Home » Posts tagged 'DONG A'
Tag Archives: DONG A
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
EVOGLIPTIN
EVOGLIPTIN
CAS: 1222102-29-5 FREE
HCL……1246960-27-9
tartare.. 1222102 -51-3
Dong-A Pharmaceutical. Co., Ltd, 동아제약 주식회사
2-Piperazinone, 4-((3R)-3-amino-1-oxo-4-(2,4,5-trifluorophenyl)butyl)-3-((1,1-dimethylethoxy)methyl)-, (3R)-
R)-4-((R)-3-Amino-4-(2,4,5-trifluorophenyl)-butanoyl)-3-(t-butoxymethyl)-piperazin-2-one
4-[3(R)-Amino-4-(2,4,5-trifluorophenyl)butyryl]-3(R)-(tert-butoxymethyl)piperazin-2-one hydrochloride
DA-1229
see…http://www.allfordrugs.com/2015/07/03/evogliptin/
DA-1229 is a dipeptidyl peptidase IV (CD26) inhibitor currently being developed in phase III clinical studies at Dong-A for the treatment of type 2 diabetes.
In 2014, Eurofarma aquired rights for product development and commercialization in Brazil.


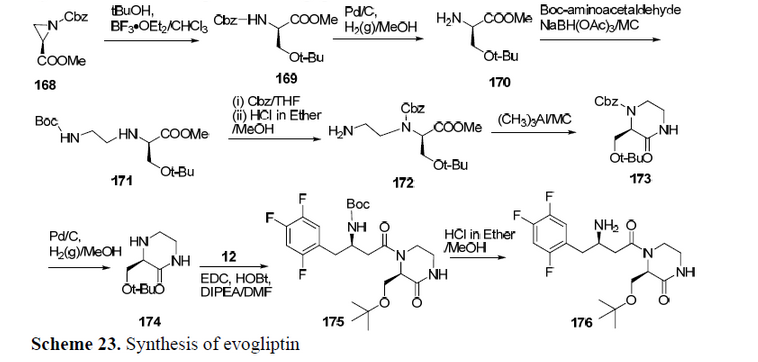
If above image is not clear then see at…….http://www.allfordrugs.com/2015/07/03/evogliptin/
86…………H. J. Kim, W. Y. Kwak, J. P. Min, J. Y. Lee, T. H. Yoon, H. D. Kim, C. Y. Shin, M. K.
Kim, S. H. Choi, H. S. Kim, E. K. Yang, Y. H. Cheong, Y. N. Chae, K. J. Park, J. M.
Jang, S. J. Choi, M. H. Son, S. H. Kim, M. Yoo and B. J. Lee, Bioorg. Med. Chem. Lett.,
2011, 21 (12), 3809-3812.
[87] …………K. S. Lim, J. Y. Cho, B. H. Kim, J. R. Kim, H. S. Kim, D. K. Kim, S. H. Kim, H. J. Yim,
S. H. Lee, S. G. Shin, I. J. Jang and K. S. Yu, Br. J. Clin. Pharmacol., 2009, 68 (6), 883-
890.
- Originator Dong-A Pharmaceutical
- Developer Dong-A ST
- Class Amides; Antihyperglycaemics; Fluorobenzenes; Piperazines; Small molecules
- Mechanism of Action CD26 antigen inhibitors
- Orphan Drug Status No
- On Fast track No
- New Molecular Entity Yes
- Available For Licensing Yes – Type 2 diabetes mellitus
Highest Development Phases
- Phase III Type 2 diabetes mellitus
Most Recent Events
- 01 Sep 2014 Phase-I clinical trials in Type-2 diabetes mellitus (In volunteers) in United Kingdom (PO)
- 31 Jul 2014 Phase-III clinical trials in Type-2 diabetes mellitus in South Korea (PO)
- 31 Jul 2014 Dong-A ST initiates enrolment in a phase I trial in patients with renal impairment in South Korea (NCT02214693)
…………………………………..
WO 2010114291
http://www.google.co.in/patents/WO2010114291A2?cl=en
Formula 1

