

Graphical abstract

PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » Posts tagged 'FEMALE VIAGRA'
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |

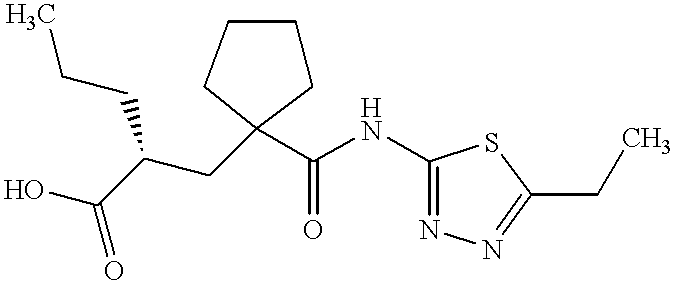
UK-414,495
Molecular Formula: C16H25N3O3S
Molecular Weight: 339.453
UK 414495
CAS 388630-36-2
OF
(-)-(2R)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid;
AND
Cyclopentanepropanoic acid, 1-[[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]-α-propyl-, (αR)-
((R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl) carbamoyl]cyclopentyl}methyl) valeric acid)
(2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
…………………………………………………
Cas 337962-93-3 RACEMIC…………2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid

…………………………………………………………………..
ITS ENANTIOMER
(+)-(2S)-2-[[1-[[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl]cyclopentyl]methyl]pentanoic acid……………337962-74-0
CAS SUMMARY
| Cas number | 388630-36-2 337962-74-0 (enantiomer) 337962-93-3 (racemate) 388630-59-9 (sodium salt) |

 desired
desired
UK-414,495 is a drug developed by Pfizer for the treatment of female sexual arousal disorder.[1] UK-414,495 acts as a potent, selective inhibitor of the enzyme neutral endopeptidase, which normally serves to break down the neuropeptide VIP. The consequent increase in VIP activity alters blood flow to the genital region leading to increased lubrication and muscle relaxation.[2][3][4]

A female equivalent of Viagra could soon be available to help women increase their sexual arousal, scientists claim.
For years they have endeavoured to create an alternative for women that mimics the effects of the male Viagra pill.
Now, the pharmaceutical company behind the original pill has created a prototype which increases blood flow to the genitalia in a similar way to Viagra.

Pfizer have come up with a prototype version of the female equivalent of Viagra
More than half of women experience sexual dysfunction at some point in their lives.
They may suffer a lack of desire, emotional or mental health problems and physical problems that mean they avoid having sex.
Pharmaceutical giant Pfizer has developed a drug, so far called only UK-414,495, which is supposed to increase sexual arousal, but will not affect desire, mood or emotional problems.
Some women take Viagra with mixed results and the drug has been used in fertility treatment to increase blood flow to the pelvis and encourage an embryo to implant in the womb.
But this is the first pill that claims to be an equivalent of the male Viagra.
The research, which involved animals, is published by the British Journal of Pharmacology, though Pfizer say they won’t develop the drug and warn that the chemical may not work the same way in humans, according to the Telegraph.
Chris Wayman, the lead researcher, said: ‘Before this work, we knew surprisingly little about the processes that control all of these changes.

Pfizer claim the tablets may help overcome female sexual arousal disorder
‘Now that we are beginning to establish the pathways involved in sexual arousal, scientists may be able to find ways of helping women who would like to overcome female sexual arousal disorder.
‘While the particular chemical compound in this research did not prove appropriate for further developments, the implications of the research could lead to the development of a product in the future.’
Viagra was originally developed as a treatment for high blood pressure and the heart condition angina, but men who took part in early trials realised the drug had an interesting side effect.
Clinical trials suggested the drug had little effect on angina and instead induced erections in men.
The drug first went on sale in 1998 and has since been prescribed to 25million men, creating a multi-billion pound global market.
The name Viagra has become so associated with men’s erectile problems that many cures are marketed as ‘herbal viagra’.
It is known by many nicknames, including Vitamin V and the Blue Pill.
Read more: http://www.dailymail.co.uk/health/article-1265842/Female-Viagra-help-women-increase-sexual-arousal.html#ixzz39lkmpSik
…………………………………

scheme
http://www.google.com/patents/US20020052370




| Ex | Prec | n | Y | Data |
| 43 | Prep 37 | 0 |
 |
1H NMR (CDCl3, 400 MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1H); LRMS: m/z 339.8 (MH+) Anal. Found: C, 56.46; H, 7.46; N, 12.36. C16H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%. |
Example 29 (2R)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0354]
desired[0355] and
Example 30 (2S)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl) methyl]pentanoic acid
[0356]
 undesired
undesired[0357] The acid from Example 4 (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 29 as a white foam, 400 mg, 99.5% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.30 (bs, 1H), LRMS: m/z 338 (MH−), [α]D=−9.0°(c=0.1, methanol),
and
the title compound of example 30 as a white foam, 386 mg, 99% ee, 1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H);
[0358] LRMS: m/z 338 (MH−); and [α]D=+3.8°(c=0.1, methanol).
[0359] Alternatively, Example 29 may be prepared as follows:
[0360] To a solution of the product from Preparation 51a (574 g, 1.45 mol) in dichloromethane (2.87 L) was added trifluoroacetic acid (1.15 L) over a period of 50 minutes with cooling at 10° C. After addition was complete, the reaction was allowed to warm to ambient temperature with stirring under a nitrogen atmosphere for 24 hours. Deionised water (2.6 L) was then added. The reaction mixture was then washed with deionised water (3×2.6 L). The dichloromethane layer was concentrated to a volume of approximately 1 L to give the crude title compound (439 g, 1.29 mol, 96% yield) as a solution in dichloromethane. A purified sample of the title compound was obtained using the following procedure. To a dichloromethane solution (2.34 L) of the crude product, that had been filtered to remove any particulate contamination, was added isopropyl acetate (1.38 L). The resultant mixture was distilled at atmospheric pressure whilst being simultaneously replaced with isopropyl acetate until the solution temperature reached 87° C. The heating was stopped and the solution was allowed to cool to ambient temperature with stirring for 14 hours to give a cloudy brown solution. The agitation rate was then increased and crystallisation commenced. The suspension was then allowed to granulate for 12 hours at ambient temperature. The resultant suspension was then cooled to 0° C. for 3.5 hours and the solid was then collected by filtration. The filter cake was then washed with isopropyl acetate (2×185 ml, then 2×90 ml) and the solid was dried under vacuum at 40-45° C. for 18 hours to give the title compound (602 g, 0.18 mol, 70% yield) as a cream coloured, crystalline solid;
m.p.: 130-136° C.;
LRMS (negative APCI): m/z [M−H]− 338;
1H-NMR (CDCl3, 300 MHz) δ: 0.92 (t, 3H), 1.27-1.52 (m, 7H), 1.52-1.89 (m, 8H), 2.11-2.27 (m, 1H), 2.27-2.37 (m, 1H), 2.42-2.55 (m, 1H), 2.65 (dd, 2H), 3.00 (q, 2H), 12.25 (bs, 1H).
[0361] Example 29 may be purified as follows:
[0362] The title product from Example 29 was disolved in methanol. To this solution was added sodium methoxide (1 equivalent) in methanol (1 ml/g of Example 29) and the mixture was stirred at room temperature for 20 minutes. The solvent was removed in vacuo and the residue was azeotoped with ethyl acetate to give a brown residue. Ethyl acetate was added and the solution filtered to give a brown solid which was washed with tert-butylmethyl ether to give the crude sodium salt of Example 29. This crude product (35 g) was partitioned between water (200 ml) and ethyl acetate (350 ml). Concentrated hydrochloric acid (˜7 ml) was added until the pH of the aqueous layer was pH2. The aqueous phase was washed with ethyl acetate (2×100 ml). The combined layers were dried using magnesium sulphate. The solvent was removed in vacuo to give a light brown solid (31 g). Ethyl acetate (64 ml, 2 ml/g) and diisopropyl ether (155 ml, 5 ml/g) were added and the mixture heated to 68° C. until a clear solution was obtained (˜30 min). Upon cooling to room temperature, crystallisation of the free acid occurred. After 30 minutes stirring at room temperature the product was collected by filtration and washed with diisopropyl ether. The product was dried in a vacuum oven at 50° C. overnight. (20.2 g, 61% recovery from the sodium salt.); m.p. 135 degC (determined using a Perkin Elmer DSC7 at a heating rate of 20° C./minute).
[0372] The title compound of Example 29 metabolysed to form (2R)-1-(2-{[(5-ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}pentyl)cyclopentanecarboxylic acid.

[0373] This compound was prepared as follows:
[0374] The product from Preparation 102 (430 mg, 1 mmol) was taken up in ethanol (5 mls) and methanol (1 ml) and hydrogenated at 30 psi hydrogen pressure at room temperature for 2 h. The mixture was then filtered through a plug of Arbocel®) and evaporated to a yellow oil. This oil was purified by column chromatography using firstly 19:1, then 9:1 DCM:MeOH as eluant to provide the product as a clear oil (120 mg, 35%); 1HNMR (400 MHz, CDCl3) 0.88 (t, 3H), 1.20-1.88 (m, 13H), 1.90-2.03 (m, 1H), 2.24-2.38 (m, 1H), 2.43-2.72 (m, 2H), 2.95 (q, 2H); LRMS m/z 340.2 (M+H).
Example 31 (R)-2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0375] and

Example 32 (S)-2-{[1-({[2-(Hydroxymethyl)-2.3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid
[0376]

[0377] 2-{[1-({[2-(Hydroxymethyl)-2,3-dihydro-1H-inden-2-yl]amino}carbonyl)-cyclopentyl]methyl}pentanoic acid (WO 9110644, Example 8) was further purified by HPLC using an AD column and hexane:isopropanol:trifluoroacetic acid (90:10:0.1) as eluant, to give the title compound of Example 31, 99% ee, [α]D=+10.40 (c=0.067, ethanol) and the title compound of Example 32, 99% ee, [α]D=−10.9° (c=0.046, ethanol).
………………..
http://www.google.com/patents/US6734186
Example 7 (+)-2-[(1-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]carbonyl}cyclopentyl)methyl]pentanoic Acid (F63)

The acid from Preparation 18 (18/ex4) (824 mg) was further purified by HPLC using an AD column and using hexane:iso-propanol:trifluoroacetic acid (85:15:0.2) as eluant to give the title compound of example 7 as a white foam, 386 mg, 99% ee,1H NMR (CDCl3, 400 MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 (m, 1H), 2.30 (m, 1H), 2.44 (m, 1H), 2.60 (m, 1H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH-); and [α]D=+3.80°(c=0.1, methanol)
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409

