 1H), 7.64 (t, J=6.7 Hz, 1H), 7.94 (d, J=2.7 Hz, 1H), 11.53 (s, 1H).
-
Example 9 Synthesis of 2-((1H-pyrrolo[2,3-b]pyridin-5-yl)oxy)-4-(4-((4′-chloro-5,5-dimethyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid (Compound (L))
-
Solution preparation: 10% KH2PO4 (aq): KH2PO4 (6 g) in water (56 g); 2:1 heptane/2-MeTHF:heptane (16 mL) in 2-MeTHF (8 mL).
-
Compound (K) (5.79 g), potassium tert-butoxide (4.89 g), 2-methyltetrahydrofuran (87 mL), and water (0.45 mL) were combined in a suitable reactor under nitrogen and heated to 55° C. until reaction completion. The reaction mixture was cooled to 22° C., washed with the 10% KH2PO4 solution (31 g) twice. The organic layer was then washed with water (30 g).
-
After removal of the aqueous layer, the organic layer was concentrated to 4 volumes (˜19 mL) and heated to no less than 50° C. Heptane (23 ml) was slowly added. The resulting suspension was cooled to 10° C. Solids were then collected by vacuum filtration with recirculation of the liquors and the filter cake washed with 2:1 heptane/2-MeTHF (24 ml). Drying of the solids at 80° C. under vacuum yielded 4.0 g of Compound (L) in approximately 85% weight-adjusted yield. 1H NMR (DMSO-d6): δ 0.91 (s, 6H), 1.37 (t, J=6.4 Hz, 2H), 1.94 (s, br, 2H), 2.15 (m, 6H), 2.71 (s, br, 2H), 3.09 (m, 4H), 6.31 (d, J=2.3 Hz, 1H), 6.34 (dd, J=3.4, 1.9 Hz, 1H), 6.7 (dd, J=9.0, 2.4 Hz, 1H), 7.02 (m, 2H), 7.32 (m, 2H), 7.37 (d, J=2.6 Hz, 1H), 7.44 (t, J=3.0 Hz, 1H), 7.72 (d, J=9.0 Hz, 1H), 7.96 (d, J=2.7 Hz, 1H) & 11.59 (m, 1H).
-
Example 10 Synthesis of 4-(4-{[2-(4-chlorophenyl)-4,4-dimethylcyclohex-1-en-1-yl]methyl}piperazin-1-yl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide (Compound (I))
-
Solution preparation prior to reaction: 10% Acetic Acid:Acetic Acid (37 mL) in water (333 g); 5% NaHCO3:NaHCO3 (9 g) in water (176 g); 5% NaCl:NaCl (9 g) in water (176 g).
-
Compound (N) (13.5 g), DMAP (10.5 g), EDAC (10.7 g) and dichloromethane (300 mL) were combined in a suitable reactor and agitated at 25° C. In a second suitable reactor was charged the Acid (Compound (L), 25 g), Et3N (8.7 g) and dichloromethane (120 mL). The resulting Acid (Compound (L)) solution was slowly charged to the initial suspension of Compound (N) and agitated until reaction completion.






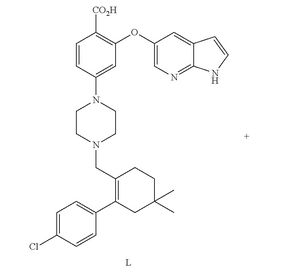
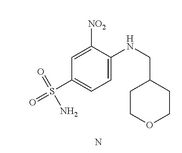
-
N,N-dimethylethylenediamine (9.4 g) was then charged to the reaction mixture with continued agitation. The reaction mixture was warmed to 35° C. and washed with 10% Acetic acid solution (185 mL) twice. The lower organic layer was diluted with more dichloromethane (75 mL) and methanol (12.5 mL). The organic, product layer was then washed with 5% NaHCO3 solution (185 mL) and then washed with 5% NaCl solution (185 mL) at 35° C. The lower, organic layer was separated and then concentrated to 8 vol (˜256 mL) diluted with methanol (26 mL) and warmed to 38° C. Ethyl Acetate (230 mL) was slowly charged. The resulting suspension was slowly cooled to 10° C. and then filtered. The wet cake was washed twice with a 1:1 mix of dichloromethane and ethyl acetate (˜2 vol, 64 mL). After drying the wet cake at 90° C., 32 g (84%) of Compound (I) was isolated.
-
1H NMR (DMSO-d6): δ 0.90 (s, 6H), 1.24 (m, 2H), 1.36 (t, J=6.4 Hz, 2H), 1.60 (m, 2H), 1.87 (m, 1H), 1.93 (s, br, 2H), 2.12 (m, 2H), 2.19 (m, 4H), 2.74 (s, br, 2H), 3.06 (m, 4H), 3.26 (m, 4H), 3.83 (m, 2H), 6.17 (d, J=2.1 Hz, 1H), 6.37 (dd, J=3.4, 1.9 Hz, 1H), 6.66 (dd, J=9.1, 2.2 Hz, 1H), 7.01 (m, 2H), 7.31 (m, 2H), 7.48 (m, 3H), 7.78 (dd, J=9.3, 2.3 Hz, 1H), 8.02 (d, J=2.61 Hz, 1H), 8.54 (d, J=2.33 Hz, 1H), 8.58 (t, J=5.9 Hz, 1H, NH), 11.65 (m, 1H).