Korea Patent Publication No. 2008-0094604 the call to the scheme, as indicated by A Ⅰ) of formula (II) beta-compound of formula 3 is already substituted heterocyclic compound having 1-hydroxy-benzotriazole group (HOBT) 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDC) and reacting with a tertiary amine to prepare a compound of formula (4) connected by peptide bonds; Ⅱ) beta comprises the step of reacting under acidic conditions a compound of the formula (4) – a method of manufacturing the heterocyclic compounds of the formula I having an amino group is disclosed.
– Scheme A]

(Wherein, PG is a protecting group.)
In this case, the beta of the formula (2) of Scheme A – a compound having an amino group is prepared in addition to the DPP-IV inhibitor International Publication represented by Formula 1 WO03 / 000181, WO03 / 004498, WO03 / 082817, WO04 / 007468, WO04 / 032836, WO05 / 011581, WO06 / 097175, WO07 / 077508, WO07 / 063928, WO08 / 028662 WO08 / it may be used for the production of different DPP-IV inhibitors according 087,560 and can be prepared in a number of ways.
To, the compound of Formula 2 is an example as shown in Scheme J. Med.Chem. 2005; 141, and Synthesis 1997; it can be produced by the known method described in 873.

Specifically, (2S) – (+) – 2,5- dihydro-3,6-dimethoxy-2-isopropyl-pyrazine 2,4,5-trifluoro-react with benzyl bromide and acid treatment, and then the amine an ester compound obtained by the protection reaction. Ester compounds are hydrolyzed to re-3- (2,4,5-trifluoro-phenyl) -2-amino-propionic acid tert such as isobutyl chloroformate, triethylamine or diisopropylethylamine to give the amine, and then using diazomethane to form a diazo ketone, and then may be prepared by reaction with silver benzoate. However, the reaction can be performed at low temperature (-78 ℃) or high alpha-amino acid to purchase and use, and may have a risk of problems such as the need to use large diazomethane.
To a different process for preparing a compound of Formula 2 as shown in scheme Tetrahedron: Asymmetry 2006; It is known in 2622; 205 or similarly Bioorganic & Medicinal Chemistry Letters 2007.

That is, a 1,1′-carbonyl-2,4,5 which the phenyl trifluoroacetic acid activated using the following imidazole mono-methyl words potassium carbonate is reacted with the beta-keto ester compound is prepared. This produced an enamine ester using ammonium acetate and ammonium solution, the ester compound chloro (1,5-cyclooctadiene) rhodium (I) dimer using a chiral ferrocenyl ligands I the reaction of the high-pressure hydrogen with a chiral primary amine with a beta-amino ester compound after production and can lead to hydrolysis to prepare a compound of formula (2). However, use of expensive metal catalyst has a problem that must be performed in high pressure hydrogenation.
The method for preparing a compound of Formula 2 is disclosed in International Publication No. WO 04/87650.

Specifically, 2,4,5-fluorophenyl reagent is oxalyl chloride, the acid activated acid with 2,2-dimethyl-1,3-dioxane-4,6-dione, and after the reaction of methanol and the resulting material at reflux to prepare a corresponding compound. With a selective reducing reagents which enantiomers (S) -BINAP-RuCl 2 and hydrogen through a reaction (S) – producing a compound having coordinated to each other, it again after the decomposition, and the singer O- benzyl hydroxyl amine and the coupling reaction and the intermediate is prepared. To do this, the resulting intermediate tree azodicarboxylate and diisopropyl azodicarboxylate presence ring condensation reaction, treated with an aqueous solution of lithium hydroxide to (R) – while having the formula (II) coordinated to the amine group protected with a benzyl-O- the compound can be produced. However, the method has a problem as a whole to be prepared by the reaction yield to be low and a long processing time to perform the reaction.
Thus, the conventional known method for producing a compound of the general formula (2) has the disadvantage of using expensive reagents, or not suitable for commercial mass-production method by a long synthesis time yield is also low.
In addition, the compound represented by General Formula (3), as described in Korea Patent Publication No. 2008-0094604 call, can be prepared by way of reaction schemes.