Female sexual arousal disorder (FSAD) is a highly prevalent sexual disorder affecting up to 40% of women. We describe herein our efforts to identify a selective neutral endopeptidase (NEP) inhibitor as a potential treatment for FSAD. The rationale for this approach, together with a description of the medicinal chemistry strategy, lead compounds, and SAR investigations are detailed. In particular, the strategy of starting with the clinically precedented selective NEP inhibitor, Candoxatrilat, and targeting low molecular weight and relatively polar mono-carboxylic acids is described. This led ultimately to the prototype development candidate R–13, for which detailed pharmacology and pharmacokinetic parameters are presented.
ACID ENTRY 13
…………………………………………..
WO 2002002513
http://www.google.com/patents/WO2002002513A1?cl=en
…………………..
WO 2002003995
http://www.google.com/patents/WO2002003995A2?cl=en
Scheme 12
LiAIHψ THF, 6hr at reflux




Example 1
( f?)-2-r(1 r(5-ethyl-1.3.4-thiadiazol-2-yl)aminolcarbonyl)cvclopentyl) methyllpentanoic acid
and
Example 2
( S)-2-r(1-fr(5-Ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)cvclopentyl)- methyllpentanoic acid
The title product from stage c) below (824mg) was further purified by HPLC using an AD column and using hexane:/sσ-propanol:trifluoroacetic acid (85:15:0.2) as elutant to give the title product from Example 1 , 400mg, 99.5% ee, 1H NMR (CDCI3, 400MHz) δ: 0.90 (t, 3H), 1.36 (m, 6H), 1.50-1.80 (m, 9H), 2.19 (m, 1 H), 2.30 (m, 1 H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.30 (bs, 1 H), LRMS : m/z 338 (MH“ ), [α]D = -9.0° (c = 0.1 , methanol), and the title product from Example 2, 386mg, 99% ee, 1H NMR (CDCl3, 400MHz) δ: 0.90 (t, 3H), 1.38 (m, 6H), 1.50-1.79 (m, 9H), 2.19 ( , 1 H), 2.30 ( , 1H), 2.44 (m, 1 H), 2.60 (m, 1 H), 2.98 (q, 2H), 12.10-12.27 (bs, 1H); LRMS: m/z 338 (MH“); and [α]D = +3.8° (c = 0.1 , methanol)
Preparation of Starting Materials a) 1 -r2-(tø/t-Butoxycarbonyl)-4-pentvπ-cvclopentane carboxylic acid
A mixture of 1 -[2-(tø t-butoxycarbonyl)-4-pentenyl]-cyclopentane carboxylic acid (EP 274234) (23g, 81.5mmol) and 10% palladium on charcoal (2g) in dry ethanol (200ml) was hydrogenated at 30psi and room temperature for 18 hours. The reaction mixture was filtered through Arbocel®, and the filtrate evaporated under reduced pressure to give a yellow oil. The crude product was purified by column chromatography on silica gel, using ethyl acetate:pentane (40:60) as the eluant, to provide the desired product as a clear oil, 21 g, 91%; 1H NMR (CDCI3, 0.86 (t, 3H), 1.22-1.58 (m, 15H), 1.64 (m, 4H), 1.78 (dd, 1H), 2.00-2.18 ( , 3H), 2.24 ( , 1H); LRMS : m/z 283 (M-HV b) tert-Butyl 2-1Ϊ1 -flT5-ethyl-1.3.4-thiadiazol-2-vnaminolcarbonyl)- cvclopentvDmethyllpentanoate.
1 -(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.21 mmol), 1 – hydroxybenzotriazole hydrate (0.2mmol), N-methylmorpholine (0.31 mmol) and 2-amino-5-ethyl-1 ,3,4-thiadiazole (0.22mmol) were added to a solution of the product from stage a) above (150mg, 0.53mmol) in N,N- dimethylformamide (3ml), and the reaction stirred at 90°C for 18 hours. The cooled solution was diluted with ethyl acetate (90ml), washed with water
(3x25ml), and brine (25ml), then dried (MgSO ) and evaporated under reduced pressure. The crude product was purified by chromatography on silica gel, using ethyl acetate:pentane (30:70) as the eluant to afford the title compound, 92%; 1H NMR (CDCI3, 300MHz) δ: 0.82 (t, 3H), 1.20-1.80 (m, 22H), 1.84 (m, 1 H), 2.20 (m, 4H), 3.04 (q, 2H), 9.10 (bs, 1 H); LRMS : m/z
396.2 (MH+).
c) . 2-r(1-H,(5-ethyl-1.3.4-thiadiazol-2-yl)amino1carbonyl)cvclopentyl) methyllpentanoic acid.
Trifluoroacetic acid (5ml) was added to a solution of the title product from stage b) above (0.31 mmol) in dichloromethane (5ml), and the solution stirred at room temperature for 4 hours. The reaction mixture was concentrated under reduced pressure and the residue azeotroped with toluene and dichloromethane to afford the title compound as a clear oil, 81 %, 1H NMR
(CDCI3, 400MHz) δ: 0.92 (t, 3H), 1.35 (t, 3H), 1.25-1.80 (m, 11 H), 2.20-2.50 (m, 4H), 2.95 (q, 2H), 12.10 (bs, 1 H); LRMS : m/z 339.8 (MH+); Anal. Found: C, 56.46; H, 7.46; N, 12.36. Cι6H25N3O3S requires C, 56.62; H, 7.44; N, 12.37%.
………………………………………

