PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Copper histidinate, sold under the brand name Zycubo, is a medication used for the treatment of Menkes disease.[1] Copper histidinate is a copper replacement therapy given by subcutaneous injection.[1][2]
The most common side effects include infections, respiratory problems, seizures, vomiting, fever, anemia and injection site reactions.[2]
Copper histidinate was approved for medical use in the United States in January 2026.[2]
Menkes disease is a neurodegenerative disorder caused by a genetic defect that impairs a child’s ability to absorb copper.[2] The disease is characterized by seizures, failure to gain weight and grow, developmental delays, and intellectual disability.[2] It leads to abnormalities of the vascular system, bladder, bowel, bones, muscles, and nervous system.[2]
SYN
A275388 — Flores-Pulido AA, Jimenez-Perez VM, Garcia-Chong NR: Sintesis y uso de histidinato de cobre en ninos con enfermedad de Menkes en Mexico. Gac Med Mex. 2019;155(2):191-195. doi: 10.24875/GMM.18004310. [PubMed:31056589]
World Health Organization (2025). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 94”. WHO Drug Information. 39 (3). hdl:10665/383022.
Clinical trial number NCT00811785 for “Molecular Bases of Response to Copper Treatment in Menkes Disease, Related Phenotypes, and Unexplained Copper Deficiency” at ClinicalTrials.gov
Baxdrostat is under investigation in clinical trial NCT06344104 (A Phase III Study to Investigate the Efficacy and Safety of Baxdrostat in Asian Participants With Uncontrolled Hypertension on Two or More Medications Including Participants With Resistant Hypertension).
In analogy to the procedures described for the preparation of intermediate A-2 [E] and for the preparation of intermediate B-1, Suzuki reaction of (+)-(R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (intermediate B-3b) with 1-methyl-6-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-3,4-dihydro-1H-quinolin-2-one (intermediate A-1) gave (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one and after subsequent reaction with propionyl chloride the title compound as colorless solid. MS: 364.2 (M+H +).
Dissolve 4-bromo-6,7-dihydroisoquinolin-8(5H)-one (1.56 g, 6.9 mmol) and (S)-tert-butylsulfenamide (2.51 g, 20.7 mmol) in 20 mL of tetrahydrofuran. Add ethyl titanate (10.08 mL, 48.28 mmol). Heat to 65°C and stir for 48 hours. Cool to room temperature, add ethyl acetate and water, stir for 15 minutes, and remove the resulting solid by filtration. Separate the liquids, dry the organic phase over anhydrous sodium sulfate, filter, and evaporate to dryness under reduced pressure to obtain the crude product (S,Z)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H)-tert-butylsulfenimide), which is used directly in the next step.
Step B
Compound (S,Z)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H)-tert-butylsulfonyl imide) (1.98 g, 6 mmol) was dissolved in 15 mL of tetrahydrofuran and cooled to -45°C. Sodium borohydride (0.34 g, 9.0 mmol) was added, and the mixture was allowed to return to room temperature and stirred for 18 hours. The mixture was quenched with ice water and extracted with dichloromethane. The resulting organic phase was washed with saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain compound (S)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonyl imide (755 mg, 38% yield). LC/MS (ESI): m/z = 331.2 [M+H] + .
Step C
To a mixture of (S)-N-(4-bromo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonimide (0.66 g, 2 mmol), pinacol diboronate (1.05 g, 2.1 mmol), and AcOK (0.578 g, 6 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.144 g, 0.2 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (15 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.45 g, 60% yield). LC/MS (ESI): m/z = 378.3 [M+H] + .
Step D
To a reaction flask, add 6-bromo-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.29 g, 1.2 mmol), (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.42 g, 1.26 mmol), bistriphenylphosphine palladium dichloride (84 mg, 0.12 mmol), cuprous iodide (38 mg, 0.2 mmol), triethylamine (1.01 g, 10.0 mmol), and 15 mL of N,N-dimethylformamide. The atmosphere was purged with nitrogen three times and the reaction was stirred at 90°C overnight. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and water, and extracted with ethyl acetate. The resulting organic phase was washed with water and saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to afford (S)-2-methyl-N-((R)-4-(1-methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)tert-butylsulfonimide (0.37 g, 74% yield) as a yellow solid. LC/MS (ESI): m/z = 411.5 [M+H] + .
Step E
Compound (S)-2-methyl-N-((R)-4-(1-methyl-2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)-5,6,7,8-tetrahydroisoquinolin-8-yl)tert-butylsulfonimide (0.33 g, 0.80 mmol) was dissolved in 1 mL of dichloromethane, and 1 mL of trifluoroacetic acid was added. The mixture was stirred and reacted for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was purified by reverse preparative column chromatography to obtain compound (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.24 g, 97% yield). LC/MS (ESI): m/z = 307.1 [M+H] + .
Step F
To a reaction flask, add (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (100 mg, 0.33 mmol), triethylamine (51 mg, 0.5 mmol), and 4 ml of tetrahydrofuran. After cooling in an ice-water bath, slowly add a solution of propionyl chloride (46.25 mg, 0.5 mmol) in 0.5 ml of tetrahydrofuran dropwise. Stirring is continued for 4 hours after addition. The reaction mixture is quenched with methanol and evaporated to dryness under reduced pressure. The residue is purified by column chromatography to obtain the target compound, Baxdrostat (46 mg, 38% yield). LC/MS(ESI):m/z=363.1[M+H]+.H NMR(400MHz, CDCl3)ppm 1.22(t,3H)1.79(s,3H)2.07(s,1H)2.28(q,2H)2.43-2.68(m,2H)2.71(t,2H)2.82-3.12(m,2H) 3.40(s,3H)5.34(d,1H)5.78(d,1H)7.05(d,1H)7.09(s,1H)7.17(d,1H)8.28(s,1H)8.49(s,1H)
Example 2
Step A
Compound (S)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H))-tert-butylsulfonylimide (1.65 g, 5 mmol) was dissolved in 20 mL of dichloromethane, and 20 mL of trifluoroacetic acid was added. The mixture was stirred and reacted for 1 hour. The reaction solution was concentrated under reduced pressure. The residue was purified by reverse-phase preparative column chromatography to obtain compound (R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (1.07 g, 94% yield). LC/MS (ESI): m/z = 226.0 [M+H] + .
Step B
To a mixture of (R)-4-bromo-5,6,7,8-tetrahydroisoquinolin-8-amine (0.86 g, 3.8 mmol), pinacol diboron (2 g, 4 mmol), AcOK (1.10 g, 11.4 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.27 g, 0.38 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (10 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (R)-8-amino-5,6,7,8-tetrahydroisoquinoline-4-boronic acid pinacol ester (0.68 g, 65% yield). LC/MS (ESI): m/z = 274.1 [M+H] + .
Step C
To a reaction flask, add 6-bromo-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.72 g, 3.0 mmol), (R)-8-amino-5,6,7,8-tetrahydroisoquinolin-4-boronic acid pinacol ester (0.99 g, 3.6 mmol), bistriphenylphosphine palladium dichloride (210 mg, 0.3 mmol), and potassium phosphate monohydrate (204 mg, 0.9 mmol). Dissolve the mixture in dioxane and water (9:1, 30 mL). Replace the atmosphere with nitrogen three times and allow the mixture to react overnight at 90°C with stirring. Cool to room temperature, dilute the reaction solution with ethyl acetate and water, and extract with ethyl acetate. The resulting organic phase is then washed with water and saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain (R)-6-(8-amino-5,6,7,8-tetrahydroisoquinolin-4-yl)-1-methyl-3,4-dihydroquinolin-2(1H)-one (0.81 g, 88% yield). LC/MS (ESI): m/z = 307.1 [M+H] + . The target compound, Baxdrostat, was then prepared using a method similar to the last step in Example 1.
Example 3
Step A
4-Bromo-6,7-dihydroisoquinolin-8(5H)-one (1.88 g, 6.9 mmol) and (S)-tert-butylsulfenamide (2.51 g, 20.7 mmol) were dissolved in 20 mL of tetrahydrofuran. Ethyl titanate (10.08 mL, 48.28 mmol) was added and the mixture was heated to 65°C with stirring for 48 hours. After cooling to room temperature, ethyl acetate and water were added and stirred for 15 minutes. The resulting solid was removed by filtration. The organic phase was separated and dried over anhydrous sodium sulfate, filtered, and evaporated to dryness under reduced pressure to obtain the crude product (S,Z)-N-(4-bromo-6,7-dihydroisoquinolin-8(5H)-tert-butylsulfenimide), which was used directly in the next step. LC/MS (ESI): m/z = 376.2 [M+H] + .
Step B
Compound (S,Z)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H)-tert-butylsulfonyl imide) (2.26 g, 6 mmol) was dissolved in 15 mL of tetrahydrofuran and cooled to -45°C. Sodium borohydride (0.36 g, 9.0 mmol) was added, and the mixture was allowed to return to room temperature and stirred for 18 hours. The mixture was quenched with ice water and extracted with dichloromethane. The resulting organic phase was washed with saturated brine, dried over anhydrous sodium sulfate, and evaporated to dryness under reduced pressure. The residue was purified by column chromatography to obtain compound (S)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonyl imide (1.04 g, 46% yield). LC/MS (ESI): m/z = 378.0 [M+H] + .
Step C
To a mixture of (S)-N-(4-iodo-6,7-dihydroisoquinoline-8(5H))-tert-butylsulfonimide (0.76 g, 2 mmol), pinacol diboronate (1.05 g, 2.1 mmol), and AcOK (0.578 g, 6 mmol) in toluene (10 mL) was added Pd(dppf)Cl 2 (0.144 g, 0.2 mmol). The mixture was degassed and stirred at 130 ° C for 3 hours. The reaction mixture was filtered and concentrated to give a residue. EtOAc (15 mL) and water (10 mL) were added to the residue. The organic phase was washed with brine (50 mL), dried over anhydrous sodium sulfate, filtered and concentrated to give a residue. The residue was purified by column chromatography (SiO 2 ) and eluted with 30-40% ethyl acetate in petroleum ether to afford (S)-N-tert-butylsulfonamido-6,7-dihydroisoquinolin-8(5H)-4-boronic acid pinacol ester (0.51 g, 68% yield). LC/MS (ESI): m/z = 378.2 [M+H] + .
The next three steps were carried out in the same manner as in Example 1 to prepare the target compound Baxdrostat.
A new treatment has been shown to significantly lower blood pressure in people whose levels stay dangerously high, despite taking several existing medicines, according to the results of a Phase III clinical trial led by a UCL Professor. Globally, around 1.3 billion people have high blood pressure (hypertension), and in around half of cases the condition is uncontrolled or treatment resistant. These individuals face a much greater risk of heart attack, stroke, kidney disease, and early death. In the UK the number of people with hypertension is around 14 million.
The international BaxHTN trial, led by Professor Bryan Williams (UCL Institute of Cardiovascular Science), assessed the new drug baxdrostat—which is taken as a tablet—with participation from nearly 800 patients across 214 clinics worldwide.
The trial results showed that, after 12 weeks, patients taking baxdrostat (1 mg or 2 mg once daily in pill form) saw their blood pressure fall by around 9-10 mmHg more than placebo—a reduction large enough to cut cardiovascular risk. About four in 10 patients reached healthy blood pressure levels, compared with fewer than two in 10 on placebo.
Principal Investigator, Professor Williams, who is presenting the results at ESC, said, “Achieving a nearly 10 mmHg reduction in systolic blood pressure with baxdrostat in the BaxHTN Phase III trial is exciting, as this level of reduction is linked to substantially lower risk of heart attack, stroke, heart failure and kidney disease.”
How baxdrostat works
Blood pressure is strongly influenced by a hormone called aldosterone, which helps the kidneys regulate salt and water balance.
Some people produce too much aldosterone, causing the body to hold onto salt and water. This aldosterone dysregulation pushes blood pressure up and makes it very difficult to control.
Addressing aldosterone dysregulation has been a key effort in research over many decades, but it has been so far difficult to achieve.
Baxdrostat works by blocking aldosterone production, directly addressing this driver of high blood pressure (hypertension).
Professor Williams, Chair of Medicine at UCL, said, “These findings are an important advance in treatment and in our understanding of the cause of difficult-to-control blood pressure.
“Around half of people treated for hypertension do not have it controlled, however this is a conservative estimate and the number is likely higher, especially as the target blood pressure we try to reach is now much lower than it was previously.
“In patients with uncontrolled or resistant hypertension, the addition of baxdrostat 1mg or 2mg once daily to background antihypertensive therapy led to clinically meaningful reductions in systolic blood pressure, which persisted for up to 32 weeks with no unanticipated safety findings.
“This suggests that aldosterone is playing an important role in causing difficult to control blood pressure in millions of patients and offers hope for more effective treatment in the future.”
Historically, higher-income Western countries were reported to have far higher levels of hypertension. However, largely due to changing diets (adding less salt to food), the numbers of people living with the condition is now far higher in Eastern and lower-income countries. More than half of those affected live in Asia, including 226 million people in China and 199 million in India.
Professor Williams added, “The results suggest that this drug could potentially help up to half a billion people globally—and as many as 10 million people in the UK alone, especially at the new target level for optimal blood pressure control.”
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Dogra S, Shah S, Gitzel L, Pusukur B, Sood A, Vyas AV, Gupta R (July 2023). “Baxdrostat: A Novel Aldosterone Synthase Inhibitor for Treatment Resistant Hypertension”. Current Problems in Cardiology. 48 (11): 101918. doi:10.1016/j.cpcardiol.2023.101918. PMID37399857. S2CID259320969.
Awosika A, Cho Y, Bose U, Omole AE, Adabanya U (October 2023). “Evaluating phase II results of Baxdrostat, an aldosterone synthase inhibitor for hypertension”. Expert Opinion on Investigational Drugs. 32 (11): 985–995. doi:10.1080/13543784.2023.2276755. PMID37883217. S2CID264517675.
Orforglipron has a half-life of 29 to 49 hours across the doses tested and is taken once per day by mouth without food or water restrictions.[3]
Safety and dosing trials showed that the incidence of adverse events in orforglipron-treated participants was 62–89%, mostly from gastrointestinal discomfort (44–70% with orforglipron, 18% with placebo) having mild to moderate severity.[6] The most common side effects of orforglipon are diarrhea, nausea, upset stomach, and constipation.[1][6]
The ability of orforglipron to reduce blood sugar levels and body weight was judged favorable compared to dulaglutide.[6]
Phase III ACHIEVE-1 trial
In April 2025, results from a Phase III clinical trial involving 559 people with type 2 diabetes who took an oral orforglipron pill, injectabledulaglutide or a placebo daily for 40 weeks showed that orforglipron produced a reduction in blood glucose levels by 1.3 to 1.6 percentage points from a starting level of 8%.[1][7]
More than 65% of participants taking the highest dose of orforglipron achieved a reduction of hemoglobin A1C level by more than or equal to 1.5 percentage points, bringing them into the non-diabetic range as defined by the American Diabetes Association.[1] People taking the highest dose of the pill lost 8% of their weight, or around 16 lb (7.3 kg), on average after 40 weeks.[1][8]
Side effects were similar to those seen with other GLP-1 agonists, and no significant liver problems were observed.[1]
^ Wharton S, Blevins T, Connery L, et al. (June 2023). “Daily Oral GLP-1 Receptor Agonist Orforglipron for Adults with Obesity”. The New England Journal of Medicine. 389 (10): 877–888. doi:10.1056/NEJMoa2302392. PMID37351564.
To treat platinum-resistant epithelial ovarian, fallopian tube, or primary peritoneal cancer after one to three prior systemic treatment regimens, at least one of which included bevacizumab
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Cushing syndrome
Phase IIICushing syndrome; Ovarian cancer; Pancreatic cancer
Phase IIFallopian tube cancer; Peritoneal cancer; Prostate cancer
Phase I/IISolid tumours
Phase IAdrenocortical carcinoma
Most Recent Events
09 Sep 2022Subgroup analysis efficacy data from a phase-II trial in Ovarian cancer presented at the 47th European Society for Medical Oncology Congress (ESMO-2022)
29 Jun 2022Phase-III clinical trials in Ovarian cancer (Combination therapy, Recurrent, Second-line therapy or greater) in USA (PO)
06 Jun 2022Corcept Therapeutics announces intentions to submit a NDA for Ovarian cancer
The drug was approved by the USFDA in 2026 for the treatment of platinum-resistant ovarian cancer.[3]
Relacorilant is an orally available antagonist of the glucocorticoid receptor (GR), with potential antineoplastic activity. Upon administration, relacorilant competitively binds to and blocks GRs. This inhibits the activity of GRs, and prevents both the translocation of the ligand-GR complexes to the nucleus and gene expression of GR-associated genes. This decreases the negative effects that result from excess levels of endogenous glucocorticoids, like those seen when tumors overproduce glucocorticoids. In addition, by binding to GRs and preventing their activity, inhibition with CORT125134 also inhibits the proliferation of GR-overexpressing cancer cells. GRs are overexpressed in certain tumor cell types and promote tumor cell proliferation.
OriginatorCorcept Therapeutics
DeveloperCorcept Therapeutics; University of Chicago
Mechanism of ActionGlucocorticoid receptor antagonists
Orphan Drug StatusYes – Pancreatic cancer; Ovarian cancer; Cushing syndrome
RegisteredFallopian tube cancer; Ovarian cancer; Peritoneal cancer
PreregistrationCushing syndrome
Phase IIIAdenocarcinoma
Phase IIProstate cancer
DiscontinuedAdrenocortical carcinoma
27 Mar 2026Discontinued – Phase-I for Adrenocortical carcinoma (Inoperable/Unresectable, Late-stage disease, Metastatic disease, Combination therapy) in USA (PO), before March 2026 (Corcept Therapeutics pipeline, March 2026)
27 Mar 2026Corcept Therapeutics plans the phase II STELLA trial for Cervical cancer (Combination therapy, Second-line therapy or greater) in first quarter of 2026
25 Mar 2026Registered for Fallopian tube cancer (Combination therapy, Second-line therapy or greater) in USA (PO) – First global approval
Relacorilant (CORT125134)118) is being developed by Corcept Therapeutics, Inc. It is an orally active, high-affinity, selective antagonist of the glucocorticoid receptor that may benefit from the modulation of cortisol activity. In structural optimization, the introduction of a trifluoromethyl group to the 4-position on the pyridyl moiety was found to increase HepG2 tyrosine amino transferase assay potency by a factor of four. Relacorilant is currently being evaluated in a phase II clinical study in patients with Cushing’s syndrome.119)
2-Bromo-4-(trifluoromethyl)pyridine (17) prepared from (E)-4-ethoxy-1,1,1-trifluorobut-3-en-2-one is employed as a key intermediate for the preparation of relacorilant as shown in Scheme 31.120)
118) H. Hunt, T. Johnson, N. Ray and I. Walters (Corcept Therapeutics, Inc.): PCT Int. Appl. WO2013/177559 (2013).
119) H. J. Hunt, J. K. Belanoff, I. Walters, B. Gourdet, J. Thomas, N. Barton, J. Unitt, T. Phillips, D. Swift and E. Eaton: Identification of the Clinical Candidate (R)-(1-(4-Fluorophenyl)-6-((1-methyl-1H-pyrazol-4-yl)sulfonyl)-4,4a,5,6,7,8-hexahydro-1H-pyrazolo[3,4-g]isoquinolin-4a-yl)(4-(trifluoromethyl)pyridin-2-yl)methanone (CORT125134): A Selective Glucocorticoid Receptor (GR) Antagonist. J. Med. Chem. 60, 3405–3421 (2017). [Abstract] [Google Scholar]
120) B. Lehnemann, J. Jung and A. Meudt (Archimica GmbH): PCT Int. Appl. WO 2007/000249 (2007).
The nonselective glucocorticoid receptor (GR) antagonist mifepristone has been approved in the U.S. for the treatment of selected patients with Cushing’s syndrome. While this drug is highly effective, lack of selectivity for GR leads to unwanted side effects in some patients. Optimization of the previously described fused azadecalin series of selective GR antagonists led to the identification of CORT125134, which is currently being evaluated in a phase 2 clinical study in patients with Cushing’s syndrome.
Cushing’s syndrome (CS) is a metabolic disorder caused by chronic hypercortisolism. CS is associated with cardiovascular, metabolic, skeletal and psychological dysfunctions and can be fatal if left untreated. The first-line treatment for all forms of CS is a surgery. However, medical therapy has to be chosen if surgical resection is not an option or is deemed ineffective. Currently available therapeutics are either not selective and have side effects or are only available as an injection (pasireotide).
Mechanism of ActionType 4 cyclic nucleotide phosphodiesterase inhibitors
RegisteredAtopic dermatitis
27 Sep 2021Registered for Atopic dermatitis (In adolescents, In children, In adults) in Japan (Topical)
11 Nov 2020Otsuka Pharmaceutical completes a phase III trial in Atopic dermatitis (In children, In adolescents, In adults) in Japan (Topical) (NCT03961529)
28 Sep 2020Preregistration for Atopic dermatitis in Japan (In children, In adolescents, In adults) (Topical)
Difamilast is under investigation in clinical trial NCT01702181 (A Safety Study to Evaluate the Use and Effectiveness of a Topical Ointment to Treat Adults With Atopic Dermatitis).
Synthesis of Oxazole Compound (Type A Crystal)
Compound (5) (white powder) was prepared in accordance with the method disclosed in Example 352 of PTL 1 (WO2007/058338).
Synthesis of difamilast commenced with the monobenzylated protocatechuic acid ethyl ester 15.1. Phenol 15.1 was first converted into the corresponding isopropyl ether, which was subsequently debenzylated under palladium-catalyzed hydrogenation conditions to generate the phenolic intermediate 15.3. Difluoromethylation of 15.3 was accomplished by introducing sodium chlorodifluoroacetate 15.4 in the presence of potassium carbonate at an elevated temperature. The decarboxylative C− O bond-forming reaction presumably proceeded via a difluorocarbene species. The difluoromethylated product was treated with acid followed by ester hydrolysis under a basic medium to furnish benzoic acid derivative 15.5. Benzoic acid 15.5 was subsequently transformed into benzamide 15.6 via a benzoyl imidazole intermediate. Condensation of benzamide 15.6 with 1-acetoxy-3-chloroacetone 15.7 produced an oxazole derivative, which was subsequently saponified and recrystallized from 50% aqueous MeOH to generate alcohol 15.8.