Specifically, the starting material D- serine methyl ester is substituted by a hydroxy group when reflux again substituted by trityl chloride as methoxy groups converted to the aziridine compound.
[Scheme 3]

<Example 3> (R)-4-[(R)-3-아미노-4-(2,4,5-트리플루오로페닐)부타노일]-3-(t-부톡시메틸)피페라진-2-온(화학식 1) Preparation of the hydrochloride
Step 1: t- butyl (R)-4-[(R)-2-(t-부톡시메틸)-3-옥소피페라진-1-일]-4-옥소 – 1-(2,4,5-트리플루오로페닐)부탄-2-일카르바메이트(화학식 Preparation of 4)
2 L flask, prepared in Example 1 (R) -3-t- butoxycarbonyl-4- (2,4,5-trifluoro-phenyl) butanoate acid (Formula 2) 10.0 g of toluene was dissolved in 450 mL of bis (2,2′-benzothiazolyl) disulfide 13.0 g, was cooled and then 10.2 g triphenylphosphine was added to the reaction solution at 0 ℃. While stirring the reaction mixture was added to a solution of 0.8 mL of triethylamine in 20 mL of toluene was stirred at room temperature for 5 hours. The reaction mixture was cooled to 0 ℃ and prepared in Example 2 (R) -3- (t- butoxymethyl) piperazin-2-one (Formula 3) was dissolved in 5.6 g of toluene and 40 mL pyridine a 2.4 mL was added slowly. After 30 minutes the reaction mixture was heated to room temperature and stirred for 1 hour. Saturated sheet to be the aqueous acid solution to a pH of 2.5 and then diluted with ethyl acetate 400 mL. Washed twice with brine and the organic layer was dehydrated with magnesium sulfate and concentrated. The residue was purified by column chromatography to give the title compound 838 mg.
1 H NMR (400 MHz, CDCl 3) δ 7.03 (m, 1H), 6.88 (m, 1H), 5.97 (m, 1H), 5.48 (m, 1H), 4.16 ~ 4.07 (m, 1H), 4.02 ~ 3.91 (m, 1H), 3.74 (m, 2H) 3.37 (m, 2H), 3.24 (m, 1H), 2.92 (m, 2H), 2.80 (m, 1H), 2.59 (m, 2H), 1.34 ( d, 9H), 1.13 (s, 9H)
Step 2: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluoro-phenyl) butane five days] -3- (t- butoxymethyl) piperazin-2- on the production of (I) hydrochloride
Prepared in Step 1 t- butyl (R)-4-[(R)-2-(t-부톡시메틸)-3-옥소피페라진-1-일]-4-옥소-1-(2,4,5-트리플루오로페닐)부탄-2-일카르바메이트 97 mg was dissolved in methanol was added 3 mL 2N- hydrochloric acid / diethyl ether 2 mL was stirred at room temperature for 3 hours. The reaction mixture was concentrated and dried under reduced pressure to give 64 mg of the title compound as a foaming solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.37 (m, 1H), 7.23 (m, 1H), 4.80 (m, 1H), 4.59 ~ 4.40 (m, 1H), 3.93 (m, 1H), 3.90 ~ 3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89 ~ 2.66 (m, 2H), 1.18 (s, 3H ), 1.11 (s, 6H)
Mass (M + 1): 402
<Example 4> (R)-4-[(R)-3-아미노-4-(2,4,5-트리플루오로페닐)부타노일]-3-(t-부톡시메틸)피페라진-2-온(화학식 1) tartaric acid salts
Step 1: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluoro-phenyl) butane five days] -3- (t- butoxymethyl) piperazin-2- Preparation of one (I)
Example 3 to give a compound of formula I in hydrochloride 60 mg 5% sodium hydrogen carbonate in dichloromethane was added to 10 mL of an aqueous solution / 2-propanol (4/1 (v / v)) was added to the mixed solution and extracted two times 10 mL The organic layer was dried under reduced pressure to give 55 mg of the title compound as a solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.27 (m, 1H), 7.14 (m, 1H), 4.56 ~ 4.39 (m, 1H), 3.96 ~ 3.81 (m, 3H), 3.70 (m, 1H) , 3.46 (m, 1H), 3.43 ~ 3.32 (m, 1H), 2.83 ~ 2.65 (m, 3H), 2.58 ~ 2.40 (m, 2H), 1.16 (s, 3H), 1.11 (s, 6H)
Mass (M + 1): 402
Step 2: (R) -4 – [(R) -3- amino-4- (2,4,5-trifluorophenyl) butanoyl] -3- (t- butoxymethyl) piperazin-2- one (I) tartaric acid salt [
Was dissolved 55 mg of the compound of step 1 in 0.56 mL of acetone, L- tartrate 26 mg ethanol / water (9/1 (v / v)) was added slowly to a solution of 0.35 mL was stirred for 30 minutes. Here was added 0.56 mL of 2-propanol was stirred for 10 minutes and re-filtered to give 77 mg of the title compound as a solid.
1 H NMR (400 MHz, CD 3 OD) δ 7.38 (m, 1H), 7.22 (m, 1H), 4.80 (m, 1H), 4.59 ~ 4.40 (m, 1H), 4.40 (s, 2H), 3.93 (m, 1H), 3.90 ~ 3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89 ~ 2.66 (m, 2H ), 1.15 (s, 3H), 1.11 (s, 6H)
Mass (M + 1): 402
………………………………
WO 2010114292
http://www.google.com/patents/WO2010114292A2?cl=en
…………………………………
Discovery of DA-1229: a potent, long acting dipeptidyl peptidase-4 inhibitor for the treatment of type 2 diabetes
Bioorg Med Chem Lett 2011, 21(12): 3809
http://www.sciencedirect.com/science/article/pii/S0960894X11004859

