PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
[4R-[4a,5b,6b(R*)]]-6-[[2-Carboxy-6-(1-hydroxyethyl)-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5 H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
5H-Pyrazolo[1,2-a][1,2,4]triazol-4-ium, 6-[[(4R,5S,6S)-2-carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-, inner salt [ACD/Index Name]
6-[[(4R,5S,6S)-2-Carboxy-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-en-3-yl]thio]-6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt
https://patents.google.com/patent/WO2014097221A1/esBiapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)- hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3 -yljthio] 6,7-dihydro-5H- pyrazolo[l,2-a][l,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.EXAMPLESExample 1 : Purification of BiapenemBiapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.Yield: 84%HPLC Purity: 99.87% Example 2: Purification of BiapenemBiapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.Yield: 81.77%HPLC Purity: 99.80% PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013150550
The present invention relates to an improved process for the preparation of carbapenem antibiotic; more particularly relates to the preparation of Ertapenem monosodium salt of formula (I) having purity greater than 98.5% and having pharmaceutically acceptable level of residual solvent and palladium content.
The US patents namely US 5,478,820 and US 5,856,321 disclose various processes for preparing Ertapenem and its sodium salt. Example 12 of US 5,478,820 discloses a process in which the Ertapenem was isolated using column purification followed by freeze-drying technique. According to Example-4 of this patent disodium salt of Ertapenem was prepared by dissolving crude product in water using NaHCO3, followed by purification using column chromatography and subsequent lyophilization.
US 6,504,027 provides a process for preparing Ertapenem in crystalline form which comprises deprotecting and extracting a polar organic solution containing a crude mono-protected Ertapenem of formula
wherein P represents protecting group and X represents charge balancing group like sodium
with C4.10 alcohol in the presence of ion-pairing reagent followed by adjusting the pH of the aqueous layer to 5.5 and crystallizing using methanol and 1-propanol to produce a crystalline compound; this patent process involves operations like
multiple extractions which is cumbersome in plant and said operation affects the overall yield.
US 7,145,002 provides a process for producing Ertapenem or its sodium salt and/or its solvate in crystalline form. This patent states (refer para 3, lines 31-41) that contact of Ertapenem sodium with water and alcoholic solvents results in the formation of crystalline solvates. The processes reported in examples- 1 & 2 provide crystalline Ertapenem monosodium which is isolated from a mixture of methanol, 1-propanol and water followed by washing with aqueous isopropyl alcohol which results in the formation of crystalline solvate of Ertapenem sodium. Applicant found the Ertapenem monosodium obtained according to this process contain higher amount of residual solvent and palladium content.
US 7,022,841 provide a process for reducing the levels of organic solvents in Ertapenem to pharmaceutically acceptable levels. This patent discloses (Refer para 1, lines 52-60) that Ertapenem sodium obtained from water/alcohol mixture according to US 7, 145,002 becomes amorphous when water content of the solid is reduced and further the organic solvent present in the solid is not readily removed. In view of this drawback, this patent provides a process wherein the water content of Ertapenem sodium is maintained between 13-25% during the washing and drying process. This patent further discloses that (Refer para 9, lines 6-14) the washing of Ertapenem sodium can be carried out using anhydrous solvents which results in the formation of amorphous solid, which is then dried using hydrated nitrogen by increasing the water content of the solid. Due to the hygroscopic and unstable nature of Ertapenem sodium when in contact with water, the above processes result in more degradation of Ertapenem. The patent further discloses in example 5 that the degradation of Ertapenem sodium is more when it takes more time for drying.
Further this patent requires repetitive washing and control of moisture content to get the desired results.
For isolation of Ertapenem sodium from the reaction mass, all the above discussed prior art patents utilize methanol and 1-propanol as crystallization solvent. The filtration of Ertapenem sodium formed by using these solvents or their mixture takes longer time duration and subsequent drying for the removal of residual solvent also takes several hours due to occlusion of solvent into Ertapenem sodium. During these operations the Ertapenem sodium degrades an results in the formation of many impurities such as several dimers, methanolysis impurity etc., and hence the reported processes is not suitable to manufacture Ertapenem sodium on commercial scale with purity greater than 98.5% and with pharmaceutically acceptable level of residual solvent content.
Methanolysis impurity Dimer-I
Dimer-II
Further the applicant found that Ertapenem monosodium isolated by following the process reported in prior art was having palladium content above the pharmaceutically acceptable level. Hence the process reported in prior art is not suitable on manufacturing scale where maintaining stringent technological condition is cumbersome and involves higher operating cost.
Thus all the reported processes suffer in terms of one or more of the following facts:
■ Filtration time of Ertapenem sodium takes several hours.
■ Drying time takes several hours due to occlusion of solvent and nature of the solid.
■ Stringent technological condition is required for maintenance of moisture content during washing & drying operation.
■ Palladium content is found to be higher (greater than 25 ppm) which is not acceptable for pharmaceutical products.
■ The isolated Ertapenem sodium is having higher amount of residual solvents.
■ The purity is reduced over to several hours of filtration & drying.
With our continued research for developing a process for the preparation of Ertapenem monosodium of formula (I) to overcome the above mentioned drawbacks, we surprisingly found that when esters of organic acid were used as solvents in place of 1-propanol, the solid obtained was easily filterable with less cycle time. Further the washing with hydrocarbon solvents containing 0-75% alcoholic solvent followed by drying results in Ertapenem having residual solvent content well below the pharmaceutically acceptable levels. The use of thiourea, thiosemicarbazide or their N-substituted derivatives in the presence of organic solvents during isolation brings down the palladium content to pharmaceutically acceptable level.
The Ertapenem or its sodium salt can be prepared according the processes provi
(I)
P’ and P” represent carboxylic protecting groups and X is H or Na
Scheme-1
The present invention is illustrated with the following examples, which should not be construed to limit the scope of the invention.
Example- I
Preparation of Ertapenem monosodium of formula (I)
Step-I:
To a stirred solution of p-nitrobenzyl (4R,5S,6S)-3-(diphenyloxy)phosphoryloxy-6-[(lR)-l-hydroxyethyl]-4-methyl-7-oxo-l-azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound II) (100 g) and (2S,4S)-2-[[(3-carboxyphenyl) amino]carbonyl]-4-mercapto-l-(4-nitrobenzyl)pyrrolidinecarboxylate (compound III) (75 g) in N,N-dimethylformamide was added Ν,Ν-diisopropylethylamine at -30 to -40° C and stirred. The reaction mass, after completion of the reaction, was quenched with a mixture of phosphate buffer solution-ethyl acetate and the pH was adjusted to 5 – 6 with phosphoric acid. The organic layer was separated, washed with water and subjected to carbon treatment. To the organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl), a solution of sodium 2-ethylhexanoate (42 g in 500 mL methanol) was added and taken to next step as such. (If required the compound of formula (IV) is isolated either as sodium salt or as free acid by following the process reported in prior art and taken further)
Step-II:
To the Step-I organic layer containing the compound of formula (IV) (wherein P’ and P” refers to p-nitrobenzyl & X is Na), 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8- 10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered to remove palladium on carbon. To the filtrate, thiourea (5 g) and tetrahydrofuran were added and stirred. The aqueous layer was separated and treated with carbon and neutral alumina at 10-15° C while degassing and filtered. The filtrate was added to methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 46 g; Purity by HPLC: 98.93%; Palladium content: 1.8 ppm by ICP MS
The HPLC purity of Ertapenem monosodium was checked using the following parameters
Column : Zorbax Eclipse plus C8, (50 mm x 4.6 mm), 1.8μ).
Mobile phase : Ammoniam acetate buffer: Acetonitile: water
Detector : UV at 250 nm
Flow rate : 0.5 mL/min
Run time : 45 min.
Example- II
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon & neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with cyclohexane (200 ml) and
dried under vacuum. Yield: 44 g; Purity by HPLC: 98.84%; Palladium content: 0.93 ppm by ICP MS
The term ICP MS method refers to the inductively coupled plasma mass spectrometry. The following parameter was used to determine the content of palladium.
The carbapenem was digested in a closed vessel system in presence of reagents Nitric acid, Hydrogen peroxide and Hydrochloric acid by using Microwave reaction system with microwave radiation power 1200 Watts. The digested sample was introduced into inductively coupled plasma mass spectrometer by help of Peltier cooled spray chamber. The sample aerosol is getting atomized then ionized in the argon plasma. The ionized Palladium was estimated by using Quadrupole mass detector. The sample was quantified against NIST traceable reference standards at mass number ! 05.
Example- III
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was separated and treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of toluene: ethanol (200 ml) and dried under vacuum. Yield: 42 g; Purity by HPLC: 99.03%
Example- IV
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the filtrate was treated with thiosemicarbazide and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C followed by the addition of ethyl acetate and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol (200 ml) and dried under vacuum. Yield: 41 g; Purity by HPLC: 99.13%; Palladium content: 1.71 ppm by ICP MS
Example- V
Preparation of Ertapenem monosodium of formula (I)
To the Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and subjected to hydrogenation using palladium on carbon at 8-10° C with 9-10 kg hydrogen pressure. The reaction mass, after completion of reaction, was filtered and the filtrate was treated with thiourea and 2-methyltetrahydrofuran and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5 – 6 using aqueous acetic acid. To the mass, a mixture of ethyl acetate containing 10% methyl acetate was added and stirred. The solid obtained was
filtered, washed with cyclohexane:ethanol and dried under vacuum. Yield: 40.5 g; Purity by HPLC: 98.77%; Palladium content: 1.43 ppm by ICP MS
Example-VI
(V ) (V I )
The diprotected Meropenem of formula (V) (where P and P’ were p-nitrobenzyl) was dissolved in tetrahydrofuran and 3-(N-morpholino)propanesulfonic acid buffer and hydrogenated using palladium on carbon at 9-10 kg hydrogen pressure. The mass was filtered and the filtrate was washed with ethyl acetate. The aqueous layer was treated with thiourea and 2-methyltetrahydrofuran. The aqueous layer was separated, treated with carbon and degassed. The carbon was filtered off and acetone was added to the filtrate to crystallize Meropenem trihydrate of formula (VI). The product was filtered and washed with aq. acetone and dried under vacuum to get Meropenem trihydrate. Purity: 99.8%; Pd content: 0.08 ppm
Reference example-I:
Preparation of Ertapenem monosodium of formula (I)
To Step-I organic layer as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered. The filtrate was treated with thiourea and tetrahydrofuran and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass ethyl acetate was added and stirred. The solid obtained was filtered, washed with ethanol (5 * 100 ml) and dried under vacuum. Yield: 31 g; Purity by HPLC: 96.76%
Reference example-II:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass , as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction was filtered and the layers separated. The aqueous layer was treated with carbon and neutral alumina at 10-15° C and filtered. The filtrate was mixed with methanol at -20° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass, ethyl acetate was added and stirred. The solid obtained was filtered, washed with a mixture of cyclohexane: ethanol and dried under vacuum. Yield: 43 g; Purity by HPLC: 98.6%; Palladium content: 35.8 ppm by ICP MS.
Reference example-HI:
Preparation of Ertapenem monosodium of formula (I)
To the Step-I reaction mass as provided in Example-I, 3-(N-morpholino)propanesulfonic acid solution was added and hydrogenated at 9-10 kg pressure using palladium on carbon at 8-10° C. The reaction mass, after completion of reaction, was filtered and the layers separated. The aqueous layer was treated with carbon, neutral alumina at 10-15° C and filtered. The filtrate was mixed with 1-propanol at -5° C and the pH was adjusted to 5.5-5.7 using aqueous acetic acid. To the mass methanol and 1-propanol were added and stirred. The solid obtained was filtered, washed with ethanol and dried under nitrogen atmosphere in vacuum. Yield: 25 g; Purity by HPLC: 97 %.: palladium content: 38.2 ppm
The following tables illustrate the advantages of the present invention over prior art process:
Table-I: Comparison of present process with prior art process
The crystallization and washing method disclosed in US 7,022,841 was followed.