First, an activation−displacement process transformed alcohol 15.8 into bromide 15.9 via a mesylate intermediate. Alkyl bromide 15.9 was then treated with potassium phthalimide to incorporate the nitrogen center via an SN2-type displacement. Methylamine-mediated phthalimide deprotection and subsequent salt formation produced amine 15.11 as a hydrochloride salt in 69% yield over 3 steps. Finally, hydrochloride salt 15.11 was treated with aqueous sodium bicarbonate to generate a free amine, which was subjected to amide bond formation with 2-ethoxybenzoic acid 15.12 to deliver difamilast after recrystallization from aqueous EtOH.
Patent Documents 1 and 2 report an oxazole compound having a specific inhibitory action on phosphodiesterase 4 (PDE4) and a method for producing the same. PDE4 is the predominant PDE in inflammatory cells, inhibition of PDE4 increases intracellular cAMP concentration, and the increase in this concentration downregulates the inflammatory response through regulation of the expression of TNF-α, IL-23, and other inflammatory cytokines. .. Elevated cAMP levels also increase anti-inflammatory cytokines such as IL-10. Therefore, it is considered that the oxazole compound is suitable for use as an anti-inflammatory agent. For example, it may be useful for controlling skin eczema and dermatitis, including atopic dermatitis. Patent Document 3 describes an ointment that stably contains an oxazole compound having a specific inhibitory effect on PDE4 and can be efficiently absorbed into the skin. The contents of Patent Documents 1 to 3 are incorporated in the present specification by reference.
[Synthesis of Oxazole Compound (Type A Crystal)]
Compound (5) (white powder) was prepared by the method described in Example 352 of Patent Document 1 (International Publication No. 2007/088383).
[Preparation of B-type crystal 2]
Using the obtained B-type crystal as a seed crystal, it was examined to further prepare a B-type crystal. Specifically,
B-type crystals were prepared as follows according to the method described in Patent Document 3 (International Publication No. 2017/115780).
[0072]
[Chem. 6]
[0073]
Compound (1) 20.00 g (66.8 mmol) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate to cool the mixture, and 11.48 g (100 mmol) of methanesulfonyl chloride was introduced into the compound (1) at 10 to 30 ° C. Stir for hours. Subsequently, 17.41 g (200 mmol) of lithium bromide was added, and the mixture was stirred at 20 to 35 ° C. for 1 hour. 100 mL of water was added to the reaction solution to separate the layers, and the organic layer was concentrated under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve it, and the mixture was concentrated again under reduced pressure. 200 mL of N, N-dimethylformamide and 17.33 g (93.6 mmol) of phthalimide potassium were added to the concentrated residue, and the mixture was reacted at 75 to 85 ° C. for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals, and the precipitated crystals were collected by filtration and dried at 80 ° C. to obtain 27.20 g (yield 95.01%) of compound (3).
[0074]
[Chem. 7]
[0075]
Compound (3) 20.00 g (46.7 mmol), 40 mL of a 40% aqueous methylamine solution, 40 mL of methanol, and 100 mL of water were mixed and reacted under reflux for 30 minutes. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75 ° C. to separate the liquids. A mixed solution of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted again to 65 to 75 ° C. to separate the liquids. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. Precipitated crystals were collected by filtration to obtain 27.58 g of wet crystals of compound (4).
[0076]
Wet crystals (46.7 mmol) of compound (4) were mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and the mixture was stirred at 20 to 30 ° C. for 1 hour. To the reaction solution, 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (WSC) were added, and 20 to 30 were added. The reaction was carried out at ° C. for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50 ° C. to separate the solutions. 60 mL of water and 6 mL of a 25%
aqueous sodium hydroxide solution were added to the organic layer, the temperature was adjusted again to 40 to 50 ° C., the liquid was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% aqueous sodium hydroxide solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. Activated carbon was removed by filtration, washed with 12 mL of ethanol, the filtrate was cooled, and 10 mg of B-type crystals (seed crystals) were added to precipitate crystals. Precipitated crystals were collected by filtration and dried at 60 ° C. to obtain 18.38 g (yield 88.18%) of crystals of compound (5).
Using the compound obtained in Example 347 and 2-bromopropane, white powdery N-[2-(4-difluoromethoxy-3-isopropoxyphenyl)oxazol-4-ylmethyl]-2-ethoxybenzamide was obtained following the procedure of Example 348.
Production Example 1: Production 1 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0146]
[Chem. 11]
[0147]
10.00 g (55.5 mmol) of compound (1a) and 9.20 g (66.6 mmol) of potassium carbonate were added to 40 ml of N,N-dimethylformamide and 6 ml of water, and the mixture was stirred until exotherm subsided. 16.92 g (111 mmol) of sodium chlorodifluoroacetate was added thereto, and the mixture was reacted at 95 to 110°C for 3 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the solution was partitioned. 80 ml of water was added again to the organic layer, followed by partitioning. 3 ml of concentrated hydrochloric acid was added to the organic layer, and the mixture was stirred at 60 to 70°C for 30 minutes. 40 ml of water and 10 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the mixture was partitioned. 5.93 g (61.1 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 22.08 g (61.0 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at a temperature of 20°C or below. The mixture was reacted at 20°C or below for 15 minutes, and 10 ml of a 25% sodium hydroxide aqueous solution was added dropwise thereto at a temperature of 20°C or below, followed by pouring in 83.95 g (66.6 mmol) of a 10% sodium sulfite aqueous solution. Additionally, 2 ml of concentrated hydrochloric acid was added and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue to dissolve the residue, and 5 ml of concentrated hydrochloric acid was added dropwise thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 11.81 g (yield: 86.4%) of compound (3) as a white powder.
Production Example 2: Production 2 of Compound (3)
Compound (3) was produced in accordance with the following reaction scheme.
[0149]
[Chem. 12]
[0150]
10.00 g (53.2 mmol) of compound (1b), 9.55 g (69.1 mmol) of potassium carbonate, and 8.50 g (69.1 mmol) of isopropyl bromide were added to 40 ml of N,N-dimethylformamide, and the mixture was reacted at 75 to 85°C for 2 hours. 80 ml of butyl acetate and 80 ml of water were added to the reaction solution, and the mixture was partitioned. 5.68 g (58.5 mmol) of sulfamic acid and 10 ml of water were added to the organic layer, and 21.15 g (58.5 mmol) of a 25% sodium chlorite aqueous solution was added dropwise thereto at 20°C or below, followed by reaction for 15 minutes. 10 ml of a 25% sodium hydroxide aqueous solution was added thereto at 20°C or below, and subsequently 80.41 g (63.8 mmol) of a 10% sodium sulfite aqueous solution was poured in. Additionally, 2 ml of concentrated hydrochloric acid was added, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 40 ml of methanol, 80 ml of water, and 10 ml of a 25% sodium hydroxide aqueous solution were added to the concentrated residue, and the residue was dissolved, followed by dropwise addition of 5 ml of concentrated hydrochloric acid to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 12.09 g (yield: 92.4%) of compound (3) as a white powder.
[0151]
Production Example 3: Production of Compound (7)
Compound (7) was produced in accordance with the following reaction scheme.
[0152]
[Chem. 13]
Production Example 4: Production of Compound (11)
Compound (11) was produced in accordance with the following reaction scheme.
[0160]
[Chem. 14]
[0161]
Synthesis of Compound (9)
20.00 g (66.8 mmol) of compound (7) and 17.28 g (134 mmol) of N,N-diisopropylethylamine were added to 300 ml of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto and reacted at 20 to 35°C for 1 hour. 100 ml of water was added to the reaction solution, and the mixture was partitioned, followed by concentration of the organic layer under reduced pressure. 300 ml of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 ml of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue and reacted at 75 to 85°C for 1 hour. 200 ml of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 25.90 g (yield: 90.5%) of compound (9) as a white powder.
Synthesis of Compound (10)
15.00 g (35.0 mmol) of compound (9) was mixed with 30 ml of a 40% methylamine aqueous solution, 30 ml of methanol, and 75 ml of water, and reacted under reflux for 30 minutes. 150 ml of cyclopentyl methyl ether (CPME) and 15 ml of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by partitioning. A mixture of 150 ml of water and 7.50 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by partitioning. 3.75 ml of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 11.95 g (yield: quant.) of compound (10) as a white powder.
Synthesis of Compound (11)
13.30 g (39.7 mmol) of compound (10) was mixed with 3.83 g (37.8 mmol) of triethylamine and 108 ml of ethyl acetate, and stirred at 20 to 30°C for 1 hour. 9.78 g (58.9 mmol) of 2-ethoxybenzoic acid and 11.28 g (58.8 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 54 ml of water and 5.4 ml of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by partitioning. 54 ml of water and 5.4 ml of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was partitioned, and the organic layer was concentrated under reduced pressure. 45 ml of ethanol, 18 ml of water, 5.4 ml of a 25% sodium hydroxide aqueous solution, and 0.54 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 11 ml of ethanol. The filtrate was cooled, and a seed crystal was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 35°C, thereby obtaining 12.88 g (72.6%) of compound (11) as a white powder.
Type B Crystal Preparation 2
Analysis was conducted to further prepare the type B crystal using the obtained type B crystal as a seed crystal. More specifically, the type B crystal was prepared as follows, in accordance with the method disclosed in PTL 3 (WO2017/115780).
[0072]
[0073]
20.00 g (66.8 mmol) of compound (1) and 17.28 g (134 mmol) of diisopropylethylamine were added to 300 mL of ethyl acetate, and the mixture was cooled. 11.48 g (100 mmol) of methanesulfonyl chloride was poured in and stirred at 10 to 30°C for 1 hour. 17.41 g (200 mmol) of lithium bromide was added thereto, and the mixture was stirred at 20 to 35°C for 1 hour. 100 mL of water was added to the reaction solution, and the mixture was separated, followed by concentration of the organic layer under reduced pressure. 300 mL of ethyl acetate was added to the concentrated residue to dissolve the residue, and the solution was again concentrated under reduced pressure. 200 mL of N,N-dimethylformamide and 17.33 g (93.6 mmol) of potassium phthalimide were added to the concentrated residue, and reacted at 75 to 85°C for 1 hour. 200 mL of water was added to the reaction solution to precipitate crystals. The precipitated crystals were collected by filtration and dried at 80°C, thereby obtaining 27.20 g (yield: 95.01%) of compound (3).
[0074]
[0075]
20.00 g (46.7 mmol) of compound (3), 40 mL of a 40% methylamine aqueous solution, 40 mL of methanol, and 100 mL of water were mixed and reacted for 30 minutes under reflux. 200 mL of cyclopentyl methyl ether (CPME) and 20 mL of a 25% sodium hydroxide aqueous solution were added to the reaction solution, and the temperature was adjusted to 65 to 75°C, followed by separation. A mixture of 100 mL of water and 20.00 g of sodium chloride was added to the organic layer, and the temperature was adjusted to 65 to 75°C again, followed by separation. 5 mL of concentrated hydrochloric acid was added to the organic layer to precipitate crystals. The precipitated crystals were collected by filtration, thereby obtaining 27.58 g of compound (4) as a wet crystal.
[0076]
The wet crystal (46.7 mmol) of compound (4) was mixed with 120 mL of ethyl acetate and 7.1 mL (51.4 mmol) of triethylamine, and stirred at 20 to 30°C for 1 hour. 10.09 g (60.7 mmol) of 2-ethoxybenzoic acid and 11.63 g (60.7 mmol) of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) were added to the reaction solution, and reacted at 20 to 30°C for 1 hour. 60 mL of water and 6 mL of concentrated hydrochloric acid were added to the reaction solution, and the temperature was adjusted to 40 to 50°C, followed by separation. 60 mL of water and 6 mL of a 25% sodium hydroxide aqueous solution were added to the organic layer, and the temperature was adjusted to 40 to 50°C again. The mixture was separated, and the organic layer was concentrated under reduced pressure. 50 mL of ethanol, 20 mL of water, 6 mL of a 25% sodium hydroxide aqueous solution, and 0.6 g of activated carbon were added to the concentrated residue, and the mixture was refluxed for 30 minutes. The activated carbon was removed by filtration, and the filtrate was washed with 12 mL of ethanol. The filtrate was cooled, and 10 mg of the type B crystal (a seed crystal) was added thereto to precipitate crystals. The precipitated crystals were collected by filtration and dried at 60°C, thereby obtaining 18.38 g (88.18%) of compound (5).
PATENT
WO2014034958A1
WO2007058338A2
WO2007058338A9
WO2007058338A3
US9181205B2
US2015239855A1
USRE46792E
US2020078340A1
US2017216260A1
US2019070151A1
US2009221586A1
US8637559B2
US2014100226A1
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID 19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26 Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo. Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.; Mukae, H. A phase 2/3 study of S-217622 in participants with SARS CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024. (26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in Japanese patients with mild-to-moderate COVID-19 or asymptomatic SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents Chemother. 2022, 66, No. 00697. (27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.; Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T. Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132− 136. (28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J. Structural basis for the inhibition of coronaviral main proteases by ensitrelvir. Structure 2023, 31, 1016. (30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.; Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al. Development of a manufacturing process toward the convergent synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023, 9, 836−843. (31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano, S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J., Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.
Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry
The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012
Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.
Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.
Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]
FDA APPROVED 2026, 4/20/2026, doravirine and islatravir, Idvynso
To treat HIV-1 infection (as a complete regimen) in adults to replace the current antiretroviral regimen in those who are virologically-suppressed on a stable antiretroviral regimen with no history of virologic treatment failure and no known substitutions associated with resistance to doravirine
Islatravir is known to be a nucleoside reverse transcriptase inhibitor, useful for treating HIV-1 and -2 infection and AIDS.
Islatravir (MK-8591, EFdA), useful for the treatment of eg HIV, AIDS and related diseases.
Merck & Co and Idenix , under license from Yamasa Shoyu , are developing islatravir, a nucleoside reverse transcriptase inhibitor, for the oral prevention and treatment of HIV-1 and HIV-2 infection; in July 2019, data from a phase IIb trial in patients with HIV-1 infection were presented.In August 2015, Merck licensed Codexis ‘ CodeEvolver® protein engineering platform technology to develop enzymes for use in the manufacture of the pharmaceutical products such as islatravir.
Islatravir (4′-ethynyl-2-fluoro-2′-deoxyadenosine, EFdA, or MK-8591) is an investigational drug for the treatment of HIV infection.[1]It is classified as a nucleoside reverse transcriptase translocation inhibitor (NRTTI).[2]Merck is developing a subdermal drug-eluting implant to administer islatravir.[3][4]
Biological activity
Islatravir has activity against HIV in animal models,[5] and is being studied clinically for HIV treatment and prophylaxis.[6] Islatravir is a nucleoside analog reverse transcriptase translocation inhibitor that unlike other such inhibitors, inhibits HIV through multiple mechanisms,[5] providing rapid suppression of the virus, when tested in macaques and mice.[7] Nevertheless, there are HIV strains resistant to islatravir and research is ongoing.[8]
PATENTS
WO2020014046 ,
PATENT
WO2020014047
PATENT
WO2020014050 (assigned to Codexis ), covering engineered phosphopentomutase (PPM) enzymes, useful in the synthesis of pharmaceutical compounds including islatravir.
4’-Ethynyl-2’-deoxy nucleoside analogs are known for activity against HIV, AIDS and related diseases.
One example of a 4’-ethynyl nucleoside analog is 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA, also known as MK-8591) which is a nucleoside reverse transcriptase translocation inhibitor that blocks HIV-l and SIV viral replication in vitro (Kawamoto, A., Kodama, E., Sarafianos S. F. et al, Int. J. Biochem. Cell Biol.; 40(l l):24lO-2O [2008]; Ohrui, H., Kohgo, S., Hayakawa, H. et al, Nucleosides, Nucleotides & Nucleic Acids, 26, 1543-1546
[2007]) and in vivo (Hattori, S., Ide, K., Nakata, H. et al. Antimicrobial. Agents and
Chemotherapy, 53, 3887-3893 [2009]). EFdA is claimed in US Patent No. 7,339,053 (referred to in the‘053 patent as 2,-deoxy-4’-C-ethynyl-2-fluoroadenosine). EFdA has the following chemical structure:
EFdA is metabolized in cells to its active triphosphate anabolite which inhibits HIV reverse transcriptase. In contrast to nucleoside reverse transcriptase inhibitors (NsRTIs) and nucleotide reverse transcriptase inhibitors (NtRTIs) currently available for the treatment of HIV infection which lack a 3′-OH group to block incorporation of incoming nucleotide, EFdA retains a 3′ OH group and acts as a chain terminator by preventing translocation of the primer template in the reverse transcriptase (RT) active site and preventing binding of incoming
deoxyribonucleotide triphosphates (dNTPs). In addition, the pucker of the modified ribose ring of EFdA is believed to contribute to inhibition of reverse transcriptase by placing the 3′-OH in a vector in which phosphotransfer from the incoming nucleotide is inefficient. (Michailidis E, et ak, Mechanism of inhibition of HIV-l reverse transcriptase by 4’-ethynyl-2-fluoro-2’-deoxyadenosine triphosphate, J Biol Chem 284:35681-35691 [2009]; Michailidis E, et ak, 4’-Ethynyl-2-fluoro-2’-deoxyadenosine (EFdA) inhibits HIV-l reverse transcriptase with multiple mechanisms, J Biol Chem 289:24533-24548 [2014] ).
In in-vitro HIV replication assays, EFdA is a potent antiretroviral and exhibits comparable antiviral activity against clinical isolates across all subtypes that have been evaluated. It is rapidly anabolized in both lymphoid derived cell lines and in peripheral blood mononuclear cells to the active triphosphate in vitro, and the intracellular half-life of EFdA Triphosphate (EFdA- TP) exceeds 72 hrs. (Stoddart, C. A., Galkina, et ak, Oral Administration of the Nucleoside EFdA (4’-Ethynyl-2-Fluoro-2’-Deoxyadenosine) Provides Rapid Suppression of HIV Viremia in Humanized Mice and Favorable Pharmacokinetic Properties in Mice and the Rhesus Macaque, Antimicrob Agents Chemother, 2015 Jul; 59(7): 4190-4198, Published online 2015 May 4).
EFdA has been shown to have efficacy in animal models of HIV infection including humanized mouse models and an SIV infected rhesus macaque model. Pharmacokinetic studies of orally administered EFdA in mouse and rhesus monkey have demonstrated rapid absorption and high plasma concentrations. A long intracellular half-life was demonstrated by the fact that isolated peripheral blood mononuclear cells from the rhesus macaque were refractory to SIV infection 24 hr after drug administration. (Ibid.)
Previous syntheses of 4’-ethynyl nucleoside analogs including EFdA suffer from modest stereoselectivity in the formation of the C-N bond between the ethynyl-deoxyribose sugar and the 2-fluoroadenine (also referred to as 2-fluoro-9H-purin-6-amine) nucleobase. The previous syntheses also require protecting groups to carry out the glycosylation reaction which reduces the efficiency of the syntheses.
The synthesis described in Kei Fukuyama, et ak, Synthesis of EFdA via a
Diastereoselective Aldol Reaction of a Protected 3-Keto Furanose, Organic Letters 2015, 17(4), pp. 828-831; DOI: 10.102 l/ol5036535) is a l4-step synthesis from D-glucose diacetonide that uses diastereoselective reactions to set the three stereocenters. The stereochemistry of the anomeric center is controlled by having a 2′-acetoxy directing group that is subsequently removed by hydrolysis and deoxygenation. This route requires 4 chromatographic purifications, and the stoichiometric use of a toxic organotin reagent for late-stage deoxygenation.
In another route (see Mark McLaughlin, et al., Enantioselective Synthesis of 4′-Ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) via Enzymatic Desymmetrization, Organic Letters 2017, 19 (4), pp. 926-929), the fully-substituted 4′- carbinol is generated stereoselectively with an enzymatic desymmetrization. The 3 ‘-stereocenter is set with a catalytic asymmetric transfer hydrogenation, and the anomeric 1 ‘-linkage is established in modest stereoselectivity using substrate control, with an upgrade in stereochemical purity achieved by crystallization of an intermediate. This process requires 15 steps, requires the use of several protecting groups and generates the glycosyl linkage between the nucleobase and sugar fragments in low
stereoselectivity (1.8: 1).
A l2-step synthesis for making EFdA from R-glyceraldehyde acetonide is described in Kageyama, M., et al., Concise Synthesis of the Anti-HIV Nucleoside EFdA, Biosci. Biotechnol. Biochem, 2012 , 76, pp. 1219 -1225; and Enantioselective Total Synthesis of the Potent Anti-HIV Nucleoside EFdA, Masayuki Kageyama, et al., Organic Letters 2011 13 (19), pp. 5264-5266 [DOL 10.1021 / ol202116k] . The syntheses use the chiral starting material to set the 3′-stereocenter with moderate diastereoselectivity. After chromatographic separation of stereoisomers, the new stereocenter is used to guide a diastereoselective alkyne addition to set the fully-substituted 4′-stereocenter. The anomeric 1 ‘-position is established with little stereocontrol and requires chromatography to separate the anomers. This route requires chromatographic separation of diastereoisomers at two different stages and starts from an expensive chiral starting material.
Kohgo, S., et al., Design, Efficient Synthesis, and Anti-HIV Activity of 4′-C-Cyano- and 4′-C-Ethynyl-2′-deoxy Purine Nucleosides, Nucleosides, Nucleotides and Nucleic Acids, 2004, 23, pp. 671-690 [ DOL 10.1081/NCN-120037508] describes a synthetic route that starts from an existing nucleoside and modifies both the sugar and nucleobase portions. It is an 18-step synthesis starting from 2-amino-2′-deoxy adenosine with a low 2.5% overall yield.
It is known that enzymes such as purine nucleoside phosphorylase (PNP, EC 2.4.2.1) can form the glycosyl linkage in nucleosides and nucleoside analogs in high stereoselectivity and without the use of protecting groups. See for example the review: New Trends in Nucleoside Biotechnology, Mikhailopulo, I. A., Miroshnikov, A.I,. Acta Naturae 2010, 2, pp. 36-58.
However, the current scope of the sugar fragments capable of undergoing reaction catalyzed by PNP has been limited to the a- 1 -phosphates of natural ribose and deoxyribose along with a small number of analogs with small H, NH2, or F substituents at the C2’ and C3’ positions and replacements of the C5’ OH group. There have been no reports of successful glycosylation catalyzed by PNP using sugars with carbon substituents on the ring or any substitution at the C4’ position.