A series of β-amino amide containing substituted piperazine-2-one derivatives was synthesized and evaluated as inhibitors of dipeptidyl pepdidase-4 (DPP-4) for the treatment of type 2 diabetes. As results of intensive SAR study of the series, (R)-4-[(R)-3-amino-4-(2,4,5-trifluorophenyl)-butanoyl]-3-(t-butoxymethyl)-piperazin-2-one (DA-1229) displayed potent DPP-4 inhibition pattern in several animal models, was selected for clinical development.
http://www.luye.cn/en/uploads//2014-07/21/_1405936452_zr21xh.pdf
Dong-A ST has licensed its new diabetes drug Evogliptin to 17 Latin American countries including Mexico, Venezuela, Argentina, Chile, Colombia, Ecuador, Peru, the Dominican Republic, and Uruguay, Jung Jae-wook, Dong-A ST’s PR manager, told Business Korea.
Dong-A ST and Eurofarma, a Brazilian pharmaceutical company, concluded the licensing contract at Dong-A ST’s headquarters on April 13 in Seoul.
Eurofarma will be responsible for Evogliptin’s product development and sales in the 17 Latin American countries, Dong-A ST said. Dong-A ST will receive royalties from Eurofarma, and export the raw material of the medicine.
Dong-A ST has been developing Evogliptin with the support of the Ministry of Health & Welfare of South Korea as an innovative new medicine research project since May 2008. Evogliptin is a DPP-4 remedy based on the inhibition mechanism which is “excellent” at reducing blood sugar, whilst “less likely” to cause weight increases and hypoglycemia, the company said.
Park Chan-il, president of Dong-A ST, said that Dong-A ST will pursue further out-licensing “over the globe,” through continuous investment in research and development.
Maurizio Billi, Eurofarma’s president, wished to expand both companies’ partnership in the innovative new remedy development sector, according to Dong-A ST.
Last July, Dong-A ST and Eurofarma concluded a contract out-licensing Evogliptin to Brazil itself, the company said.
see gliptins at…..http://drugsynthesisint.blogspot.in/p/gliptin-series.html
Dong-A Pharm. Co., Ltd, Yongin-si, Gyeonggi-do, Republic of Korea.