The above table indicates that the use of ethyl acetate as crystallization solvent results with improved yield and high purity with less filtration and drying time thereby increasing the productivity significantly on manufacturing scale. Further the use of thiourea or thiosemicarbazide as reagents in the present process results in the pharmaceutically acceptable level of palladium content.
Table-II: Comparison of solvents for washing Ertapenem monosodium
The above table indicates that the use of hydrocarbon solvents containing 0-75% of alcoholic solvent helps in washing to remove the residual solvent content in shorter duration and with single run wash. On the other hands the use of ethanol alone results in Ertapenem monosodium having less yield and purity requiring repetitive washing.
Table-IH: Effect of different reagent in reduction of palladium content
Reagent : thiourea, thiosemicarbazide or its N-substituted derivatives
Advantages of the process of the present invention:
> The use of ester of an organic acid for the crystallization of Ertapenem sodium results in fast filtration and reduced cycle time, thereby increasing the productivity.
> Washing of Ertapenem sodium with hydrocarbon solvent optionally containing alcohol results in improved physical nature of Ertapenem sodium resulting in reduced washing and drying time thereby avoid the degradation of Ertapenem and providing Ertapenem sodium with purity greater than 98.5% by HPLC.
Use of thiourea, thiosemicarbazide or their N-substituted derivatives in the process results in Ertapenem sodium having pharmaceutically acceptable level of palladium content.
Ia’A hydrogenator is charged with 63 g of 10% Pd on carbon catalyst (dry weight) in 1.8 L of water. The vessel is placed under hydrogen then vented and placed under nitrogen. Sodium hydroxide (68 g, 50%) is charged adjusting the pH to about 7.5 with carbon dioxide.The enol phosphate (170 g) and the thiol (86 g) are dissolved in 1.3‘L of N- ethylpyrrolidinone (NEP). The mixture is cooled to below -40°C and 1,1,3,3- tetramethylguanidine (109 g) is added. After 3 hours, the reaction mixture is quenched into the hydrogenator at below 15°C adjusting the pH to about 8 with carbon dioxide. The vessel is placed under hydrogen. When the reaction is complete, the hydrogen is vented and the reaction mixture is treated with activated carbon and filtered. The filtrate is extracted with iso-amyl alcohol containing diphenylphosphoric acid (240 g) and 50% NaOH (44 g). The resulting aqueous solution is further extracted with iso-amyl alcohol to give an aqueous solution containing at least 90 mg/mL of the product. Both extractions are performed using two CINC centrifugal separators set in series for countercurrent extraction. The pH is adjusted to 5.5 with acetic acid. The product is crystallized by adding equal volumes of methanol and 1- propanol at below -5°C and isolated by filtration. The solid is washed with a mixture of 2-propanol and water (85: 15 v/v) then dried to yield a compound of formula la’.While certain preferred embodiments of the invention have been described herein in detail, numerous alternative embodiments are contemplated as falling within the scope of the appended claims. Consequently the invention is not to be limited thereby.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleUS4866171A1987-04-111989-09-12Lederle (Japan), Ltd.(1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazolium-6-yl)]thio-6-[R-1-hydroxyethyl]-1-methyl-carbapenum-3-carboxylateUS5241073A1990-10-121993-08-31Lederle (Japan)Process for preparing (1R,5S,6S)-2-[(6,7-dihydro-5H-pyrazolo [1,2-a][1,2,4]triazolium-6-yl)]thio-6-[(R)-1-hydroxyethyl]-1-methyl-carbapenem-3-carboxylate and starting materials thereofUS5286856A1991-09-201994-02-15Takeda Chemical Industries, Ltd.Production of crystalline penemWO2002057266A1 *2001-01-162002-07-25Merck & Co., Inc.Improved process for carbapenem synthesisWO2009047604A1 *2007-10-082009-04-16Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of carbapenem antibioticCN102268025A *2011-07-152011-12-07海南美兰史克制药有限公司一种比阿培南化合物及其制法
Biapenem is chemically known as 6-[[2(4R,5S,6S)-carboxy-6-[(lR)-hydroxy ethyl] -4-methyl-7-oxo- 1 -azabicyclo [3.2.0]hept-2-en-3-yljthio] 6,7-dihydro-5H-pyrazolo[1,2-a][1,2,4]triazol-4-ium inner salt, and is represented by Formula 1. It is indicated for the treatment of bacterial infection and sepsis.
Formula 1
U.S. Patent No. 4,866,171, in Example 6, discloses the purification of biapenem using chromatography and/or lyophilization techniques. This patent also describes a process for the conversion of amorphous biapenem into a crystalline form by dissolving the amorphous biapenem in water while heating, followed by cooling, then washing the obtained crystals with a 50% aqueous ethanol solution.
U.S. Patent No. 5,241,073 describes a process for the purification of biapenem involving column chromatography and crystallization with ethanol.
U.S. Patent No. 5,286,856 describes a process for the crystallization of biapenem from an aqueous solution, comprising maintaining the temperature of the aqueous solution from eutectic temperature (-10°C to -2°C) to a temperature lower than 0°C, followed by lyophilization.
The Journal of Organic Chemistry, 63(23):8145-8149 (1998) describes the purification of biapenem involving resin chromatography.
The present invention provides an alternate process for the purification of biapenem that avoids making use of tedious techniques like chromatography and lyophilization. At the same time, it results in a high yield and high purity of the final product. Advantageously, the crystalline biapenem of this invention can be directly isolated from the reaction mixture. Further, the process of the present invention involves fewer steps, is easily scalable, and industrially advantageous.
EXAMPLES
Example 1 : Purification of Biapenem
Biapenem (12 g) was added into water (300 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.6 g) was added to the reaction mixture and stirred for 10 minutes to 15 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (36 mL). The filtrate obtained was passed through a 0.45 micron filter, and its pH was adjusted to 5.5 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (336 mL) was added to the reaction mixture at 5°C to 10°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (60 mL). The solid was dried under reduced pressure (720 mmHg) at 30°C to 35°C to obtain the title product as white crystals.
Yield: 84%
HPLC Purity: 99.87%
Example 2: Purification of Biapenem
Biapenem (18 g) was added into water (450 mL) at 65°C, stirred for 5 minutes, and cooled to 30°C within 10 minutes. Enoantichromos carbon (0.9 g) was added to the reaction mixture and stirred for 30 minutes at 25°C to 30°C. The reaction mixture was filtered through a hyflo bed and washed with water (54 mL). The filtrate obtained was passed through a 0.45 micron filter and its pH was adjusted to 4.9 using 5% aqueous sodium hydroxide solution at 10°C to 15°C. Acetone (504 mL) was added to the reaction mixture at 10°C to 15°C. The resultant slurry was stirred for 3 hours at 5°C to 10°C, filtered, and the obtained solid was washed with acetone (90 mL). The solid was dried under reduced pressure (720 mmHg) at 35°C to 40°C to obtain the title product as white crystals.
Background of the Invention Biapenem is a synthetic broad-spectrum carbapenem antibiotic which suppresses bacterial growth by inhibiting the enzymes responsible for bacterial cell wall synthesis, and shows broad-spectrum antibacterial activity both against gram-positive bacteria and gram-negative bacteria. Biapenem is chemically known as (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][ 1,2,4] triazol-8-ium-6-ylsulfanyl)-6-( 1 -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo [3.2.0]hept-2-ene-2-carboxylate and marketed in Japan as OMEGACIN®.Various methods are reported in the prior art for the preparation of Biapenem of formula (I) which includes the condensation of compound of formula (II) with compound of formula (III) and subsequent deprotection of the protecting group as shown in scheme-1. wherein R1 is hydrogen or hydroxy protecting group such as tert-butyl dimethyl silyl and the like, R2 is hydrogen or carboxyl protecting group such as p-nitrobenzyl, p-methoxy benzyl, allyl and the like, A is an activating group such as P(0)(OR)2, SO2R and the like wherein R is selected from substituted or unsubstituted C1-6 alkyl, aralkyl or aryl to form the compound of formula (II). The X” in compound of formula (III) is halogen selected from Br or CI.Biapenem was first disclosed in US 4,866,171 and the said patent also discloses a process for the preparation of the same. US 5,241,073 disclosed the method for the preparation of compound of formula (III) followed by condensation with compound of general formula (II) using base such as N-ethyldiisopropylamine and subsequent deprotection yields Biapenem which was isolated by column chromatography followed by crystallization from ethanol.EP 0289801 discloses a process for the preparation of crystalline Biapenem wherein Biapenem was dissolved in water and lyophilized to get amorphous compound. The amorphous compound was dissolved in water at 40° C followed by cooling to get crystalline product. This patent further provides the PXRD values of the crystalline Biapenem. The Biapenem obtained according to the process provided in this patent takes longer time for reconstitution and hence not suitable.US 5,286,856 and US 5,424,069 provide a process for the crystallization of Biapenem which utilizes freeze-drying technique and vial lyophillisation method respectively. These patents disclose (refer para 1, lines 10-33 of US’ 856) that the process provided in EP 0289801 results with Biapenem crystals which take relatively longer time for dissolution during use. To overcome the above issues, these patents utilize the freeze-drying and vial lyophillisation methods. The said methods involve freezing of the solution containing Biapenem followed by raising the temperature and repeating the cooling and heating process followed by lyophillisation to get the crystalline product. Lyophillisation and related process are capital intensive techniques and uneconomical in commercial scale operations.All the above said prior arts utilize either the lyophillisation technique or preparing the amorphous material and crystallizing it from water to get crystalline Biapenem.Biapenem is available as powder for injection which needs to be reconstituted with water or saline solution before injection. The process of preparing a solution having an appropriate concentration of an active ingredient for the administration is called “reconstitution”. The reconstitution time (RCT) plays a critical role in injectable powders. Short reconstitution time is preferable for both a member of medical center and patients. If the reconstitution time is too long, it will increase the preparation time thus making it difficult to administrate it to many patients at the same, which will eventually lower the competitiveness of the drug. The problem before the applicants is to find economic and robust process for the preparation of Biapenem with high purity and yield which should dissolve in water in less than 25 seconds (reconstitution time). With our continued intensive and diligent research for developing a process for the preparation of Biapenem having high purity and yield with reconstitution time of less than 25 seconds, we have identified an improved process which is commercially viable and eliminates the issues associated with reconstitution time. The process of this invention is simple and obviates the use of freeze crystallization. Further the present invention fulfils the need for a process for the manufacture of Biapenem which is convenient to operate in commercial scale
Objectives of the inventionThe main objective of the present invention is to provide a simple and commercially viable, industrially scalable process for the crystallization of Biapenem of formula (I) with high purity and good yield.Yet another objective of the present invention is to provide a simple and commercially suitable process for the preparation of Biapenem of formula (I) with reconstitution time less than 25 seconds. The reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Summary of the inventionAccordingly the primary aspect of the present invention is to provide an improved process for the preparation of Biapenem of formula (I) the said process comprises;(i) obtaining a solution of Biapenem in water containing co-solvent; and(ii) adding anti-solvent in to the solution of step (i) or vice-versa to crystallize Biapenem followed by filtration. Detailed Description In an embodiment of the present invention, the co-solvent used in step (i) is selected from alcoholic solvents consisting of methanol, ethanol, isopropyl alcohol, n-propanol, n-butanol and iso-butanol or mixtures thereof; preferably methanol, ethanol and isopropyl alcohol; more preferably methanol.In another embodiment of the present invention the anti-solvent used in step (ii) is selected from acetone, methyl ethyl ketone, methyl isobutyl ketone, ethyl acetate, methyl acetate, butyl acetate, tetrahydrofuran or mixtures thereof; preferably acetone. In yet another embodiment of the present invention, the solution of Biapenem in step (i) can be obtained by (a) dissolving Biapenem in water followed by addition of co-solvent (b) dissolving Biapenem in water containing the co-solvent (c) the aqueous solution containing Biapenem can be obtained directly from the reaction mass followed by addition of co-solvent (d) the aqueous solution of Biapenem containing co-solvent can be obtained directly from the reaction mass. The said solutions, if necessary can be subjected to sterile filtration before the addition of anti-solvent. Thus the present invention provided a process for the preparation of sterile Biapenem having reconstitution time less than 25 seconds, more preferably less than 15 seconds.The prior art lyophillisation process for the preparation of Biapenem requires capital investment and high operating cost due to the involvement of repetitive heating and cooling process which is tedious technology in commercial scale operations. The reported prior art process for the crystallization of Biapenem of formula (I) from water results in the formation of crystalline powder which takes longer time for dissolution in water or saline solution (reconstitution time). Surprisingly, applicant found that the use of co-solvents during the crystallization of Biapenem results with Biapenem having reconstitution time of less than 25 seconds. This constitutes the novelty of the present invention.In this present invention the Biapenem of formula (I) is obtained as crystalline solid with purity above 99.0 % by HPLC with good stability and further can be easily filled in vials.