PATENT
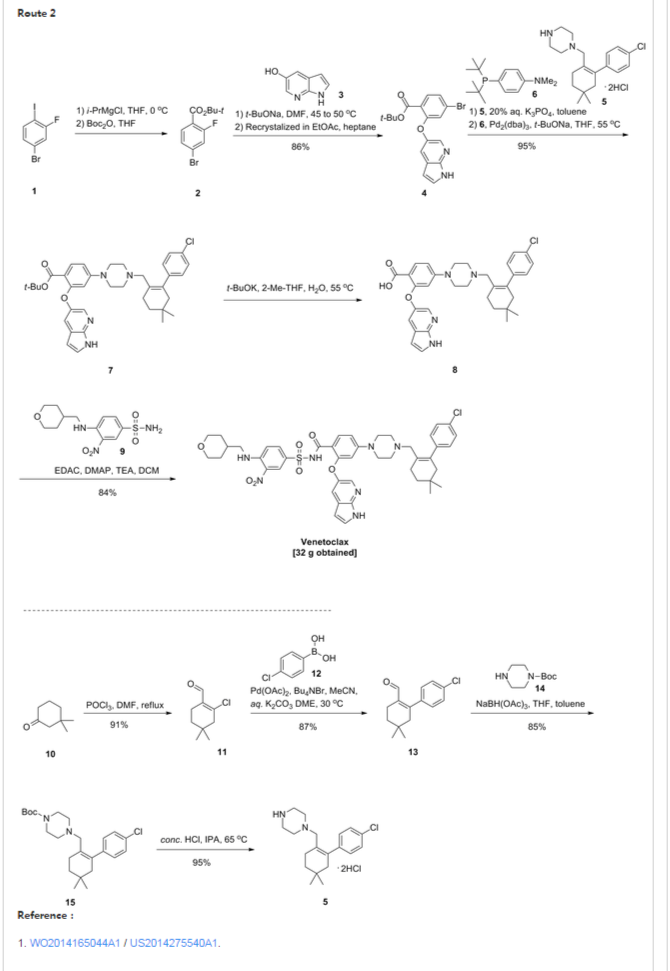
| Patent | Submitted | Granted |
|---|---|---|
| APOPTOSIS-INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2014275082] | 2014-02-10 | 2014-09-18 |
| Processes For The Preparation Of An Apoptosis-Inducing Agent [US2014275540] | 2014-03-12 | 2014-09-18 |
| APOPTOSIS INDUCING AGENTS FOR THE TREATMENT OF CANCER AND IMMUNE AND AUTOIMMUNE DISEASES [US2010305122] | 2010-12-02 | |
| Panel of micrornas that silence the MCL-1 gene and sensitize cancer cells to ABT-263 [US8742083] | 2010-12-23 | 2014-06-03 |
| Treatment Of Cancers Using PI3 Kinase Isoform Modulators [US2014377258] | 2014-05-30 | 2014-12-25 |
| METHODS OF TREATMENT USING SELECTIVE BCL-2 INHIBITORS [US2012129853] | 2011-11-22 | 2012-05-24 |
| INHIBITION OF MCL-1 AND/OR BFL-1/A1 [US2015051249] | 2013-03-14 | 2015-02-19 |
| COMBINATION THERAPY OF A TYPE II ANTI-CD20 ANTIBODY WITH A SELECTIVE BCL-2 INHIBITOR [US2014248262] | 2013-09-06 | 2014-09-04 |
References
- New Drugs Online Report for venetoclax
- Hard-to-Treat CLL Yields to Investigational Drug. ASH Dec 2015 refs: Roberts AW, et al “Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia” N Engl J Med 2015; DOI: 10.1056/NEJMoa1513257.
- Phase 2 Study of Venetoclax in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia with 17p Deletion Meets Primary Endpoint
- ABT-199 BH-3 Mimetic Enters Phase Ia Trial For Chronic Lymphocytic Leukemia. 2011
- For Refractory CLL, Venetoclax’s Complete Response Rate Is Tops. 2015
External links
- ABT-199 inc formula and structure
|
References |
1: Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW. ABT-199, a potent and selective BCL-2
inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013 Jan 6. doi: 10.1038/nm.3048. [Epub ahead of print] PubMed PMID: 23291630.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
4-(4-{[2-(4-Chlorophenyl)-4,4-dimethyl-1-cyclohexen-1-yl]methyl}-1-piperazinyl)-N-({3-nitro-4-[(tetrahydro-2H-pyran-4-ylmethyl)amino]phenyl}sulfonyl)-2-(1H-pyrrolo[2,3-b]pyridin-5-yloxy)benzamide
|
|
| Identifiers | |
| CAS Number | 1257044-40-8 |
| PubChem | CID: 49846579 |
| ChemSpider | 29315017 |
| Chemical data | |
| Formula | C45H50ClN7O7S |
| Molecular mass | 868.44 g/mol |
/////////
CC1(CCC(=C(C1)c2ccc(cc2)Cl)CN3CCN(CC3)c4ccc(c(c4)Oc5cc6cc[nH]c6nc5)C(=O)NS(=O)(=O)c7ccc(c(c7)[N+](=O)[O-])NCC8CCOCC8)C
OR
CC1(CCC(=C(C1)C2=CC=C(C=C2)Cl)CN3CCN(CC3)C4=CC(=C(C=C4)C(=O)NS(=O)(=O)C5=CC(=C(C=C5)NCC6CCOCC6)[N+](=O)[O-])OC7=CN=C8C(=C7)C=CN8)C
