
Home » Posts tagged 'discovery'
Tag Archives: discovery
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
BMS-852927

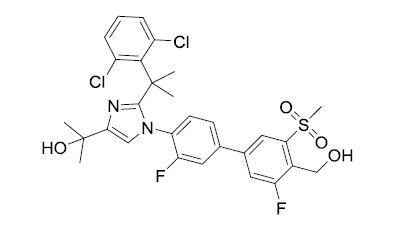
BMS-852927
CAS 256918-39-4
609.51 MW
C29 H28 Cl2 F2 N2 O4 S MF
2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol
1H-Imidazole-4-methanol, 2-[1-(2,6-dichlorophenyl)-1-methylethyl]-1-[3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)[1,1′-biphenyl]-4-yl]-α,α-dimethyl-
Treat metabolic syndrome

Brett Busch, Ph.D.
https://www.linkedin.com/in/brettbbusch

| Brett B. Busch, William C. Stevens, Jr., Ellen K. Kick, Haiying Zhang, Venkataiah Bollu,Richard Martin, Raju Mohan | |
| Applicant | Exelixis, Inc. |
| Brett B. Busch, William C. Stevens, JR., Ellen K. Kick, Haiying Zhang, Venkataiah Bollu,Richard Martin, Raju Mohan | |
| Bristol-Myers Squibb Company, Exelixis Patent Company Llc |
- Originator Exelixis
- Developer Bristol-Myers Squibb
- Class Antihyperlipidaemics; Small molecules
- Mechanism of Action Liver X receptor modulators
- Discontinued Atherosclerosis; Hypercholesterolaemia
Most Recent Events
- 04 Jun 2014 BMS 852927 is still in phase I trials for atherosclerosis and in preclinical development for hypecholesterolaemia in USA
- 02 Aug 2013 Bristol-Myers Squibb terminates the planned phase I trial for Hypercholesterolaemia in Germany, Canada and Switzerland (NCT01651273)
- 06 Jul 2012 Bristol-Myers Squibb plans a phase I trial for Hypercholesterolaemia in Germany, Canada and Switzerland (NCT01651273)
1H-NMR (DMSO-d6, 400 MHz) δ 7.94 (m, 2H), 7.63 (dd, 1H, J = 11.29, 1.51 Hz), 7.34 (d, 1H, J = 9.54
Hz), 7.14 (m, 3H), 7.05 (m, 1H), 6.83 (s, 1H), 5.58 (t, 1H, J = 5.27 Hz), 4.96 (d, 2H, J = 4.27 Hz), 4.70
(s, 1H), 3.46 (s, 3H), 1.96 (s, 6H), 1.45 (s, 6H); MS m/e 609.16 (M+H+);
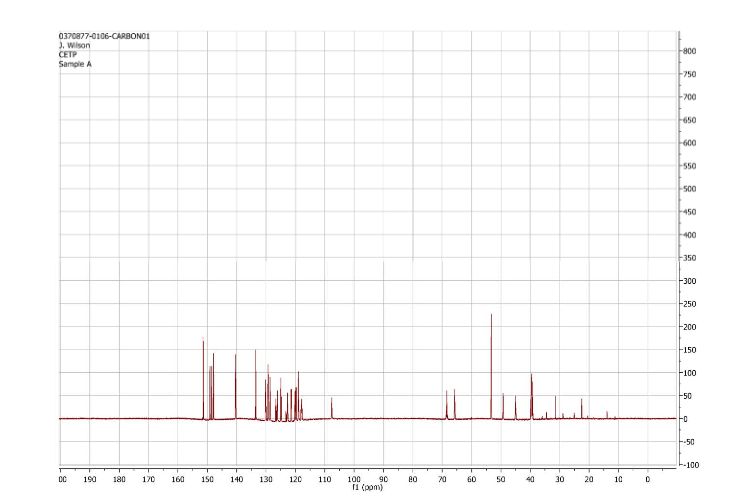
13CNMR (DMSO-d6, 400MHz) 161.42 (d, J=249.49 Hz), 156.85 (d, J=250.25 Hz), 153.18, 148.39, 141.69 (d, J=3.05 Hz), 139.45 (dd, J=9.16, 1.53 Hz), 139.32 (dd, J=8.39, 1.53 Hz), 138.58, 134.68, 131.39, 129.96, 128.40,
127.12 (d, J=17.55 Hz), 125.72 (d, J=12.97 Hz), 123.15 (d, J=2.29 Hz), 122.49 (d, J=3.05 Hz), 119.04
(d, J=25.18 Hz), 116.30, 114.52 (d, J=22.13 Hz), 68.11, 51.97 (d, J=5.34 Hz ), 45.53, 44.78, 44.29,
31.01, 30.53.
19F-NMR (JEOL 500 MHz, CDCl3) -113.55, -116.73.
HPLC (XBridge 5μ C18 4.6x50mm, 4 mL/min, Solvent A: 10 % MeOH/water with 0.2 % H3PO4, Solvent B: 90 % MeOH/water with0.2 % H3PO4, gradient with 0-100 % B over 4 minutes): 2.56 minutes, Purity, 99.7%.
HRMS (m/z,Obs.): 609.12065 [M+H]+; (Calc.): 609.11877. Formula: C29H29Cl2F2N2O4S. Anal. Calcd. for
C29H28N2O4SCl2F2•0.10 C2H6O•0.10 C4H5O2: C, 57.05; H, 4.75; Cl, 11.42; F, 6.10; N, 4.50; S, 5.15.
Found: C, 57.14; H, 4.54; Cl, 11.57; F, 5.94; N, 4.36; S, 5.07. The residual solvents, ethyl acetate (1.39
weight %), ethanol (0.74 weight %), dichloromethane (0.05 weight %), and heptane (< 0.05 weight %)
were identified in the sample by GC/MS and the retention times were matched with the reference standards.