The following examples are provided by way of illustration only and should not be construed to limit the scope of the invention.
Crystallization of (4R,5S,6S)-3-(6,7-dihvdro-5H-pyrazolo[l,2-al 11,2,41 triazol-8-ium-6-vlsulfanvl)-6-(l-hydroxvethvl)-4-methvl-7-oxo-l-azabicyclo [3.2.01hept-2-ene-2-carboxvlate [Biapenem of formula (1)1:Example -1:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. Activated carbon and EDTA were added to the clear solution and filtered through hi-flow bed, washed with water followed by filtration through micron filters in sterile area. To the filtrate, methanol (600 mL) was added followed by acetone under stirring. To the reaction mass, Biapenem seed material was added and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 85 g Purity by HPLC: 99.5% Reconstitution time (RCT): < 15 seconds
Example -2:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. To the filtrate, isopropyl alcohol (500 ml) was added followed by acetone under stirring. The mass was cooled and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 83 g Purity by HPLC: 99.6% Reconstitution time: < 15 seconds
Example -3;To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through micron filters. To the filtrate, ethanol (600 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem.Yield: 84 g Purity by HPLC: 99.5% Reconstitution time : < 15 seconds
Example -4:To water (4 lit), Biapenem (100 g) was added at 40° C and dissolved to get a clear solution. The solution was filtered through hi-flow bed, washed with water followed by filtration through micron filters. To the filtrate, methanol (450 ml) was added followed by acetone and stirred. The crystallized product was filtered, washed with aqueous acetone and dried under vacuum to get crystalline Biapenem. Yield: 87 g Purity by HPLC: 99.4% Reconstitution time (RCT): < 15 seconds
Reference example -1:Preparation of Biapenem (Non-Sterile)Step-I: Preparation of p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pvrazolofl,2-al[l,2,41triazol-8-ium-6-vlsulfanvn-6-(l-hvdroxvethyl)-4-methvI-7-oxo-l-azabicvclo[3.2.01hept-2-ene-2-carboxylate [Compound of formula (IV)1To a mixture of acetonitrile and DMF, P-Nitrobenzyl (4R,5S,6S)-3-(dipheny loxy)phosphory loxy-6- [(1R)-1 -hydroxyethy 1] -4-methy 1-7-oxo-1 -azabicyclo[3,2,0]hept-2-ene-2-carboxylate (compound of formula II) and 6,7-dihydro-6-mercapto-5H-pyrazolo[l,2-a] [1,2,4] triazole chloride (compound of formula III) were added and cooled to 0-5° C. To this mixture, N-ethyldiisopropyl amine was added and stirred till the completion of the reaction, followed by the addition of dichloromethane to crystallize the p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l -hydroxyethyl)-4-methyl-7-oxo-1 -azabicyclo[3.2.0] hept-2-ene-2-carboxylate which was filtered and dried under nitrogen.
Step-II: Preparation of BiapenemTo a solution of MOPS buffer and THF, p-Nitrobenzyl (4R,5S,6S)-3-(6,7-dihydro-5H-pyrazolo[l,2-a][l,2,4]triazol-8-ium-6-ylsulfanyl)-6-(l-hydroxy ethyl)-4-methyl-7-oxo-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylate (Compound of formula-IV) was added at pH 7-8 and cooled to 5-10° C. The mixture was hydrogenated using palladium on carbon as catalyst. The catalyst was filtered and the filtrate was treated with activated carbon and filtered. The filtrate was extracted with dichloromethane and the layers separated. The aqueous layer was degassed. To the aqueous layer, acetone was added to crystallize Biapenem at 20-25° C. The product was filtered, washed with aqueous acetone and dried under vacuum to get Biapenem (Non-Sterile).
Reference example -2: Crystallization of Biapenem
Example -1 was repeated without the addition of methanol.Yield: 84 g Purity by HPLC: 99.5%Reconstitution time : > 90 secondsThe reconstitution time is calculated by the time taken to dissolve 300 mg of Biapenem in 100 ml of water or saline solution.Table-1: Comparative Data:The comparative data provided in the table-1 clearly indicates that the addition of co-solvent during crystallization provides Biapenem with reconstitution time less than 25 seconds.
Serdexmethylphenidate is a derivative of dexmethylphenidate created by pharmaceutical company KemPharm. The compound is under investigation for the treatment of ADHD in children, adolescents, and adults as of 2020.[2] The drug was approved for medical use by the FDA in March, 2021. Serdexmethylphenidate is a prodrug which has a delayed onset of action and a prolonged duration of effects compared to dexmethylphenidate, its parent compound.
SCHEME
WO2021173533
US20200237742
WO2019241019
US20190381017
NEW DRUG APPROVALS
ONE TIME
$10.00
Formulations
Serdexmethylphenidate/dexmethylphenidate (Azstarys), a co-formulation of serdexmethylphenidate and dexmethylphenidate, was approved by the Food and Drug Administration (FDA) in March 2021, for the treatment of ADHD in those above six years of age. Co-formulation of serdexmethylphenidate with dexmethylphenidate allows for a more rapid onset of action while still retaining up to 13 hours of therapeutic efficacy.[3][4]
Due to serdexmethylphenidate’s delayed onset and prolonged duration of effects, several dosage forms containing serdexmethylphenidate have been investigated for use as long-acting psychostimulants in the treatment of ADHD. Under the developmental codename KP484, serdexmethylphenidate has been investigated as a “super-extended duration” psychostimulant, with therapeutic efficacy lasting up to 16 hours following oral administration. In 2011, MonoSol Rx entered into a partnership with KenPharm to develop oral films containing KP415.[5]
Abuse potential
The abuse potential of serdexmethylphenidate is theorized to be lower than other psychostimulants because serdexmethylphenidate is an inactive prodrug of dexmethylphenidate, and must undergo enzymatic metabolism prior to exerting any stimulant effects.[6] Common routes of administration used during the abuse of psychostimulants such as insufflation and intravenous injection have little impact on the pharmacokinetics and metabolism of serdexmethylphenidate and do not result in a faster onset of action.[7]
SYN
SYN
US 20200237742
Title(EN) Serdexmethylphenidate Conjugates, Compositions And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
Following deprotection (for example, but not limited to, deprotection methods as illustrated by Scheme 6 or Scheme 7) of a protected serdexmethylphenidate intermediate (for example, but not limited to, the serdexmethylphenidate intermediate prepared according to Scheme 4 or Scheme 5) , crude serdexmethylphenidate can be purified by several methods, including, but not limited to, the method according to Scheme 8.
An alternative embodiment for preparing serdexmethylphenidate is shown in FIG. 1.
Novel intermediates are produced during the process of synthesizing serdexmethylphenidate (i.e., process intermediates). These process intermediates may be isolated or form in situ, and include, but are not limited to, 3-(((S)-2-(tert-butoxy)-1-carboxyethyl)carbamoyl)-1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium; tert-butyl O-(tert-butyl)-N-nicotinoyl-L-serinate; chloromethyl (R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carboxylate; and 3-(((S)-1,3-di-tert-butoxy-1-oxopropan-2-yl)carbamoyl)-1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium.
Novel metabolites and/or novel degradants are produced during the breakdown of serdexmethylphenidate in vitro and/or in vivo. These metabolites and/or degradants include, but are not limited to, 1-((((R)-2-((R)-carboxy(phenyl)methyl)piperidine-1-carbonyl)oxy)methyl)-3-(((S)-1-carboxy-2-hydroxyethyl)carbamoyl)pyridin-1-ium; and 3-carboxy-1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium; nicotinic acid (niacin); and nicotinoyl-L-serine.
In certain embodiments of synthesizing serdexmethylphenidate other compounds may be produced including, but not limited to, dichloromethyl (R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carboxylate; 3-((1-carboxy-2-(((1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium-3-carbonyl)-L-seryl)oxy)ethyl)carbamoyl)-1-((((S)-2-((S)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium; N,N-diethyl-N-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)ethanaminium; 1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)-2,6-dimethylpyridin-1-ium; (((S)-1,3-di-tert-butoxy-1-oxopropan-2-yl)amino)methyl (R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carboxylate; ((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidin-1-yl)methyl (R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carboxylate; 3-(((R)-1-carboxy-2-chloroethyl)carbamoyl)-1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium; and 3-(((S)-3 -hydroxy-1-isopropoxy-1-oxopropan-2-yl)carbamoyl)-1-((((R)-2-((R)-2-methoxy-2-oxo-1-phenylethyl)piperidine-1-carbonyl)oxy)methyl)pyridin-1-ium.
Title(EN) Compositions Comprising Serdexmethylphenidate Conjugates And Methods Of Use Thereof
Abstract
(EN)
The present technology is directed to one or more compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more compositions and oral formulations comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology also relates to one or more methods of using compositions comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof. The present technology additionally relates to one or more pharmaceutical kits containing a composition comprising serdexmethylphenidate conjugates and unconjugated d-methylphenidate and/or a pharmaceutically acceptable salt thereof.
ClassAcetamides; Butyric acids; Hepatoprotectants; Small molecules; Sulfones; Thiazepines
Mechanism of Action Sodium-bile acid cotransporter inhibitors
Orphan Drug Status Yes – Primary biliary cirrhosis; Biliary atresia; Intrahepatic cholestasis; Alagille syndrome
New Molecular Entity Yes
Phase III Biliary atresia; Intrahepatic cholestasis
Phase II Alagille syndrome; Cholestasis; Primary biliary cirrhosis
No development reported Non-alcoholic steatohepatitis
22 Jul 2020 Albireo initiates an expanded-access programme for Intrahepatic cholestasis in USA, Canada, Australia and Europe
14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Belgium (PO) after July 2020 (EudraCT2019-003807-37)
14 Jul 2020 Phase-III clinical trials in Biliary atresia (In infants, In neonates) in Germany, France, United Kingdom, Hungary (PO) (EudraCT2019-003807-37)
UPDATE Bylvay, FDA APPROVED2021/7/20 AND EMA 2021/7/16
A-4250 (odevixibat) is a selective inhibitor of the ileal bile acid transporter (IBAT) that acts locally in the gut. Ileum absorbs glyco-and taurine-conjugated forms of the bile salts. IBAT is the first step in absorption at the brush-border membrane. A-4250 works by decreasing the re-absorption of bile acids from the small intestine to the liver, whichreduces the toxic levels of bile acids during the progression of the disease. It exhibits therapeutic intervention by checking the transport of bile acids. Studies show that A-4250 has the potential to decrease the damage in the liver cells and the development of fibrosis/cirrhosis of the liver known to occur in progressive familial intrahepatic cholestasis. A-4250 is a designated orphan drug in the USA for October 2012. A-4250 is a designated orphan drug in the EU for October 2016. A-4250 was awarded PRIME status for PFIC by EMA in October 2016. A-4250 is in phase II clinical trials by Albireo for the treatment of primary biliary cirrhosis (PBC) and cholestatic pruritus. In an open label Phase 2 study in children with cholestatic liver disease and pruritus, odevixibat showed reductions in serum bile acids and pruritus in most patients and exhibited a favorable overall tolerability profile.