………………………………..
Original Research Article
SEE
The discovery of small molecule inhibitors of neutral endopeptidase. Structure-activity studies on functionalized glutaramides
Chem Biol Drug Des 2006, 67(1): 74
Optimization of oral pharmacokinetics in the discovery of clinical candidates for the treatment of sexual dysfunction
237th ACS Natl Meet (March 22-26, Salt Lake City) 2009, Abst MEDI 173
Novel selective inhibitors of neutral endopeptidase for the treatment of female sexual arousal disorder. Synthesis and activity of functionalized glutaramides
J Med Chem 2006, 49(14): 4409
Bioorganic & Medicinal Chemistry (2007), 15(1), 142-159
Journal of Medicinal Chemistry (2007), 50(24), 6165-6176.
|
5-7-2004
|
Treatment of sexual dysfunction
|
|
|
11-15-2002
|
Treatment of sexual dysfunction
|
|
|
5-3-2002
|
Cyclopentyl-substituted glutaramide derivatives as inhibitors of neutral endopeptidase
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| (R)-2-({1-[(5-ethyl-1,3,4-thiadiazol-2-yl)carbamoyl]cyclopentyl}methyl)valeric acid | |
| Clinical data | |
| Legal status | ? |
| Identifiers | |
| CAS number | 337962-93-3 |
| ATC code | ? |
| PubChem | CID 9949799 |
| Chemical data | |
| Formula | C16H25N3O3S |
| Mol. mass | 339.452 g/mol |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US6734186 * | Nov 8, 2000 | May 11, 2004 | Pfizer Inc. | Phosphodiesterase 2 inhibitor |
| US7956195 * | Dec 21, 2007 | Jun 7, 2011 | Abbott Laboratories | reacting arylboronic acids with a cycloalkanone, in the presence of a rhodium catalyst or BINAP, to form a substituted arylcycloalkanone, then formin of a hydantoin, alkylation of the hydantoin, resolution, hydrolysis of the hydantoin to the amino acids and esterification of acids; chemical intermediates |
| WO2005007166A1 * | Jul 12, 2004 | Jan 27, 2005 | Alasdair Mark Naylor | Treatment of sexual dysfunction |
Female Sexual Response
The female sexual response phase of arousal is not easily distinguished from the phase of desire until physiological changes begin to take place in the vagina and clitoris as well as other sexual organs. Sexual excitement and pleasure are accompanied by a combination of vascular and neuromuscular events which lead to engorgement of the clitoris, labia and vaginal wall, increased vaginal lubrication and dilatation of the vaginal lumen (Levin, 1980; Ottesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Masters et al., 1996; Berman et al., 1999).
Vaginal engorgement enables transudation to occur and this process is responsible for increased vaginal lubrication. Transudation allows a flow of plasma through the epithelium and onto the vaginal surface, the driving force for which is increased blood flow in the vaginal capillary bed during the aroused state. In addition engorgement leads to an increase in vaginal length and luminal diameter, especially in the distal ⅔ of the vaginal canal. The luminal dilatation of the vagina is due to a combination of smooth muscle relaxation of its wall and skeletal muscle relaxation of the pelvic floor muscles. Some sexual pain disorders such as vaginismus are thought to be due, at least in part, by inadequate relaxation preventing dilatation of the vagina; it has yet to be ascertained if this is primarily a smooth or skeletal muscle problem. (Levin, 1980; Oltesen, 1983; Levin, 1991; Levin, 1992; Sjoberg, 1992; Wagner, 1992; Schiavi et al., 1995; Master et al., 1996; Berman et al., 1999).
The vasculature and micro vasculature of the vagina are innervated by nerves containing neuropeptides and other neurotransmitter candidates. These include calcitonin gene-related peptide (CGRP), neuropeptide Y (NPY; Sequence No. 4), nitric oxide synthase (NOS), substance P and vasoactive intestinal peptide (VIP; Sequence No. 8) (Hoyle et al., 1996). Peptides that are present in the clitoris are discussed infra. Nitric oxide synthase, which is often colocalised with VIP (Sequence No. 8), displays a greater expression, immunologically, in the deep arteries and veins rather than in the blood vessels of the propria (Hoyle et al., 1996).
Female Sexual Dysfunction
It is known that some individuals can suffer from female sexual dysfunction (FSD). FSD is best defined as the difficulty or inability of a woman to find satisfaction in sexual expression. FSD is a collective term for several diverse female sexual disorders (Leiblum, 1998, Berman et al., 1999). The woman may have lack of desire, difficulty with arousal or orgasm, pain with intercourse or a combination of these problems. Several types of disease, medications, injuries or psychological problems can cause FSD.
Studies investigating sexual dysfunction in couples reveals that up to 76% of women have complaints of sexual dysfunction and that 30-50% of women in the USA experience FSD.
Sub-types of FSD include hypoactive sexual desire disorder, female sexual arousal disorder, orgasmic disorder and sexual desire disorder.
Treatments in development are targeted to treat specific subtypes of FSD, predominantly desire and arousal disorders.
The categories of FSD are best defined by contrasting them to the phases of normal female sexual response: desire, arousal and orgasm (Leiblum 1998). Desire or libido is the drive for sexual expression—and manifestations often include sexual thoughts either when in the company of an interested partner or when exposed to other erotic stimuli. In contrast, sexual arousal is the vascular response to sexual stimulation, an important component of which is vaginal lubrication and elongation of the vagina. Thus, sexual arousal, in contrast to sexual desire, is a response relating to genital (e.g. vaginal and clitoral) blood flow and not necessarily sensitivity. Orgasm is the release of sexual tension that has culminated during arousal. Hence, FSD typically occurs when a woman has an inadequate or unsatisfactory response in any of these phases, usually desire, arousal or orgasm. FSD categories include hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorders and sexual pain disorders.
Hypoactive sexual desire disorder is present if a woman has no or little desire to be sexual, and has no or few sexual thoughts or fantasies. This type of FSD can be caused by low testosterone levels, due either to natural menopause or to surgical menopause. Other causes include illness, medications, fatigue, depression and anxiety.
Female sexual arousal disorder (FSAD) is characterised by inadequate genital response to sexual stimulation. The genitalia (e.g. the vagina and/or the clitoris) do not undergo the engorgement that characterises normal sexual arousal. The vaginal walls are poorly lubricated, so that intercourse is painful. Orgasms may be impeded. Arousal disorder can be caused by reduced oestrogen at menopause or after childbirth and during lactation, as well as by illnesses, with vascular components such as diabetes and atherosclerosis. Other causes result from treatment with diuretics, antihistamines, antidepressants eg SSRIs or antihypertensive agents. FSAD is discussed in more detail infra.
Sexual pain disorders (which include dyspareunia and vaginismus) are characterised by pain resulting from penetration and may be caused by medications which reduce lubrication, endometriosis, pelvic inflammatory disease, inflammatory bowel disease or urinary tract problems.
The prevalence of FSD is difficult to gauge because the term covers several types of problem, some of which are difficult to measure, and because the interest in treating FSD is relatively recent. Many women’s sexual problems are associated either directly with the female ageing process or with chronic illnesses such as diabetes and hypertension.
There are wide variations in the reported incidence and prevalence of FSD, in part explained by the use of differing evaluation criteria, but most investigators report that a significant proportion of otherwise healthy women have symptoms of one or more of the FSD subgroups. By way of example, studies comparing sexual dysfunction in couples reveal that 63% of women had arousal or orgasmic dysfunction compared with 40% of men have erectile or ejaculatory dysfunction (Frank et al., 1978).
However, the prevalence of female sexual arousal disorder varies considerably from survey to survey. In a recent National Health and Social Life Survey 19% of women reported lubrication difficulties whereas 14% of women in an outpatient gynaecological clinic reported similar difficulties with lubrication (Rosen et al., 1993).
Several studies have also reported dysfunction with sexual arousal in diabetic women (up to 47%), this included reduced vaginal lubrication (Wincze et al., 1993). There was no association between neuropathy and sexual dysfunction.
Numerous studies have also shown that between 11-48% of women overall may have reduced sexual desire with age. Similarly, between 11-50% of women report problems with arousal and lubrication, and therefore experience pain with intercourse. Vaginismus is far less common, affecting approximately 1% of women.
Studies of sexually experienced women have detailed that 5-10% have primary anorgasmia. Another 10% have infrequent orgasms and a further 10% experience them inconsistently (Spector et al., 1990).
Because FSD consists of several subtypes that express symptoms in separate phases of the sexual response cycle, there is not a single therapy. Current treatment of FSD focuses principally on psychological or relationship issues. Treatment of FSD is gradually evolving as more clinical and basic science studies are dedicated to the investigation of this medical problem. Female sexual complaints are not all psychological in pathophysiology, especially for those individuals who may have a component of vasculogenic dysfunction (eg FSAD) contributing to the overall female sexual complaint. There are at present no drugs licensed for the treatment of FSD. Empirical drug therapy includes oestrogen administration (topically or as hormone replacement therapy), androgens or mood-altering drugs such as buspirone or trazodone. These treatment options are often unsatisfactory due to low efficacy or unacceptable side effects.
Since interest is relatively recent in treating FSD pharmacologically, therapy consists of the following:- psychological counselling, over-the-counter sexual lubricants, and investigational candidates, including drugs approved for other conditions. These medications consist of hormonal agents, either testosterone or combinations of oestrogen and testosterone and more recently vascular drugs, that have proved effective in male erectile dysfunction. None of these agents has been demonstrated to be very effective in treating FSD.
Female Sexual Arousal Disorder (FSAD)
The sexual arousal response consists of vasocongestion in the pelvis, vaginal lubrication and expansion and swelling of the external genitalia. The disturbance causes marked distress and/or interpersonal difficulty. Studies investigating sexual dysfunction in couples reveals that there is a large number of females who suffer from sexual arousal dysfunction; otherwise known as female sexual arousal disorder (FSAD).
The Diagnostic and Statistical Manual (DSM) IV of the American Psychiatric Association defines Female Sexual Arousal Disorder (FSAD) as being:
“a persistent or recurrent inability to attain or to maintain until completion of the sexual activity adequate lubrication-swelling response of sexual excitement. The disturbance must cause marked distress or interpersonal difficulty.”
FSAD is a highly prevalent sexual disorder affecting pre-, peri- and post menopausal (±HRT) women. It is associated with concomitant disorders such as depression, cardiovascular diseases, diabetes and UG disorders.
The primary consequences of FSAD are lack of engorgement/swelling, lack of lubrication and lack of pleasurable genital sensation. The secondary consequences of FSAD are reduced sexual desire, pain during intercourse and difficulty in achieving an orgasm.
It has recently been hypothesised that there is a vascular basis for at least a proportion of patients with symptoms of FSAD (Goldstein et al., 1998) with animal data supporting this view (Park et al., 1997).
Drug candidates for treating FSAD, which are under investigation for efficacy, are primarily erectile dysfunction therapies that promote circulation to the male genitalia. They consist of two types of formulation, oral or sublingual medications (Apomorphine, Phentolamine, Sildenafil), and prostaglandin (PGE1-Alprostadil) that are injected or administered transurethrally in men, and topically to the genitalia in women.
The present invention seeks to provide an effective means of treating FSD, and in particular FSAD.
SUMMARY
The present invention is based on findings that FSAD is associated with reduced genital blood flow—in particular reduced blood flow in the vagina and/or the clitoris. Hence, treatment of women with FSAD can be achieved by enhancement of genital blood flow with vasoactive agents. In our studies, we have shown that cAMP mediates vaginal and clitoral vasorelaxation and that genital (e.g. vaginal and clitoral) blood flow can be enhanced/potentiated by elevation of cAMP levels. This is a seminal finding.
In this respect, no one has previously proposed that FSAD can be treated in such a way—i.e. by direct or indirect elevation of cAMP levels. Moreover, there are no teachings in the art to suggest that FSAD was associated with a detrimental modulation of cAMP activity and/or levels or that cAMP is responsible for mediating vaginal and clitoral vasorelaxation. Hence, the present invention is even further surprising.
In addition, we have found that by using agents of the present invention it is possible to increase genital engorgement and treat FSAD—e.g. increased lubrication in the vagina and increased sensitivity in the vagina and clitoris. Thus, in a broad aspect, the present invention relates to the use of a cAMP potentiator to treat FSD, in particular FSAD.
The present invention is advantageous as it provides a means for restoring a normal sexual arousal response—namely increased genital blood flow leading to vaginal, clitoral and labial engorgement. This will result in increased vaginal lubrication via plasma transudation, increased vaginal compliance and increased genital (e.g. vaginal and clitoral) sensitivity. Hence, the present invention provides a means to restore, or potentiate, the normal sexual arousal response.
More particularly, the present invention relates to:
A pharmaceutical composition for use (or when in use) in the treatment of FSD, in particular FSAD; the pharmaceutical composition comprising an agent capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
The use of an agent in the manufacture of a medicament (such as a pharmaceutical composition) for the treatment of FSD, in particular FSAD; wherein the agent is capable of potentiating cAMP in the sexual genitalia of a female suffering from FSD, in particular FSAD.
A method of treating a female suffering from FSD, in particular FSAD; the method comprising delivering to the female an agent that is capable of potentiating cAMP in the sexual genitalia; wherein the agent is in an amount to cause potentiation of cAMP in the sexual genitalia of the female; wherein the agent is optionally admixed with a pharmaceutically acceptable carrier, diluent or excipient.
An assay method for identifying an agent that can be used to treat FSD, in particular FSAD, the assay method comprising: determining whether an agent can directly or indirectly potentiate cAMP; wherein a potentiation of cAMP in the presence of the agent is indicative that the agent may be useful in the treatment of FSD, in particular FSAD.

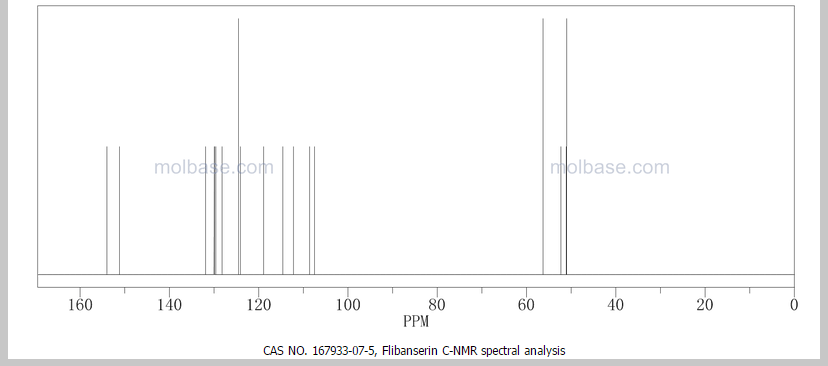
Flibanserin, girosa
167933-07-5 cas no
147359-76-0 (monoHCl)
Women with low libido in the US will have to wait even longer for approval of the first ever treatment for the condition after regulators requested more data on the forerunner flibanserin, delaying its submission until later this year.
The US Food and Drug Administration has asked manufacturer Sprout Pharmaceuticals for data on how flibanserin interacts with other medicines and also how it affects driving ability, after around 10% of patients experienced sleepiness while on the drug
Read more at: http://www.pharmatimes.com/Article/14-02-11/Female_Viagra_now_on_track_for_Q3_filing_in_USA.aspx#ixzz2tAWxwzRD
December 11, 2013 – Sprout Pharmaceuticals today announced that it has received and appealed the Food and Drug Administration’s (FDA) Complete Response Letter (CRL) for flibanserin through the Formal Dispute Resolution process.
Flibanserin is an investigational, once-daily treatment for Hypoactive Sexual Desire Disorder, or HSDD, in premenopausal women. HSDD is the most commonly reported form of female sexual dysfunction
read all here
A new drug being developed by Boehringer Ingelheim could give a boost to the sex drive of women with low libido. The drug, known as flibanserin, has been shown in clinical trials to increase their sexual desire when taken once a day at bedtime.
The results from four pivotal Phase III clinical trials on women with hypoactive sexual desire disorder (HSDD) were presented this week at the European Society for Sexual Medicine’s congress in Lyon, France. The trials showed that participants taking flibanserin had a significant improvement in their sexual desire compared to those given a placebo. They also experienced less of the distress associated with sexual dysfunction.
The drug was initially being investigated as a treatment for depression, and acts on the serotonin receptors in the brain – it is both a 5-HT1A receptor agonist and a 5-HT2A receptor antagonist. It is also a partial agonist at the dopamine D4 receptor.