Access to the ribose and deoxyribose a- 1 -phosphate substrates for the PNP-catalyzed glycosylation has been demonstrated by translocation of the phosphate group from the 5’-hydroxyl to G -hydroxyl position with the enzyme phosphopentomutase (PPM, EC 5.4.2.7) (see Mikhailopulo, I. A., et al. supra). However, the scope of the sugars for which PPM is capable of catalyzing this reaction has been limited to ribose, arabinose, 2-deoxyribose, and 2,3-dideoxyribose. No examples have been reported of successful reaction with sugar phosphates containing any additional substituents.
Deoxyribose phosphate aldolase (DERA, EC 4.1.2.4) enzymes are known to catalyze the aldol addition of acetaldehyde to other short-chain aldehydes (see review: Stephen M. Dean, et al., Recent Advances in Aldolase-Catalyzed Asymmetric Synthesis, Adv. Synth. Catal. 2007, 349, pp. 1308 – 1320; DOI: 10. l002/adsc.200700115). However, no examples have been reported with aldehydes bearing a fully substituted carbon a to the aldehyde.
ETS Patent 7,229, 797 describes the formation of deoxyribonucleosides from the natural unsubstituted deoxyribose 1 -phosphate by use of purine nucleoside phosphorylase (PNP) and additionally using enzymes such as sucrose phosphorylase to remove the inorganic phosphate byproduct and drive the equilibrium. It does not disclose enzyme engineering for the creation of PNP enzymes that can generate nucleosides from the unnatural 4-ethynyl-D-2-deoxyribose 1-phosphate, nor that through engineering of PPM and DERA enzymes to act on unnatural substrates, 4-ethynyl-D-2-deoxyribose 1 -phosphate can be generated.
In view of the difficult and lengthy synthetic options developed to date for producing 4’-ethynyl nucleoside analogs, it would be desirable to develop an improved enzymatic synthesis for 4’-ethynyl nucleoside analogs such as EFdA that reduces the number of process steps, minimizes the use of protecting groups, improves the stereoselectivity of glycosylation and avoids the use of toxic materials.
Surprisingly, it has been found that PPM enzymes have some activity with the 3-atom ethynyl substituent at the 4’ position on ribose and that the PPM enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a reaction for
isomerization of
4-ethynyl-D-2-deoxyribose 5-phosphate (6) to 4-ethynyl-D-2-deoxyribose 1 -phosphate (6.5) catalyzed by PPM to enable a more efficient method for production of 4’-ethynyl-2’-deoxy nucleosides.
Additionally, PNP enzymes have also been found to have some activity with the 3-atom ethynyl substituent at the 4 position on deoxyribose and that the PNP enzyme activity could be improved by introducing mutations into the enzymes to successfully develop a glycosylation reaction catalyzed by PNP to enable a more efficient method for production of 4’ -ethynyl -2’-deoxy nucleosides.
Even further improvement to the overall synthetic method came from the finding that
DERA enzymes, particularly the DERA from Shewanella halifaxensis, have activity for aldol reaction with 2-ethynyl-glyceraldehyde 3-phosphate which has a fully substituted a-carbon. This discovery allowed for the efficient synthesis of 4-ethynyl-D-2-deoxyribose 5-phosphate, a precursor to 4’-ethynyl-2’-deoxy nucleoside analogs, e.g., including EFdA.
SUMMARY OF THE INVENTION
The present invention involves the use of engineered enzymes in a novel enzymatic synthesis of 4’-ethynyl-2’-deoxy nucleoside analogs, including EFdA, that eliminates the use of protecting groups on intermediates, improves the stereoselectivity of glycosylation and greatly reduces the number of process steps needed to make said compounds compared to prior methods, among other process improvements. It further relates to novel intermediates which are an integral part of the enzymatic process.
The overall process is summarized in the following Scheme 1 and Scheme 2; the latter scheme provides an alternative method for making compound 5:
Scheme 1
kinase
p p y
Scheme 1A
kinase galactose oxidase
3 2X+ 9
2
p p y
It has been discovered that 4’-ethynyl-2’-deoxy nucleoside analogs such as EFdA can be synthesized employing a final step one-pot process by combining 4-ethynyl-D-2-deoxyribose 5-phosphate (6) with two enzymes, phosphopentomutase (PPM) [for example but not limited to SEQ ID NO.: 8] and purine nucleoside phosphorylase (PNP) [for example but not limited to SEQ ID NO.: 9, SEQ ID NO.: 15], as shown in Scheme 2.
Scheme 2
Scheme 2A
Several upstream intermediates used in the present process for the synthesis of the final product 4’-ethynyl-2’-deoxy nucleosides and analogs thereof are also made using enzymatic reaction methods as shown in Scheme 3; Scheme 3 A and Scheme 3B
Scheme 3
Scheme 3A
o2
pTsOH
deoxyribose
aldolase
Scheme 3B
Experimental Procedures
Preparation of 2-ethynyl-2-hvdroxypropane-l,3-diyl diacetate 12)
Method A:
To a -35 °C solution of diacetoxyacetone (1) (159 g, 914.0 mmol) in THF (1000 mL) was added 1600 mL of a 0.5 M solution of ethynyl magnesium chloride in THF maintaining the temperature below -20 °C. After the reaction reached completion, acetic acid (78 mL) in 400 mL methyl tert-butyl ether (MTBE) was added dropwise keeping the temperature below -20 °C. MTBE (800 mL) was then added and the mixture was warmed to room temp. Saturated NaCl in water (1000 mL) was added followed by saturated NH4CI solution in water (1050 mL). The organic layer was separated, dried over Na2SC>4 and evaporated to give compound (2) as an oil (160 g, 88%). 1H NMR (CDCI3, 500 MHz): d 4.26 (dd, 4 H), 2.55 (s, 1H), 2.14 (s, 6H).
Preparation of 2-ethynyl-propane-l,2,3-triol 13)
Method B:
To a solution of 2-ethynyl-2-hydroxypropane-l,3-diyl diacetate (2) (70 g, 350 mmol) in ethanol was added a 0.5M solution of sodium methoxylate in methanol (69.9 mL, 35.0 mmol) at room temperature (rt). The reaction was stirred at rt for 2 hours (h) to reach completion. The solvents were evaporated and the residue was re-dissolved in 100 mL water and extracted with 3 x 50 mL MTBE. The aqueous layer was sparged with nitrogen to remove residual solvents to give a 40.9% solution of 2-ethynyl-propane-l,2,3-triol (3) (108 g , 100% yield) as determined by nuclear magnetic resonance (NMR) (maleic acid as internal standard). lH NMR (D2O, 500 MHz): d 3.60 (dd, 4 H), 2.85 (s, 1H).
Alternate Preparations o ethynyl-glvcer aldehyde 14)
Method Cl:
In a stirred reactor, 2-ethynyl-propane-l,2,3-triol (3) (1.1 g, 9.47 mmol) in sodium phosphate buffer (30 mL, 100 mM, pH 7.0) containing antifoam 204 (Sigma A6426, 1 drop ~ 20 pL) was warmed to 30 °C with air sparging at 12.5 seem. Galactose oxidase (GOase, SEQ ID NO.: 1) (250 mg), Horseradish Peroxidase* (Type I, 5 mg) and bovine catalase** (5 mg) dissolved in sodium phosphate buffer (5 mL 100 mM, pH 7.0) were added to the reactor, followed by the addition of CuS04 aq. solution (100 mM, 150 pL). The reaction mixture was stirred at 600 rpm with air sparging for 47h to give (f?)-2-ethynyl-glyceraldehyde (4) in 47% conversion (by NMR) and 72% e.e. . (The product was not isolated). lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish Type I, commercially available from SIGMA (P8125), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345)
Method C2:
In a stirred 100 L jacketed reactor charged with deionized water (56.2 kg), sodium dihydrogen phosphate (1.212 kg, 10 moles) was added. The pH was adjusted to 7.02 using 10 N sodium hydroxide solution (852.6 g) at 25 °C. The reactor was charged with Antifoam 204 (A6426, 10 mL), followed CuS04*5H20 (6.5 g). Galactose oxidase (451.2 g) (SEQ ID NO.: 10) was added and stirred for 15 min while sparged with air. Horseradish peroxidase* (200.2 g) and catalase** (502.6 g) were added and the reactor was rinsed with water (2.0 kg). Next 2-ethynyl-propane- 1,2, 3 -triol (3) solution in water (9.48%, 30.34 kg, 24.72 mol) was added followed by an additional portion of Antifoam 204 (A6426, 10 mL). The reaction was sparged with air and
stirred overnight to give 94.0 kg of (A)-2-ethynyl-glyceraldehyde (4) in 66% conversion (by NMR) and 84% e.e. Assay yield 60%: 1H NMR (D20, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
* Horse Radish Peroxidase: wild type peroxidase from horseradish purified, commercially available from Toyobo (PEO-301), isolated from horseradish roots (Amoracia rusticana).
** Bovine catalase: heme-dependent catalase from bovine source, commercially available from Sigma (C1345).
The above reaction was also performed using the galactose oxidase (SEQ ID NO.: 11) and the product (4) was obtained in 67% conversion (by NMR) and 88% e.e. and assay yield 59%: 1H NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H).
Method C3:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 0.5 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase (SEQ ID NO.: 17, 450 mg enzyme powder) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (Cl 345, Sigma-Aldrich, 150 mg, 2000-5000 U/mg, 0.75 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg,
130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (25 wt%, 12 mL, 25.8 mmol). The reaction mixture was stirred at 30 °C with aeration at 125 seem and sampled using EasySampler over 20h to give 70% conversion and form compound (4) ((A)- 2-ethynyl-glyceraldehyde) in 58% assay yield and 99% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C4: Oxidation with immobilized galactose oxidase
Galactose
Oxidase
immobilized
3
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (16 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 160 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution. In a vessel evolved galactose oxidase (SEQ ID NO.: 17, 2.00 g) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (50 mL) and the resin. The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 160 mL) and potassium PIPES buffer (10 column volumes, 160 mL; 50 mM, pH 7.5) and it was used directly in a reaction. Reaction procedure:
In a 100 mL Easy Max vessel equipped with sparger and flow controller, water (82 mL) and PIPES potassium buffer (5mL, 1 M) were charged. The pH was adjusted to 7.5 using 5 M KOH solution at 25 °C. Antifoam 204 (200 pL) was added, followed by evolved galactose oxidase immobilized on the resin (SEQ ID NO.: 17, 750 mg enzyme powder per 6 mL resin) and copper(II) sulfate pentahydrate (100 pL, 100 mM). The reaction mixture was sparged with air at 125 standard cubic centimeters per minute (seem) for 15 min. Bovine catalase (C1345, Sigma-Aldrich, 210 mg, 2000-5000 U/mg, 1.05 MU) was charged, followed by horseradish peroxidase (HRP, Toyobo PEO-301, 100 mg, 130 U/mg, 1.3 kU) and the aqueous solution of 2-ethynyl-propane- 1,2, 3 -triol (3) (25 wt%, 13 mL, 29.4 mmol). The reaction mixture was stirred at 25 °C with aeration at 125 seem. After 22h the reaction reached 91% conversion to give 200 mM (//)-2-ethynyl-glyceraldehyde (4) solution (100 mL, 68% assay yield, 97% e.e. lH NMR (D2O, 500 MHz): d 4.29 (s, 1H), 3.65 (dd, 2H), 2.83 (s, 1H). The crude reaction stream was carried directly into the subsequent phosphorylation step.
Method C5: Optional Isolation of aldehyde via formation of aminal (8)
Step 1: Preparation of (S)-2-( \ .3-dibenzylimidazolidin-2-yl )but-3-yne- l 2-diol
A 100 L jacketed cylindrical vessel equipped with nitrogen bubbler, mechanical stirrer and thermocouple was charged with crude oxidase reaction stream containing (f?)-2-ethynyl-glyceraldehyde ((4), 26.0 kg, 1.85 wt% aldehyde, 3.64 mol) and inerted with N2 atmosphere. The aqueous solution was warmed to 20 °C and Af,A-di methyl dodecan- 1 -ami ne oxide (DDAO) (30 wt% in water, 798 g, 0.96 mol;) was added, followed by MTBE (55.3 kg, 76 L) and N,N -dibenzylethane-l, 2-diamine (1.55 kg, 6.43 mol). The brown, biphasic mixture was stirred overnight at 20 °C under nitrogen atmosphere. After 17 hours the stirring was stopped and the organic phase was removed and discarded. A light brown MTBE solution of fV)-2-( l ,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (56.5 kg, 2.02 wt% aminal, 3.39 mmol, 93% assay yield) was obtained.
Six similar MTBE solutions were processed together in a single distillation and crystallization step (in total 374.4 kg of solution, containing 7.91 kg aminal).
A 50 L jacketed cylindrical vessel equipped with mechanical stirrer, distillation head (condenser at -20 °C) and thermocouple was charged with aminal solution (45 L). Vacuum was applied to the vessel (65-95 torr) and the jacket was set to 40 °C. Solvent was removed by distillation until a volume of 35 L had been reached. At this point, the internal temperature was 6.1 °C and an off-white solid had begun to crystallize. The remaining MTBE solution was slowly added, maintaining a constant volume of 35-40 L and an internal temperature of 0-10 °C. Once all the MTBE solution had been added the volume was decreased to 25 L. Distillation was halted, the vessel was inerted with nitrogen and the jacket temperature was decreased to 10 °C. The resulting pale yellow suspension was aged at this temperature for 2 hours and the solids were collected by filtration. The filter cake was washed with cold (-2 °C) MTBE (12.7 kg) and then dried under nitrogen flow for 7 hours. (5)-2-(l,3-dibenzylimidazolidin-2-yl)-but-3-yne-l,2-diol was obtained as an off-white crystalline solid (5.75 kg) lff NMR (500 MHz, DMSO-i¾) d 7.42 – 7.35 (m, 4H), 7.32 (td, J= 7.5, 1.6 Hz, 4H), 7.27 – 7.21 (m, 2H), 5.10 (t, J= 5.6 Hz, 1H), 5.03 (s, 1H), 4.28 (d, J= l3.3Hz, 1H), 4.16 (d, J= 13.3 Hz, 1H), 3.76 (s, 1H), 3.70 – 3.58 (m, 4H), 3.21 (d, J= 0.9 Hz, 1H), 2.90 – 2.80 (m, 2H), 2.60 – 2.51 (m, 2H).13C NMR (126 MHz, DMSO-i¾) d 140.0, 140.0, 128.5, 128.3, 128.2, 128.1, 126.8, 126.8, 88.6, 86.9, 75.0, 74.0, 66.4, 60.7, 60.5, 50.4, 50.3, 39.5. HR-MS (ESI) Aminal (M + H+) C21H25N202+ calculated 337.1911; found 337.1922.
Step 2: Prep l (8)
A 4 L jacketed cylindrical vessel equipped with nitrogen bubbler and mechanical stirrer was charged with of TsOH»H20 (12.0 g, 63.1 mmol), water (60 mL), (ri)-2-(l,3-dibenzylimidazolidin-2-yl)but-3-yne-l,2-diol (110 g, 327 mmol) and MTBE (1700 mL). The biphasic mixture was placed under nitrogen and the jacket temperature was set to 15 °C. A solution of TsOH»H20 (114 g, 599.3 mmol) in water (600 mL) was added dropwise over 1.5 hours with overhead stirring (200 rpm). After addition had completed, the jacket temperature was lowered to 5 °C and the resulting slurry was aged for 1 hour. The solids were removed by filtration and washed with cold water (270 mL). The biphasic solution was transferred to a separating funnel and the organic phase was removed and discarded. The aqueous phase was treated with DOWEX™ MARATHON™ A resin (hydroxide form, 11.0 g) and AMBERLYST® 15 resin (hydrogen form, 11.0 g) while sparging with N2 at a rate of 200 seem for 24 hours to remove residual MTBE. The resins were removed by filtration to give a colorless aqueous solution of (f?)-2-hydroxy-2-(hydroxymethyl)but-3-ynal (774 g, 4.6 wt% aldehyde, 82% yield). lH MR (500 MHz, D2O) d 5.01 (s, 1H), 3.77 (d, J= 11.7 Hz, 1H), 3.73 (d, J= 11.7 Hz, 1H), 2.92 (s, 1H). 13C NMR (126 MHz, D2O) d 129.4, 125.4, 90.3, 81.0, 76.0, 73.9, 65.3. HRMS
Alternate Preparations o ethvnyl-glvceraldehvde 3-phosphate (5):
Method Dl: Acetate kinase: ATP -regeneration system
Pantothenate kinase PanK
ATP
Acetate kinase
4 Acetate phosphate
5
In a stirred reactor, to a solution of adenosine diphosphate disodium salt (40 mg, 0.087 mmol) and magnesium chloride (38 mg, 0.400 mmol) in HEPES buffer (66 mM, pH 7.5, 30 mL) was added (i?)-2-ethynyl-glyceraldehyde (4) (1.9 mL, 210 g/L solution in water, 3.51 mmol), followed by acetate kinase (SEQ ID NO.: 3) (40 mg), and pantothenate kinase (SEQ ID NO.: 2) (120 mg). The reaction mixture was warmed to 25 °C and a solution of acetyl phosphate lithium potassium salt (1.3 g, 7.01 mmol) in HEPES buffer (50 mM, pH 7.5, 10 mL) was added dropwise over 4 hours, with pH maintained at 7.5 using 5M sodium hydroxide. The reaction was stirred for 18 hours to give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 85% conversion (by HPLC) (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method D2: Pyruvate oxidase ATP -regeneration system
Pan
Pyruvate oxidase
Pyruvate
Phosphate
02
In a stirred reactor, a solution of sodium pyruvate (3.11 g, 28 mmol) and phosphoric acid (0.523 mL, 7.71 mmol) in 76 mL water pH 7.5 was charged with (i?)-2-ethynyl-glyceraldehyde (4) (3.8 mL, 210 g/L solution in water, 7.01 mmol), adenosine diphosphate disodium salt (80 mg, 0.174 mmol), thiamine pyrophosphate (40 mg, 0.086 mmol), flavin adenine dinucleotide disodium salt hydrate (64 mg, 0.077 mmol), and magnesium chloride (400 pL, 1 M solution in water, 0.4 mmol). The pH was re-adjusted to 7.5 with 5M aq sodium hydroxide and the reaction volume was re-adjusted to 80 mL with water. Acetate kinase (SEQ ID NO.: 3) (80 mg), pyruvate oxidase (SEQ ID NO.: 4) (80 mg, lyophilized cell free extract), pantothenate kinase (SEQ ID NO.: 2) (400 mg), and catalase (800 pL, ammonium sulfate suspension CAT-101, Biocatalytics) were added. The reaction was stirred at 500 rpm and 30 °C with air sparging for 72 hours to give (//)-2-ethynyl-glyceraldehyde 3 -phosphate 5 in 95% conversion (by HPLC) (The product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
The above reaction was also performed using the pantothenate kinase (SEQ ID NO.: 13) and the product 5 was obtained in 66% conversion. (The product was not isolated). iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H).
Method D3: Acetate kinase: ATP -regeneration system using immobilized enzymes
Panth
Acetate phosphate
Enzyme immobilization procedure:
NUVIA™ Immobilized Metal-ion Affinity Chromatography (IMAC) nickel-charged resin (168 mL based on settled volume) was added to a filter funnel and washed with binding buffer (1.6 L; 500 mM sodium chloride, 50 mM sodium phosphate, pH 8.0). In a vessel, pantothenate kinase
(8.4 g) (SEQ ID NO.: 12) and acetate kinase (2.8 g) (SEQ ID NO.: 3) were dissolved in binding buffer (500 mL). The washed resin was charged to the vessel and the solution was stirred for 4 hours at 20 °C. The resin was filtered and washed first with binding buffer (1.6 L) followed by piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (840 mL; 50 mM, pH 6.5). The washed resin was used directly in the next step.
Reaction procedure:
To a 1 L reactor, a solution of (f?)-2-ethynyl-glyceraldehyde (4) in water (608.7 g, 4.6 wt%, 212 mmol) was charged and cooled to 5 °C. To the cooled solution piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (32.7 mL, 1 M, pH 6.5, 32.7 mmol), magnesium chloride (9.33 mL, 1 M, 9.33 mmol), acetyl phosphate diammonium salt (51.8 g, 265 mmol), adenosine diphosphate disodium salt hydrate (1.17 g, 2.12 mmol), and water (192 mL) were added. The solution was allowed to stir and pH was adjusted to 6.4 using 5 N KOH. The reaction was warmed to 20 °C and 168 mL of resin with co-immobilized pantothenate kinase (SEQ ID NO.: 12) and acetate kinase (SEQ ID NO.: 3) was added. The reaction was stirred for 10 hours with 5 N KOH used to maintain a pH of 6.4 to give (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in
92% conversion (by HPLC) and 91% yield (by 3 lp NMR with tetraphenylphosphonium chloride as internal standard) (the product was not isolated). lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Preparation of 4-ethynyl-D-2-deoxyribose 5-phosphate 16)
Method E:
To a solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (5, 20 mL, 5.3 mmol) in water, a solution of acetaldehyde in water (40 wt.%, 2.02 mL, 15.9 mmol) was added at room
temperature, followed by the addition of Deoxyribose-phosphate aldolase (DERA) (SEQ ID NO. : 6), 25 mg solution in triethanolamine hydrochloride buffer (1 mL, 1 M, pH 7.0). The reactor was sealed and the mixture was stirred overnight at 30 °C and 600 rpm to give 4-ethynyl-D-2-deoxyribose 5-phosphate (6) in 99% conv. and 99% e.e., 99% d.e. as a 1 : 1 anomer mixture (The product was not isolated) a-anomer: lH NMR (D2O, 600 MHz) 5 5.31 (t, 1H), 4.13 (t, 1H), 3.81-3.72 (m, 2H), 2.89 (s, 1H), 2.42-2.34 (m, 1H), 1.87-1.79 (m, 1H); 13c NMR (D2O, 151 MHz) 5 97.7 (s), 81.4 (d), 79.4 (s), 78.9 (s), 71.1 (s), 67.7 (d), 39.6 (s). b-anomer: 1H NMR
Alternate Preparations of (2 ?,3A,5 ?)-5-(6-amino-2-fluoro-9H-purin-9-yl)-2-ethynyl-2-(hydroxymethyl)tetrahydrofuran-3-ol monohydrate (7) [alternative name 4’-ethynyl-2-fluoro- 2’-deoxvadenosine or EFdAI
Method FI:
Ammonium ((2f?,3ri)-2-ethynyl-3,5-dihydroxytetrahydrofuran-2-yl)m ethyl hydrogen phosphate (1.00 g, 3.91 mmol) was dissolved in 10 mL of pH 7.5 buffer (100 mM triethanolamine ΉO containing 5 mM MnCl2). The solution pH was adjusted to 7.3 with 5 N NaOH. To the solution was added 2-fluoroadenine (0.599 g, 3.91 mmol) and sucrose (2.68 g, 7.82 mmol). The enzyme solution was prepared by dissolving phosphopentomutase (SEQ ID NO. : 8) (100 mg), purine nucleoside phosphorylase (SEQ ID NO.: 9) (50 mg), and sucrose phosphorylase (SEQ ID NO. :
7) (10 mg) in 10 mL of the pH 7.5 buffer. The enzyme solution was added to the reagent mixture and the resulting suspension was shaken at 40 °C. After 20 h, the suspension was cooled to 0 °C and filtered, rinsing with cold water. The solid was suction dried to give the title compound (1.12 g, 92%) as a single isomer.