Odevixibat is a highly potent, non-systemic ileal bile acid transport inhibitor (IBATi) that has has minimal systemic exposure and acts locally in the small intestine. Albireo is developing odevixibat to treat rare pediatric cholestatic liver diseases, including progressive familial intrahepatic cholestasis, biliary atresia and Alagille syndrome.
With normal function, approximately 95 percent of bile acids released from the liver into the bile ducts to aid in liver function are recirculated to the liver via the IBAT in a process called enterohepatic circulation. In people with cholestatic liver diseases, the bile flow is interrupted, resulting in elevated levels of toxic bile acids accumulating in the liver and serum. Accordingly, a product capable of inhibiting the IBAT could lead to a reduction in bile acids returning to the liver and may represent a promising approach for treating cholestatic liver diseases.
The randomized, double-blind, placebo-controlled, global multicenter PEDFIC 1 Phase 3 clinical trial of odevixibat in 62 patients, ages 6 months to 15.9 years, with PFIC type 1 or type 2 met its two primary endpoints demonstrating that odevixibat reduced serum bile acids (sBAs) (p=0.003) and improved pruritus (p=0.004), and was well tolerated with a low single digit diarrhea rate. These topline data substantiate the potential for odevixibat to be first drug for PFIC patients. The Company intends to complete regulatory filings in the EU and U.S. no later than early 2021, in anticipation of regulatory approval, issuance of a rare pediatric disease priority review voucher and launch in the second half of 2021.
Odevixibat is being evaluated in the ongoing PEDFIC 2 open-label trial (NCT03659916) designed to assess long-term safety and durability of response in a cohort of patients rolled over from PEDFIC 1 and a second cohort of PFIC patients who are not eligible for PEDFIC 1.
Odevixibat is also currently being evaluated in a second Phase 3 clinical trial, BOLD (NCT04336722), in patients with biliary atresia. BOLD, the largest prospective intervention trial ever conducted in biliary atresia, is a double-blind, randomized, placebo-controlled trial which will enroll approximately 200 patients at up to 75 sites globally to evaluate the efficacy and safety of odevixibat in children with biliary atresia who have undergone a Kasai procedure before age three months. The company also anticipates initiating a pivotal trial of odevixibat for Alagille syndrome by the end of 2020.
The odevixibat PFIC program, or elements of it, have received fast track, rare pediatric disease and orphan drug designations in the United States. In addition, the FDA has granted orphan drug designation to odevixibat for the treatment of Alagille syndrome, biliary atresia and primary biliary cholangitis. The EMA has granted odevixibat orphan designation, as well as access to the PRIority MEdicines (PRIME) scheme for the treatment of PFIC. Its Paediatric Committee has agreed to Albireo’s odevixibat Pediatric Investigation Plan for PFIC. EMA has also granted orphan designation to odevixibat for the treatment of biliary atresia, Alagille syndrome and primary biliary cholangitis.
A solution of 1,1-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-[N-((R)-α-carboxy-4-hydroxybenzyl)carbamoylmethoxy]-2,3,4,5-tetrahydro-1,2,5-benzothiadiazepine (Example 18; 0.075 g, 0.114 mmol), butanoic acid, 2-amino-, 1,1-dimethylethyl ester, hydrochloride, (2S)-(0.031 g, 0.160 mmol) and Ν-methylmorpholine (0.050 ml, 0.457 mmol) in DMF (4 ml) was stirred at RT for 10 min, after which TBTU (0.048 g, 0.149 mmol) was added. After 1h, the conversion to the ester was complete. M/z: 797.4. The solution was diluted with toluene and then concentrated. The residue was dissolved in a mixture of DCM (5 ml) and TFA (2 ml) and the mixture was stirred for 7h. The solvent was removed under reduced pressure. The residue was purified by preparative HPLC using a gradient of 20-60% MeCΝ in 0.1M ammonium acetate buffer as eluent. The title compound was obtained in 0.056 g (66 %) as a white solid. ΝMR (400 MHz, DMSO-d6): 0.70 (3H, t), 0.70-0.80 (6H, m), 0.85-1.75 (14H, m), 2.10 (3H, s), 3.80 (2H, brs), 4.00-4.15 (1H, m), 4.65 (1H, d(AB)), 4.70 (1H, d(AB)), 5.50 (1H, d), 6.60 (1H, s), 6.65-7.40 (11H, m), 8.35 (1H, d), 8.50 (1H, d) 9.40 (1H, brs).
The compound l,l-dioxo-3,3-dibutyl-5-phenyl-7-methylthio-8-(A/-{(R)-a-[A/-((S)-l-carboxypropyl) carbamoyl]-4-hydroxybenzyl}carbamoylmethoxy)-2,3,4,5-tetrahydro-l,2,5-benzothiadiazepine (odevixibat; also known as A4250) is disclosed in WO 03/022286. The structure of odevixibat is shown below.
As an inhibitor of the ileal bile acid transporter (IBAT) mechanism, odevixibat inhibits the natural reabsorption of bile acids from the ileum into the hepatic portal circulation. Bile acids that are not reabsorbed from the ileum are instead excreted into the faeces. The overall removal of bile acids from the enterohepatic circulation leads to a decrease in the level of bile acids in serum and the liver. Odevixibat, or a pharmaceutically acceptable salt thereof, is therefore useful in the treatment or prevention of diseases such as dyslipidemia, constipation, diabetes and liver diseases, and especially liver diseases that are associated with elevated bile acid levels.
According to the experimental section of WO 03/022286, the last step in the preparation of odevixibat involves the hydrolysis of a tert-butyl ester under acidic conditions. The crude compound was obtained by evaporation of the solvent under reduced pressure followed by purification of the residue by preparative HPLC (Example 29). No crystalline material was identified.
Amorphous materials may contain high levels of residual solvents, which is highly undesirable for materials that should be used as pharmaceuticals. Also, because of their lower chemical and physical stability, as compared with crystalline material, amorphous materials may display faster
decomposition and may spontaneously form crystals with a variable degree of crystallinity. This may result in unreproducible solubility rates and difficulties in storing and handling the material. In pharmaceutical preparations, the active pharmaceutical ingredient (API) is for that reason preferably used in a highly crystalline state. Thus, there is a need for crystal modifications of odevixibat having improved properties with respect to stability, bulk handling and solubility. In particular, it is an object of the present invention to provide a stable crystal modification of odevixibat that does not contain high levels of residual solvents, that has improved chemical stability and can be obtained in high levels of crystallinity.
Example 1
Preparation of crystal modification 1
Absolute alcohol (100.42 kg) and crude odevixibat (18.16 kg) were charged to a 250-L GLR with stirring under nitrogen atmosphere. Purified water (12.71 kg) was added and the reaction mass was stirred under nitrogen atmosphere at 25 ± 5 °C for 15 minutes. Stirring was continued at 25 ± 5 °C for 3 to 60 minutes, until a clear solution had formed. The solution was filtered through a 5.0 m SS cartridge filter, followed by a 0.2 m PP cartridge filter and then transferred to a clean reactor.
Purified water (63.56 kg) was added slowly over a period of 2 to 3 hours at 25 ± 5 °C, and the solution was seeded with crystal modification 1 of odevixibat. The solution was stirred at 25 ± 5 °C for 12 hours. During this time, the solution turned turbid. The precipitated solids were filtered through centrifuge and the material was spin dried for 30 minutes. The material was thereafter vacuum dried in a Nutsche filter for 12 hours. The material was then dried in a vacuum tray drier at 25 ± 5 °C under vacuum (550 mm Hg) for 10 hours and then at 30 ± 5 °C under vacuum (550 mm Hg) for 16 hours. The material was isolated as an off-white crystalline solid. The isolated crystalline material was milled and stored in LDPE bags.
An overhydrated sample was analyzed with XRPD and the diffractogram is shown in Figure 2.
Another sample was dried at 50 °C in vacuum and thereafter analysed with XRPD. The diffractogram of the dried sample is shown in Figure 1.
The diffractograms for the drying of the sample are shown in Figures 3 and 4 for 2Q ranges 5 – 13 ° and 18 – 25 °, respectively (overhydrated sample at the bottom and dry sample at the top).
A Double-Blind, Randomized, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Odevixibat (A4250) in Children with Biliary Atresia Who Have Undergone a Kasai Hepatoportoenterostomy (BOLD)
An Open-label Extension Study to Evaluate Long-term Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 2)
A Double-Blind, Randomized, Placebo-Controlled, Phase 3 Study to Demonstrate Efficacy and Safety of A4250 in Children with Progressive Familial Intrahepatic Cholestasis Types 1 and 2 (PEDFIC 1)
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt
It is well established that agents which inhibit the 20 transport of bile acids across the ileum can also cause a decrease in the level of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255- 25287, discusses biochemistry, physiology, and known active agents affecting bile acids and cholesterol.
A class of ileal bile acid transport-inhibiting compounds which was recently discovered to be useful for influencing the level of blood serum cholesterol is 30 tetrahydrobenzothiepine-l,l-dioxides (THBDO compounds). (U.S. Patent Application No. 08/816,065)
Some classes of compounds show enhanced potency as pharmaceutical therapeutics after they have been enantiomerically-enriched (see, for example, Richard B. Silverman, The Organic Chemistry of Drug Design and Drug Action, Academic Press, 1992, pp. 76-82) . Therefore, THBDO compounds that have been enantiomerically-enriched are of particular interest.
A class of chemistry useful as intermediates in the preparation of racemic THBDO compounds is tetrahydrobenzothiepine-1-oxides (THBO compounds) . THBDO compounds and THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring. A method of preparing enantiomerically-enriched samples of another phenyl/seven-member fused ring system, the benzothiazepines, is described by Higashikawa (JP 59144777) , where racemic benzothiazepine derivatives are optically resolved on a chromatographic column containing chiral crown ethers as a stationary phase. Although optical resolution is achieved, the Higashikawa method is limited to producing only small quantities of the enantiomerically-enriched benzothiazepine derivatives. Giordano (CA 2068231) reports the cyclization of (2S, 3S) -aminophenylthiopropionates in the presence of a phosphonic acid to produce (2S, 3S) -benzothiazepin-4-ones . However, that preparation is constrained by the need to use enantiomerically-enriched starting materials rather than racemic starting materials. In addition, the Giordano method controls the stereochemistry of the seven-member ring of the benzothiazepin-4-one only at the 2- and 3 -positions. The 4- and 5-positions of the seven-member ring of the benzothiazepin-4-one are not asymmetric centers, and the stereochemistry at these sites therefore cannot be controlled by the Giordano method. A method by which enantiomerically-enriched 1,5- benzothiazepin-3-hydroxy-4 (5H) -one compounds have been produced is through the asymmetric reduction of 1,5- benzothiazepin-3,4 (2H, 5H) -dione compounds, reported by Yamada, et al . (J“. Org. Chem. 1996, 61 (24), 8586-8590). The product is obtained by treating the racemic 1,5- benzothiazepin-3,4 (2H, 5H) -dione with the reaction product of an optically active alpha-amino acid and a reducing agent, for example sodium borohydride. Although a product with high optical purity was achieved, the method is limited by the use of a relatively expensive chemical reduction step.