Neurotransmitters such as serotonin are believed to be involved in sexual function, and antidepressants are commonly associated with a loss of libido, so this was an obvious side-effect to look out for during clinical trials in depression. But far from suppressing the libido in women, it appeared to have the opposite effect, so trials in women with HSDD were initiated.
Hormone replacement can improve the libido of women who have had their ovaries removed, but there is no available drug to treat those who have not. There have been accusations that pharma companies invent new diseases like HSDD in order to sell more medicines, but according to Kathleen Segraves, an assistant professor at Case Western Reserve University in the US who has worked in the field of sexual functioning for many years, this is not the case here. HSDD is a very real disorder, she says, and the potential for a treatment for these women is very exciting.

Flibanserin (code name BIMT-17; proposed trade name Girosa) is a drug that was investigated by Boehringer Ingelheim as a novel, non-hormonal treatment for pre-menopausal women with Hypoactive Sexual Desire Disorder (HSDD).[1][2] Development was terminated in October 2010 following a negative report by the U.S. Food and Drug Administration.[3]
HSDD is the most commonly reported female sexual complaint and characterized by a decrease in sexual desire that causes marked personal distress and/or personal difficulties. According to prevalence studies about 1 in 10 women reported low sexual desire with associated distress, which may be HSDD.[4] The neurobiological pathway of female sexual desire involves interactions among multiple neurotransmitters, sex hormones and various psychosocial factors. Sexual desire is modulated in distinct brain areas by a balance between inhibitory and excitatory neurotransmitters, serotonin acting as an inhibitor while dopamine and norepinephrine act as a stimulator of sexual desire.[5][6]Flibanserin is a 5-HT1A receptor agonist and 5-HT2A receptor antagonist that had initially been investigated as an antidepressant. Preclinical evidence suggested that flibanserin targets these receptors preferentially in selective brain areas and helps to restore a balance between these inhibitory and excitatory effects.[6] HSDD has been recognized as a distinct sexual function disorder for more than 30 years.
The proposed mechanism of action refers back to the Kinsey dual control model. Several sex steroids, neurotransmitters, and hormones have important excitatory or inhibitory effects on the sexual response. Among the neurotransmitters, the excitatory activity is driven by dopamine and norepinephrine, while the inhibitory activity is driven by serotonin. The balance between these systems is relevant for a healthy sexual response. By modulating these neurotransmitters in selective brain areas, flibanserin, a 5-HT1A receptoragonist and 5-HT2A receptor antagonist, is likely to restore the balance between these neurotransmitter systems.[6]
Several large pivotal Phase III studies with Flibanserin were conducted in the USA, Canada and Europe. They involved more than 5,000 pre-menopausal women with generalized acquired Hypoactive Sexual Desire Disorder (HSDD). The results of the Phase III North American Trials demonstrated that
Although the two North American trials that used the flibanserin 100 mg qhs dose showed a statistically significant difference between flibanserin and placebo for the endpoint of [satisfying sexual events], they both failed to demonstrate a statistically significant improvement on the co-primary endpoint of sexual desire. Therefore, neither study met the agreed-upon criteria for success in establishing the efficacy of flibanserin for the treatment of [Hypoactive Sexual Desire Disorder].
These data were first presented on November 16, 2009 at the congress of the European Society for Sexual Medicine in Lyon, France. The women receiving Flibanserin reported that the average number of times they had “satisfying sexual events” rose from 2.8 to 4.5 times a month. However, women receiving placebo reported also an increase of “satisfying sexual events” from 2.7 to 3.7 times a month.
Evaluation of the overall improvement of their condition and whether the benefit was meaningful to the women, showed a significantly higher rate of a meaningful benefit in the flibanserin-treated patient group versus the placebo group.The onset of the Flibanserin effect was seen from the first timepoint measured after 4 weeks of treatment and maintained throughout the treatment period.
The overall incidence of adverse events among women taking flibanserin was low, the majority of adverse events being mild to moderate and resolved during the treatment. The most commonly reported adverse events included dizziness, nausea, fatigue, somnolence and insomnia.

On June 18, 2010, a federal advisory panel to the U.S. Food and Drug Administration (FDA) unanimously voted against recommending approval of Flibanserin.
Earlier in the week, a FDA staff report also recommended non-approval of the drug. While the FDA still might approve Flibanserin, in the past, negative panel votes tended to cause the FDA not to approve.
On October 8, 2010, Boehringer Ingelheim announced that it would discontinue its development of flibanserin in light of the FDA advisory panel’s recommendation.
On June 27, 2013, Sprout Pharmaceuticals confirmed they had resubmitted flibanserin for FDA approval.
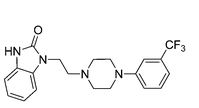
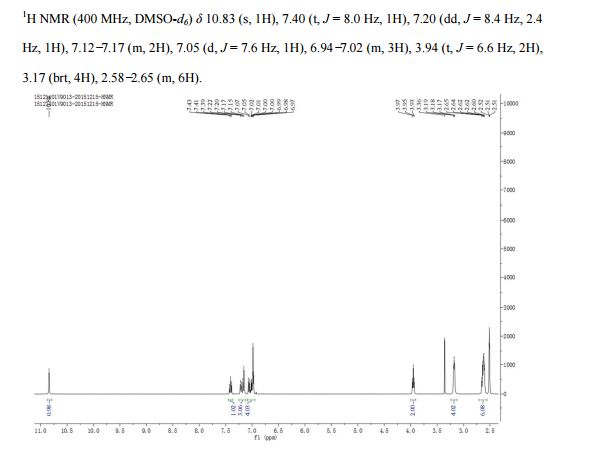
Flibanserin, chemically 1 -[2-(4-(3-trifluoromethylphenyl)piperazin-1 – yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one was disclosed in form of its hydrochloride in European Patent No. 526,434 (‘434) and has the following chemical structure:

Process for preparation of flibanserin were disclosed in European Patent No. ‘434, U.S. Application Publication No. 2007/0032655 and Drugs of the future 1998, 23(1): 9-16.
According to European Patent No. ‘434 flibanserin is prepared by condensing 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethyl phenyl piperazine. According to U.S. Application Publication No. 2007/0032655 flibanserin is prepared by condensing 1-[(3-trifluoromethyl)phenyl]-4-(2- chloroethyl)piperazine with 1 -(2-propenyl)-1 ,3-dihydro-benzimidazol-2H-one.
According to Drugs of the future 1998, 23(1): 9-16 flibanserin is prepared by reacting 1-(2-chloroethyl)-2,3-dihydro-1 H-benzimidazol-one with m- trifluoromethylphenylpiperazine.
PATENT
1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1H-benzimidazol-2-one
Compound 3
Hydrochloride salt (isopropanol) M.p. 230-231°C
Analysis

¹H NMR (DMSO-d₆/CDCL₃ 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (8H), 4.36 (t, 2H), 4.1-3.1 (10H)
CLIP

The compound 1-[2-(4-(3-trifluoromethyl-phenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazol-2-one (flibanserin) is disclosed in form of its hydrochlorid in European Patent Application EP-A-526434 and has the following chemical structure:

Flibanserin shows affinity for the 5-HTιA and 5-HT2-receptor. It is therefore a promising therapeutic agent for the treatment of a variety of diseases, for instance depression, schizophrenia, Parkinson, anxiety, sleep disturbances, sexual and mental disorders and age associated memory impairment.

EXAMPLE……… EP1518858A1
375 kg of 1-[(3-trifluoromethyl)phenyl]-4-(2-cloroethyl)piperazin are charged in a reactor with 2500 kg of water and 200 kg of aqueous Sodium Hydroxide 45%. Under stirring 169.2 kg of 1-(2-propenyl)-1,3-dihydro-benzimidazol-2H-one, 780 kg of isopropanol, 2000 kg of water and 220 kg of aqueous Sodium Hydroxide 45% are added. The reaction mixture is heated to 75-85° C. and 160 kg of concentrated hydrochloric acid and 200 kg of water are added.
The reaction mixture is stirred at constant temperature for about 45 minutes. After distillation of a mixture of water and Isopropanol (about 3000 kg) the remaining residue is cooled to about 65-75° C. and the pH is adjusted to 6.5-7.5 by addition of 125 kg of aqueous Sodium Hydroxide 45%. After cooling to a temperature of 45-50° C., the pH value is adjusted to 8-9 by addition of about 4 kg of aqueous Sodium Hydroxide 45%. Subsequently the mixture is cooled to 30-35° C. and centrifuged. The residue thus obtained is washed with 340 l of water and 126 l of isopropanol and then with water until chlorides elimination.
The wet product is dried under vacuum at a temperature of about 45-55° C. which leads to 358 kg of crude flibanserin polymorph A. The crude product thus obtained is loaded in a reactor with 1750 kg of Acetone and the resulting mixture is heated under stirring until reflux. The obtained solution is filtered and the filtrate is concentrated by distillation. The temperature is maintained for about 1 hour 0-5° C., then the precipitate solid is isolated by filtration and dried at 55° C. for at least 12 hours.
The final yield is 280 kg of pure flibanserin polymorph A.
CLIP
Flibanserin may be prepared by reacting 1-(phenylvinyl)-2,3-dihydro-1H-benzimidazol-2-one (I) with 1,2-dichloroethane (II) in the presence of NaH in warm dimethylformamide. The resulting 1-(2-chloroethyl)-2,3-dihydro-1H-benzimidazol-one (III) is in turn coupled with commercially available m-trifluoromethylphenylpiperazine hydrochloride (IV) in the presence of sodium carbonate and catalytic potassium iodide in refluxing ethanol. The crude flibanserin hydrochloride (V) is then dissolved in aqueous ethanol and the pure base is precipitated upon addition of sodium hydroxide.
PICK UP INTERMEDIATES FROM CHEM24H.COM
1-(1-phenylvinyl)-1,3-dihydro-2H-benzimidazol-2-one (I)
1,2-dichloroethane (II)
1-(2-chloroethyl)-1,3-dihydro-2H-benzimidazol-2-one (III)
1-[3-(trifluoromethyl)phenyl]piperazine; N-[3-(trifluoromethyl)phenyl]piperazine (IV)
1-(2-[4-[3-(trifluoromethyl)phenyl]piperazino]ethyl)-1,3-dihydro-2H-benzimidazol-2-one (V)
PATENT


wherein R2 is as defined in formula X; with a compound of formula Xl:

According to another aspect of the present invention there is provided a novel compound or a salt thereof selected from the compounds of formula I, IV and VII:


Wherein R is hydrogen or an amino protecting group.
Preferable the amino protecting groups are selected from butyl, 1 ,1- diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl.
R1 is independently selected from chlorine, bromine, iodine, methanesulphonate, trifluoromethanesulphonate, paratoluenesulphonate or benzenesulphonate. Preferable R1 is independently selected from chlorine, bromine or iodine and more preferable R1 is chlorine.
Wherein R2 is hydrogen or an amino protecting group.
The amino protecting group may be any of the groups commonly used to protect the amino function such as alkyl, substituted alkyl, hetero substituted alkyl, substituted or unsubstituted unsaturated alkyl, alkyl substituted hetero atoms, substituted or unsubstituted phenyl, substituted or unsubstituted benzyl, alkyoxy carbonyl groups and aryloxy carbonyl groups.
Preferable the amino protecting groups are selected from butyl, 1 ,1 – diphenylmethyl, methoxymethyl, benzyloxymethyl, trichloroethoxymethyl, pyrrolidinomethyl, cyanomethyl, pivaloyloxymethyl, allyl, 2-propenyl, t- butyldimethylsilyl, methoxy, thiomethyl, phenylvinyl, 4-methoxyphenyl, benzyl, A- methoxybenzyl, 2,4-dimethoxybenzyl, 2-nitrobenzyl, t-butoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, 4-chlorophenoxycarbonyl, A- nitrophenoxycarbonyl, methoxycarbonyl and ethoxycarbonyl. Still more preferable protecting groups are selected from t- butoxycarbonyl, ethoxycarbonyl, methoxycarbonyl, benzyloxycarbonyl, phenoxycarbonyl, phenylvinyl and 2-propenyl. The following examples are given for the purpose of illustrating the present invention and should not be considered as limitations on the scope and spirit of the invention.
EXAMPLES Example 1
A mixture of sodium hydroxide (47 gm) and i-(α-methylvinyl) benzimidazol-2-one (100 gm) in dimethylformamide (400 ml) was .stirred for 1 hour at room temperature. Dibromoethane (217 gm) was slowly added to the mixture and stirred at 1 hour 30 minutes. The resulting solution after addition water (500 ml) was extracted with ethyl acetate. The combined ethyl acetate extract washed with water. After drying the solvent was removed under vacuum to yield 132 gm of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H-benzimidazol- 2-one as a yellow oily liquid.
Example 2 A mixture of 1 ,3-dihydro-1-(2-bromoethyl)-3-isopropenyl-2H- benzimidazol-2-one (100 gm), diethanolamine (175 ml), sodium carbonate (40 gm) and potassium iodide (10 gm) was heated to 90 to 95 deg C and stirred for 2 hours. The reaction mass was cooled to room temperature and added water (500 ml). The resulting mixture extracted into ethyl acetate and the organic layer washed with water. After drying the solvent was removed under vacuum to yield 105 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3-isopropenyl- 2H-benzimidazol-2-one as a thick yellow oily liquid.
Example 3
To the mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 2 and chloroform (300 ml), thionyl chloride (95 ml) was slowly added. The mixture was heated to reflux and stirred for 2 hours. The excess thionyl chloride and chloroform was distilled off to yield 98 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2- chloroethyl)amino]ethyl]-3-isopropenyl-2H-benzimidazol-2-one as a brown coloured sticky residue.
Example 4
1 ,3-dihydro-1-[2-[N-[bis-(2-chloroethyl)amino]ethyl]-3-isopropenyl-2H- benzimidazol-2-one (98 gm) obtained as in example 3 was added to water (500 ml) and concentrated hydrochloric acid (200 ml) mixture. The mixture was heated to 60 to 65 deg C and stirred for 1 hour. The contents of the flask cooled to room temperature and pH of the solution adjusted to 9 – 10 with 10% sodium hydroxide solution. The resulting solution extracted with ethyl acetate and washed the organic layer with water. Evaporate the solvent under reduced pressure to yield 82 gm of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]- 2H-benzimidazol-2-one as a dark brown coloured oily liquid
Example 5
A mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-chloroethyl)amino]ethyl]-1,2-H- benzimidazol-2-one (82 gm) obtained as in example 4, xylene (300 ml) and m- trifluoromethyl aniline (58 gm) was refluxed for 64 hours. The reaction mass was cooled to room temperature and filtered to obtain 1-[2-(4-(3- thfluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H-benzimidazole-2-one hydrochloride (Flibanserin hydrochloride) as a light brown coloured solid.
The crude flibanserin hydrochloride was purified in isopropyl alcohol to give 85 gm of pure flibanserin hydrochloride as off white solid.
Example 6
Piperazine (12 gm), toluene(60 ml) and tetra butyl ammonium bromide (1 gm) mixture was heated to 60 deg C, added 1 ,3-dihydro-1-(2-bromoethyl)-3- isopropenyl-2H-benzimidazol-2-one (10 gm) and stirred for 4 hours at 90 to 95 deg C. The mixture was cooled to 60 deg C and added water (50 ml). The separated toluene layer distilled under vacuum to give 8.5 gm of 1 ,3-dihydro-1- (2-piperazinyl)ethyl-3-isopropenyl-2H-benzimidazol-2-one as a white solid.
Example 7
To the mixture of concentrated hydrochloric acid (20 ml) and water (100 ml) was added 1 ,3-dihydro-1-(2-piperazinylethyl)-3-isopropenyl-2H- benzimidazol-2-one (10 gm) obtained as in example 6 and heated to 60 to 65 deg C 1 hour. The mixture was cooled to room temperature and pH of the solution was adjusted to 9 – 10 with 10% sodium hydroxide solution, extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield 8.5 gm of 1 ,3-dihydro-1-(2- piperazinyl ethyl)-2H-benzimidazol-2-one as a white solid.
Example 8
3-trifluoromethylaniline (40 gm) and hydrobromic acid (85 ml; 48- 50%w/w) mixture was cooled to 0 to 5 deg C. To this mixture added sodium nitrite solution (18.5 gm in 25 ml of water) at 5 to 10 deg C and copper powder (1 gm). The temperature was slowly raised to 50 to 55 deg C and stirred for 30 minutes. Added water (200 ml) to reaction mass and applied steam distillation, collected m-trifluoromethylbromobenzene as oily liquid. The oily liquid washed with sulfuric acid for two times (2 X 10 ml) followed by washed with water (2 X 20 ml) and dried the liquid with sodium sulphate to give 22 gm of m- trifluoromethylbromobenzene.
Example 9
To a mixture of 1 ,3-dihydro-1-(2-piperazinyl ethyl)-2H-benzimidazol-2- one (10 gm) obtained as in example 7, m-trifluoromethylbromobenzene (9 gm) obtained as in example 8, sodium tert-butoxide (5.5 gm), palladium acetate (4.5 mg) and xylene (80 ml) was added tri-tert.-butylphosphine (0.2 ml). The mixture was heated to 120 deg C and stirred for 3 hours. The reaction mass was cooled, added water (100 ml) and extracted with ethyl acetate and the organic layer was washed with water. After drying the solvent was removed under vacuum to yield
10 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]-2,3-dihydro-1 H- benzimidazole-2-one (Flibanserin).
Example 10
To a mixture of 1 ,3-dihydro-1-[2-[N-bis-(2-hydroxyethyl)amino]ethyl]-3- isopropenyl-2H-benzimidazol-2-one (100 gm) obtained as in example 3, cyclohexane (400 ml) and sodium carbonate (35 gm) was added benzene sulfonyl chloride (116 gm) at room temperature. The mixture was heated to 80 to
85 deg C and stirred for 8 hours . The contents were cooled to room temperature and added water (500 ml). Distilled the organic layer to give 182 gm of 1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one.
Example 11
1 ,3-dihydro-1 -[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzitηidazol-2-one (100 gm) obtained as in example 10, dimethylformamide (500 ml) and sodium corbonate (18 gm) was mixed and heated to 70 deg C. To the mixture was added m-trifluoromethyl aniline (27 gm) and heated to 80 to 85 deg C, stirred for 5 hours. The reaction mass was cooled and added water (2000 ml), filtered the solid to yield 1 ,3-dihydro-1-[2-[4-(3- trifluoromethylphenyl)piperazinyl]ethyl]-3-isopropenyl-2H benzimidazol-2-one. Example 12
1 ,3-dihydro-1-[2-[N-[bis-(2-benzenesulfonyloxy)- ethyl]amino]ethyl]-3- isopropenyl- 2H-benzimidazol-2-one (100 gm) obtained as in example 11 added to the mixture of water (500 ml) and concentrated hydrochloric acid (200 ml), heated to 65 deg C and stirred for 1 hour. The reaction mass was cooled to room temperature and pH adjusted to 10 to 10-5 with 10% sodium hydroxide solution. The resulting mixture was extracted with ethyl acetate and the organic
■ layer was washed with water. After drying the solvent was removed under vacuum to yield 87 gm of 1-[2-(4-(3-trifluoromethylphenyl)piperazin-1-yl)ethyl]- 2,3-dihydro-1 H-benzimidazole -2-one (Flibanserin).
Paper
Journal of Pharmaceutical and Biomedical Analysis, v.57, 2012 Jan 5, p.104(5)
Isolation and structural elucidation of flibanserin as an adulterant in a health supplement used for female sexual performance enhancement
Low, Min-Yong et al
http://www.sciencedirect.com/science/article/pii/S0731708511004833
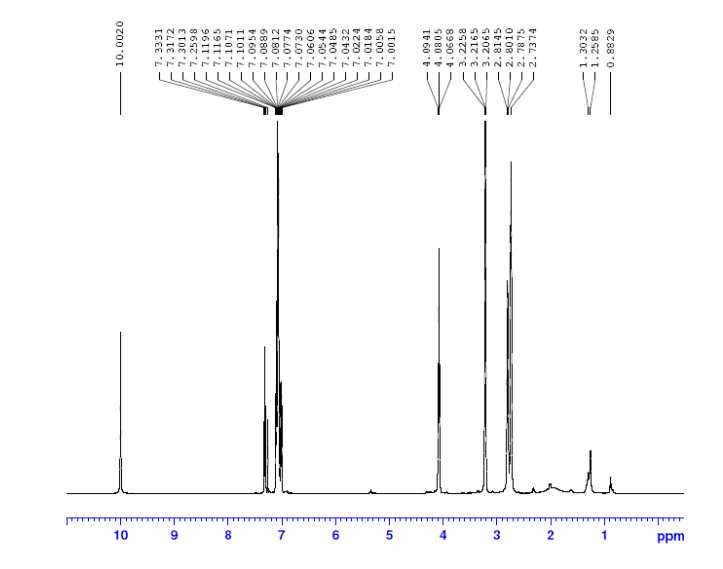
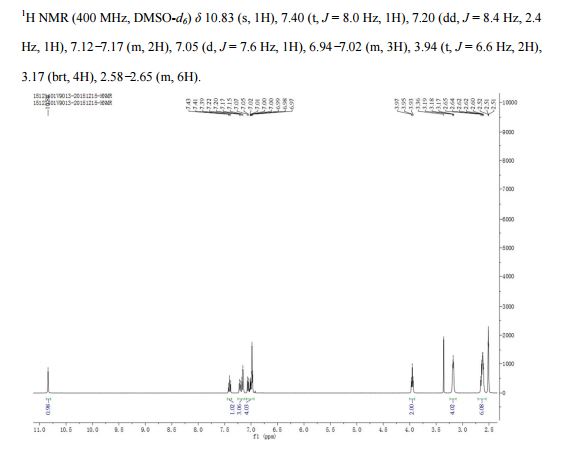
This proposed formula and structure was further confirmed by 1H and 13C NMR data which indicated the presence of 20 carbon atoms and 21 protons.
1H NMR
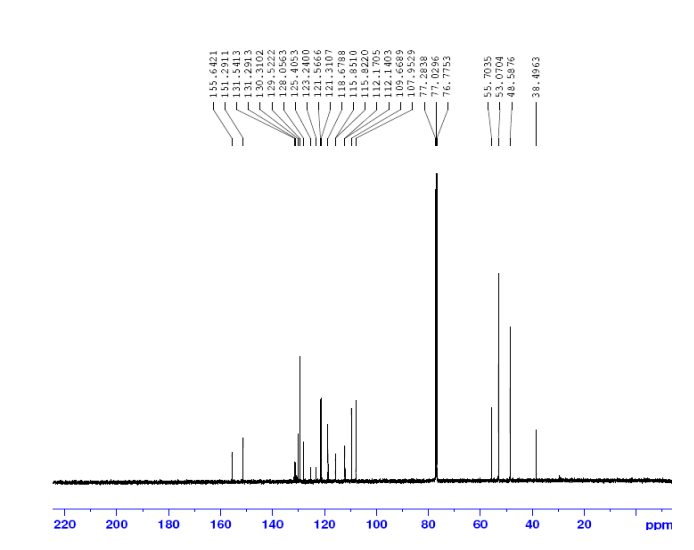
13C NMR
1D and 2DNMR data were used to assign the protons and carbon atoms.
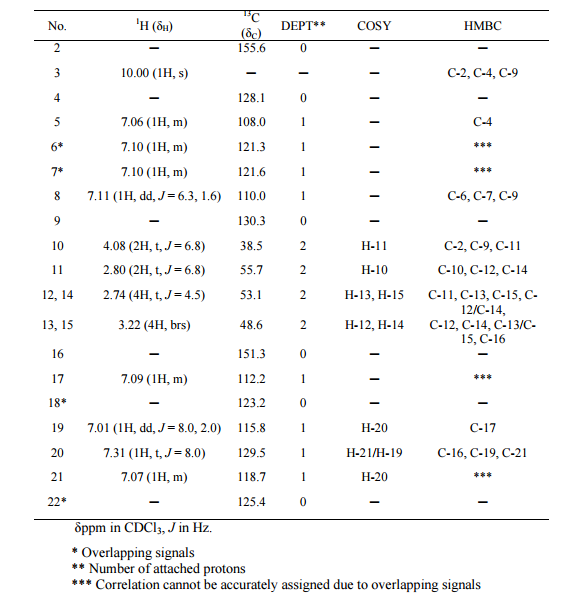
In the1H NMR spectrum , a sharp singlet at 10.00 ppm integrating for one
proton is a typical proton attached to nitrogen. HMBC correlated this proton to C-2, C-4, and C-9 suggesting that it was H-3.
Complex signals were observedbetween 7.00 to 7.31 ppm, integrating for eight protons. A triplet at 7.31 ppm,integrating for a proton has a coupling constant of 8.0 Hz. HMBC correlated thisproton with C-16, C-19, and C-21 suggesting that it was H-20.
A double-doubletsplitting pattern at chemical shift 7.11 ppm, integrating for a proton, has couplingconstants of 6.3 Hz and 1.6 Hz.
HMBC correlated this proton to C-6, C-7, and C-9 showing that it was H-8. Overlapped signals were observed from 7.04 ppm to7.10 ppm, integrating for five protons. A double-doublet splitting pattern at 7.01ppm with coupling constant 8.0 Hz and 2.0 Hz, integrating for a proton was
observed.
HMBC correlated this proton to C-17 suggesting that it was either H-19or H-21. Four triplet signals were also observed from 2.73 ppm to 4.08 ppm,integrating for a total of twelve protons.
Two of these triplet signals at 2.74 ppmand 3.22 ppm integrated for four protons each, suggesting overlapping signals ofmethylene protons. This was further confirmed by 13C and DEPT NMR.
13C and DEPT NMR data showed the signals of four methylene, eight methineand six quaternary carbon atoms. The DEPT signals at 53.1 ppm and 48.6 ppmhave intensities which were double of those from the rest of the methylene carbonsignals, suggesting two methylene carbon atoms each contributing to the signal at 53.1 ppm and 48.6 ppm.
DEPT