The PPM and PNP enzymes used in this step were each derived from mutations starting from the enzymes from E. coli ( Escherichia coli). The sucrose phosphorylase (SP) used in this step was derived from Alloscardovia omnicolens ; SP derived from other organisms could also be used.
Method F2:
To an aqueous solution of (f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) (950 mL, 157 mmol) containing piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at a pH from about 5.5 to 6.0 was added triethanolamine (7.09 g, 47.5 mmol). The pH of the solution was adjusted from 7.1 to 7.6 using potassium hydroxide (8 mL, 8M). Manganese(II) chloride hydrate (0.592 g, 4.70 mmol) was added followed by sucrose (161 g, 470 mmol), giving a pH of 7.5 To the solution
was added the following enzymes: deoxyribose-phosphate aldolase (SEQ ID NO. : 14) (461 mg), sucrose phosphorylase (SEQ ID NO. : 7) (494 mg), phosphopentomutase (SEQ ID NO.: 8)(2.63 g), and purine nucleoside phosphorylase (SEQ ID NO. : 15) (659 mg). Once the enzymes were dissolved, 2-fluoroadenine (19.80 g, 125 mmol) was added. The reaction was heated to 35 °C and acetaldehyde was added (40 wt% in isopropyl alcohol, 29.8 mL, 235 mmol). After reacting for 2h, the mixture was seeded with EFdA crystalline product (0.96 g, 2 mol%). After reacting over 26 h at 35 °C, the slurry was cooled to 0 °C, and the solids were collected by filtration, washing with water two times (40 mL ea.). The solids were dried under a nitrogen sweep. Yield 43.2 g, 92 wt%, 96.2% corrected. ¾ NMR: (300 MHz, DMSO-d6, ppm): d 7.68 (br s, 2H), 7.32 (d, J = 2.0 Hz, 1H), 6.44 (t, J = 5.8 Hz, 1H), 5.52 (d, J = 5.6 Hz, 1H), 5.27 (t, J = 6.0 Hz, 1H), 4.44 (q, J = 6.4 Hz, 1H), 3.60 (q, J = 6.0 Hz, 1H), 3.53 (q, J = 6.4 Hz, 1H), 3.48 (s, 1H), 2.48-2.41 (m, 1H), 2.37-2.30 (m, 1H). 13C NMR (150.92 MHz, DMSO-d6, ppm) d 158.5 (d, JCF = 203.5), 157.6 (d, JCF = 21.2), 150.2 (d, JCF = 20.2), 139.7 (d, JCF = 2.4), 117.4 (d, JCF = 4.0), 85.1, 82.0, 81.4, 78.7, 70.1, 64.2, 38.1. LC-MS: (ES, m/z): calculated for C12H12FN5O3 (M+Na): 316.0822; found 316.0818.
Alternate Preparations of -2-ethvnyl-propane-l,2,3-triol 1 1-phosphate 19) :
Method Gl: Acetate kinase: ATP-regeneration system using enzymes SEQ. ID No.: 2 and SEQ. ID No.: 3
Panthotenate kinase PanK
ATP
Acetate kinase
Acetate phosphate
A 50 mL reactor was charged with a solution of 2-ethynyl-propane-l,2,3-triol (3) in water (9.29 g, 9.46 wt%, 7.57 mmol) potassium PIPES buffer (1.02 mL, 1 M, pH 6.5, 1.02 mmol), magnesium chloride (292 pL, 1 M, 0.292 mmol), acetyl phosphate diammonium salt (1.851 g, 89 wt%, 9.46 mmol), adenosine diphosphate disodium salt hydrate (ADP, 42 mg, 0.076 mmol, 0.01 eq), and water (28 mL). The pH was adjusted to 6.4 using 5 M KOH, the solution was warmed to 20 °C and evolved pantothenate kinase PanK SEQ. ID No.: 2 (264 mg) and acetate kinase AcK SEQ. ID No. : 3 (88 mg) were added. The reaction was stirred for 16 hours with pH maintained at 6.4 using 5 N KOH. The final reaction contents provided C.V)-2-ethynyl -propane- 1 ,2,3-triol 1-phosphate (9) in >95% e.e. and 99% conversion (by 31P NMR). The product was not isolated. ¾ NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
Method G2: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
To a jacketed reactor aqueous solution 2-ethynyl-propane-l,2,3-triol (3) (11.47 kg, 8.7% wt, 8.61 mol) and water (7.5kg) was charged, followed by 1M BIS-TRIS methane buffer pH 6.5 (1L) and magnesium chloride (41.4 g). ATP (48g, 0.086 mol, 0.01 equivalent) and diammonium acetyl phosphate (2.021 kg, 89%, 10.33 mmol) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using KOH (270.4 g). Evolved pantothenate kinase SEQ. ID No.: 20 (20.4 g) and evolved acetate kinase SEQ. ID No.: 21 (3 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which pH dropped to 5.5.
Quantitative conversion of 2-ethynyl-propane-l,2,3-triol (3) was obtained as judged by ‘H and 31P NMR. Such prepared (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) solution (397 mM, 22.5 kg, 98% yield) was used in subsequent oxidation step without any further purification. ‘H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H),
2.93 (s, 1H).
Method G3: Acetate kinase: ATP-regeneration system using enzyme SEQ. ID No.: 20 and enzyme SEQ. ID No.: 21 with deuterated compound (3) to assign absolute stereochemistry and demonstrate desymmetrizing phosphorylation.
Acetate phosphate
Z-d2, 95:5 er
Evolved pantothenate kinase SEQ. ID No. : 20 (100 pL of 10 g/L solution in water ) and evolved acetate kinase SEQ. ID No. : 21 (100 pL of 2g/L solution in water) were added to a solution containing diammonium acetyl phosphate (41 mg), 2-ethynyl-propane-l, l-72-l,2,3-triol ((A)- 3-d2, 20 mg, 170 pmol), magnesium chloride (10 pL of 1 M solution in water), ADP (10 pL of 100 g/L solution in water), and sodium phosphate buffer (10 pL of 1 M solution in water) in water (800 pL) at pH 6.5. The reaction was incubated for 24h at rt to give deuterated 2-ethynyl-propane-l,2,3-triol l-phosphate analogs (S)-9-(3,3-d2) and (S)-9-(l,l-d2) in 95:5 ratio and 99% overall yield. The ratio of phosphorylated compounds was determined by 31P NMR to be -95:5, confirming stereoselective phosphorylation of the 2-ethynyl-propane-l,2,3-triol (3) at the pro-(S) hydroxyl group (i.e. a desymmetrizing phosphorylation). 1H NMR (D2O, 500 MHz) d 3.89 (m, 2H), 3.72 (d, 7= 11.6 Hz, 1 H), 3.65 (d, J= 11.6 Hz, 1H), 2.93 (s, 1H). 13C NMR (D20, 126 MHz) d 82.9 (s), 75.1 (s), 71.0 (d, J= 6.9 Hz), 67.0 (d, J= 4.5 Hz), 64.7 (s).
Method G4: Acetate kinase: ATP-regeneration system using immobilized enzymes SEQ. ID No. : 20 and enzyme SEQ. ID No. : 21
Panthotenate kinase PanK
– – ATP
Acetate kinase
Acetate phosphate
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (75 mL based on settled volume) was added to a filter funnel and washed with water (9 column volumes, 3 x 225 mL) and binding buffer (1 column volume, 75mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0). In a vessel pantothenate kinase (SEQ ID NO. : 20, 6.0 g) lyophilized powder was resuspended in binding buffer (200 mL) and the washed resin was added. The solution was mixed using rotating mixer at 25 °C for 6h. The resin was filtered and washed with binding buffer (6 column volumes, 6 x 225 mL) and BIS-TRIS buffer (8 column volumes, 600 mL; 50 mM, pH 6.2).
Reaction procedure:
An aqueous solution of 2-ethynyl-propane-l,2,3-triol (3) (574 g, 8.7% wt, 0.430 mol) and water (350 mL) was charged to a jacketed reactor, followed by 1M BIS-TRIS methane buffer pH 6.5 (50 mL) and magnesium chloride (2.033 g, 0.01 mol). ATP (2.37g, 0.0043 mol, 0.01 equivalent) and diammonium acetyl phosphate (101 g, 89%, 0.530 mmol, 1.2 eq) were added, the solution was warmed up to 20 °C and the pH of the solution was re-adjusted to 6.8 using 5 M KOH.
Resin with immobilized pantothenate kinase SEQ. ID No. : 20 (25 mL) and evolved acetate kinase SEQ. ID No. : 21 (0.15 g) were then charged as solids. The reaction was stirred for at 20 °C for l6h during which the pH dropped to 5.5. Quantitative conversion of 2-ethynyl-propane- I,2,3-triol (3) to (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9) was obtained as judged by ¾ and 31P NMR. ¾ NMR (D20, 500 MHz) d 3.89 (m, 2H), 3.72 (d, J= 11.6 Hz, 1 H), 3.65 (d, J =
I I .6 Hz, 1H), 2.93 (s, 1H).
Alternate Preparations of (i?V2-ethvnyl-glvceraldehvde 3-phosphate 15):
Method HI: Immobilized galactose oxidases SEP ID No.: 16
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 16 g of washed resin. In a vessel evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the glycosylation reaction.
Reaction procedure:
The resin with immobilized galactose oxidase SEQ ID NO.: 16 (3.0 g) was added to a solution of S)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (f?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the(f?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used
directly in the glycosylation reaction. iH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H2: Immobilized galactose oxidases SEP ID No.: 17
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (10 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 100 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give l6g of washed resin. In a vessel, evolved galactose oxidase (SEQ ID NO.: 16, 750 mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 5.00 mL), followed by addition of binding buffer (20 mL) and the washed resin (3.0g). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 100 mL) and BIS-TRIS methane buffer (10 column volumes, 100 mL; 50 mM, pH 7.5) and it was used directly in the reaction.
Reaction procedure:
The resin with immobilized evolved galactose oxidase SEQ ID NO.: 17 (3.0 g) was added to a solution of (ri)-2-ethynyl-propane-l,2,3-triol l-phosphate (9, 5.4 mmol, 270 mM, 20 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), followed by addition of copper (II) sulphate solution in water (30 pL, 100 mM) and horseradish peroxidase (PEO-301, 18 mg) and bovine catalase (C1345, 120 mg) resuspended in water (600 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 22 °C for 4 days to reach final conversion of 77% and give (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) in 95% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was used directly in the glycosylation reaction. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
Method H3: Immobilized galactose oxidases SEQ ID No.: 18
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 18, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution ((9), 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 90% conversion and the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H),
4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (MΉ): 193.1; found 193.0.
Method H4: Immobilized galactose oxidases SEP ID No.: 19
Enzyme immobilization procedure:
Nuvia IMAC Ni-charged resin (3 mL based on settled volume) was added to a filter funnel and washed with binding buffer (10 column volumes, 30 mL; 500 mM sodium chloride, 50 mM sodium phosphate, 15 mM imidazole, pH 8.0) to remove the resin storage solution and give 2.4 g of washed resin. In a vial evolved galactose oxidase (SEQ ID NO.: 19, 75mg) lyophilized powders were resuspended in copper (II) sulphate solution (100 mM; 1.00 mL), followed by addition of binding buffer (5 mL) and the washed resin (400 mg). The solution was mixed using rotating mixer at 20 °C for 5h. The resin was filtered and washed with binding buffer (10 column volumes, 4 mL) and BIS-TRIS methane buffer (10 column volumes, 4 mL; 50 mM, pH 7.5) and it was used directly in a reaction.
Reaction procedure:
Immobilized evolved GOase SEQ ID NO.: 18 was added (400 mg) to a solution of (5)-2-ethynyl-propane-l,2,3-triol l-phosphate solution (9, 5.4 mmol, 270 mM, 1 mL) in BIS-TRIS methane buffer (35 mM, pH adjusted to 7.2), , followed by addition of horseradish peroxidase (PEO-301, 1 mg) and catalase from Corynebacterium glutamicum (Roche, lyophilizate, #11650645103, 3 mg) resuspended in water (100 pL). The reaction was sealed with gas permeable membrane and shaken vigorously at 30 °C for 48h. Final conversion after 2 days reached 100% conversion and (i?)-2-ethynyl-glyceraldehyde 3 -phosphate (5) was obtained in >99% e.e. The enzyme resin was filtered off and the solution of the (i?)-2-ethynyl-glyceraldehyde 3-phosphate (5) was used directly without further purification. lH NMR (D2O, 400 MHz): d 5.02 (s, 1H), 4.00 (dq, 2 H), 2.88 (s, 1H). LC-MS: (ES, m/z): calculated for C5H7O6P (M-H): 193.1; found 193.0.
PATENT
CA 2502109
WO 2017053216
US 20200010834
US 20200010868
PAPER
Organic letters (2017), 19(4), 926-929.
Organic Letters (2017), 19(4), 926-929.
Journal of medicinal chemistry (2018), 61(20), 9218-9228.
Bioscience, Biotechnology, and Biochemistry (2020), 84(2), 217-227.
PAPER
Organic letters (2011), 13(19), 5264-6.
A concise enantioselective total synthesis of 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA), an extremely potent anti-HIV agent, has been accomplished from (R)-glyceraldehyde acetonide in 18% overall yield by a 12-step sequence involving a highly diastereoselective ethynylation of an α-alkoxy ketone intermediate.
Processes for preparing islatravir and its analogs comprising the reaction of a substituted tetrahydrofuran compound with purine nucleoside phosphorylase and a nucleobase, followed by stereochemical synthesis, glycosylation, reduction, oxidation and isolation are claimed. Also claimed are novel intermediates of islatravir and processes for their preparation and their use for the preparation of islatravir.
(2R,3S,5R)-5-(6-Amino-2-fluoropurin-9-yl)-2-ethynyl-2-(hydroxymethyl)- tetrahydrofuran-3-ol (1). To a stirred solution of 16 (66.2 mg, 0.115 mmol) in MeOH/CH2Cl2 (2:1, 1.5 mL) was added NH4F (85.1 mg, 2.30 mmol) at room temperature. After 16 h, MeOH (0.5 mL) was added, and the resulting mixture was stirred for an additional 27 h. To the mixture was added 10% NaOH in MeOH (1.5 mL) to adjust the pH of the mixture to ca. 10. After 10 min, Dowex 50W×8 (200– 400 mesh (H)) was added until the pH of the mixture reached ca. 4. To the resulting mixture was added CaCO3 (259 mg, 2.59 mmol), and the mixture was stirred for 30 min. The mixture was filtered through a pad of Celite, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography (CHCl3/MeOH = 10:1) to give 29.3 mg (87%) of 1. Mp: 220.0–221.4 °C (dec.); [α] 25 D +12.4 (c 0.97, MeOH); IR: νmax 3315 (br m), 3179 (br m), 1690 (vs), 1356 (vs); 1 H NMR (600 MHz, DMSO-d6): δ 2.43 (1H, ddd, J = 13.2, 7.3, 7.3 Hz), 2.70 (1H, ddd, J = 13.2, 6.8, 5.1 Hz), 3.52 (1H, s), 3.54 (1H, dd, J = 11.7, 6.4 Hz), 3.65 (1H, dd, J = 11.7, 5.0 Hz), 4.57 (1H, m), 5.32 (1H, m), 5.60 (1H, m), 6.24 (1H, dd, J = 7.2, 5.1 Hz), 7.82 (1H, br s), 7.92 (1H, br s), 8.31 (1H, s); 13C NMR (150 MHz): δ 38.3, 64.4, 70.3, 79.2, 81.7, 82.2, 85.4, 117.6, 140.0, 150.4 (d, JCF = 20.7 Hz), 157.8 (d, JCF = 21.2 Hz), 158.8 (d, JCF = 203.4 Hz); HRMS (FAB): m/z calcd for C12H13FN5 O3, 294.1002; found, 294.1000 ([M+H]+ ).
EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine), a nucleoside reverse transcriptase inhibitor with extremely potent anti-HIV activity, was concisely synthesized from (R)-glyceraldehyde acetonide in an 18% overall yield by a 12-step sequence involving highly diastereoselective ethynylation of an α-alkoxy ketone intermediate. The present synthesis is superior, both in overall yield and in the number of steps, to the previous one which required 18 steps from an expensive starting material and resulted in a modest overall yield of 2.5%.
PAPER
Bioscience, Biotechnology, and Biochemistry (2012), 76(6), 1219-1225.
Making small-molecule drugs usually goes something like this: set up a reaction, purify the intermediate, change a solvent, and repeat, repeat, repeat to get the final product. But there’s a lot of waste involved, which is why chemists stress the environmental benefits of an alternate approach: biocatalysis. Engineering enzymes to make reactions happen saves a lot of materials, minimizes chemical and hazardous waste, and even uses less plasticware and glassware. And not having to isolate intermediates saves time.
Some pharmaceutical companies are investigating biocatalysis at different points in their drug development pipelines, but mostly at one or two steps into the making of a small molecule. Scientists at Merck & Co. have taken this further—they are reporting an entire drug synthesis using a chain of nine enzymes, five of which had been engineered, to produce an experimental HIV drug at high yield in just a few steps (Science 2019, DOI: 10.1126/science.aay8484).
This biocatalytic cascade is turning heads. For the most part, scientists aren’t using biocatalysis to manufacture a compound so much as to develop it, says Princeton University chemist Todd Hyster. The Merck process stitches together nine enzymes to get good yields of the final product, which Hyster says is no small feat.
It literally took my breath away.
Alison Narayan, assistant professor, University of Michigan
“I was blown away,” Hyster says of the first time he saw Merck scientists talk about this work. “It’s something that was very complicated.”
Mark Huffman, a chemist who led the work at Merck with Anna Fryszkowska, says they turned to biocatalysis in order to overcome a couple of key hurdles in synthesizing some molecules. One is stereochemistry. Islatravir is a nucleoside that blocks the HIV enzyme reverse transciptase and traditionally, in medicinal chemistry, it’s been hard to get the stereochemistry of nucleosides right, Huffman says. But this is something enzymes are designed by nature to do. The other is preventing unwanted side reactions. A number of steps in the traditional chemical synthesis of islatravir put the compound’s functional groups at risk of being lopped off, so they must be protected. Huffman says enzymes are specific in the types of reactions they catalyze, so there’s little to no risk of an unwanted side reaction.
Scientists at Merck & Co. use nine enzymes, five of which are engineered, to biocatalytically make an HIV drug called islatravir without having to purify intermediates. Enzymes not engineered by Merck are not pictured.
On top of that, Huffman says, they are doing these reactions at neutral pH, in aqueous solvents, and at room temperature, which cuts down on electricity and the need for multiple bioreactors running under different conditions. Islatravir normally takes between 12 and 18 steps to make. With biocatalysis, the team has cut this down to three.
“You don’t have rigorous equipment requirements,” he says. “You’re usually running [these reactions] under much milder conditions.”
To run the cascade, the team started with 2-ethynylglycerol, and added a mixture of three enzymes to run one group of reactions. They then added more enzymes to drive a second set of reactions. Then, they remove the enzymes from the solution, which are immobilized and easy to filter out, and use four more enzymes to drive the final reactions that lead to islatravir. There are no intermediate purification steps. The overall yield is about 51% using biocatalysis, compared to yields of 7% and 15% using two more traditional syntheses.
To make their biocatalysts, the team surveyed natural enzymes, mostly from microbes, that interacted with the different intermediates in islatravir production. One of the reasons why Huffman says islatravir is an ideal small molecule to produce using biocatalysis is that most organisms have to make and break down nucleosides, so there are several natural enzymes found across multiple species. This gave the team a lot of starting material from which to alter amino acids and build the enzymes they needed to do their syntheses. By making adjustments to active sites and other areas of the enzymes, the team built five of the nine enzymes needed to make islatravir biochemically.
Huffman says that while islatravir is a good molecule to show that scientists can build large biocatalytic cascades, Merck is also looking at biocatalysis to make other small molecules and biologic drugs.
Alison Narayan, a biocatalysis chemist at the University of Michigan, calls Merck “bold” for putting the time, money, and people behind this change in production—it takes a lot of resources to try an entire synthesis via biocatalysis. And, she says, they’ve succeeded spectacularly. “It literally took my breath away,” Narayan says of her first exposure to this project in 2018. “I think it’s a huge accomplishment.”
She says that Merck’s islatravir work shows that industry is starting to appreciate what biocatalysis can do for their drug pipelines and their financial bottom lines. Alongside Merck, companies like GlaxoSmithKline and Pfizer are also exploring biocatalysis at different points in drug development and manufacturing.
“It’s an important proof of concept,” Narayan says. “This is a practical way to build molecules, and this will be the way that people will build molecules when you take into consideration efficiency, green-ness, and constructing an effective synthesis. Biocatalysis has a lot to offer.”
An investigational drug targeting the HIV virus is synthesized with nine enzymes
By Elaine O’Reilly and James Ryan
Natural biosynthesis assembles a vast array of complex natural products starting from a limited set of building blocks, under physiological conditions, and in the presence of numerous other biomolecules. Organisms rely on the extraordinary selectivity of enzymes and their ability to operate under similar reaction conditions, meaning that these catalysts are perfectly adapted to mediate cascade reactions. In these multistep processes, the product of one biocatalytic step becomes the substrate for the next transformation (Display footnote number:1-3). On page 1255 of this issue, Huffman et al. (Display footnote number:4) report the development of an impressive nine-enzyme biocatalytic cascade for the synthesis of the investigational drug islatravir for the treatment of human HIV.
This study represents a partnership between scientists from Merck and Codexis. These two companies have a history of successfully collaborating to develop biocatalysts for the synthesis of important pharmaceuticals. Almost a decade ago, they developed a chemoenzymatic route for the synthesis of the type 2 diabetes drug sitagliptin (Januvia), relying on a key enzyme-catalyzed transamination with a highly engineered (R)-selective transaminase (Display footnote number:5). The work was considered a landmark example of directed evolution and functioned to highlight the potential application of biocatalysis to revolutionize industrial chemical processes.