The microbial reduction of racemic 1, 5-benzothiazepin- 3 , 4 (2H, 5H) -dione compounds to produce enantiomerically- enriched 1, 5-benzothiazepin-3-hydroxy-4 (5H) -one compounds is reported by Patel et al . , U.S. Patent 5,559,017. This method is limited by the inherent problems of maintaining a viable and pure bacterial culture of the appropriate species and variety. In addition, that method is limited in scale, producing only microgram quantities of the desired product. Until now, there have been no reported processes for preparing enantiomerically-enriched THBDO compounds or enantiomerically-enriched THBO compounds. Furthermore, there have been no reported processes for controlling the stereochemistry at the 4- and 5-positions of the seven- member rings of THBDO compounds or THBO compounds
FDA Grants Breakthrough Designation to Shire’s Rare GI Therapies
Tue, 06/14/2016
Shire announced that the U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy Designation for two investigational products for rare diseases: SHP621 (budesonide oral suspension, or BOS) for eosinophilic esophagitis (EoE), and SHP625 (maralixibat) for progressive familial intrahepatic cholestasis type 2 (PFIC2).
“Receiving Breakthrough Therapy Designation on two pipeline products this past week reflects the potential of our strong and innovative pipeline of more than 60 programs,” said Flemming Ornskov, M.D., MPH, and CEO, Shire. “Shire is committed to bringing innovation to the rare and specialty areas we focus on. We persevere to see compounds through the many stages of development through their challenges and successes, and always keep patients with unmet needs top of mind.”
EoE is a serious, chronic and rare disease that stems from an elevated number of eosinophils, a type of white blood cell, that infiltrate the walls of the esophagus. EoE is characterized by an inflammation of the esophagus that may lead to difficulty swallowing (dysphagia). The diagnosed prevalence of EoE ranges from approximately 15-55 cases per 100,000 persons, with high-end estimates reported by studies in Western regions.
PFIC refers to a group of autosomal-recessive liver disorders of childhood that disrupt bile formation and present with cholestasis. The symptoms of PFIC include severe itching of the skin (pruritus), and jaundice. PFIC is estimated to affect 1 in 50,000 to 1 in 100,000 births. PFIC2 is the most common type of PFIC, accounting for around half of cases.
According to the FDA, Breakthrough Therapy Designation is granted to a therapy that is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on one or more clinically significant endpoints over current standard of care. Under the designation, the FDA provides intensive guidance, organizational commitment involving senior managers, and eligibility for rolling and priority review of the application; this process helps ensure patients have access to therapies as soon as possible, pending approval. Breakthrough Therapy Designation does not guarantee that FDA will ultimately approve BOS for EoE or maralixibat for PFIC2, and the timing of any such approval is uncertain.
“On behalf of patients in the United States with EoE and PFIC2, we are so pleased that the FDA has granted Breakthrough Therapy Designation to BOS and maralixibat,” said Philip J. Vickers, Ph.D., Head of R&D, Shire. “We look forward to working with the agency to continue their development and, pending FDA approval, deliver these therapeutic options to the patients who need them most.”
It is well established that agents which inhibit the transport of bile acids across the tissue of the ileum can also cause a decrease in the levels of cholesterol in blood serum. Stedronski, in “Interaction of bile acids and cholesterol with nonsystemic agents having hypocholesterolemic properties,” Biochimica et Biophysica Acta, 1210 (1994) 255-287 discusses biochemistry, physiology, and known active agents surrounding bile acids and cholesterol. Bile acids are actively transported across the tissue of the ileum by an apical sodium co-dependent bile acid transporter (ASBT), alternatively known as an ileal bile acid transporter (IBAT). A class of ASBT-inhibiting compounds that was recently discovered to be useful for influencing the level of blood serum cholesterol comprises tetrahydrobenzothiepine oxides (THBO compounds, PCT Patent Application No. WO 96/08484). Further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 97/33882. Additional THBO compounds useful as ASBT inhibitors are described in U.S. Patent No. 5,994,391. Still further THBO compounds useful as ASBT inhibitors are described in PCT Patent Application No. WO 99/64409. Included in the THBO class are tetrahydrobenzo-thiepine-l -oxides and tetrahydrobenzothiepine- 1,1 -dioxides. THBO compounds possess chemical structures in which a phenyl ring is fused to a seven-member ring.
Published methods for the preparation of THBO compounds include the synthesis through an aromatic sulfone aldehyde intermediate. For example l-(2,2-dibutyl-3-oxopropylsulfonyl)-2-((4-methoxyphenyl)methyl)benzene (29) was cyclized with potassium t-butoxide to form tetrahydrobenzothiepine- 1,1 -dioxide (svn-24) as shown in Eq. 1.
Compound 29 was prepared by reacting 2-chloro-5-nitrobenzoic acid chloride with anisole in the presence of aluminum trichloride to produce a chlorobenzophenone compound; the chlorobenzophenone compound was reduced in the presence of trifluoromethanesulfonic acid and triethylsilane to produce a chlorodiphenylmethane compound; the chlorodiphenylmethane compound was treated with lithium sulfide and 2,2-dibutyl-3-(methanesulfonato)propanal to produce l-(2,2-dibutyl-3-oxopropylthio)-2-((4-methoxyphenyl)methyl)-4-dimethylaminobenzene (40); and 40 was oxidized with m-chloroperbenzoic acid to produce 29. The first step of that method of preparing compound 29 requires the use of a corrosive and reactive carboxylic acid chloride that was prepared by the reaction of the corresponding carboxylic acid with phosphorus pentachloride. Phosphorus pentachloride readily hydrolyzes to produce volatile and hazardous hydrogen chloride. The reaction of 2,2-dibutyl-3-(methanesulfonato)propanal with the lithium sulfide and the chlorodiphenylmethane compound required the intermediacy of a cyclic tin compound to make the of 2,2-dibutyl-3-(methanesulfonato)propanal. The tin compound is expensive and creates a toxic waste stream. In WO 97/33882 compound syn-24 was dealkylated using boron tribromide to produce the phenol compound 28. Boron tribromide is a corrosive and hazardous material that generates hydrogen bromide gas and requires special handling. Upon hydrolysis, boron tribromide also produces borate salts that are costly and time-consuming to separate and dispose of.
An alternative method of preparing THBO compounds was described in WO 97/33882, wherein a 1,3-propanediol was reacted with thionyl chloride to form a cyclic sulfite compound. The cyclic sulfite compound was oxidized to produce a cyclic sulfate compound. The cyclic sulfate was condensed with a 2-methylthiophenol that had been deprotonated with sodium hydride. The product of the condensation was a (2-methylphenyl) (3′-hydroxypropyl)thioether compound. The thioether compound was oxidized to form an thioether aldehyde compound. The thioether aldehyde compound was further oxidized to form an aldehyde sulfone compound which in turn was cyclized in the presence of potassium t-butoxide to form a 4-hydroxytetrahydrobenzothiepine 1,1 -dioxide compound. This cyclic sulfate route to THBO compounds requires an expensive catalyst. Additionally it requires the use of SOCI2, which in turn requires special equipment to handle. PCT Patent Application No. WO 97/33882 describes a method by which the phenol compound 28 was reacted at its phenol hydroxyl group to attach a variety of functional groups to the molecule, such as a quaternary ammonium group. For example, (4R,5R)-28 was reacted with l,4-bis(chloromethyl)benzene (?,??’-dichloro-p-xylene) to produce the chloromethyl benzyl- ether (4R,5R)-27. Compound (4R,5R)-27 was treated with diazabicyclo[2.2.2]octane (DABCO) to produce (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro-4-hydroxy-l , 1 -dioxido-1 -benzothiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza-l-azomabicyclo[2.2.2]octane chloride (41). This method suffers from low yields because of a propensity for two molecules of compound (4R,5R)-28 to react with one molecule of l,4-bis(chloromethyl)benzene to form a bis(benzothiepine) adduct. Once the bis-adduct forms, the reactive chloromethyl group of compound (4R,5R)-27 is not available to react with an amine to form the quaternary ammonium product.
A method of preparing enantiomerically enriched tetrahydrobenzothiepine oxides is described in PCT Patent Application No. WO 99/32478. In that method, an aryl-3- hydroxypropylsulfide compound was oxidized with an asymmetric oxidizing agent, for example (lR (->(8,9-dichloro-10-camphorsulfonyl)oxaziridine, to yield a chiral aryl-3-hydroxypropylsulfoxide. Reaction of the aryl-3-hydroxypropylsulfoxide with an oxidizing agent such as sulfur trioxide pyridine complex yielded an aryl-3-propanalsulfoxide. The aryl- 3-propanalsulfoxide was cyclized with a base such as potassium t-butoxide to enantioselectively produce a tetrahydrobenzothiepine- 1 -oxide. The tetrahydrobenzothiepine- 1 -oxide was further oxidized to produce a tetrahydrobenzothiepine- 1 , 1 -dioxide. Although this method could produce tetrahydrobenzothiepine- 1,1 -dioxide compounds of high enantiomeric purity, it requires the use of an expensive asymmetric oxidizing agent. Some 5-amidobenzothiepine compounds and methods to make them are described in
PCT Patent Application Number WO 92/18462. In Svnlett. 9, 943-944(1995) 2-bromophenyl 3-benzoyloxy-l-buten-4-yl sulfone was treated with tributyl tin hydride and AIBN to produce 3-benzoyloxytetrahydrobenzothiepine-1,1 -dioxide. In addition to forming the desired ASBT inhibitors, it is also desirable to form such
ASBT inhibitors of higher purity and having lower levels of residual solvent impurities. This is especially so with respect to ASBT inhibitors having a positively charged substituent, for example, the compounds designated as 41 (supra) and 60 (infra). It is further desirable to provide methods for making such high purity ASBT inhibitors.
( 4R, 5R) -26 A 1000 mL 4 neck jacketed Ace reactor flask was fitted with a mechanical stirrer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask was purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). To this was added 4.2 grams of 50% sodium hydroxide. The mixture was heated to 50°C and stiπed for 15 minutes. To the flask was added 8.3 grams of 55 dissolved in 10 mL of DMAC, all at once. The temperature was held at 50°C for 24 hrs. To the flask was added 250 mL of toluene followed by 125 mL of dilution water. The mixture was stiπed for 15 minutes and the layers were then allowed to separate at 50°C. The flask was then charged with 125 mL of saturated sodium chloride solution and stiπed 15 minutes. Layers separated cleanly in 30 seconds at 50°C. Approximately half of the solvent was distilled off under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
( 4R, 5R) -27 Toluene was charged back to the reaction mixture of Step 1 and the mixture was cooled to 35°C. To the mixture was then added 7.0 grams of thionyl chloride over 5 minutes. The reaction was exothermic and reached 39°C. The reaction turned cloudy on first addition of thionyl chloride, partially cleared then finally remained cloudy. The mixture was stirred for 0.5 hr and was then washed with 0.25N NaOH. The mixture appeared to form a small amount of solids that diminished on stirring, and the layers cleanly separated. The solvent was distilled to a minimum stir volume under vacuum at 50°C. The residual reaction mixture contained (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 was charged with 350 mL of methyl ethyl ketone (MEK) followed by 10.5 mL water and 6.4 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 10 mL of MEK. The mixture was heated to reflux, and HPLC showed <0.5% of (4R,5R)-27. The reaction remained homogenous initially then crystallized at the completion of the reaction. An additional 5.3 mL of water was charged to the flask to redissolve product. Approximately 160 mL of solvent was then distilled off at atmospheric pressure. The mixture started to form crystals after 70 mL of solvent was distilled. Water separated out of distillate indicating a ternary azeotrope between toluene, water and methyl ethyl ketone (MEK). The mixture was then cooled to 25°C. The solids were filtered and washed with 150 mL MEK, and let dry under vacuum at 60°C. Isolated 29.8.0 g of off-white crystalline 4 Example 11a. Alternate Preparation of (4R,5R)-l-((4-(4-(3,3-dibutyl-7-(dimemylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a 28 mm Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 100 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 4.02 grams of 50% sodium hydroxide. The mixture is stiπed for 30 minutes. To the flask is added 8.7 grams of 55 dissolved in 12.5 mL of DMAC, all at once. The charge vessel is washed with 12.5 mL DMAC and the wash is added to the reactor. The reactor is stiπed for 3 hours. To the reactor is added 0.19 mL of 49.4% aq. NaOH and the mixture is stirred for 2 hours. To the mixture is added 0.9 g DABCO dissolved in 12.5 mL DMAC. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of dilution water. The mixture is stiπed for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stiπed 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (225 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 30°C. To the mixture is then added 6.7 grams of thionyl chloride over 30 to 45 minutes. The temperature is maintained below 35°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is stiπed for 0.5 hr and is then charged with 156.6 mL of 4% NaOH wash over a 30 minute period. The addition of the wash is stopped when the pH of the mixture reaches’ 8.0 to 10.0. The bottom aqueous layer is removed at 30°C and any rag layer is retained with the organic layer. To the mixture is charged 175 mL of saturated NaCl wash with agitation. The layers are separated at 30°C and the bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-27.