HMQC results further indicated that these two methylene carbon signals at 53.1 ppm and 48.6 ppm were correlated to the protons signal at 2.73 ppm and 4.08 ppm respectively, which corresponded to four protons each. The finding confirmed overlapping methylene carbon signals (at 53.1 ppm and 48.6 ppm) and methylene proton signals (at 2.73 ppm and 4.08 ppm). Hence, the unknown compound has six methylene carbon atoms with a total of twelve methylene protons.
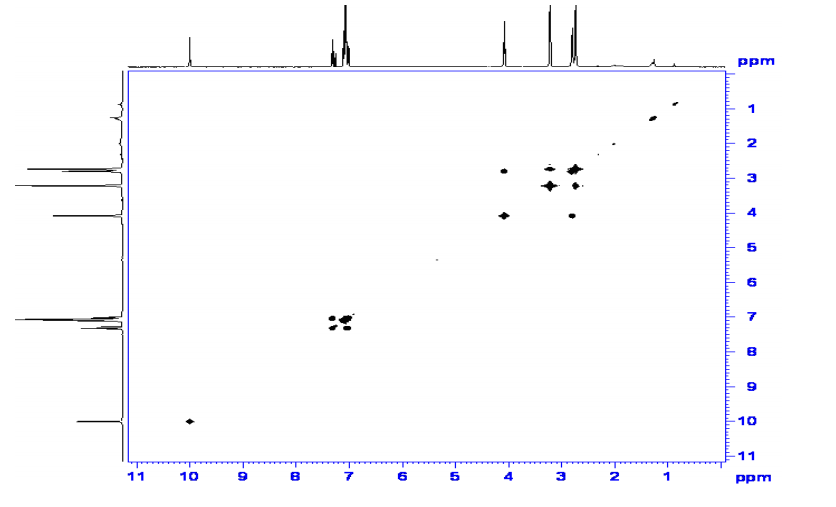
The chemical shifts of the twelve methylene protons suggested that they were attached to relatively electronegative atoms. It was speculated that the six methylene groups were attached to the nitrogen atoms and the electron withdrawing effect of these electronegative nitrogen atoms resulted in the deshielding of the protons. HMBC and COSY correlations were used to assign the rest of the protons
HMBC