The cascade for synthesizing islatravir was inspired by the bacterial nucleoside salvage pathway, which recycles precious nucleosides by using three key enzymes: a purine nucleoside phosphorylase (PNP), a phosphopentomutase (PPM), and a deoxyribose-5-phosphate aldolase (DERA) (see the figure). However, to achieve the synthesis of the target molecule, Huffman et al. required the natural nucleoside degradative cascade to run in reverse. The reversible nature of enzymes is central to the design of this cascade and is one of the important features that sets biocatalysts apart from the majority of traditional chemical catalysts.
The success of the cascade developed by the team also relied on all three enzymes accepting non-natural substrates bearing a fully substituted carbon at the C-4 position of the 2-deoxyribose ring. The authors reconstructed the reverse nucleoside salvage pathway from a PNP and PPM found in Escherichia coli and a DERA from Shewanella halifaxensis. The native E. coli enzymes required engineering to improve their activity. The DERA displayed existing high activity and stereoselectivity for the formation of the desired sugar phosphate enantiomer, but it required engineering to improve its ability to operate at high substrate concentration.
One of the many advantages of performing biocatalytic cascade reactions is the effective displacement of unfavorable reaction equilibria that can be achieved through product removal. However, despite performing the PNP and PPM steps in tandem, the reaction proceeded with poor conversion, and the inorganic phosphate by-product inhibits the enzymes. An elegant solution to these issues was the inclusion of an auxiliary sucrose phosphorylase, along with its sugar substrate, which removed free phosphate and effectively displaced the reaction equilibrium toward product formation.
Having assembled enzymes for the three key steps in the cascade, Huffman et al. sought to develop a biocatalytic route for the synthesis of the DERA substrate 2-ethynylglyceraldehyde 3-phosphate. Extensive screening of a broad range of kinases resulted in the identification of pantothenate kinase (PanK) from E. coli, which displayed low levels of activity (∼1% conversion) toward the (R)-enantiomer of the target aldehyde. Despite the modest initial activity, directed evolution was successfully used to substantially improve the productivity and stability of this enzyme. Finally, after 12 rounds of evolution, the authors reversed the enantioselectivity and improved the activity, stability, and expression of a galactose oxidase variant for the desymmetrization of the starting substrate, 2-ethynylglycerol.
Opens in modal lightbox
GRAPHIC: A. KITTERMAN/SCIENCE
Viewable Image – engineering a biocatalytic cascade
Image Caption
GRAPHIC: A. KITTERMAN/SCIENCE
Advancements in protein engineering, rapid gene sequencing, and the availability of low-cost DNA synthesis have made it possible to alter the properties of enzymes and fine-tune them for biocatalytic applications (Display footnote number:6-8). The work by Huffman et al. is a milestone in cascade design, largely because of the number of biocatalysts operating in tandem and the engineering feat required to optimize five of the nine enzymes involved in the synthesis. It also highlights how biosynthetic or degradative pathways can be a source of inspiration for the design of efficient biocatalytic cascades and how sequences can be reconstituted using enzymes recruited from multiple sources—in this case, of bacterial, fungal, plant, and mammalian origin. The diverse role that biocatalysts can play is also exemplified in this work, where five engineered enzymes are directly involved in the synthesis of the target molecule, and four additional enzymes function to recycle coenzyme, remove inhibitory by-products, and maintain the correct oxidation state of the copper cofactor.
Previous approaches reported for the synthesis of islatravir relied on multistep syntheses and require protecting group manipulations and intermediate purification steps (Display footnote number:9, 10). The incorporation of a key biocatalytic step or steps has the potential to revolutionize synthetic design strategies by making possible transformations that are not accessible using solely chemical approaches (Display footnote number:11, 12). The application of enzymes in industry and the development of chemoenzymatic routes to complex molecules is now well established. However, multistep syntheses exclusively comprising biocatalytic transformations are rare (Display footnote number:13), and this contribution sets a new standard for the synthesis of complex molecules with enzymatic cascades.
School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. Email: elaine.oreilly@ucd.ie.
REFERENCES AND NOTES
1. S. P. France, L. J. Hepworth, N. J. Turner, S. L. Flitsch, ACS Catal.7, 710 (2017).
2. S. Gandomkar, A. Żadło-Dobrowolska, W. Kroutil, ChemCatChem11, 225 (2019).
3. P. Both et al., Angew. Chem. Int. Ed.55, 1511 (2016).
4. M. A. Huffman et al., Science366, 1255 (2019).
5. C. K. Savile et al., Science329, 305 (2010).
6. F. H. Arnold, Angew. Chem. Int. Ed.57, 4143 (2018).
7. C. Zeymer, D. Hilvert, Annu. Rev. Biochem.87, 131 (2018).
8. C. A. Denard, H. Ren, H. Zhao, Curr. Opin. Chem. Biol.25, 55 (2015).
9. M. McLaughlin et al., Org. Lett.19, 926 (2017).
10. M. Kageyama, T. Nagasawa, M. Yoshida, H. Ohrui, S. Kuwahara, Org. Lett.13, 5264 (2011).
11. N. J. Turner, E. O’Reilly, Nat. Chem. Biol.9, 285 (2013).
12. M. Hönig, P. Sondermann, N. J. Turner, E. M. Carreira, Angew. Chem. Int. Ed. 56, 8942 (2017).
13. S. Wu et al., Nat. Commun.7, 11917 (2016).
ACKNOWLEDGMENTS
J.R. acknowledges the School of Chemistry, University College Dublin, for support.
References
^Kawamoto, A; Kodama, E; Sarafianos, SG; Sakagami, Y; Kohgo, S; Kitano, K; Ashida, N; Iwai, Y; Hayakawa, H; Nakata, H; Mitsuya, H; Arnold, E; Matsuoka, M (2008). “2′-deoxy-4′-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants”. The International Journal of Biochemistry & Cell Biology. 40 (11): 2410–20. doi:10.1016/j.biocel.2008.04.007. PMID18487070.
Tebipenem (brand name: Orapenem) is a broad-spectrum orally-administered antibiotic, from the carbapenem subgroup of β-lactam antibiotics. It was developed as a replacement drug to combat bacteria that had acquired antibiotic resistance to commonly used antibiotics.[1][2] Tebipenem is formulated as the estertebipenem pivoxil due to the better absorption and improved bioavailability of this form.[3] It has performed well in clinical trials for ear infection and looks likely to be further developed in future.[4] It is only marketed in Japan.[5] Tebipenem is the first carbapenem whose prodrug form, the pivalyl ester, is orally available.[6]
Tebipenem pivoxil, an oral carbapenem prodrug, was launched in Japan in 2009 by Meiji Seika Pharma for the treatment of bacterial infection in children. The drug candidate was originally developed at Wyeth Pharmaceuticals (now Pfizer) and was subsequently licensed to Meiji Seika.
Tebipenem pivoxil was approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Apr 22, 2009. It was developed and marketed as Orapenem® by Meiji Seika in Japan.
In 2017, the product was licensed to Spero Therapeutics by Meiji Seika Pharma for worldwide development and commercialization, except in Japan and certain Asian countries, where Meiji will retain rights.
In 2017, the FDA granted the drug qualified infectious disease product designation for complicated urinary tract infections (cUTI), diabetic foot infections (DFI) and community acquired bacterial pneumonia (CABP)
Tebipenem pivoxil is a broad-spectrum orally-administered antibiotic, from the carbapenem subgroup of β-lactam antibiotics. Carbapenems are a class of beta-lactam antibiotics, which act by inhibiting the synthesis of the peptidoglycan layer of bacterial cell walls. It is used to treat otorhinolaryngological infection, otitis media and bacterial pneumonia.
Orapenem® is available as granules for oral use, containing 100 mg Tebipenem pivoxil/g granules. According to the weight of children, 4 mg/kg, and twice a day after dinner.
Alternatively Bipei Nan ester (Tebipenempivoxil) (I), chemical name: (+) – (4R, 5S, 6S) -6 – [(lR) -l- hydroxyethyl] -4-methyl-7 – oxo-3 [[1- (2-thiazol-2-yl) -3-azetidinyl] thio] -1-azabicyclo [3. 2.0] hept-2-ene 2-carboxylic acid methyl -2- pivaloyl), of the formula:
[0003]
[0004] developed by the American company Pfizer, for Bipei Nan ester fine granules developed by the Japanese company Meiji, in February 2009 was approved in Japan, and listed in April 2009.Alternatively Bipei Nan prodrug esters are for Bipei Nan, the lower the water after oral administration of the parent drug esterase for Bipei Nan, penicillin-binding protein binding to bacteria (the PBP), inhibition of bacterial cell wall synthesis, and is the only can oral carbapenem antibiotics.
[0005] Alternatively esters Bipei Nan structural characteristics, is a C3-side chain is thiazolyl substituted azetidinyl group, while in the C2 position by matching volts carboxylic ester forming a prodrug, increased oral absorbability; its oral absorption is better than in most β_ lactam antibiotics already on the market now.Bipei Nan for a broad spectrum antibiotic; especially for the PRSP in recent years, mainly due to the infection caused by children (penicillin-resistant Streptococcus pneumoniae), MRSP (erythromycin-resistant Streptococcus pneumoniae) and Haemophilusinfluenzae (Haemophilus influenzae) showed a very strong antibacterial effect.Alternatively Bipei Nan as prodrugs compared to for Bipei Nan horses volts for Bipei Nan ester having a better absorption kinetics, has good stability.
[0006] TakeshiIsoda like literature SynthesesandPharmacokineticStudiesof ProdrugEstersfortheDevelopmentofOralCarbapenem, L-084 (TheJournalof Antibiotics (2006) 59, 241 – 247; doi:. 10 · 1038 / ja 2006. 34) discloses a method for the synthesis of ester Bipei Nan : the Bipei Nan Alternatively, benzyltriethylammonium chloride, and chloromethyl pivalate was dissolved in N, N- dimethylformamide was added a solution of N, N- diisopropylethylamine, in the reaction was stirred for 4h at 45 ° C, the reaction was cooled to complete 5 ° C, was added ethyl acetate and water, the mixture was adjusted with aqueous citric acid I.OM PH = 4, the organic phase was discarded, the aqueous phase was adjusted with potassium bicarbonate to PH = 7. 6, the mixture extracted with ethyl acetate, the organic phase was washed with water and aqueous sodium chloride, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure, the residue was subjected to silica gel column to give a yellow solid which was slurried with ethyl acetate to give colorless crystals.The column chromatography method requires not only consume a large amount of organic solvent together IJ, and a long time, so this method is not suitable for industrial production. In addition, the resulting large solid slurried with ethyl acetate, a solid dispersion is not easy, affect the uniformity of the product.
[0007] Patent US5534510, EP0632039, JP10-195076 discloses a method for synthesizing Bipei Nan ester is: dissolved after lyophilization for Bipei Nan aqueous sodium bicarbonate, lyophilized solid was dissolved in N, N- two dimethylformamide, adding a specific acid, methyl iodide, LH stirred at room temperature, ethyl acetate was added the reaction was completed, the organic phase was washed with saturated aqueous sodium bicarbonate, washed with brine, dried over anhydrous magnesium sulfate, the solvent was removed, the residue was subjected Alternatively Bipei Nan silica gel column to give the ester.The Mr. Fang Fati Bipei Nan for Bipei Nan into the sodium salt of pivalic acid with methyl iodide, prior to lyophilization to remove water required for the reaction, or affect the subsequent reaction with methyl iodide pivalate, lyophilized difficult when enlarged and the operation will take longer, the method further column chromatography operations shall therefore not suitable for industrial production.
[0008] Chinese patent CN102633801A disclosed for the preparation of esters Bipei Nan, including the following steps of: for Bipei Nan I. 37g, N, N- 11 ml of dimethylformamide, potassium carbonate 0 · 5g, tetrabutylphosphonium bromide 0 · 03g, reaction at 0 · 5h -KTC, Stuttgart dropwise at this temperature, methyl iodide 〇.88g acid, ethyl acetate was added the reaction was completed Ilml insolubles were filtered off, the filtrate was washed with water 22ml, water Ilml phase was extracted once with ethyl acetate, the combined ethyl acetate, washed with water and ethyl acetate are added 11 ml of water, adjusted to 3.5 with aqueous citric acid, phases were separated, the aqueous phase washed with ethyl acetate Ilml with the aqueous phase is added acetic acid 22ml ethyl ester, was adjusted to 7.5 with aqueous sodium bicarbonate, phase separation, washed with water and 22ml ethyl acetate, the ethyl acetate phase over anhydrous sodium sulfate was added 0. 5g, dried decolorizing charcoal, the filtrate was concentrated to a volume, stirring crystallization, cooling to 〇~5 ° C followed by stirring crystallization, filtration, and dried to give a white solid 〇.54g.Pivalate methyl iodide using the method of low-temperature reaction, post-treatment operation complicated, solvent volume, increasing both cost and environmental pollution, and methyl iodide pivalate unstable, expensive, not suitable for industrial production.
Example 1
[0029] Alternatively the Bipei Nan 18g, N, N- dimethylformamide 162mL, potassium 6. 54g, tetrabutylammonium bromide (λ45g, the reaction 60min, was added dropwise at this temperature at 25 ° C for chloromethyl pivalate 8. 93g, after the completion of the reaction, water was added and stirred for 10 minutes, 320ml, 160ml ethyl acetate was added to extract the aqueous phase extracted with ethyl acetate and once with 160ml ethyl acetate were combined, washed with water, phase separation, ethyl acetate washed with water 640ml paint ester, the ethyl acetate phase over anhydrous sodium sulfate was added 60g, decolorizing charcoal and dried, and the filtrate was concentrated, was added dropwise 25 ° C under stirring for crystallization 162mL isopropyl ether, filtered and dried to give a white solid 17. 64g. purity by HPLC 99.87%, the yield was 89.7%.
[0030] TBPN1HNMR data (CDCl3):. 5 989-5 978ppm (1H, d, 5 5, H-13.), 5 858-5 847ppm (1H, d, 5 5, Η-13.)… , 4 · 436-4. 390ppm (2H, m, H-22e, H-24e), 4. 232-4. 217ppm (2H, m, H-2, H-9), 4. 173-4. 146ppm (1H, m, H-21), 4. 039-3. 960ppm (4H, m, H-22a, H-24a, H-28), 3. 402-3. 372ppm (2H, t, 7. 5 , H-27), 3. 243-3. 230ppm (1H, m, H-3), 3. 199-3. 167ppm (1H, m, H-8), I. 348-1. 337ppm (3H, d, 5. 5, H-1), I. 24ppm (12H, m, H-17, H-18, H-19, H-10) ·
Abe, T.; Hayashi, K.; Mihira, A.; Satoh, C.; Tamai, S.; Yamamoto, S.; Hikida, M.; Kumagai, T.; Kitamura, M.
L-084, a new oral carbapenem: Synthesis and structure-activity relationships of C2-substituted 1beta-methylcarbapenems
38th Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 24-27, San Diego) 1998, Abst F-64
CLIP
EP 0632039; EP 0717042; JP 1996053453; US 5534510; US 5659043; US 5783703
Halogenation of allylamine (I) with either bromine or sulfuryl chloride produced the corresponding (halomethyl)aziridines (II). Subsequent treatment of (II) with n-butyllithium at -78 C yielded 1-azabicyclobutane (III). Opening of the bicyclic system of (III) with formic acid followed by acid hydrolysis provided 3-hydroxyazetidine (IV). This was condensed with 2-(methylsulfanyl)thiazoline (V) to give thiazolinylazetidine (VI). Alternatively, 3-hydroxyazetidine (IV) was condensed with 2-chloroethyl isothiocyanate (VII) to give the intermediate thiourea (VIII), which cyclized to the thiazoline (VI). Conversion of the hydroxyl group of (VI) into the thioacetate (IX) was carried out by either coupling with thioacetic acid under Mitsunobu conditions or by conversion to mesylate (X) followed by displacement with potassium thioacetate. The required thiol (XI) was then obtained from (IX) by basic hydrolysis of the thioacetate ester.
1-azabicyclobutane (III) was opened with thioacetic acid with concomitant N-acetylation yielding (XII). Further acid hydrolysis of (XII) gave 3-mercaptoazetidine (XIII). Condensation of (XIII) with either 2-(methylthio)thiazoline (V) or 2-chloroethyl isothiocyanate (VII) then produced thiazolinylazetidine (XI).
A further procedure consisted in the opening of 1-azabicyclobutane (III) with 2-mercaptothiazoline (XIV) to give (XV). Subsequent rearrangement of (XV) in the presence of methanesulfonic acid produced thiazolinyl azetidine (XI).
Condensation of (phosphoryloxy)carbapenem (XVI) with 3-mercapto-1-(1,3-thiazolin-2-yl)azetidine (XI) gave thioether (XVII). The p-nitrobenzyl ester group of (XVII) was then deprotected with Zn powder to afford the target carboxylic acid.
Jump up^Kato, K.; Shirasaka, Y.; Kuraoka, E.; Kikuchi, A.; Iguchi, M.; Suzuki, H.; Shibasaki, S.; Kurosawa, T.; Tamai, I. (2010). “Intestinal Absorption Mechanism of Tebipenem Pivoxil, a Novel Oral Carbapenem: Involvement of Human OATP Family in Apical Membrane Transport”. Molecular Pharmaceutics. 7 (5): 1747–1756. doi:10.1021/mp100130b. PMID20735088.
Jump up^Sugita, R. (2013). “Good transfer of tebipenem into middle ear effusion conduces to the favorable clinical outcomes of tebipenem pivoxil in pediatric patients with acute otitis media”. Journal of Infection and Chemotherapy. 19 (3): 465–471. doi:10.1007/s10156-012-0513-5. PMID23393013.
Jump up^Rossi, S, ed. (7 August 2014). “Tebipenem Pivoxil”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 6 April 2015.
Mechanism of Action Sodium-bile acid cotransporter-inhibitors
Highest Development Phases
Phase II Primary biliary cirrhosis; Pruritus; Type 2 diabetes mellitus
Phase I Cholestasis
Most Recent Events
01 Jan 2017 Phase-II clinical trials in Pruritus in USA (PO) (NCT02966834)
14 Nov 2016 GlaxoSmithKline completes a phase I trial for Cholestasis in Healthy volunteers in Japan (PO, Tablet) (NCT02801981)
11 Nov 2016 Efficacy, safety and pharmacodynamic data from a phase II trial in Primary biliary cirrhosis and Pruritus presented at The Liver Meeting® 2016: 67th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD-2016)
GSK2330672 , an ileal bile acid transport (iBAT) inhibitor indicated for diabetes type II and cholestatic pruritus, is currently under Phase IIb evaluation in the clinic. The API is a highly complex molecule containing two stereogenic centers, one of which is quaternary
GSK-2330672 is highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor for treatment of type 2 diabetes.
More than 200 million people worldwide have diabetes. The World Health Organization estimates that 1 .1 million people died from diabetes in 2005 and projects that worldwide deaths from diabetes will double between 2005 and 2030. New chemical compounds that effectively treat diabetes could save millions of human lives.
Diabetes refers to metabolic disorders resulting in the body’s inability to effectively regulate glucose levels. Approximately 90% of all diabetes cases are a result of type 2 diabetes whereas the remaining 10% are a result of type 1 diabetes, gestational diabetes, and latent autoimmune diabetes of adulthood (LADA). All forms of diabetes result in elevated blood glucose levels and, if left untreated chronically, can increase the risk of macrovascular (heart disease, stroke, other forms of cardiovascular disease) and microvascular [kidney failure (nephropathy), blindness from diabetic retinopathy, nerve damage (diabetic neuropathy)] complications.
Type 1 diabetes, also known as juvenile or insulin-dependent diabetes mellitus (IDDM), can occur at any age, but it is most often diagnosed in children, adolescents, or young adults. Type 1 diabetes is caused by the autoimmune destruction of insulin-producing beta cells, resulting in an inability to produce sufficient insulin. Insulin controls blood glucose levels by promoting transport of blood glucose into cells for energy use. Insufficient insulin production will lead to decreased glucose uptake into cells and result in accumulation of glucose in the bloodstream. The lack of available glucose in cells will eventually lead to the onset of symptoms of type 1 diabetes: polyuria (frequent urination), polydipsia (thirst), constant hunger, weight loss, vision changes, and fatigue. Within 5-10 years of being diagnosed with type 1 diabetes, patient’s insulin-producing beta cells of the pancreas are completely destroyed, and the body can no longer produce insulin. As a result, patients with type 1 diabetes will require daily administration of insulin for the remainder of their lives.
Type 2 diabetes, also known as non-insulin-dependent diabetes mellitus (NIDDM) or adult-onset diabetes, occurs when the pancreas produces insufficient insulin and/or tissues become resistant to normal or high levels of insulin (insulin resistance), resulting in excessively high blood glucose levels. Multiple factors can lead to insulin resistance including chronically elevated blood glucose levels, genetics, obesity, lack of physical activity, and increasing age. Unlike type 1 diabetes, symptoms of type 2 diabetes are more salient, and as a result, the disease may not be diagnosed until several years after onset with a peak prevalence in adults near an age of 45 years. Unfortunately, the incidence of type 2 diabetes in children is increasing.
The primary goal of treatment of type 2 diabetes is to achieve and maintain glycemic control to reduce the risk of microvascular (diabetic neuropathy, retinopathy, or nephropathy) and macrovascular (heart disease, stroke, other forms of cardiovascular disease) complications. Current guidelines for the treatment of type 2 diabetes from the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD) [Diabetes Care, 2008, 31 (12), 1 ] outline lifestyle modification including weight loss and increased physical activity as a primary therapeutic approach for management of type 2 diabetes. However, this approach alone fails in the majority of patients within the first year, leading physicians to prescribe medications over time. The ADA and EASD recommend metformin, an agent that reduces hepatic glucose production, as a Tier 1 a medication; however, a significant number of patients taking metformin can experience gastrointestinal side effects and, in rare cases, potentially fatal lactic acidosis. Recommendations for Tier 1 b class of medications include sulfonylureas, which stimulate pancreatic insulin secretion via modulation of potassium channel activity, and exogenous insulin. While both medications rapidly and effectively reduce blood glucose levels, insulin requires 1 -4 injections per day and both agents can cause undesired weight gain and potentially fatal hypoglycemia. Tier 2a recommendations include newer agents such as thiazolidinediones (TZDs pioglitazone and rosiglitazone), which enhance insulin sensitivity of muscle, liver and fat, as well as GLP-1 analogs, which enhance postprandial glucose-mediated insulin secretion from pancreatic beta cells. While TZDs show robust, durable control of blood glucose levels, adverse effects include weight gain, edema, bone fractures in women, exacerbation of congestive heart failure, and potential increased risk of ischemic cardiovascular events. GLP-1 analogs also effectively control blood glucose levels, however, this class of medications requires injection and many patients complain of nausea. The most recent addition to the Tier 2 medication list is DPP-4 inhibitors, which, like GLP-1 analogs, enhance glucose- medicated insulin secretion from beta cells. Unfortunately, DPP-4 inhibitors only modestly control blood glucose levels, and the long-term safety of DPP-4 inhibitors remains to be firmly established. Other less prescribed medications for type 2 diabetes include a-glucosidase inhibitors, glinides, and amylin analogs. Clearly, new medications with improved efficacy, durability, and side effect profiles are needed for patients with type 2 diabetes.