Liver X receptors (LXRs) belong to a family of nuclear hormone receptors that are endogenously activated by cholesterol and its oxidized derivatives to mediate transcription of genes involved in maintaining glucose, cholesterol, and fatty acid metabolism. LXRa is found predominantly in the liver, with low levels found in kidney, intestine, spleen, and adrenal tissue. LXRp is ubiquitous in mammals and was found in nearly all tissues examined. Given the intricate link between lipid metabolism and cancer cell growth, the ubiquitous expression of LXRp in some types of cancer is unlikely to be coincidental, allowing cancer cells to synthesize lipids and lipoprotein particles to sustain their growth. At the same time, however, such stable basal expression levels make LXRp an ideal therapeutic target.

Examples of LXR agonists reported in the literature
PATENT
WO 2010138598
PATENT
WO 2012135082
PATENT
WO 2014028461
PATENT
WO 2016100619
PATENT
https://www.google.com/patents/US8618154?cl=enIt

Example 9 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol

Example 9a Preparation of 2-(2,6-dichlorophenyl)-2-methylpropanenitrile
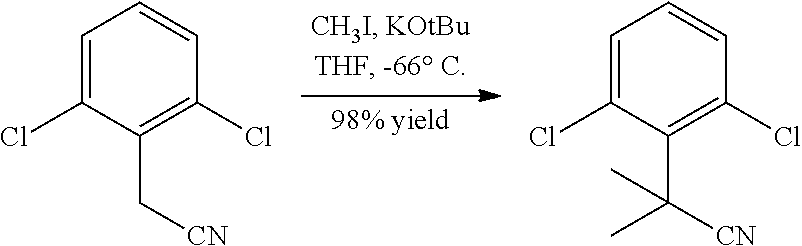
To a 1 M solution of potassium tert-butoxide (403 mL, 403 mmol) at −66° C. (acetone/dry ice) was slowly added 2-(2,6-dichlorophenyl)acetonitrile (25.0 g, 134 mmol) in anhydrous THF (150 mL). The mixture was stirred at −66° C. for 20 minutes. Then, iodomethane (33.6 mL, 538 mmol) was added drop-wise over 25 minutes at −66° C. At this stage, it was exothermic and a large amount of light yellow precipitate was observed. The suspension was stirred at −60° C. for 30 minutes. The reaction mixture was quenched with 200 mL ice water, and extracted with ether (3×150 mL). The organics were combined, washed with 150 mL brine, dried over Na2SO4, and concentrated on a rotary evaporator. The crude product (30 g, yellow oil) was purified by column chromatography (ISCO, 330 g silica, 20% EtOAc in hexanes) to afford 2-(2,6-dichlorophenyl)-2-methylpropanenitrile (28.2 g, 132 mmol, 98% yield) as a light yellowish oil. 1H-NMR (CDCl3, 400 MHz) δ 7.35 (d, 2H, J=8.03 Hz), 7.16 (t, 1H, J=8.0 Hz), 2.09 (s, 6H); 13C-NMR (CDCl3, 126 MHz) δ134.6, 133.8, 131.4, 129.0, 124.1, 38.6, 29.2; MS m/e 214.10 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 3.16 minutes.
Example 9b Preparation of N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide

2-(2,6-Dichlorophenyl)-2-methylpropanenitrile (20 g, 93 mmol) and 4-bromo-2-fluoroaniline (28.4 g, 149 mmol) were dissolved in anhydrous o-xylene (200 mL) and heated to 100° C. under N2. Trimethylaluminum (2 M) in toluene (140 mL, 280 mmol) was added drop-wise (˜0.9 mL per minute) over 2.5 hours while the reaction mixture was stirred at 100° C. After addition, the reaction mixture was stirred at 100° C. for 30 minutes, and then cooled to −5° C. The reaction mixture was very carefully quenched with potassium sodium tartrate (20 g in 100 mL water) (Caution: gas and heat formation). The reaction mixture was filtered through Celite 545. The filtrate was washed with 1N HCl (4×70 mL). The aqueous was neutralized with 2N NaOH and extracted with EtOAc (4×100 mL). The organics were combined, washed with brine, dried with Na2SO4, and concentrated on a rotary evaporator to afford 24 g of crude product. The crude product was recrystallized with 72 mL of MTBE and 240 mL of hexane to give N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide (17.5 g, 43.3 mmol, 46.4% yield) as a white solid (purity: 99%). 1H-NMR (MeOD, 400 MHz) δ 7.42 (d, 2H, J=8.0 Hz), 7.30 (m, 2H), 7.16 (t, 1H, J=8.0 Hz), 6.93 (t, 1H, J=8.0 Hz), 2.11 (s, 6H); 13C-NMR (DMSO-d6, 100 MHz) δ 166.5, 156.1, 153.7, 140.6, 138.5, 135.9, 131.4, 128.6, 128.0, 125.7, 119.5, 112.9, 50.0, 29.2; MS m/e 403.09 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.32 minutes.
Example 9c Preparation of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate

To a mixture of N-(4-bromo-2-fluorophenyl)-2-(2,6-dichlorophenyl)-2-methylpropanimidamide (48.0 g, 119 mmol), K2CO3(41.0 g, 297 mmol) in toluene (180 mL) and THF (180 mL) at 55° C. was added slowly a solution of ethyl 3-bromo-2-oxopropanoate (23.3 mL, 166 mmol) in 24 mL of THF over 50 minutes. The reaction mixture was kept at 55° C. for 1.5 hours. A white slurry was observed. The reaction mixture was cooled to 5° C. HCl (0.5N, 450 mL) was added drop-wise (end point pH=9˜10). After addition, the suspension was cooled to 0° C. The solid was collected by filtration, washed with water (2×50 mL), and then dried in a vacuum oven at 60° C. overnight. Ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate (59 g, 114 mmol, 96% yield) was obtained as a white solid. 1H-NMR (CDCl3, 400 MHz) δ 7.11 (m, 3H), 6.96 (m, 2H), 6.72 (t, 1H, J=8.28 Hz), 4.35 (m, 2H), 4.25 (d, 1H, J=10.5 Hz), 3.80 (d, 1H, J=10.8 Hz), 1.98 (s, 3H), 1.93 (s, 3H), 1.38 (t, 3H, J=7.03 Hz); 13C-NMR (CDCl3, 126 MHz) δ 173.0, 171.5, 159.8, 157.8, 137.3, 135.7, 132.1, 131.1, 128.1, 127.4, 125.6, 122.2, 120.1, 93.5, 62.5, 45.5, 30.2, 14.0; MS m/e 517.05 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.74 minutes.
Example 9d Preparation of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazole,4-carboxylate