HMQC
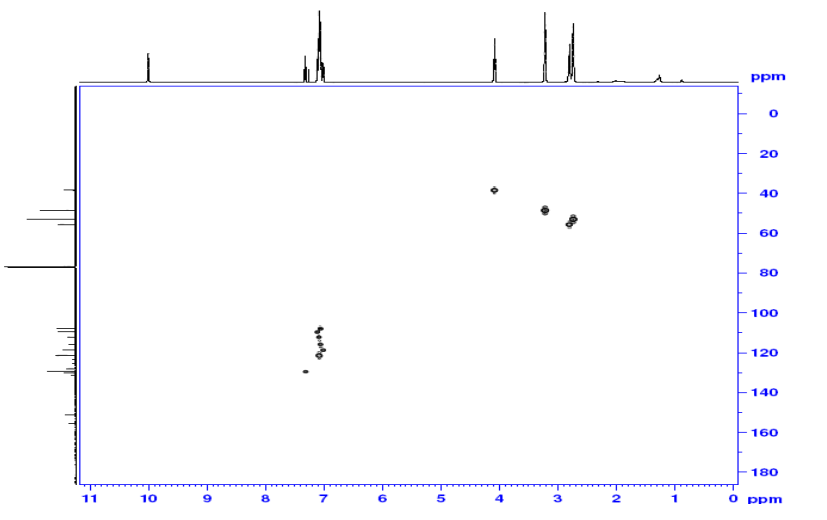
COSY
The 13C NMR data showed that there were two quaternary carbon at
155.6 ppm and 151.3 ppm. The carbon with chemical shift 155.6 ppm was C-2. Inthe structure of imidazolone, carbonyl carbon C-2 was attached to two nitrogenatoms which helped to withdraw electrons from oxygen to C-2. Hence, C-2 wasless deshielded as compared to a normal carbonyl carbon which has chemical shiftabove 170 ppm.
Eight methine carbons and two quaternary carbons with chemicalshifts above 108 ppm suggested the presence of two aromatic rings. Thequaternary carbon with chemical shift 125.4 ppm was C-22 which was attached tothree fluorine atoms. Due to the strong electron withdrawing effect of the fluorineatoms, C-22 was highly deshielded and had a high chemical shift.
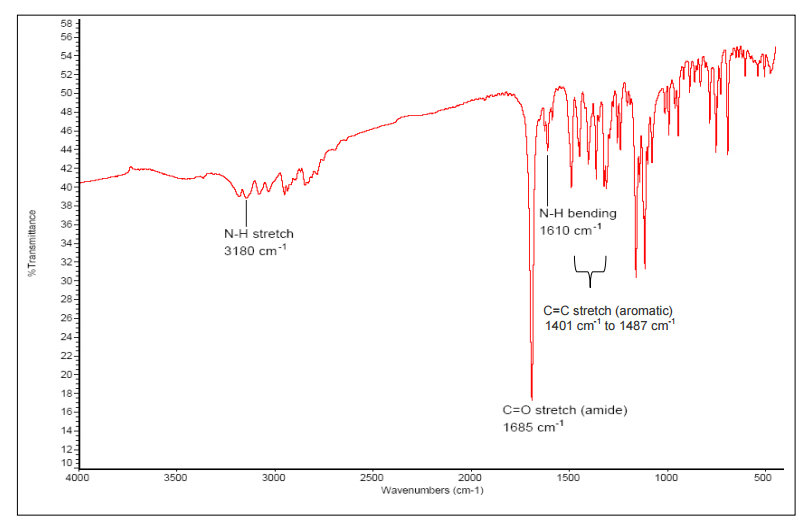
The IR spectrum of the isolated compound showed absorption bands of amide (νC=O 1685 cm-1, νN-H (stretch) 3180 cm-1, νN-H (bending) 1610 cm-1), alkyl fluoride (νC-F1077 cm-1, 1112 cm-1, 1158 cm-1), aromatic ring (ν Ar-H 3028 cm-1, 3078 cm-1 andνC=C 1401 cm-1, 1446 cm-1, 1453 cm-1, 1468 cm-1, 1487 cm-1) and alkane (νC-H2891 cm-1, 2930 cm-1 2948 cm-).
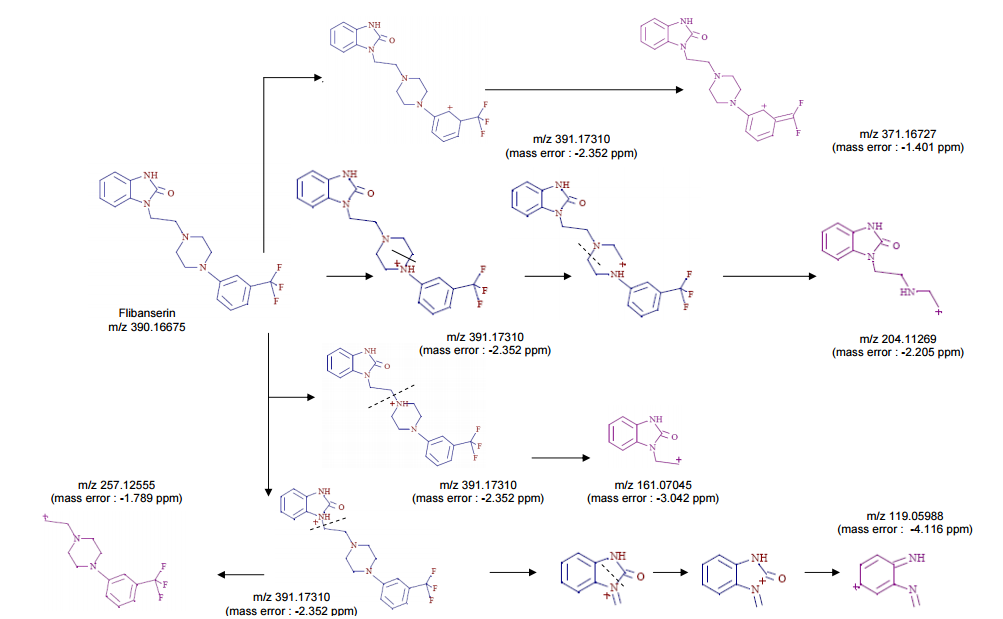
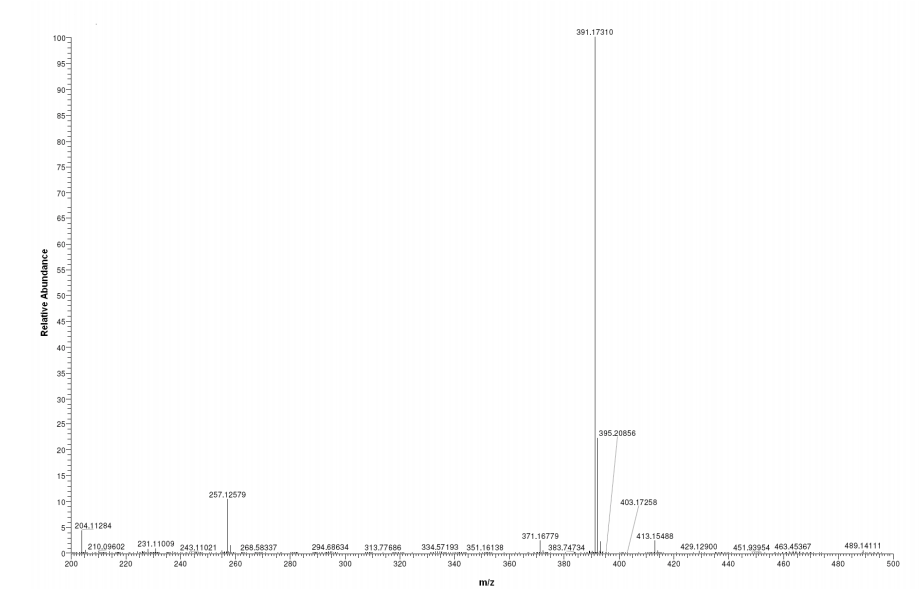
FOR MASS, HMBC ETC SEE………http://orgspectroscopyint.blogspot.in/2015/06/flibanserin.html
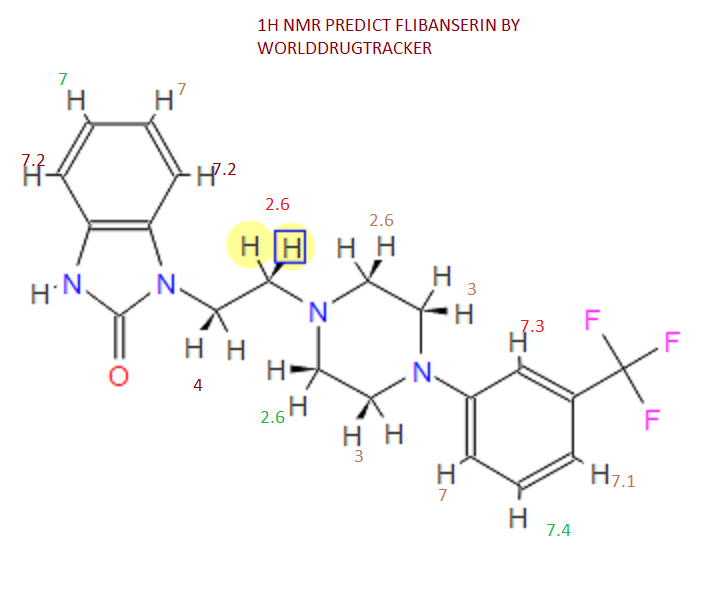
NMR PREDICT
1H NMR PREDICT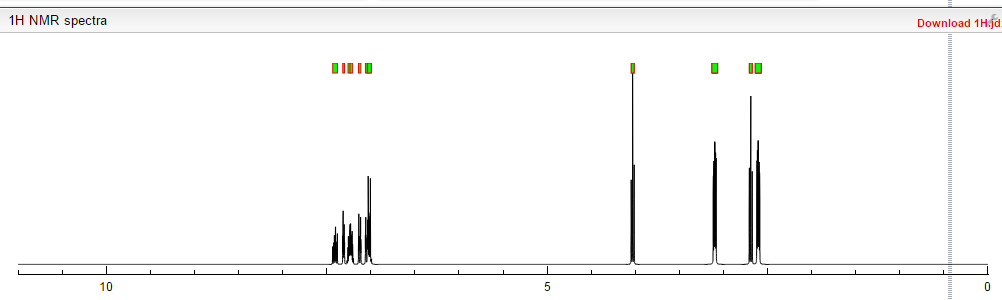
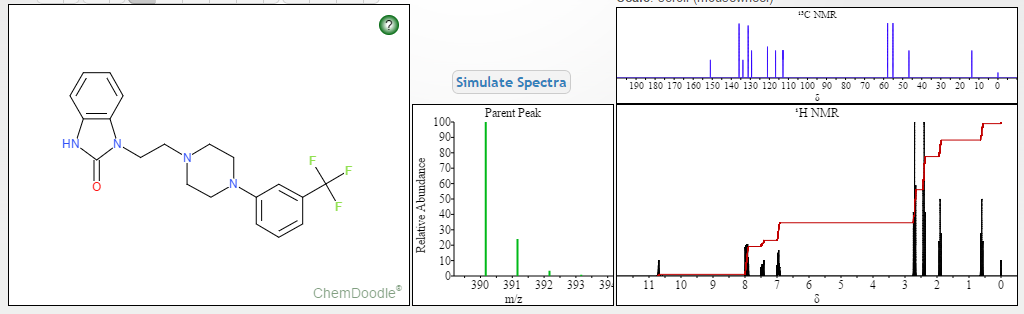
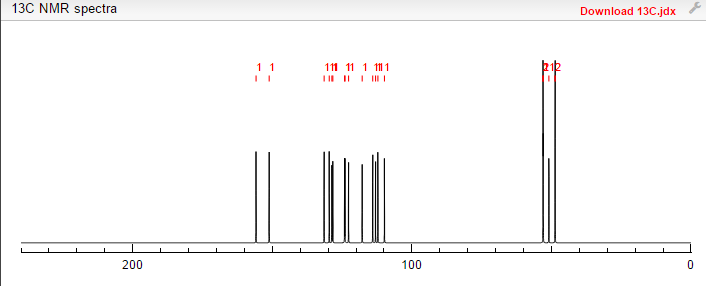
13C NMR PREDICT
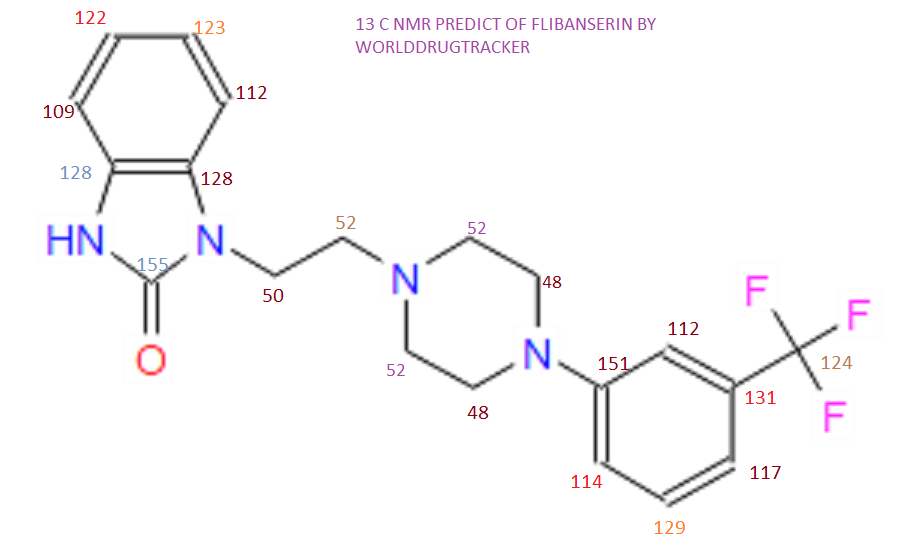
COSY PREDICT
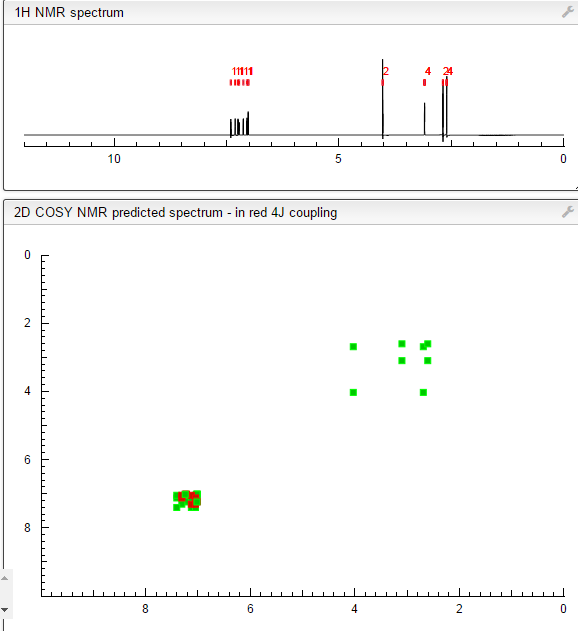
NMR PREDICT FROM MOLBASE
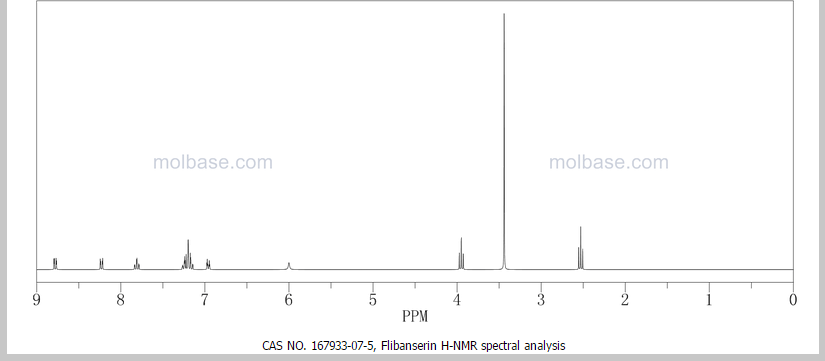
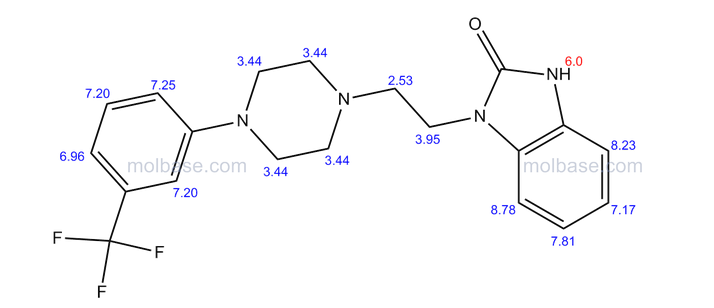