GLP-1 and GIP are peptides, known as incretins, that are secreted by L and K cells, respectively, from the gastrointestinal tract into the blood stream following ingestion of nutrients. This important physiological response serves as the primary signaling mechanism between nutrient (glucose/fat) concentration in the
gastrointestinal tract and other peripheral organs. Upon secretion, both circulating peptides initiate signals in beta cells of the pancreas to enhance glucose-stimulated insulin secretion, which, in turn, controls glucose concentrations in the blood stream (For reviews see: Diabetic Medicine 2007, 24(3), 223; Molecular and Cellular Endocrinology 2009, 297(1-2), 127; Experimental and Clinical Endocrinology & Diabetes 2001 , 109(Suppl. 2), S288).
The association between the incretin hormones GLP-1 and GIP and type 2 diabetes has been extensively explored. The majority of studies indicate that type 2 diabetes is associated with an acquired defect in GLP-1 secretion as well as GIP action (see Diabetes 2007, 56(8), 1951 and Current Diabetes Reports 2006, 6(3), 194). The use of exogenous GLP-1 for treatment of patients with type 2 diabetes is severely limited due to its rapid degradation by the protease DPP-4. Multiple modified peptides have been designed as GLP-1 mimetics that are DPP-4 resistant and show longer half-lives than endogenous GLP-1 . Agents with this profile that have been shown to be highly effective for treatment of type 2 diabetes include exenatide and liraglutide, however, these agents require injection. Oral agents that inhibit DPP-4, such as sitagliptin vildagliptin, and saxagliptin, elevate intact GLP-1 and modestly control circulating glucose levels (see Pharmacology & Therapeutics 2010, 125(2), 328; Diabetes Care 2007, 30(6), 1335; Expert Opinion on Emerging Drugs 2008, 13(4), 593). New oral medications that increase GLP-1 secretion would be desirable for treatment of type 2 diabetes.
Bile acids have been shown to enhance peptide secretion from the
gastrointestinal tract. Bile acids are released from the gallbladder into the small intestine after each meal to facilitate digestion of nutrients, in particular fat, lipids, and lipid-soluble vitamins. Bile acids also function as hormones that regulate cholesterol homeostasis, energy, and glucose homeostasis via nuclear receptors (FXR, PXR, CAR, VDR) and the G-protein coupled receptor TGR5 (for reviews see: Nature Drug Discovery 2008, 7, 672; Diabetes, Obesity and Metabolism 2008, 10, 1004). TGR5 is a member of the Rhodopsin-like subfamily of GPCRs (Class A) that is expressed in intestine, gall bladder, adipose tissue, liver, and select regions of the central nervous system. TGR5 is activated by multiple bile acids with lithocholic and deoxycholic acids as the most potent activators {Journal of Medicinal Chemistry 2008, 51(6), 1831 ). Both deoxycholic and lithocholic acids increase GLP-1 secretion from an enteroendocrine STC-1 cell line, in part through TGR5
{Biochemical and Biophysical Research Communications 2005, 329, 386). A synthetic TGR5 agonist INT-777 has been shown to increase intestinal GLP-1 secretion in vivo in mice {Cell Metabolism 2009, 10, 167). Bile salts have been shown to promote secretion of GLP-1 from colonic L cells in a vascularly perfused rat colon model {Journal of Endocrinology 1995, 145(3), 521 ) as well as GLP-1 , peptide YY (PYY), and neurotensin in a vascularly perfused rat ileum model {Endocrinology 1998, 139(9), 3780). In humans, infusion of deoxycholate into the sigmoid colon produces a rapid and marked dose responsive increase in plasma PYY and enteroglucagon concentrations (Gi/M993, 34(9), 1219). Agents that increase ileal and colonic bile acid or bile salt concentrations will increase gut peptide secretion including, but not limited to, GLP-1 and PYY.
Bile acids are synthesized from cholesterol in the liver then undergo conjugation of the carboxylic acid with the amine functionality of taurine and glycine. Conjugated bile acids are secreted into the gall bladder where accumulation occurs until a meal is consumed. Upon eating, the gall bladder contracts and empties its contents into the duodenum, where the conjugated bile acids facilitate absorption of cholesterol, fat, and fat-soluble vitamins in the proximal small intestine (For reviews see: Frontiers in Bioscience 2009, 74, 2584; Clinical Pharmacokinetics 2002,
41(10), 751 ; Journal of Pediatric Gastroenterology and Nutrition 2001 , 32, 407). Conjugated bile acids continue to flow through the small intestine until the distal ileum where 90% are reabsorbed into enterocytes via the apical sodium-dependent bile acid transporter (ASBT, also known as iBAT). The remaining 10% are deconjugated to bile acids by intestinal bacteria in the terminal ileum and colon of which 5% are then passively reabsorbed in the colon and the remaining 5% being excreted in feces. Bile acids that are reabsorbed by ASBT in the ileum are then transported into the portal vein for recirculation to the liver. This highly regulated process, called enterohepatic recirculation, is important for the body’s overall maintenance of the total bile acid pool as the amount of bile acid that is synthesized in the liver is equivalent to the amount of bile acids that are excreted in feces.
Pharmacological disruption of bile acid reabsorption with an inhibitor of ASBT leads to increased concentrations of bile acids in the colon and feces, a physiological consequence being increased conversion of hepatic cholesterol to bile acids to compensate for fecal loss of bile acids. Many pharmaceutical companies have pursued this mechanism as a strategy for lowering serum cholesterol in patients with dyslipidemia/hypercholesterolemia (For a review see: Current Medicinal Chemistry 2006, 73, 997). Importantly, ASBT-inhibitor mediated increase in colonic bile acid/salt concentration also will increase intestinal GLP-1 , PYY, GLP-2, and other gut peptide hormone secretion. Thus, inhibitors of ASBT could be useful for treatment of type 2 diabetes, type 1 diabetes, dyslipidemia, obesity, short bowel syndrome, Chronic Idiopathic Constipation, Irritable bowel syndrome (IBS), Crohn’s disease, and arthritis.
Certain 1 ,4-thiazepines are disclosed, for example in WO 94/18183 and WO 96/05188. These compounds are said to be useful as ileal bile acid reuptake inhibitors (ASBT).
Patent publication WO 201 1/137,135 dislcoses, among other compounds, the following compound. This patent publication also discloses methods of synthesis of the compound.
The preparation of the above compound is also disclosed in J. Med. Chem, Vol 56, pp5094-51 14 (2013).
Patent publication WO 201 1/137,135 dislcoses general methods for preparing the compound. In addition, a detailed synthesis of the compound is disclosed in Example 26. J. Med. Chem, Vol 56, pp5094-51 14 (2013) also discloses a method for synthesising the compound.
The present invention discloses an improved synthesis of the compound.
The synthetic scheme of the present invention is depicted in Scheme 1 .
Treatment of 2-methoxyphenyl acetate with sulfur monochloride followed by ester hydrolysis and reduction with zinc gave rise to thiophenol (A). Epoxide ring opening of (+)-2-butyl-ethyloxirane with thiophenol (A) and subsequent treatment of tertiary alcohol (B) with chloroacetonitrile under acidic conditions gave chloroacetamide (C), which was then converted to intermediate (E) by cleavage of the chloroacetamide with thiourea followed by classical resolution with dibenzoyl-L-tartaric acid.
Benzoylation of intermediate (E) with triflic acid and benzoyl chloride afforded intermediate (H). Cyclization of intermediate (H) followed by oxidation of the sulfide to a sulphone, subseguent imine reduction and classical resolution with (+)-camphorsulfonic acid provided intermediate (G), which was then converted to intermediate (H). Intermediate (H) was converted to the target compound using the methods disclosed in Patent publication WO 201 1/137,135.
Scheme 1
Dibenzoyl-L-tataric acid
The present invention also discloses an alternative method for construction of the quaternary chiral center as depicted in Scheme 2. Reaction of intermediate (A) with (R)-2-ammonio-2-ethylhexyl sulfate (K) followed by formation of di-p-toluoyl-L-tartrate salt furnished intermediate (L).
The present invention also discloses an alternative synthesis of intermediate (H) as illustrated in Scheme 3. Acid catalyzed cyclization of intermediate (F) followed by triflation gave imine (M), which underwent asymmetric reduction with catalyst lr(COD)2BArF and ligand (N) to give intermediate (O). Oxidation of the sulfide in intermediate (O) gave sulphone intermediate (H).
The present invention differs from the synthesis disclosed in WO 201 1/137,135 and J. Med. Chem, Vol56, pp5094-51 14 (2013) in that intermediate (H) in the present invention is prepared via a new, shorter and more cost-efficient synthesis while the synthesis of the target compound from intermediate (H) remains unchanged.
Intermediate A: 3-Hydroxy-4-methoxythiophenol
A reaction vessel was charged with 2-methoxyphenyl acetate (60 g, 0.36 mol), zinc chloride (49.2 g, 0.36 mol) and DME (600 mL). The mixture was stirred and S2CI2 (53.6 g, 0.40 mol) was added. The mixture was stirred at ambient temperature for 2 h. Concentrated HCI (135.4 mL, 1 .63 mol) was diluted with water (60 mL) and added slowly to the rxn mixture, maintaining the temperature below 60 °C. The mixture was stirred at 60 °C for 2 h and then cooled to ambient
temperature. Zinc dust (56.7 g, 0.87 mol) was added in portions, maintaining the temperature below 60 °C. The mixture was stirred at 20-60 °C for 1 h and then concentrated under vacuum to -300 mL. MTBE (1 .2 L) and water (180 mL) were added and the mixture was stirred for 10 min. The layers were separated and the organic layer was washed twice with water (2x 240 mL). The layers were separated and the organic layer was concentrated under vacuum to give an oil. The oil was distilled at 1 10-1 15 °C/2 mbar to give the title compound (42 g, 75%) as colorless oil, which solidified on standing to afford the title compound as a white solid. M.P. 41 -42 °C. 1 H NMR (500 MHz, CDCI3)$ ppm 3.39 (s, 1 H), 3.88 (s, 3H), 5.65 (br. S, 1 H), 6.75 (d, J – 8.3 Hz, 1 H), 6.84 (ddd, J – 8.3, 2.2, 0.6 Hz, 1 H), 6.94 (d, J – 2.2 Hz).
Intermediate E: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt
A reaction vessel was charged with 3-hydroxy-4-methoxythiophenol (5.0 g, 25.2 mmol), (+)-2-butyl-2-ethyloxirane (3.56 g, 27.7 mmol) and EtOH (30 mL). The mixture was treated with a solution of NaOH (2.22 g, 55.5 mmol) in water (20 mL), heated to 40 °C and stirred at 40 °C for 5 h. The mixture was cooled to ambient temperature, treated with toluene (25 mL) and stirred for 10 min. The layers were separated and the organic layer was discarded. The aqueous layer was neutralized with 2N HCI (27.8 mL, 55.6 mmol) and extracted with toluene (100 mL). The organic layer was washed with water (25 mL), concentrated in vacuo to give an oil. The oil was treated with chloroacetonitrile (35.9 mL) and HOAc (4.3 mL) and cooled to 0 °C. H2SO4 (6.7 mL, 126 mmol, pre-diluted with 2.3 mL of water) was added at a rate maintaining the temperature below 10 °C. After stirred at 0 °C for 0.5 h, the reaction mixture was treated with 20% aqueous Na2CO3 solution to adjust the pH to
7-8 and then extracted with MTBE (70 ml_). The extract was washed with water (35 ml_) and concentrated in vacuo to give an oil. The oil was then dissolved in EOH (50 ml_) and treated with HOAc (10 ml_) and thiourea (2.30 g, 30.2 mmol). The mixture was heated at reflux overnight and then cooled to ambient temperature. The solids were filtered and washed with EtOH (10 ml_). The filtrate and the wash were combined and concentrated in vacuo, treated with MTBE (140 ml_) and washed successively with 10% aqueous Na2C03 and water. The mixture was concentrated in vacuo to give an oil. The oil was dissolved in MeCN (72 ml_), heated to -50 °C and then dibenzoyl-L-tartaric acid (9.0 g, 25.2 mmol) in acetonitrile (22 ml_) was added slowly. Seed crystals were added at -50 °C. The resultant slurry was stirred at 45-50 °C for 5 h, then cooled down to ambient temperature and stirred at ambient temperature overnight. The solids were filtered and washed with MeCN (2x 22 ml_). The wet cake was treated with MeCN (150 ml_) and heated to 50 °C. The slurry was stirred at 50 °C for 5 h, cooled over 1 h to ambient temperature and stirred at ambient temperature overnight. The solids were collected by filtration, washed with MeCN (2 x 20 ml_), dried under vacuum to give the title compound (5.5 g, 34% overall yield, 99.5% purity, 93.9% ee) as a white solid. 1 H NMR (500 MHz, DMSO-d6) δ ppm 0.78 (m, 6H), 1 .13 (m, 4H), 1 .51 (m, 2H), 1 .58 (q, J – 7.7 Hz, 2H), 3.08 (s, 2H), 3.75 (s, 3H), 5.66 (s, 2H), 6.88 (m, 2H), 6.93 (m, 1 H), 7.49 (m, 4H), 7.63 (m, 2H), 7.94 (m, 4H). EI-LCMS m/z 284 (M++1 of free base).
A suspension of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, dibenzoyl-L-tartrate salt (29 g, 45.2 mmol) in DCM (435 mL) was treated with water (1 16 mL) and 10% aqueous Na2C03 solution (1 16 mL). The mixture was stirred at ambient temperature until all solids were dissolved (30 min). The layers were separated. The organic layer was washed with water (2 x 60 mL), concentrated under vacuum to give (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) as an off-white solid (13.0 g, quantitative). A vessel was charged with TfOH (4.68 ml, 52.9 mmol) and DCM (30 mL) and the mixture was cooled to 0 °C. 5 g (17.6 mmol) of (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol (free base) was dissolved in DCM (10 mL) and added at a rate maintaining the temperature below 10 °C. Benzoyl chloride (4.5 mL, 38.8 mmol) was added at a rate maintaining the temperature below 10 °C. The mixture was then heated to reflux and stirred at reflux for 48 h. The mixture was cooled to 30 °C. Water (20 mL) was added and the mixture was concentrated to remove DCM. EtOH (10 mL) was added. The mixture was heated to 40 ° C, treated with 50% aqueous NaOH solution (10 mL) and stirred at 55 °C. After 1 h, the mixture was cooled to ambient temperature and the pH was adjusted to 6-7 with cone. HCI. The mixture was concentrated in vacuo to remove EtOH. EtOAc (100 mL) was added. The mixture was stirred for 5 min and the layers were separated. The organic layer was washed successively with 10% aqueous Na2CO3 (25 mL) and water (25 mL) and then concentrated in vacuo. The resultant oil was treated with DCM (15 mL). The resultant thick slurry was further diluted with DCM (15 mL) followed by addition of Hexanes (60 mL). The slurry was stirred for 5 min, filtered, washed with DCM/hexanes (1 :2, 2 x 10 mL) and dried under vacuum to give the title compound (7.67 g, 80%) as a yellow solid. 1 NMR (500 MHz, DMSO-d6) δ ppm 0.70 (t, 7.1 Hz, 3 H), 0.81 (t, 7.1 Hz, 3H), 1 .04-1 .27 (m, 8H), 2.74 (s, 2H), 3.73 (s, 3H), 6.91 (s, 1 H), 7.01 (s, 1 H), 7.52 (dd, J – 7.8, 7.2 Hz, 2H), 7.63 (t, J = 7.2 Hz, 1 H), 7.67 (d, J = 7.8 Hz, 2H). EI-LCMS m/z 388 (M++1 ).
Intermediate G: (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt
A vessel was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (1 .4 g, 3.61 mmol), toluene (8.4 ml_) and citric acid (0.035 g, 0.181 mmol, 5 mol%). The mixture was heated to reflux overnight with a Dean-Stark trap to remove water. The mixture was concentrated under reduced pressure to remove solvents. Methanol (14.0 ml_) and oxone (2.22 g, 3.61 mmol, 1 .0 equiv) were added. The mixture was stirred at gentle reflux for 2 h. The mixture was cooled to ambient temperature, and filtered to remove solids. The filter cake was washed with small amount of Methanol. The filtrate was cooled to 5 °C, and treated with sodium borohydride (0.410 g, 10.84 mmol, 3.0 equiv.) in small portions. The mixture was stirred at 5 °C for 2 h and then concentrated to remove the majority of solvents. The mixture was quenched with Water (28.0 ml_) and extracted with EtOAc (28.0 ml_). The organic layer was washed with brine, and then concentrated to remove solvents. The residue was dissolved in MeCN (14.0 ml_) and concentrated again to remove solvents. The residue was dissolved in MeCN (7.00 ml_) and MTBE (7.00 ml_) at 40 °C, and treated with (+)-camphorsulfonic acid (0.839 g, 3.61 mmol, 1 .0 equiv.) at 40 °C for 30 min. The mixture was cooled to ambient temperature, stirred for 2 h, and filtered to collect solids. The filter cake was washed with MTBE/MeCN (2:1 , 3 ml_), and dried at 50 °C to give the title compound (0.75 g, 32% overall yield, 98.6 purity, 97% de, 99.7% ee) as white solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.63 (s, 3H), 0.88 (t, J – 6.9 Hz, 3H), 0.97 (m, 6H), 1 .29-1 .39 (m, 5H), 1 .80-1 .97 (m, 6H), 2.08-2.10 (m, 1 H), 2.27 (d, J – 17.3 Hz, 1 H), 2.38-2.44 (m, 3H), 2.54 (b, 1 H), 2.91 (b, 1 H), 3.48 (d, J – 15.4 Hz, 1 H), 3.79 (s, 3H), 4.05 (d, J – 17.2 Hz, 1 H), 6.45 (s, 1 H), 6.56 (s, 1 H), 7.51 -7.56 (m, 4H), 7.68 (s, 1 H), 7.79 (b, 2H), 1 1 .46 (b, 1 H). EI-LCMS m/z 404 (M++1 of free base).
Method 1 : A mixture of (3R,5R)-3-butyl-3-ethyl-8-hydroxy-7-methoxy-5-phenyl-2,3,4,5-tetrahydrobenzo[f][1 ,4]thiazepine 1 ,1 -dioxide, (+)-camphorsulfonate salt (0.5 g, 0.786 mmol), EtOAc (5.0 mL), and 10% of Na2C03 aqueuous solution (5 mL) was stirred for 15 min. The layers were separated and the aqueous layer was discarded. The organic layer was washed with dilute brine twice, concentrated to remove solvents. EtOAc (5.0 mL) was added and the mixture was concentrated to give a pale yellow solid free base. 1 ,4-Dioxane (5.0 mL) and pyridine (0.13 mL, 1 .57 mmol) were added. The mixture was cooled to 5-10 °C and triflic anhydride (0.199 mL, 1 .180 mmol) was added while maintaining the temperature below 15 °C. The mixture was stirred at ambient temperature until completion deemed by HPLC (1 h). Toluene (5 mL) and water (5 mL) were added. Layers were separated. The organic layer was washed with water, concentrated to remove solvents. Toluene (1 .0 mL) was added to dissolve the residue followed by Isooctane (4.0 mL). The mixture was stirred at rt overnight. The solids was filtered, washed with Isooctane (4.0 mL) to give the title compound (0.34 g, 81 %) as slightly yellow solids. 1 NMR (400 MHz, CDCI3) δ ppm 0.86 (t, J – 7.2 Hz, 3H), 0.94 (t, J – 7.6 Hz, 3H), 1 .12-1 .15 (m, 1 H), 1 .22-1 .36 (m, 3H), 1 .48-1 .60 (m, 2H), 1 .86-1 .93 (m, 2H), 2.22 (dt, J = 4.1 Hz, 12 Hz, 1 H), 3.10 (d, J – 14.8 Hz, 1 H), 3.49 (d, J – 14.8 Hz, 1 H), 3.64 (s, 3H), 6.1 1 (s, 1 H), 6.36 (s, 1 H), 7.38-7.48 (m, 5), 7.98 (s, 1 H).
Method 2: A mixture of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl trifluoromethanesulfonate (0.5 g, 0.997 mmol), ligand (N) (0.078 g, 0.1 10 mmol) and lr(COD)2BArF (0.127 g, 0.100 mmol) in DCM (10.0 mL) was purged with nitrogen three times, then hydrogen three times. The mixture was shaken in Parr shaker under 10 Bar of H2 for 24 h. The experiment described above was repeated with 1 .0 g (1 .994 mmol) input of (R)-3-butyl-3-ethyl-7-methoxy-5-phenyl-2,3-dihydrobenzo[f][1 ,4]thiazepin-8-yl
trifluoromethanesulfonate. The two batches of the reaction mixture were combined,
concentrated to remove solvents, and purified by silica gel chromatography
(hexanes:EtOAc =9:1 ) to give the sulfide (O) as slightly yellow oil (0.6 g, 40% yield, 99.7% purity). The oil (0.6 g, 1 .191 mmol) was dissolved in TFA (1 .836 mL, 23.83 mmol) and stirred at 40 °C. H202 (0.268 mL, 2.62 mmol) was added slowly over 30 min. The mixture was stirred at 40 °C for 2 h and then cooled to room temperature. Water (10 mL) and toluene (6.0 mL) were added. Layers were separated and the organic layer was washed successively with aqueous sodium carbonate solution and wate, and concentrated to dryness. Toluene (6.0 mL) was added and the mixture was concentrated to dryness. The residue was dissolved in toluene (2.4 mL) and isooctane (7.20 mL) was added. The mixture was heated to reflux and then cooled to room temperature. The mixture was stirred at room temperature for 30 min. The solid was filtered and washed with isooctane to give the title compound (0.48 g, 75%).