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 325 mL of methyl ethyl ketone (MEK) and 13 mL water. Next, the reactor is charged 6.2 grams of diazabicyclo[2.2.2]octane (DABCO) dissolved in 25 mL of MEK. The mixture is heated to reflux and held for 30 minutes. Approximately 10% of solvent volume is then distilled off. The mixture starts to form crystals during distillation. The mixture is then cooled to 20°C for 1 hour. The off-white crystalline 41 (Form U) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example lib. Alternate Preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form II of 41
A 1000 mL 4 neck jacketed Ace reactor flask is fitted with a mechanical stiπer, a nitrogen inlet, an addition funnel or condenser or distilling head with receiver, a thermocouple, four internal baffles and a Teflon turbine agitator. The flask is purged with nitrogen gas and charged with 25.0 grams of (4R,5R)-28 and 125 mL of N,N-dimethylacetamide (DMAC). The mixture is heated to 50°C and to it is added 7.11 grams of 30% sodium hydroxide over a period of 15 to 30 minutes with agitation. The mixture is stiπed for 30 minutes. To the flask is added 9.5 grams of solid 55. The reactor is stiπed for 3 hours. To the mixture is added 1.2 g of solid DABCO. The mixture is stiπed 30 to 60 minutes at 50°C. To the flask is added 225 mL of toluene followed by 125 mL of water. The mixture is stirred for 15 minutes and the layers are then allowed to separate at 50°C. The bottom aqueous layer is removed but any rag layer is retained with the organic layer. The flask is then charged with 175 mL of 5% hydrochloric acid solution and stirred 15 minutes. Layers are separated at 50°C to remove the bottom aqueous layer, discarding any rag layer with the aqueous layer. The flask is then charged with 225 mL of water and stirred 15 minutes. The layers are allowed to separate at 50°C. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. Approximately half of the solvent is distilled off under vacuum at a maximum pot temperature of 80°C. The residual reaction mixture contains (4R,5R)-26.
Step 2. Preparation of (4R.5RV27.
Toluene (112.5 mL) is charged back to the reaction mixture of Step 1 and the mixture is cooled to 25°C. To the mixture is then added 7.3 grams of thionyl chloride over 15 to 45 minutes. The temperature of the mixture is maintained above 20°C and below 40°C. The reaction turns cloudy on first addition of thionyl chloride, then at about 30 minutes the layers go back together and form a clear mixture. The mixture is then charged with 179.5 mL of 4% NaOH wash over a 30 minute period. The mixture is maintained above 20°C and below 40°C during this time. The addition of the wash is stopped when the pH of the mixture reaches 8.0 to 10.0. The mixture is then allowed to separate at 40°C for at least one hour.
The bottom aqueous layer is removed and any rag layer is retained with the organic layer. To the mixture is charged 200 mL of dilution water. The mixture is stiπed for 15 minutes and then allowed to separate at 40°C for at least one hour. The bottom aqueous layer is removed, discarding any rag layer with the aqueous layer. The solvent is distilled to a minimum stir volume under vacuum at 80°C. The residual reaction mixture contains (4R,5R)-2 .
Step 3. Preparation of 41. To the reaction mixture of Step 2 is charged 350 mL of methyl ethyl ketone (MEK) and 7 mL water. The mixture is stiπed for 15 minutes and the temperature of the mixture is adjusted to 25°C. Next, the reactor is charged with 6.7 grams of solid diazabicyclo[2.2.2]octane (DABCO). The mixture is maintained at 25°C for three to four hours. It is then heated to 65°C and maintained at that temperature for 30 minutes. The mixture is then cooled to 25°C for 1 hour. The off-white crystalline 41 (Form II) is filtered and washed with 50 mL MEK, and let dry under vacuum at 100°C.
Example 12. Alternate preparation of (4R,5R)-1 -((4-(4-(3,3-dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro- 4-hydroxy- 1 , 1 -dioxido- 1 -benzithiepin-5-yl)phenoxy)methyl)phenyl)methyl-4-aza- 1 – azoniabicyclo[2.2.2]octane chloride, Form I of 41
(4R,5R)-27 (2.82 kg dry basis, 4.7 mol) was dissolved in MTBE (9.4 L). The solution of (4R,5R)-22 was passed through a 0.2 mm filter cartridge into the feeding vessel. The flask and was rinsed with MTBE (2 x 2.5 L). The obtained solution as passed through the cartridge filter and added to the solution of (4R,5R)-2 in the feeding vessel. DABCO (diazabicyclo[2.2.2]octane, 0.784 kg, 7.0 mol) was dissolved in MeOH (14.2 L). The DABCO solution was passed through the filter cartridge into the 100 L nitrogen-flushed reactor. The Pyrex bottle and the cartridge filter were rinsed with MeOH (7.5 L) and the solution was added to the reactor. The (4R,5R)-22 solution was added from the feeding vessel into the reactor at 37°C over a period of 10 min, while stirring. Methanol (6.5 L) was added to the Pyrex bottle and via the cartridge filter added to the feeding vessel to rinse the remaining (4R,5R)-2 into the reactor. The reaction mixture was brought to 50-60°C over 10-20 min and stiπed at that temperature for about 1 h. The mixture was cooled to 20-25°C over a period of 1 h. To the reaction mixture, methyl t-butyl ether (MTBE) (42 L) was added over a period of 1 h and stiπed for a minimum of 1 h at 20 – 25°C. The suspension was filtered through a Buchner funnel. The reactor and the filter cake were washed with MTBE (2 x 14 L). The solids were dried on a rotary evaporator in a 20 L flask at 400 – 12 mbar, 40°C, for 22 h. A white crystalline solid was obtained. The yield of 4 . (Form I) was 3.08 kg (2.97 kg dry, 93.8 %) and the purity 99.7 area % (HPLC; Kromasil C 4, 250 x 4.6 mm column; 0.05% TFA in H2O/0.05% TFA in ACN gradient, UV detection at 215 nm).
Example 12a. Conversion of Form I of Compound 41 into Form II of Compound 41.
To 10.0 grams of Form I of 4 . in a 400 mL jacketed reactor is added 140 mL of MEK. The reactor is stirred (358 φm) for 10 minutes at 23 °C for 10 minutes and the stirring rate is then changed to 178 φm. The suspension is heated to reflux over 1 hour using a programmed temperature ramp (0.95°C/minute) using batch temperature control (cascade mode). The delta Tmaχ is set to 5°C. The mixture is held at reflux for 1 hour. The mixture is cooled to
25°C. After 3 hours at 25°C, a sample of the mixture is collected by filtration. Filtration is rapid (seconds) and the filtrate is clear and colorless. The white solid is dried in a vacuum oven (80°C, 25 in. Hg) to give a white solid. The remainder of the suspension is stirred at 25°C for 18 hours. The mixture is filtered and the cake starts to shrink as the mother liquor reaches the top of the cake. The filtration is stopped and the reactor is rinsed with 14 mL of MEK. The reactor stirrer speed is increased from 100 to 300 φm to rinse the reactor. The rinse is added to the filter and the solid is dried with a rapid air flow for 5 minutes. The solid is dried in a vacuum oven at 25 in. Hg for 84 hours to give Form II of 4
Department of Discovery Chemistry and Department of Cardiovascular Disease, Pharmacia, 700 Chesterfield Parkway W, Chesterfield, Missouri 63017, Office of Science and Technology, Chemical Science Division, Pharmacia, 800 Lindbergh Boulevard, Creve Coeur, Missouri 63167, Department of Pharmaceutical Sciences, Pharmacia, Skokie, Illinois, and Department of Chemistry, University of Missouri, St. Louis, Missouri
In the preceding paper several compounds were reported as potent apical sodium-codependent bile acid transporter (ASBT) inhibitors. Since the primary site for active bile acid reabsorption is via ASBT, which is localized on the luminal surface of the distal ileum, we reasoned that a nonsystemic inhibitor would be desirable to minimize or eliminate potential systemic side effects of an absorbed drug. To ensure bioequivalency and product stability, it was also essential that we identify a nonhygroscopic inhibitor in its most stable crystalline form. A series of benzothiepines were prepared to refine the structure−activity relationship of the substituted phenyl ring at the 5-position of benzothiepine ring and to identify potent, crystalline, nonhygroscopic, and efficacious ASBT inhibitors with low systemic exposure.
compd
R
IC50 (nM)b
hygroscp I wt gain (%)c
hygroscp II % wt gain (%)d
crystallinitye
74
OCH2C6H4(p)CH2(N+)DB
0.28
1.59
2.1
yes
(4R–cis)-1-[[4-[[4-[3,3-Dibutyl-7-(dimethylamino)-2,3,4,5-tetrahydro-4-hydroxy-1,1-dioxido-1-benzothiepin-5-yl]phenoxy]methyl]phenyl]methyl]-4-aza-1-azoniabicyclo[2.2.2]octane Chloride Salt(74). Following a similar procedure as in General Method B, the title compound 74 was prepared from the corresponding chloromethyl benzyl ether and DABCO as a white solid, mp 223−230 °C (dec); 1H NMR (CDCl3) δ 0.89 (m, 6H), 1.27−1.52 (br m, 10H), 1.63 (m, 1H), 2.20 (m, 1H), 2.81 (s, 6H), 3.06 (ABq, JAB = 15.1 Hz, J = 43.3 Hz, 2H), 3.16 (s, 6H), 3.76 (s, 6H), 4.11 (d, J = 7.7 Hz, 1H), 5.09 (s, 2H), 5.14 (s, 2H), 5.48 (s, 1H), 5.96 (s, 1H), 6.49 (d, J = 8.9 Hz, 1H), 6.99 (d, J = 8.0 Hz, 2H), 7.26 (m, 1H), 7.44 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 7.4 Hz, 2H), 7.68 (d, J = 7.4 Hz, 2H), 7.87 (d, J = 8.9 Hz, 1H). HRMS calcd for C40H56N3O4S: 674.3992; found, 674.4005. Anal. Calcd for C40H56N3O4S: ‘ C, 67.62; H, 7.95; N, 5.92; S, 4.51. Found: C, 67.48; H, 8.32; N, 5.85; S, 4.60.