PATENT
US5576318, 1996
1 H NMR (DMSO-d6 /CDCL3 5:2) 11.09 (b, 1H), 11.04 (s, 1H), 7.5-6.9 (SH), 4.36 (t, 2H), 4.1-3.1 (10 H)
UPDATES………..

REFERENCES
| EP0200322A1 * | Mar 18, 1986 | Nov 5, 1986 | H. Lundbeck A/S | Heterocyclic compounds |
| BE904945A1 * | Title not available | |||
| GB2023594A * | Title not available | |||
| US3472854 * | May 29, 1967 | Oct 14, 1969 | Sterling Drug Inc | 1-((benzimidazolyl)-lower-alkyl)-4-substituted-piperazines |
| US4954503 * | Sep 11, 1989 | Sep 4, 1990 | Hoechst-Roussel Pharmaceuticals, Inc. | 3-(1-substituted-4-piperazinyl)-1H-indazoles |
update………..
1-(2-(4-(3-(Trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1H-benzo[d]imidazol-2(3H)-one (1)

A novel and efficient route of synthesis for making flibanserin via 2-ethoxy-1H-benzo[d]imidazole (12) was described with excellent yield. This protocol provided a more facile approach toflibanserin.
http://pubs.acs.org/doi/abs/10.1021/acs.oprd.6b00108

aReagents and conditions: (a) ethyl benzoylacetate, 200 °C; (b) dichloroethane, NaH, DMF; (c) conc HCl (aq); (d) 1-(3-(trifluoromethyl)phenyl)piperazine hydrochloride, Na2CO3, KI, EtOH; (e)

aReagents and conditions: (a) ethyl acetoacetate, KOH, EtOH, xylene, reflux, 56%; (b) 1,2-dibromoethane, K2CO3, DMF, 50 °C, 50%; (c) K2CO3, CH3CN, 70 °C, 80%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), rt, 72% over two steps.

aReagents and conditions: (a) tetraethyl orthocarbonate, AcOH, 70 °C, 94%; (b) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 75%; (c) K2CO3, NaI, H2O, reflux, 92%; (d) conc. HCl (aq), isopropanol, 70 °C; (e) NaOH (aq), 68% over two steps.
//////////////
![]()


Route to Benzimidazol-2-ones via Decarbonylative Ring Contraction of Quinoxalinediones: Application to the Synthesis of Flibanserin, A Drug for Treating Hypoactive Sexual Desire Disorder in Women and Marine Natural Product Hunanamycin Analogue
INTRODUCTION
Benzimidazol-2-ones 1 are an important class of heterocycles and a privileged scaffold in medicinal chemistry. They consist of cyclic urea fused with the aromatic backbone, which can potentially interact in a biological system by various noncovalent interactions such as hydrogen bonding and π stacking. Benzimidazolone derivatives exhibit a wide range of biological activities, and they are useful in treating various diseases including cancer, type II diabetes, central nervous system disorders, pain management, and infectious disease.1 Selected compounds embedded with a benzimidazol-2-one moiety along with their use are captured in Figure 1. It is worth mentioning that oxatomide drug with a benzimidazol-2-one core was approved for marketing a few years ago.2a Very recently, US Food and Drug Administration approved a new drug called flibanserin for the treatment of hypoactive sexual desire disorder (HSDD) in females, which contains benzimidazol-2- one motif.2b
CONCLUSIONS
We have developed a mild and new protocol for the synthesis of benzimidazol-2-ones from quinoxalinediones through decarbonylation. The present methodology can be an addition to the toolbox to prepare benzimidazolones, and it will be useful in medicinal chemistry, particularly, late-stage functionalization of natural products, drug scaffolds, or an intermediate containing quinoxaline-2,3-diones. As direct application of this method, we have successfully developed a new route for the synthesis of recently approved drug flibanserin and a urea analogue of antibiotic natural product hunanamycin A. Later application demonstrates the utility of the present method in late-stage functionalization
Synthesis of 1-(2-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)ethyl)-1,3-dihydro-2Hbenzo[d]imidazol-2-one (Flibanserin)
Flibanserin hydrochloride as white solid.
1H NMR (400MHz ,DMSO-d6) 11.06 (s, 1 H), 10.93 (br. s., 1 H), 7.54 – 7.41 (t, J = 7.9 Hz, 1 H), 7.36 – 7.22 (m, 3 H), 7.15 (d, J = 7.6 Hz, 1 H), 7.09 – 7.01 (m, 3 H), 4.30 (t, J = 6.7 Hz, 2 H), 4.01 (d, J = 11.6 Hz, 2 H), 3.75 (d, J = 10.4 Hz, 2 H), 3.54 – 3.43 (d, J = 4.2 Hz 2 H), 3.31 – 3.10 (m, 4 H);
HRMS (ESI): m/z calculated for C20H22ON4F3[M+H]+ 391.1740 found 391.1743;
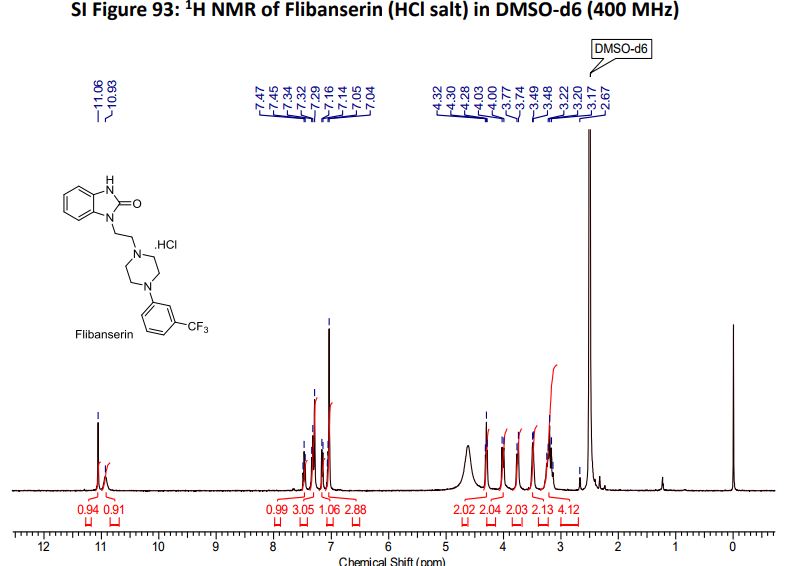
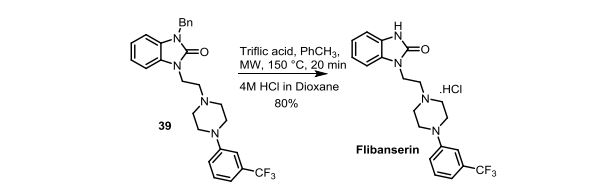

Scheme 4. Synthesis of Flibanserin through Ring Contraction
The same methodology was applied for the synthesis of flibanserin, also known as “female viagra”, which is the first approved medication for treating HSDD in women and is classified as a multifunctional serotonin agonist antagonist.(14, 15) Our synthesis of flibanserin commenced with 1-benzyl-1,4-dihydroquinoxaline-2,3-dione 36,(16) which was reacted with known chloride 37(17) under the basic condition in DMF to give the desired product 38 in good yield. Compound 38 was subjected for the decarbonylative cyclization under the optimized condition to afford the product 39 in 59% yield. Finally, the benzyl group was deprotected using trifluoromethanesulfonic acid in toluene under microwave irradiation,(8b, 18) which gave flibanserin in excellent yield (Scheme 4). The final product was isolated as HCl salt, and all of the spectral data are in agreement with the published data.(15c)

Rahul D. Shingare completed his M.Sc (Chemistry) from Fergusson College, Pune in 2008. He worked as a research associate in Ranbaxy and Lupin New drug discovery center, Gurgaon and Pune respectively until 2012 and currently pursuing his doctoral research in NCL – Pune from 2012.
Current Research Interests: Antibacterial Natural Product Hunanamycin A: Total Synthesis, SAR and Related Chemistry.
e-mail: rd.shingare@ncl.res.in

Akshay Kulkarni completed his M.Sc. from Ferguson College, Pune University in the year 2015 and joined our group as a Project Assistant in the month of October, 2015.
Current research interest: Synthesis of silicon incorporated biologically active antimalerial compounds.
e-mail : as.kulkarni@ncl.res.in
Dr.D. Srinivasa Reddy
Organic Chemistry Division
CSIR-National Chemical Laboratory
See, previous synthesis of Flibanserin:
(a) Bietti, G.; Borsini, F.; Turconi, M.; Giraldo, E.; Bignotti, M. For treatment of central nervous system disorders. U.S. Patent 5,576,318, 1996.
(b) Mohan, R. D.; Reddy, P. K.;Reddy, B. V. Process for the preparation of Flibanserin involving novel intermediates. WO2010128516 A2,2010.
(c) Yang, F.; Wu, C.; Li, Z.; Tian, G.; Wu, J.; Zhu, F.; Zhang, J.; He, Y.; Shen, J. A Facile route of synthesis for making Flibanserin Org. Process Res. Dev. 2016, 20, 1576 DOI: 10.1021/acs.oprd.6b00108
 ], [CAS]
], [CAS]Xueong, X. Preparation method of Flibanserin. CN104926734 A, 2015.
//////////