Intermediate L: (R)-5-((2-amino-2-ethylhexyl)thio)-2-methoxyphenol, di-p-toluoyl-L-tartrate salt
To a mixture of (R)-2-amino-2-ethylhexyl hydrogen sulfate (1 1 .1 g, 49.3 mmol) in water (23.1 mL) was added NaOH (5.91 g, 148 mmol). The mixture was stirred at reflux for 2 h. The mixture was cooled to room temperature and extracted with MTBE (30.8 mL). The extract was washed with brine (22 mL), concentrated under vacuum and treated with methanol (30.8 mL). The mixture was stirred under nitrogen and treated with 3-hydroxy-4-methoxythiophenol (7.70 g, 49.3 mmol). The mixture was stirred under nitrogen at room temperature for 1 h. The mixture was concentrated under vacuum, treated with acetonitrile (154 mL) and then heated to 45 °C. To the stirred mixture was added (2R,3R)-2,3-bis((4-methylbenzoyl)oxy)succinic acid (19.03 g, 49.3 mmol). The resultant slurry was
stirred at 45 °C. After 2 h, the slurry was cooled to room temperature and stirred for 5 h. The solids were filtered, washed twice with acetonitrile (30 mL) and dried to give the title compound (28.0 g, 85%) as white solids. 1 NMR (400 MHz, DMSO-d6) δ (ppm): 0.70-0.75 (m, 6H), 1 .17 (b, 4H), 1 .46-1 .55 (m, 4H), 2.30 (s, 6H), 3.71 (s, 3H), 5.58 (s, 2H), 6.84 (s, 2H), 6.89 (s, 1 H), 7.24 (d, J – 1 1 .6 Hz, 4H), 7.76 (d, J – 1 1 .6 Hz, 4H).
A flask was charged with (R)-(2-((2-amino-2-ethylhexyl)thio)-4-hydroxy-5-methoxyphenyl)(phenyl)methanone (3.5 g, 9.03 mmol), citric acid (0.434 g, 2.258 mmol), 1 ,4-Dioxane (17.50 mL) and Toluene (17.50 mL). The mixture was heated to reflux with a Dean-Stark trap to distill water azetropically. The mixture was refluxed for 20 h and then cooled to room temperature. EtOAc (35.0 mL) and water (35.0 mL) were added and layers were separated. The organic layer was washed with aqueous sodium carbonate solution and concentrated to remove solvents to give crude imine as brown oil. The oil was dissolved in EtOAc (35.0 mL) and cooled to 0-5 °C. To the mixture was added triethylamine (1 .888 mL, 13.55 mmol) followed by slow addition of Tf2O (1 .831 mL, 10.84 mmol) at 0-5 °C. The mixture was stirred at room temperature for 1 h. Water was added and layers were separated. The organic layer was washed with brine, dried over Na2SO4 and concentrated under vacuum. The crude triflate was purified by silica gel chromatography
Journal of Medicinal Chemistry (2013), 56(12), 5094-5114.
The apical sodium-dependent bile acid transporter (ASBT) transports bile salts from the lumen of the gastrointestinal (GI) tract to the liver via the portal vein. Multiple pharmaceutical companies have exploited the physiological link between ASBT and hepatic cholesterol metabolism, which led to the clinical investigation of ASBT inhibitors as lipid-lowering agents. While modest lipid effects were demonstrated, the potential utility of ASBT inhibitors for treatment of type 2 diabetes has been relatively unexplored. We initiated a lead optimization effort that focused on the identification of a potent, nonabsorbable ASBT inhibitor starting from the first-generation inhibitor 264W94 (1). Extensive SAR studies culminated in the discovery of GSK2330672 (56) as a highly potent, nonabsorbable ASBT inhibitor which lowers glucose in an animal model of type 2 diabetes and shows excellent developability properties for evaluating the potential therapeutic utility of a nonabsorbable ASBT inhibitor for treatment of patients with type 2 diabetes.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Example 26: 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid
Method 1 , Step 1 : To a solution of (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (683 mg, 1 .644 mmol) in 1 ,2-dichloroethane (20 mL) was added diethyl 3- aminopentanedioate (501 mg, 2.465 mmol) and acetic acid (0.188 mL, 3.29 mmol). The reaction mixture was stirred at room temperature for 1 hr then treated with NaHB(OAc)3 (697 mg, 3.29 mmol). The reaction mixture was then stirred at room temperature overnight and quenched with aqueous potassium carbonate solution. The mixture was extracted with DCM. The combined organic layers were washed with H2O, saturated brine, dried (Na2SO4), filtered, and concentrated under reduced pressure to give diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioate (880 mg, 88%) as a light yellow oil: MS-LCMS m/z 603 (M+H)+.
Method 1 , Step 2: To a solution of diethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7- (methyloxy)-l ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (880 mg, 1 .460 mmol) in a 1 :1 :1 mixture of
THF/MeOH/H2O (30 mL) was added lithium hydroxide (175 mg, 7.30 mmol). The reaction mixture was stirred at room temperature overnight then concentrated under reduced pressure. H2O and MeCN was added to dissolve the residue. The solution was acidified with acetic acid to pH 4-5, partially concentrated to remove MeCN under reduced pressure, and left to stand for 30 min. The white precipitate was collected by filtration and dried under reduced pressure at 50°C overnight to give the title compound (803 mg, 100%) as a white solid: 1 H NMR (MeOH-d4) δ ppm 8.05 (s, 1 H), 7.27 – 7.49 (m, 5H), 6.29 (s, 1 H), 6.06 (s, 1 H), 4.25 (s, 2H), 3.60 – 3.68 (m, 1 H), 3.58 (s, 3H), 3.47 (d, J = 14.8 Hz, 1 H), 3.09 (d, J = 14.8 Hz, 1 H), 2.52 – 2.73 (m, 4H), 2.12 – 2.27 (m, 1 H), 1 .69 – 1 .84 (m, 1 H), 1 .48 – 1 .63 (m, 1 H), 1 .05 – 1 .48 (m, 5H), 0.87 (t, J = 7.4 Hz, 3H), 0.78 (t, J = 7.0 Hz, 3H); ES-LCMS m/z 547 (M+H) Method 2: A solution of dimethyl 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-
1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioate (~ 600 g) in THF (2.5 L) and MeOH (1 .25 L) was cooled in an ice-bath and a solution of NaOH (206 g, 5.15 mol) in water (2.5 L) was added dropwise over 20 min (10-22°C reaction temperature). After stirring 20 min, the solution was concentrated (to remove THF/MeOH) and acidified to pH~4 with concentrated HCI. The precipitated product was aged with stirring, collected by filtration and air dried overnight. A second 600g batch of dimethyl 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioate was saponified in a similar fashion. The combined crude products (~2 mol theoretical) were suspended in CH3CN (8 L) and water (4 L) and the stirred mixture was heated to 65°C. A solution formed which was cooled to 10°C over 2 h while seeding a few times with an authentic sample of the desired crystalline product. The resulting slurry was stirred at 10°C for 2 h, and the solid was collected by filtration. The filter cake was washed with water and air-dried overnight. Further drying to constant weight in a vacuum oven at 55°C afforded crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid as a white solid (790 g).
Method 3: (3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro- 1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 -dioxide (1802 grams, 4.336 moles) and dimethyl 3-aminopentanedioate (1334 grams, 5.671 moles) were slurried in iPrOAc (13.83 kgs). A nitrogen atmosphere was applied to the reactor. To the slurry at 20°C was added glacial acetic acid (847 ml_, 14.810 moles), and the mixture was stirred until complete dissolution was observed. Solid sodium triacetoxyborohydride (1424 grams, 6.719 moles) was next added to the reaction over a period of 7 minutes. The reaction was held at 20°C for a total of 3 hours at which time LC analysis of a sample indicated complete consumption of the (3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepine-8-carbaldehyde 1 ,1 – dioxide. Next, water (20.36 kgs) and brine (4.8 kgs) were added to the reactor. The contents of the reactor were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. A previously prepared, 10% (wt/wt) aqueous solution of sodium bicarbonate (22.5 L) was added to the reactor. The contents were stirred for 10 minutes and then settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. To the reactor was added a second wash of 10% (wt/wt) aqueous, sodium bicarbonate
(22.5 L). The contents of the reactor were stirred for 10 minutes and settled for 10 minutes. The bottom, aqueous layer was then removed and sent to waste. The contents of the reactor were then reduced to an oil under vacuum distillation. To the oil was added THF (7.15 kgs) and MeOH (3.68 kgs). The contents of the reactor were heated to 55°C and agitated vigorously until complete dissolution was observed. The contents of the reactor were then cooled to 25°C whereupon a previously prepared aqueous solution of NaOH [6.75 kgs of water and 2.09 kgs of NaOH (50% wt wt solution)] was added with cooling being applied to the jacket. The contents of the reactor were kept below 42°C during the addition of the NaOH solution. The temperature was readjusted to 25°C after the NaOH addition, and the reaction was stirred for 75 minutes before HPLC analysis indicated the reaction was complete. Heptane (7.66 kgs) was added to the reactor, and the contents were stirred for 10 minutes and then allowed to settle for 10 minutes. The aqueous layer was collected in a clean nalgene carboy. The heptane layer was removed from the reactor and sent to waste. The aqueous solution was then returned to the reactor, and the reactor was prepared for vacuum distillation. Approximately 8.5 liters of distillate was collected during the vacuum distillation. The vacuum was released from the reactor, and the temperature of the contents was readjusted to 25°C. A 1 N HCI solution (30.76 kgs) was added to the reactor over a period of 40 minutes. The resulting slurry was stirred at 25°C for 10 hours then cooled to 5°C over a period of 2 hours. The slurry was held at 5°C for 4 hours before the product was collected in a filter crock by vacuum filtration. The filter cake was then washed with cold (5°C) water (6 kgs). The product cake was air dried in the filter crock under vacuum for approximately 72 hours. The product was then transferred to three drying trays and dried in a vacuum oven at 50°C for 79 hours. The temperature of the vacuum oven was then raised to 65°C for 85 additional hours. The product was off-loaded as a single batch to give 2568 grams (93.4% yield) of intermediate grade 3-({[(3R,5R)-3- butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4- benzothiazepin-8-yl]methyl}amino)pentanedioic acid as an off-white solid.
Intermediate grade 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5- phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid was dissolved (4690 g) in a mixture of glacial acetic acid (8850 g) and purified water (4200 g) at 70°C. The resulting solution was transferred through a 5 micron polishing filter while maintaining the temperature above 30°C. The reactor and filter were rinsed through with a mixture of glacial acetic acid (980 g) and purified water (470 g). The solution temperature was adjusted to 50°C. Filtered purified water (4230 g) was added to the solution. The cloudy solution was then seeded with crystalline 3-({[(3 5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3 ,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (10 g). While maintaining the temperature at 50°C, filtered purified water was charged to the slurry at a controlled rate (1 1030 g over 130 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (20740 g over 100 minutes). A final charge of filtered purified water (3780 g) was made to the slurry. The slurry was then cooled to 10°C at a linear rate over 135 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered ethyl acetate (17280 g) then the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 50°C for 23 hours. The temperature was then increased to 60°C and drying was continued for an additional 24 hours to afford crystalline 3-({[(3R,5R)-3-butyl-3-ethyl- 7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3740 g, 79.7% yield) as a white solid.
To a slurry of this crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 – dioxido-5-phenyl-2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8- yl]methyl}amino)pentanedioic acid (3660 g) and filtered purified water (3.6 L) was added filtered glacial acetic acid (7530 g). The temperature was increased to 60°C and full dissolution was observed. The temperature was reduced to 55°C, filtered, and treated with purified water (3.2 L). The solution was then seeded with crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl-2,3,4,5- tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (18 g) to afford a slurry. Filtered purified water was charged to the slurry at a controlled rate (9 L over 140 minutes). Additional filtered purified water was then added to the slurry at a faster controlled rate (18 L over 190 minutes). The slurry was then cooled to
10°C at a linear rate over 225 minutes. The solids were filtered over sharkskin filter paper to remove the mother liquor. The cake was then rinsed with filtered purified water (18 L), and the wash liquors were removed by filtration. The resulting wetcake was isolated into trays and dried under vacuum at 60°C for 18.5 hours to afford a crystalline 3-({[(3R,5R)-3-butyl-3-ethyl-7-(methyloxy)-1 ,1 -dioxido-5-phenyl- 2,3,4,5-tetrahydro-1 ,4-benzothiazepin-8-yl]methyl}amino)pentanedioic acid (3330 g, 90.8% yield) as a white solid which was analyzed for crystallinity as summarized below.
Paper
Cowan, D. J.; Collins, J. L.; Mitchell, M. B.; Ray, J. A.; Sutton, P. W.; Sarjeant, A. A.; Boros, E. E.Enzymatic- and Iridium-Catalyzed Asymmetric Synthesis of a Benzothiazepinylphosphonate Bile Acid Transporter InhibitorJ. Org. Chem.2013, 78 ( 24) 12726– 12734, DOI: 10.1021/jo402311e
A synthesis of the benzothiazepine phosphonic acid 3, employing both enzymatic and transition metal catalysis, is described. The quaternary chiral center of 3 was obtained by resolution of ethyl (2-ethyl)norleucinate (4) with porcine liver esterase (PLE) immobilized on Sepabeads. The resulting (R)-amino acid (5) was converted in two steps to aminosulfate 7, which was used for construction of the benzothiazepine ring. Benzophenone 15, prepared in four steps from trimethylhydroquinone 11, enabled sequential incorporation of phosphorus (Arbuzov chemistry) and sulfur (Pd(0)-catalyzed thiol coupling) leading to mercaptan intermediate 18. S-Alkylation of 18 with aminosulfate 7 followed by cyclodehydration afforded dihydrobenzothiazepine 20. Iridium-catalyzed asymmetric hydrogenation of 20 with the complex of [Ir(COD)2BArF] (26) and Taniaphos ligand P afforded the (3R,5R)-tetrahydrobenzothiazepine 30 following flash chromatography. Oxidation of 30 to sulfone 31 and phosphonate hydrolysis completed the synthesis of 3 in 12 steps and 13% overall yield.
Paper
Scheme 1. Current Route to Chiral Intermediate 4 in the Synthesis of GSK2330672
Development of an Enzymatic Process for the Production of (R)-2-Butyl-2-ethyloxirane
†Synthetic Biochemistry, Advanced Manufacturing Technologies, ‡API Chemistry, ∥Protein and Cellular Sciences, GlaxoSmithKline, Medicines Research Centre, Gunnels Wood Road, Stevenage SG1 2NY, United Kingdom
§API Chemistry, ⊥Synthetic Biochemistry, Advanced Manufacturing Technologies, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, Pennsylvania 19406, United States
# Biotechnology and Environmental Shared Service, Global Manufacturing and Supply, GlaxoSmithKline, Dominion Way, Worthing BN14 8PB, United Kingdom
∇ Molecular Design, Computational and Modeling Sciences, GlaxoSmithKline, 1250 S. Collegeville Road, Collegeville, Pennsylvania 19426, United States
An epoxide resolution process was rapidly developed that allowed access to multigram scale quantities of (R)-2-butyl-2-ethyloxirane 2 at greater than 300 g/L reaction concentration using an easy-to-handle and store lyophilized powder of epoxide hydrolase from Agromyces mediolanus. The enzyme was successfully fermented on a 35 L scale and stability increased by downstream processing. Halohydrin dehalogenases also gave highly enantioselective resolution but were shown to favor hydrolysis of the (R)-2 epoxide, whereas epoxide hydrolase from Aspergillus nigerinstead provided (R)-7 via an unoptimized, enantioconvergent process.
REFERENCES
1: Nunez DJ, Yao X, Lin J, Walker A, Zuo P, Webster L, Krug-Gourley S, Zamek-Gliszczynski MJ, Gillmor DS, Johnson SL. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab. 2016 Jul;18(7):654-62. doi: 10.1111/dom.12656. Epub 2016 Apr 21. PubMed PMID: 26939572.
2: Wu Y, Aquino CJ, Cowan DJ, Anderson DL, Ambroso JL, Bishop MJ, Boros EE, Chen L, Cunningham A, Dobbins RL, Feldman PL, Harston LT, Kaldor IW, Klein R, Liang X, McIntyre MS, Merrill CL, Patterson KM, Prescott JS, Ray JS, Roller SG, Yao X, Young A, Yuen J, Collins JL. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem. 2013 Jun 27;56(12):5094-114. doi: 10.1021/jm400459m. Epub 2013 Jun 6. PubMed PMID: 23678871.
Form I may be prepared according to the procedures in WO2011/137135, Example 26.
Method 2
Form I of linerixibat was prepared according to the following procedure at a large scale (> 500 g). All charges were based on input linerixibat.
An intermediate grade linerixibat was dissolved in acetonitrile / Water (12 vols / 8 vols) at reflux (~76 °C). The solution was seeded at 70°C with Form I (2% w/w), cooled to 60 °C over 15 mins and aged at 60 °C for 2 hrs. Water (14 vols) was added over 8 hrs and aged for 1 hr. The suspension was cooled to 20 °C over 1 hr and aged for >30 mins. The slurry was filtered, washed with acetonitrile: water (6:11 v/v) (3.5 vols), then twice with water (2 vols) and blown down with nitrogen 8-18 hrs. Drying was carried out under vacuum at 40-50 °C without agitation until the Karl Fisher measurement (KF) was < 10% w/w. The batch was agitated at 4 rpm for 2 mins every 3 hrs until the KF <1 % w/w to provide Form I.
Form I can also be prepared by the above procedure without the step of seeding. Method 3
Form I of linerixibat was prepared according to the following procedure at a large scale (> 50 kg). A reactor (Reactor 1) was charged with 55.84 kg GSK2330672B (1.0 wt) intermediate grade (IG) followed by acetonitrile (12 vol) and purified water (8 vol). The mixture was heated to reflux (74-79°C), and held until complete dissolution is observed. The solution was then transferred to a reactor (Reactor 2) that had been pre-heated to 74-79°C via filter (0.22pm pipe-line filter). Reactor 1 was rinsed with acetonitrile (MeCN) (0.3 vol) and purified water (0.2 vol), and the solution in Reactor 1 was transferred to Reactor 2 via filter (0.22pm pipe-line filter). The contents of Reactor 2 were held until complete dissolution is observed. The solution in Reactor 2 was cooled to 69-72°C, then seeded with 2 w/w% (based on pure GSK2330672B input). The suspension was cooled to 58-62°C within 10-20 min. The suspension was held at 58-62°C for 2h. Purified water (14 vol) was added over 8 hrs. After the addition was complete, the slurry was held at 58-62°C for 60 min, then cooled to 18-25°C over 50-70 min. The slurry was stirred at 18-25°C for not less than 30 min, then the suspension filtered under vacuum. The reactor was rinsed with MeCN/water (6/11 v:v, 3.5 vol) and the rinse used to wash the cake. The cake was washed twice with water (2 vol). The cake was blown down with nitrogen and dried at 60°C under vacuum to give 46.35 kg GSK2330672B Form 1 solid.