a All compounds were prepared using method B in Scheme 3.b Taurocholate is transported across the baby hamster kidney cell membrane.c % weight gain in a 25 °C, 57% humidity chamber for 2 weeks.d % weight gain in a 40 °C, 80% humidity chamber for 2 weeks.e Crystallinity as determined by X-ray powder diffraction analysis.f (N+)DB is a DABCO terminal group with the quaternary ammonium attached to the linke
Example 10. Preparation of enantiomerically-enriched (4R.5R)-1- r.4- r _4- .3.3 -Dibutyl-7- (dimethylamino) -2.3 ,4.5- tetrahydro-4-hydroxy-1, l-dioxido-l-benzothiepin-5- yl] henoxy] ethyl] phenyl1methyl] -4-aza-l- azoniabicyclo [2.2.2] octane chloride ( (4R,5R) -XXVII) ♦
( (4R,5R) -XXVII) * = chiral center
Step 1. Preparation of 4-flUoro-2- ( (4- methoxyphenyl) methyl) -phenol To a stirred solution of 23.66 g of 95% sodium hydride (0.94 mol) in 600 mL of dry toluene was added 100.0 g of 4- fluorophenol (0.89 mol) at 0°C. The mixture was stirred at 90°C for 1 hour until gas evolution stopped. The mixture was cooled down to room temperature and a solution of 139.71 g of 3 -methoxybenzyl chloride (0.89 mol) in 400 mL of dry toluene was added. After refluxing for 24 hours, the mixture was cooled to room temperature and quenched with 500 mL of water. The organic layer was separated, dried over MgS04, and concentrated under high vacuum. The remaining starting materials were removed by distillation. The crude dark red oil was filtered through a layer of 1 L of silica gel with neat hexane to yield 53.00 g (25.6%) of the product as a pink solid: *H NMR (CDC13) d 3.79 (s, 3H) , 3.90 (s, 2H) , 4.58 (s, IH) , 6.70-6.74 (m, IH) , 6.79-6.88 (m, 4H) , 7.11-7.16 (m, 2H) .
Step 2. Preparation of 4-fluoro-2- ( (4- methoxyphenyl) methyl) -thiophenol
Step 2a. Preparation of thiocarbamate To a stirred solution of 50.00 g (215.30 mmol) of 4- fluoro-2- ( ( -methoxyphenyl) methyl) -phenol in 500 mL of dry DMF was added 11.20 g of 60% sodium hydride dispersion in mineral oil (279.90 mmol) at 2°C. The mixture was allowed to warm to room temperature and 26.61 g of dimethylthiocarbamoyl chloride (215.30 mmol) was added. The reaction mixture was stirred at room temperature overnight. The mixture was quenched with 100 mL of water in an ice bath. The solution was extracted with 500 mL of diethyl ether. The ether solution was washed with 500 mL of water and 500 mL of brine. The ether solution was dried over MgS04 and stripped to dryness. The crude product was filtered through a plug of 500 mL silica gel using 5% ethyl acetate/hexane to yield 48.00 g (69.8%) of the product as a pale white solid: XH NMR (CDC13) d 3.21 (s, 3H) , 3.46 (s, 3H) , 3.80 (s, 3H) , 3.82 (s, 2H) , 6.78-6.86 (m, 3H) , 6.90- 7.00 (m, 2H) , 7.09 (d, J = 8.7 Hz, 2H) .
Step 2b. Rearrangement and hydrolysis of thiocarbamate to 4-fluoro-2- ( (4 -methoxyphenyl) methyl) -thiophenol A stirred solution of 48.00 g (150.29 mmol) of thiocarbamate (obtained from Step 2a) in 200 mL of diphenyl ether was refluxed at 270°C overnight. The solution was cooled down to room temperature and filtered through 1 L of silica gel with 2 L of hexane to remove phenyl ether. The rearrangement product was washed with 5% ethyl acetate/hexane to give 46.00 g (95.8%) of the product as a pale yellow solid: XH NMR (CDC13) d 3.02 (s, 3H) , 3.10 (s, 3H) , 3.80 (s, 3H) , 4.07 (s, 2H) , 6.82-6.86 (m, 3H) , 6.93 (dt, J = 8.4 Hz, 2.7 Hz, IH) , 7.08 (d, J = 8.7 Hz, 2H) , 7.49 (dd, J = 6.0 Hz, 8.7 Hz, IH) . To a solution of 46.00 g (144.02 mmol) of the rearrangement product (above) in 200 mL of methanol and 200 mL of THF was added 17.28 g of NaOH (432.06 mmol) . The mixture was refluxed under nitrogen overnight . The solvents were evaporated off and 200 mL of water was added. The aqueous solution was washed with 200 mL of diethyl ether twice and placed in an ice bath. The aqueous mixture was acidified to pH 6 with concentrated HCl solution. The solution was extracted with 300 mL of diethyl ether twice. The ether layers were combined, dried over MgS04 and stripped to dryness to afford 27.00 g (75.5%) of the product as a brown oil: XH NMR (CDC13) d 3.24 (s, IH) , 3.80 (s, 3H) , 3.99 (s, 2H) , 6.81-6.87 (m, 4H) , 7.09 (d, J = 8.7 Hz, 2H) , 7.27- 7.33 (m, IH) .
Step 3. Preparation of dibutyl cyclic sulfate
Step 3a. Preparation of 2 , 2-dibutyl-l, 3-propanediol . To a stirred solution of di-butyl-diethylmalonate (Aldrich) (150g, 0.55 mol in dry THF (700ml) in an acetone/dry ice bath was added LAH (1 M THF) 662 ml (1.2 eq. , 0.66 mol) dropwise maintaining the temperature between -20 to 0°C. The reaction was stirred at RT overnight. The reaction was cooled to -20°C and 40 ml of water, and 80 mL of 10% NaOH and 80 ml of water were added dropwise. The resulting suspension was filtered. The filtrate was dried over sodium sulphate and concentrated in vacuo to give diol 598.4 g (yield 95%) as an oil. MS spectra and proton and carbon NMR spectra were consistent with the product.
Step 3b. Preparation of dibutyl cyclic sulfite
A solution of 2 , 2-dibutyl-l, 3-propanediol (103 g, 0.548 0 mol, obtained from Step 3a) and triethylamine (221 g, 2.19 mol) in anhydrous methylene chloride (500 ml) was stirred at 0°C under nitrogen. To the mixture, thionyl chloride (97.8* g, 0.‘82 mol) was added dropwise and within 5 min the solution turned yellow and then black when the addition was 5 completed within half an hour. The reaction mixture was stirred for 3 hrs. at 0°C. GC showed that there was no starting material left. The mixture was washed with ice water twice then with brine twice . The organic phase was dried over magnesium sulfate and concentrated under vacuum 0 to give 128 g (100%) of the dibutyl cyclic sulfite as a black oil. Mass spectrum (MS) was consistent with the product .
Step 3c. Oxidation of dibutyl cyclic sulfite to 5 dibutyl cyclic sulfate
To a solution of the dibutyl cyclic sulfite (127.5 g , 0.54 mol, obtained from Step 3b) in 600 ml acetonitrile and 500 ml of water cooled in an ice bath under nitrogen was added ruthenium (III) chloride (1 g) and sodium periodate 0 (233 g, 1.08 mol) . The reaction was stirred overnight and the color of the solution turned black. GC showed that there was no starting material left. The mixture was extracted with 300 ml of ether and the ether extract was washed three times with brine. The organic phase was dried over magnesium sulfate and passed through celite. The filtrate was 5 concentrated under vacuum and to give 133 g (97.8%) of the dibutyl cyclic sulfate as an oil. Proton and carbon NMR and MS were consistent with the product.
Step 4. Preparation of aryl-3-hydroxypropylsulfide
10 To a stirred solution of 27.00 g (108.73 mmol) of 4- fluoro-2- ( (4-methoxyphenyl) methyl) thiophenol (obtained from Step 2) in 270 mL of diglyme was added 4.35 g of 60% sodium-, hydride dispersion in mineral oil (108.73 mmol) at 0°C. After gas evolution ceased, 29.94 g (119.60 mmol) of the
15 dibutyl cyclic sulfate (obtained from Step 3c) was added at 0°C and stirred for 10 minutes. The mixture was allowed to warm up to room temperature and stirred overnight. The solvent was evaporated and 200 mL of water was added. The solution was washed with 200 mL of diethyl ether and added
2025 mL of concentrated sulfuric acid to make a 2.0 M solution that was refluxed overnight. The solution was extracted with ethyl acetate and the organic solution was dried over MgS04 and concentrated in vacuo. The crude aryl-3 – hydroxypropylsulfide was purified by silica gel
25 chromatography (Waters Prep 500) using 8% ethyl acetate/hexane to yield 33.00 g (72.5%) of the product as a light brown oil: E NMR (CDC13) d 0.90 (t, J = 7.1 Hz, 6H) , 1.14-1.34 (m, 12H) , 2.82 (s, 2H) , 3.48 (s, 2H) , 3.79 (s, 3H) , 4.10 (s, 2H) , 6.77-6.92 (m, 4H) , 7.09 (d, J = 8.7 Hz,
To a stirred solution of 20.00 g (47.78 mmol) of aryl- 53 -hydroxypropylsulfide (obtained from Step 4) in 1 L of methylene chloride was added 31.50 g of 96% (12?) – ( -) – (8 , 8- dichloro-10-camphor-sulfonyl) oxaziridine (100.34 mmol, Aldrich) at 2°C. After all the oxaziridine dissolved the mixture was placed into a -30 °C freezer for 72 hours. The
10 solvent was evaporated and the crude solid was washed with 1 L of hexane. The white solid was filtered off and the hexane solution was concentrated in vacuo. The crude oil was purified on a silica gel column (Waters Prep 500) using 15% ethyl acetate/hexane to afford 19.00 g (95%) of the
207.00 (d, J = 8.1 Hz, 2H) , 7.18-7.23 (m, IH) , 7.99-8.04 (m, IH) . Enantiomeric excess was determined by chiral HPLC on a (2?,2?) -Whelk-0 column using 5% ethanol/hexane as the eluent. It showed to be 78% e.e. with the first eluting peak as the major product.
25
Step 6. Preparation of enantiomerically-enriched aryl-3- propanalsulfoxide
To a stirred solution of 13.27 g of triethylamine (131.16 mmol, Aldrich) in 200 mL dimethyl sulfoxide were
30 added 19.00 g (43.72 mmol) of enantiomerically-enriched aryl-3 -hydroxypropylsulfoxide (obtained from Step 5) and 20.96 g of sulfur trioxide-pyridine (131.16 mmol, Aldrich) at room temperature. After the mixture was stirred at room temperature for 48 hours, 500 mL of water was added to the mixture and stirred vigorously. The mixture was then 5 extracted with 500 mL of ethyl acetate twice. The ethyl acetate layer was separated, dried over MgS04, and concentrated in vacuo. The crude oil was filtered through 500 mL of silica gel using 15% ethyl acetate/hexane to give 17.30 g (91%) of the enantiomerically-enriched aryl-3-
Step 7. Preparation of the enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R, 5R)
20 To a stirred solution of 17.30 g (39.99 mmol) of enantiomerically-enriched aryl-3 -propanalsulfoxide (obtained from Step 6) in 300 mL of dry THF at -15°C was added 48 mL of 1.0 M potassium t-butoxide in THF (1.2 equivalents) under nitrogen. The solution was stirred at -15°C for 4 hours.
25 The solution was then quenched with 100 mL of water and neutralized with 4 mL of concentrated HCl solution at 0°C. The THF layer was separated, dried over MgS04, and concentrated in vacuo. The enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (4R,5R) was purified by
To a stirred solution of 13.44 g (31.07 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1-oxide (obtained from Step 7) in 150 mL of methylene chloride was added 9.46 g of 68% m-chloroperoxybenzoic acid (37.28 mmol,
15 Sigma) at 0 °C. After stirring at 0 °C for 2 hours, the mixture was allowed to warm up to room temperature and stirred for 4 hours. 50 mL of saturated Na2S03 was added into the mixture and stirred for 30 minutes. The solution was then neutralized with 50 mL of saturated NaHC03 solution.
20 The methylene chloride layer was separated, dried over MgS04, and concentrated in vacuo to give 13.00 g (97.5%) of the enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (4R,5R) as a light yellow solid: ‘H NMR (CDC13) d 0.89-0.95 (m, 6H) , 1.09-1.42 (m, 12H) , 2.16-2.26 (m, IH) ,
30 Step 9. Preparation of enantiomerically-enriched 7-
(dimethylamino) tetrahydrobenzothiepine-1 , 1-dioxide (4R.5R) – To a solution of 13.00 g (28.98 mmol) of enantiomerically-enriched tetrahydrobenzothiepine-1, 1- dioxide (obtained from Step 8) in 73 mL of dimethylamine (2.0 M in THF, 146 mmol) in a Parr Reactor was added ca . 20 5 mL of neat dimethylamine . The mixture was sealed and stirred at 110 °C overnight, and cooled to ambient temperature. The excess dimethylamine was evaporated. The crude oil was dissolved in 200 mL of ethyl acetate and washed with 100 mL of water, dried over MgS04 and
10 concentrated in vacuo. Purification on a silica gel column (Waters Prep 500) using 20% ethyl acetate/hexane gave 12.43 g (90.5%) of the enantiomerically- enriched 7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (4R, 5R) as a colorless solid: *H NMR (CDC13) d 0.87-0.93 (m, 6H) ,
20 IH) . The product was determined to have 78% e.e. by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. Recrystallization of this solid from ethyl acetate/hexane gave 1.70 g of the racemic product. The remaining solution was concentrated and recrystallized to
25 give 9.8 g of colorless solid. Enantiomeric excess of this solid was determined by chiral HPLC on a Chiralpak AD column using 5% ethanol/hexane as the eluent. It showed to have 96% e.e with the first eluting peak as the major product.
30 Step 10: Demethylation of 5- (4 ‘ -methoxyphenyl) -7-
(dimethylamino) tetrahydrobenzothiepine-1.1-dioxide (4R, 5R) To a solution of 47 g (99 mmol) of enantiomeric- enriched (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (obtained from Step 9) in 500 mL of methylene chloride at -10 °C was added dropwise a solution of boron tribromide (297 mL, 1M in methylene chloride, 297 mmol), and the resulting solution was stirred cold (-5 °C to 0 °C) for 1 hour or until the reaction was complete. The reaction was cooled in an acetone-dry ice bath at -10 °C, and slowly quenched with 300 mL of water. The mixture was warmed to 10 °C, and further diluted with 300 mL of saturated sodium bicarbonate solution to neutralize the mixture. The aqueous layer was separated and extracted with 300 mL of methylene chloride, and the combined extracts were washed with 200 mL of water, brine, dried over MgS04 and concentrated in vacuo. The residue was dissolved in 500 mL of ethyl acetate and stirred with 50 mL of glacial acetic acid for 30 minutes at ambient temperature. The mixture was washed twice with 200 mL of water, 200 mL of brine, dried over MgS04 and concentrated in vacuo to give the crude 4-hydroxyphenyl intermediate. The solid residue was recrystallized from methylene chloride to give 37.5 g (82%) of the desired (4R, 5R) -5- (4′ – hydoxyphenyl) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1- dioxide as a white solid: *H NMR (CDC13) d 0.84-0.97 (m, 6H) , 1.1-1.5 (m, 10H) , 1.57-1.72 (m, IH) , 2.14-2.28 (m, IH) , 2.83 (s, 6H) , 3.00 (d, J = 15.3 Hz, IH) , 3.16 (d, J – 15.3 Hz, IH) , 4.11 (s, 2H) , 5.48 (s, IH) , 6.02 (d, J – 2.4 Hz, IH) , 6.55 (dd, J“ = 9, 2.4 Hz, IH) , 6.88 (d, 8 , 7 Hz , 2H) , 7.38 (d, J – 8.7 Hz, 2H) , 7.91 (d, J“ = 9 Hz, 2H) .
Step 11: Preparation of enantiomerically-enriched chlorobenzyl intermediate Treat a solution of enantiomerically-enriched (4R,5R)- 5- (4′ -hydoxypheny1) -7- (dimethylamino) tetrahydrobenzothiepine-1, 1-dioxide (5.0 g, 10.9 mmol, obtained from Step 10) in acetone (100 mL) at 25 °C under N2 with powdered 5 K2C03 (2.3 g, 16.3 mmol, 1.5 eq.) and a, a’ -dichloro-p-xylene (6.7 g, 38.1 mmol, 3.5 eq.) . Stir the resulting solution at 65 °C for about 48 hours. Cool the reaction mixture to 25 °C and concentrate to 1/5 of original volume. Dissolve the residue in EtOAc (150 mL) and wash with water (2 x 150 mL) .
10 Extract the aqueous layer with EtOAc (2 x 150 mL) and wash the combined organic extracts with saturated aqueous NaCI (2 x 150 mL. Dry the combined extracts with MgS04 and concentrate in vacuo to provide the crude product . Purification by flash chromatography (5.4 x 45 cm silica,
1525-40% EtOAc/hexane) will afford the enantiomerically- enriched chlorobenzyl intermediate .
Step 12: Preparation of enantiomerically-enriched (4R.5R)- 1- r [4- [ [4- [3 , 3-Dibutyl-7- (dimethylamino) -2,3 , 4 , 5-tetrahvdro-
Treat a solution of the enantiomerically-enriched chlorobenzyl intermediate (4.6 g, 7.7 mmol, obtained from
25 above in Step 11) in acetonitrile (100 mL) at 25 °C under N2 with diazabicyclo [2.2.2] -octane (DABCO, 0.95 g, 8.5 mmol, 1.1 eq.) and stir at 35 °C for 2 hours. Collect the precipitated solid and wash with CH3CN. Recrystallization from CH3OH/Et20 will give the desired title compound (XXVII) .
ANY ERROR, EMAIL amcrasto@gmail.com, +919323115463

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....












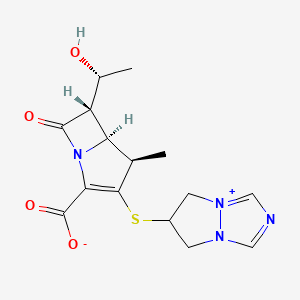











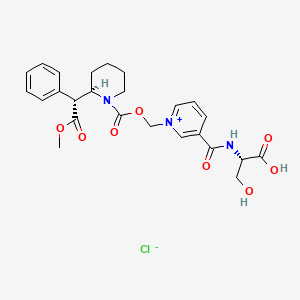

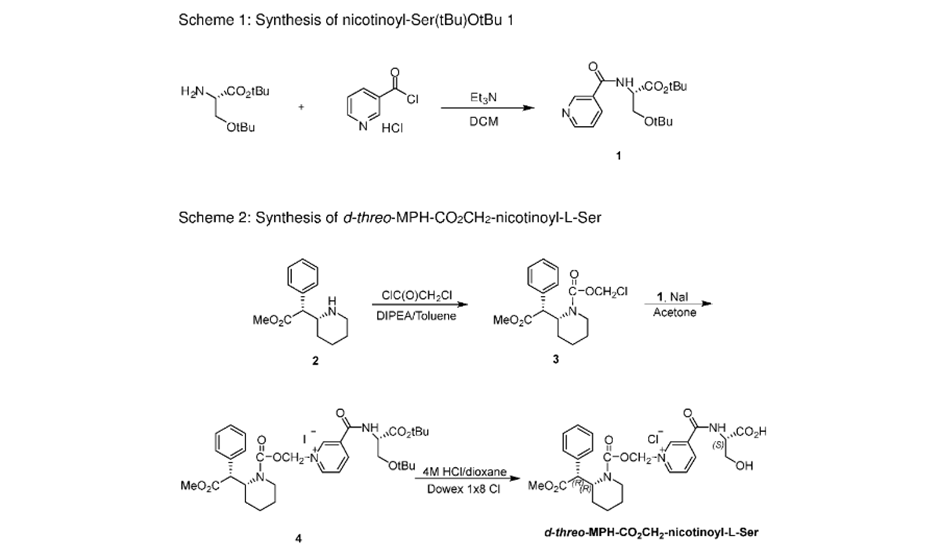







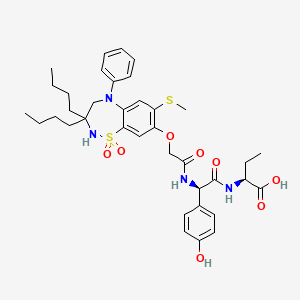
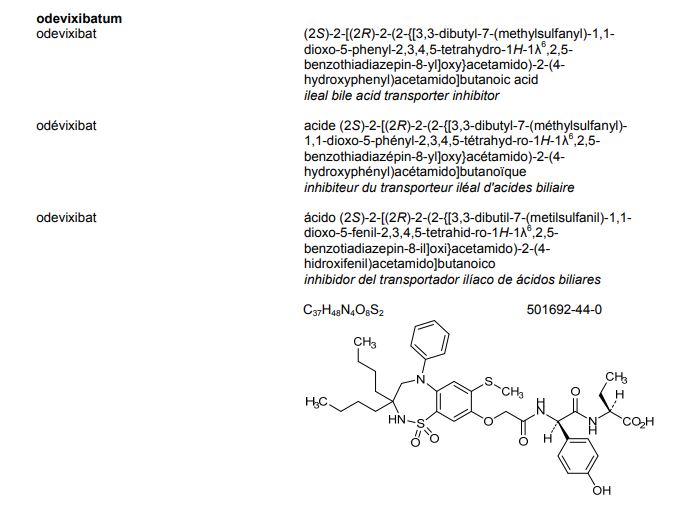



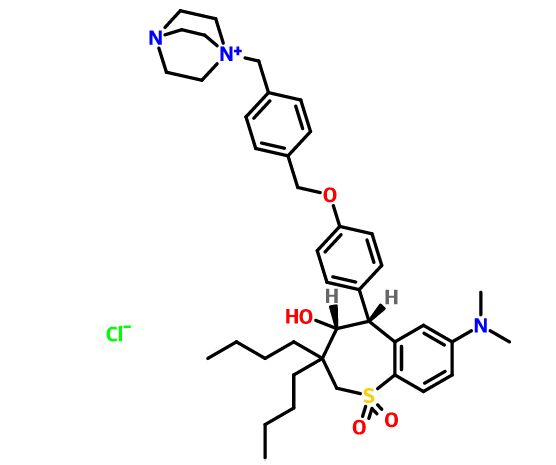



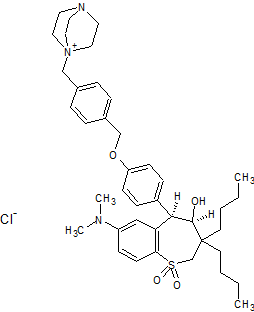
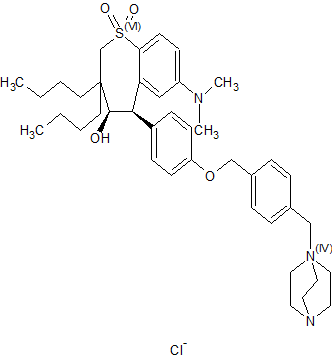















{kind=link}