Method 4
10.94g of GSK2330672B was charged and washed into a vessel using 131mL of acetonitrile (MeCN) and 88mL of water. The slurry was then heated to reflux. 4mL of 3:2v/v MeCN/water was charged followed by 5.5mL of 3:2v/v MeCN/water. The contents were cooled to 70 °C, then cooled to 60°C over 15 minutes and held stirring for 2 hours. 153 mL water was added over 8 hours, the contents were cooled to 20°C over 1 hour, and held stirring for 1 hour. The product was isolated, washed with 38mL of 6:11 MeCN/water, then washed twice with 22mL of water. After deliquoring, the product was dried at 45°C under vacuum, to give 9.35g (85.5%w/w) Form I GSK2330672B.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











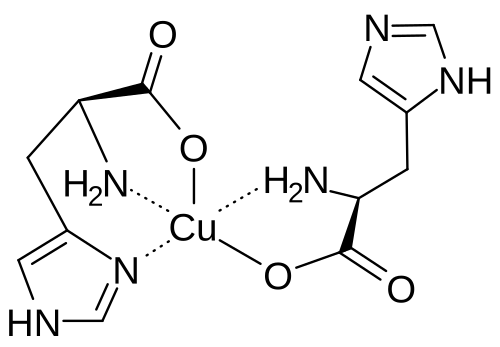
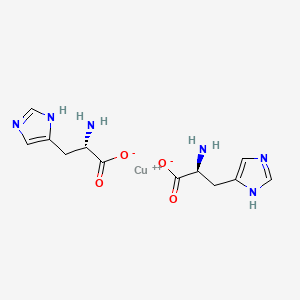
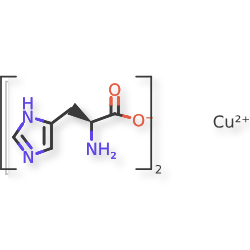






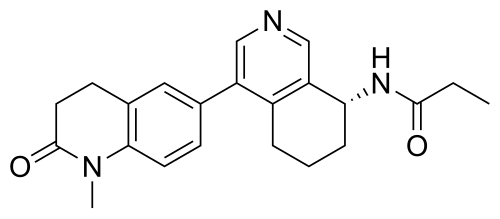
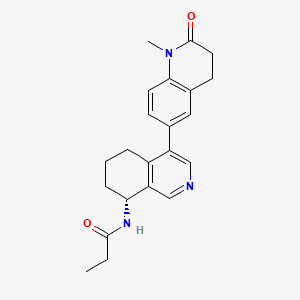
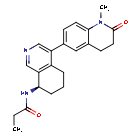
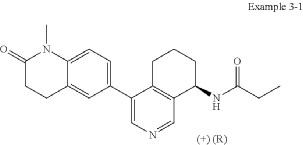
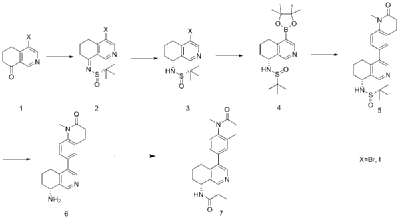
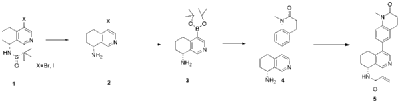
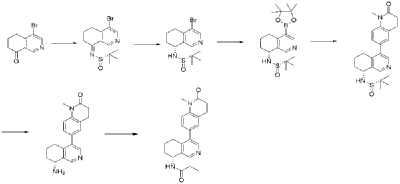
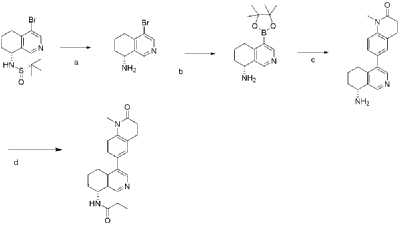
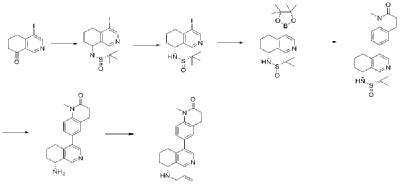

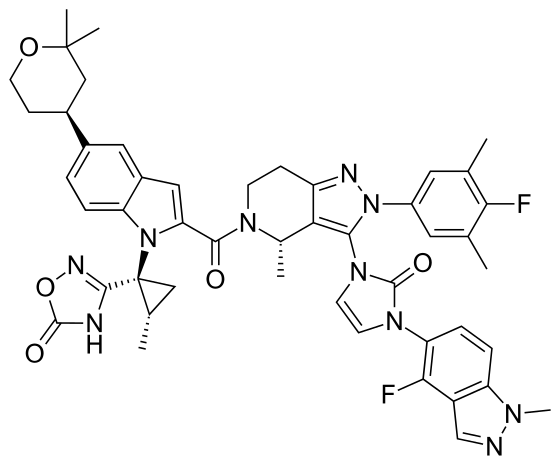
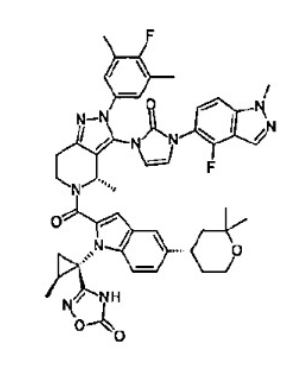
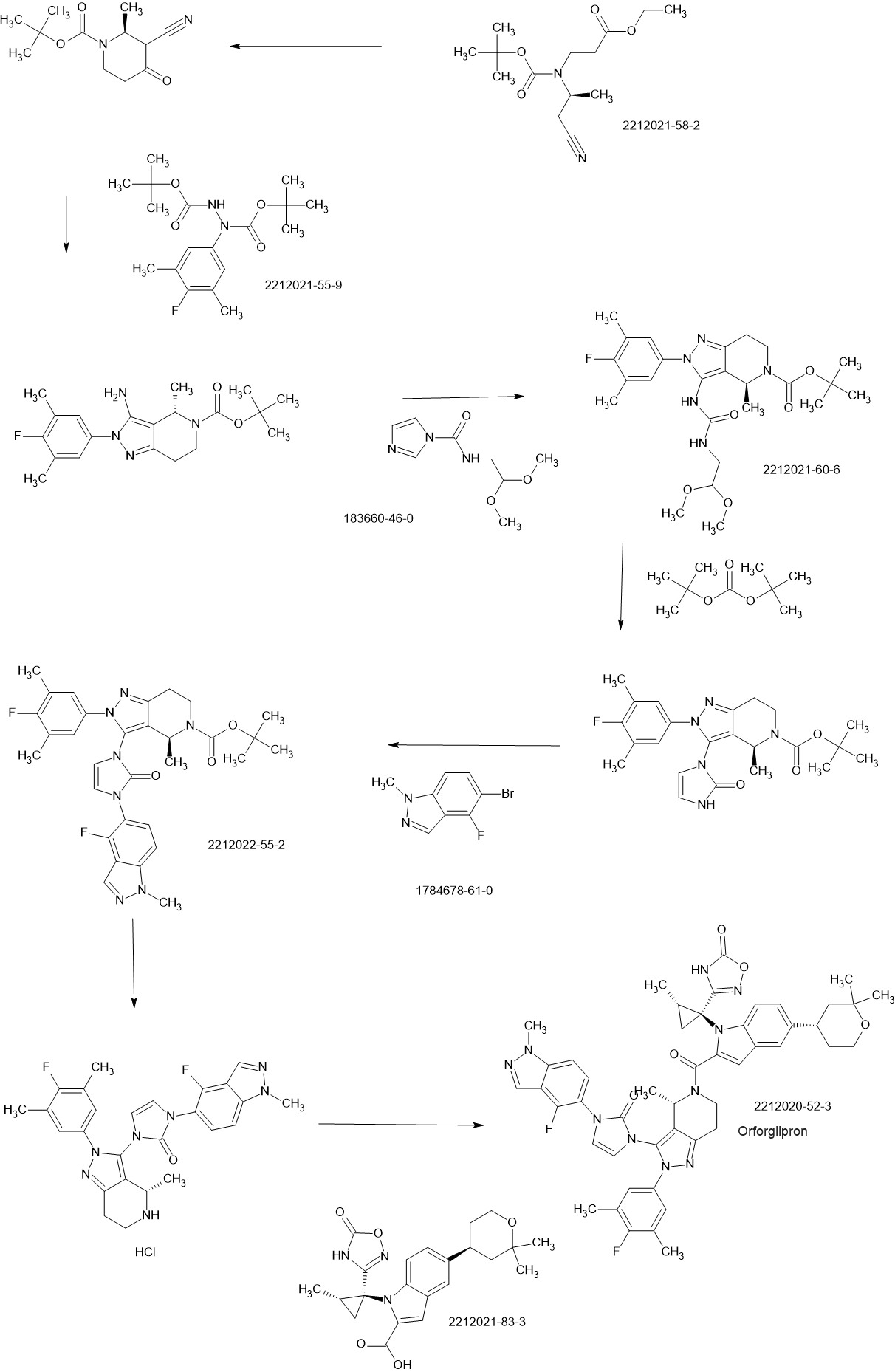
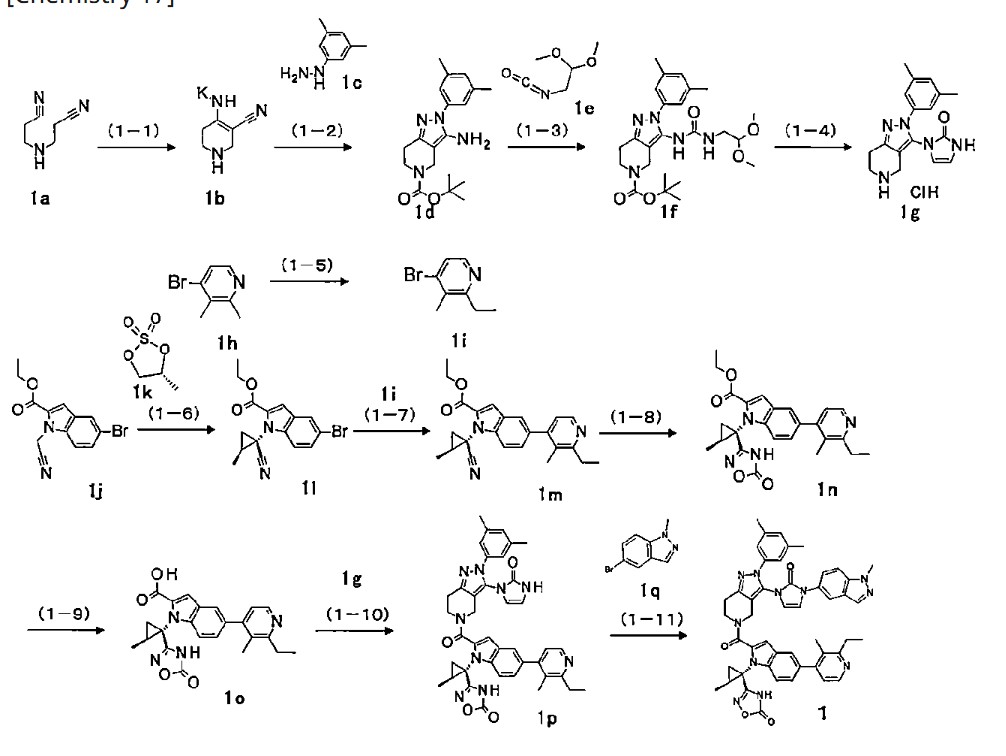
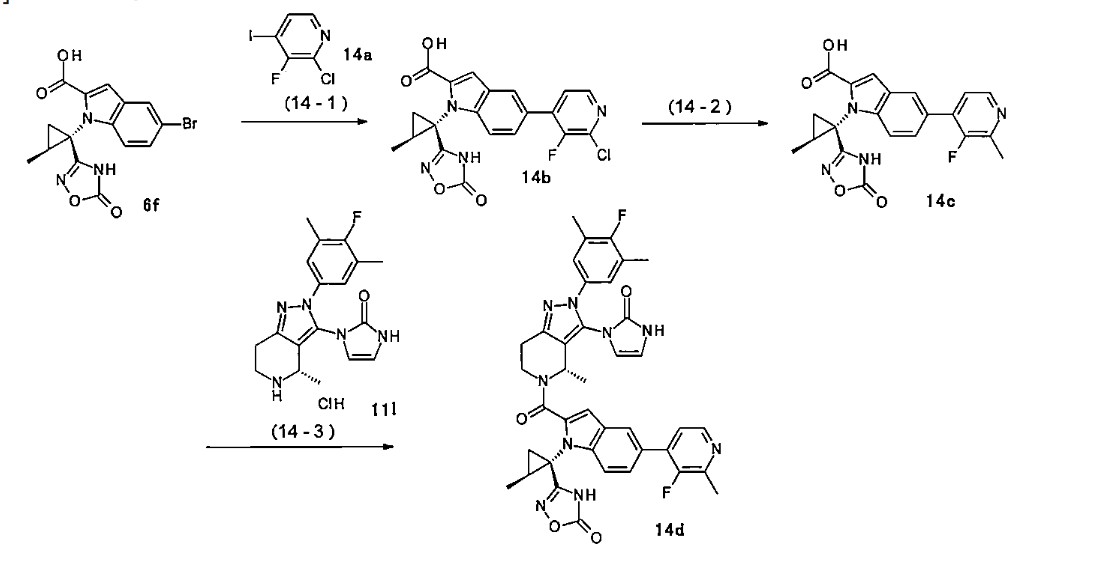
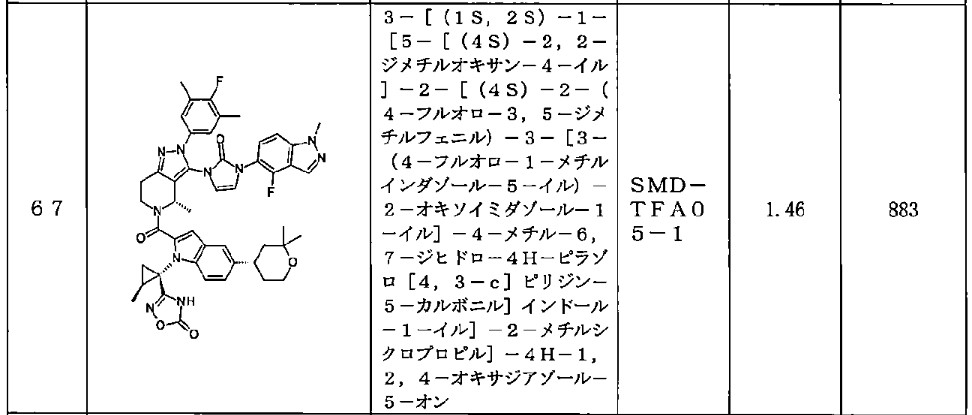
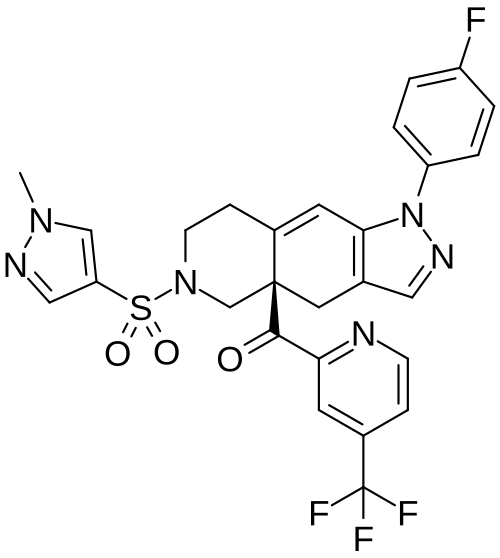
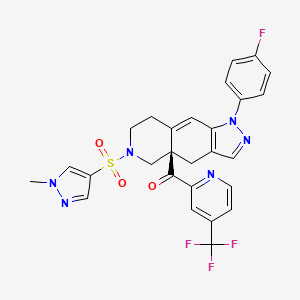






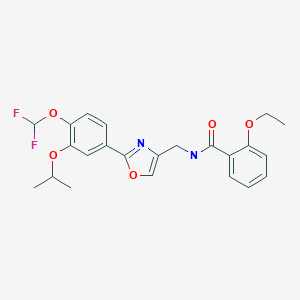

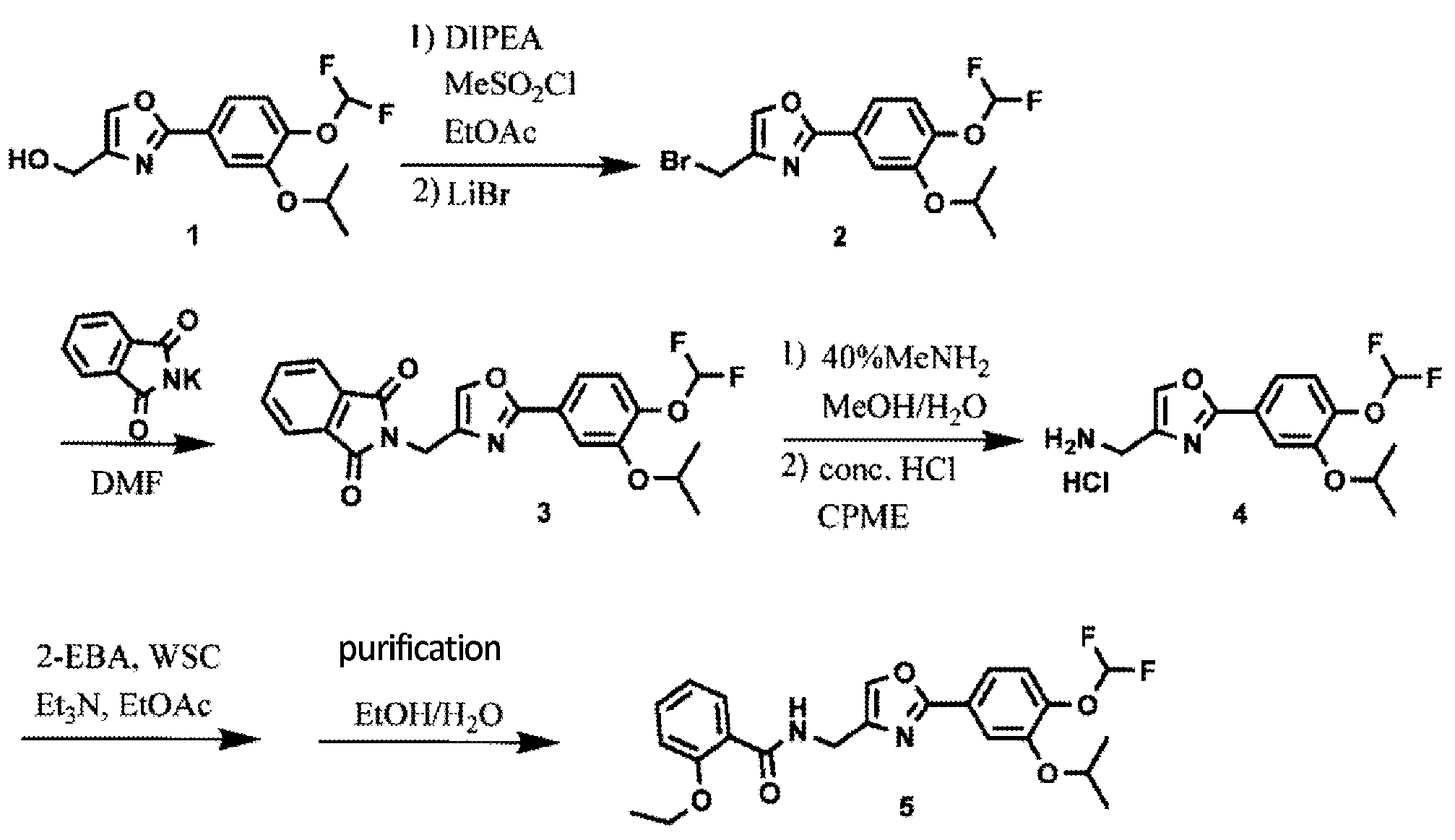
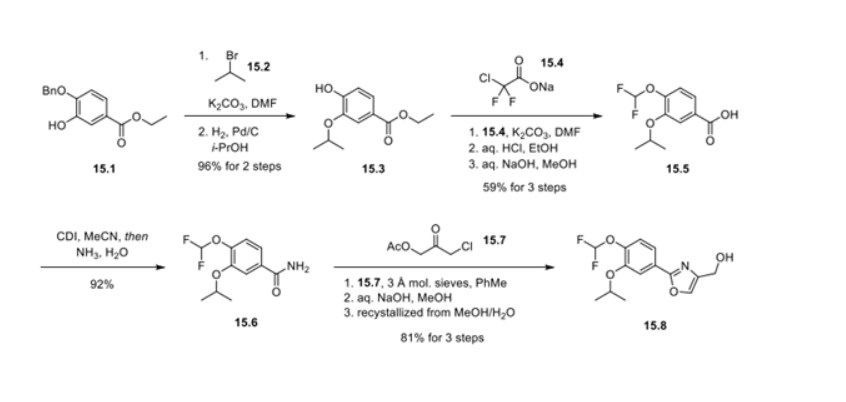
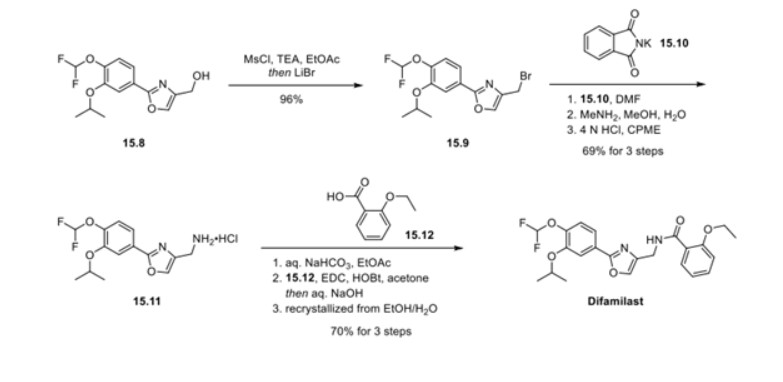









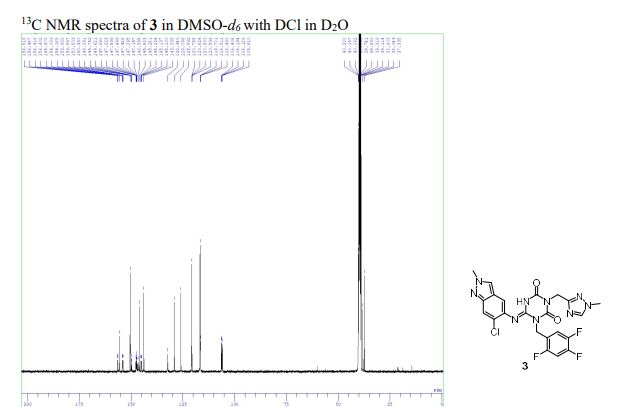






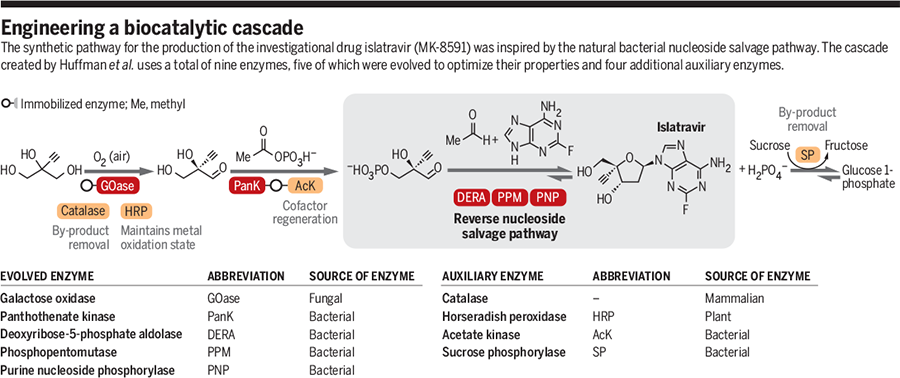





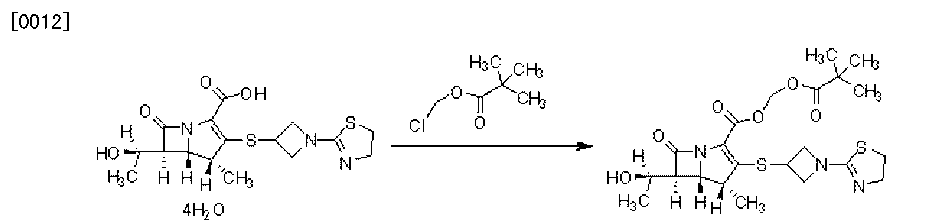
 EP 0632039; EP 0717042; JP 1996053453; US 5534510; US 5659043; US 5783703
EP 0632039; EP 0717042; JP 1996053453; US 5534510; US 5659043; US 5783703 1-azabicyclobutane (III) was opened with thioacetic acid with concomitant N-acetylation yielding (XII). Further acid hydrolysis of (XII) gave 3-mercaptoazetidine (XIII). Condensation of (XIII) with either 2-(methylthio)thiazoline (V) or 2-chloroethyl isothiocyanate (VII) then produced thiazolinylazetidine (XI).
1-azabicyclobutane (III) was opened with thioacetic acid with concomitant N-acetylation yielding (XII). Further acid hydrolysis of (XII) gave 3-mercaptoazetidine (XIII). Condensation of (XIII) with either 2-(methylthio)thiazoline (V) or 2-chloroethyl isothiocyanate (VII) then produced thiazolinylazetidine (XI).
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}