To a mixture of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-4-hydroxy-4,5-dihydro-1H-imidazole-4-carboxylate (38 g, 73 mmol) in EdOH (200 mL) was added TFA (25.0 g, 220 mmol). The mixture was stbsequently heated tn 95° C. HPLC analysis after 2.5 hours showed <1% of alcohol intermediate remaining The mixture was diluted with 300 mL of CH2Cl2 and cooled to approximately 5° C. with an ice bath. The mixture was neutralized with 1N NaOH (120 mL) and the organic layer was separated. The aqueous layer was dxtracted with CH2Cl2 (2×100 mL). The combined organic layers were concentrated on a rotary evaporator to give crude material. Recrystallization in EtOH (5 mL/1 g) provided 32 g of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophdnyl)propan-2-yl)-1H-imidazole-4-carboxylate as `n off-white solhd (86% yield). 1H-NMR (DMSO-d6, 400 MHz) δ 7.92 (s, 1H), 7.16 (d, 1H, J=8.0 Hz), 7.22 (m, 3H), 7.11 (m, 1H), 7.04 (t, 1H, J=12.0 Hz), 4.25 (q, 2H, J=8.0 Hz), 1.94 (s, 6H(, 1.27 (t, 3H, J=8.0 Hz); MS m/e 502.68 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 3.87 minutes.
Example 9e Prepar`tion of 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol

To a mixture of methylmagnesium bromide (60.0 mL, 180 mmol, 3M in ether) in 120 ml, of THF cooled with an ice/salt bath (−15 to −17° C.) was added slowly a solution of ethyl 1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazole-4-carboxylate (30 g, 60 mmol) in 65 mL of CH2Cl2 and 87 mL of THF over 45 minutes. The internal temperature was carefully kept below 0° C. A further 2×20 mL of CH2Cl2 was used to wash forward the residual material. The reaction mixture temperature was maintained below 0° C. for 1 hour with stirring. Then the reaction mixture was diluted with 100 mL of CH2Cl2, and saturated NH4Cl was added slowly. The resulting mixture was extracted with CH2Cl2 (2×80 mL). Organics were combined, washed with brine, dried with Na2SO4, and concentrated on a rotary evaporator to afford 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (28.5 g, 58.6 mmol, 98% yield) as a white solid. 1H-NMR (CDCl3, 400 MHz) δ 7.13 (dd, 1H, J=9.03, 2.01 Hz), 7.09 (s, 1H), 7.07 (s, 1H), 6.93 (m, 2H), 6.75 (t, 1H, J=8.16 Hz), 6.55 (s, 1H), 3.18 (s, 1H), 2.00 (s, 6H), 1.58 (s, 6H); 13C-NMR (CDCl3, 126 MHz) δ 158.1, 156.1, 154.5, 147.8, 139.3, 135.7, 131.3, 130.3, 127.8, 126.9, 122.7, 119.8, 115.1, 68.7, 44.8, 31.1, 29.9; MS m/e 485.05 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.78 minutes.
Example 9 Preparation of 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol

To a 1 L 3-necked round bottom flask under nitrogen was added 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (12.0 g, 24.7 mmol), [2-fluoro-6-methanesulfonyl-4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-phenyl]-methanol (9.78 g, 29.6 mmol), K2CO3 (10.2 g, 74 mmol), DME (120 mL) and water (12 mL). The mixture was heated to 60° C., and then 1,1′-bis(diphenylphosphino)ferrocene palladium (II) chloride complex (4.06 g, 4.94 mmol) was added under nitrogen. The reaction mixture was heated to 80° C. for 30 minutes. The resulting darkly colored mixture was cooled with an ice bath, and partitioned in 200 mL of CH2Cl2 and 200 mL of water. The organic layers were combined and dried with Na2SO4. After concentration, the crude product was purified by flash chromatography (ISCO, 330 g silica, 0% to 100% EtOAc in hexanes) to afford 12.79 g of crude product (85% yield) as a light yellow solid.
Recrystallization was carried out by dissolving 9.5 g of crude product in acetone (80 mL) at 65° C. The resulting solution was cooled slowly to 25° C. over 5 hours, and then cooled to 0° C. for an additional 30 minutes. Crystals began to form at 45° C. The solid was collected by filtration and rinsed with cold acetone. After drying in an oven at 45° C. under vacuum for 14 hours, 4.9 g of pure product was obtained. To recover additional crystalline product, the mother liquid was concentrated to approximately 10 mL and passed through a silica pad. EtOAc (100 mL) was used to elute the compound. The filtrate was concentrated under vacuum to give a crude solid. The crude solid was recrystallized in acetone following the procedure above to afford an additional 2.5 g of product. The combined recovery for the two crops after recrystallization was a 78% yield. 1H-NMR (DMSO-d6, 400 MHz) δ 7.94 (m, 2H), 7.63 (dd, 1H, J=11.29, 1.51 Hz), 7.34 (d, 1H, J=9.54 Hz), 7.14 (m, 3H), 7.05 (m, 1H), 6.83 (s, 1H), 5.58 (t, 2H, J=5.27 Hz), 4.96 (d, 2H, J=4.27 Hz), 4.70 (s, 1H), 3.46 (s, 3H), 1.96 (s, 6H), 1.45 (s, 6H); MS m/e 609.16 (M+H+); HPLC (XBridge 5μ C18 4.6×50 mm, 4 mL/min, Solvent A: 10% MeOH/water with 0.2% H3PO4, Solvent B: 90% MeOH/water with 0.2% H3PO4, gradient with 0-100% B over 4 minutes): 2.56 minutes.
Alternatively, Example 9 was prepared as follows:
To a 1 L 3-necked round bottom flask under nitrogen was added methyltetrahydrofuran (“MeTHF”, 6.9 kg), 2-(1-(4-bromo-2-fluorophenyl)-2-(2-(2,6-dichlorophenyl)propan-2-yl)-1H-imidazol-4-yl)propan-2-ol (1.994 kg, 4.1 moles) and (2-fluoro-6-(methylsulfonyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanol (1.38 kg, 4.19 moles). The mixture was agitated at 23° C. for 15 min until all the solids dissolved. At the conclusion of this period, (oxydi-2,1-phenylene)bis(diphenylphosphine) (0.022 kg, 0.041 moles) and Pd(OAc)2 (0.01 kg, 0.045 moles) were added as a slurry via a subsurface line. Upon completion of addition, the mixture was rinsed with additional MeTHF (1.65 kg). The resulting mixture was evacuated to less than 80 Torr and backfilled with nitrogen. This process was repeated two more times. After completion of the degassing sequence, the reaction mixture was agitated for at least 15 min and a clear, golden color was observed. In a separate reaction vessel, a solution of potassium hydroxide (0.352 kg) in water (10.00 kg) was prepared and degassed by sparging the solution with nitrogen gas for at least 15 min prior to use. The KOH solution (10.35 kg) was transferred into the reactor by vacuum. The reaction temperature exhibited a known exotherm from 20° C. to 29° C. Upon completion of addition, the resulting biphasic mixture was degassed by a series of pressure swings. The mixture was warmed to between 45-50° C. where it was stirred for at least 2 h. After this time, the reaction mixture was analyzed by HPLC, which indicated the reaction was complete. The reaction mixture was cooled to 23° C. and the stirring was stopped. The mixture was allowed to separate for 30 min and the lower spent KOH stream was removed. The product rich organic was passed through a column of thiourea functionalized silica gel (0.782 kg) (Silicycle) at ˜0.1 kg per min to remove the palladium. The product rich organic phase was washed with a 5% NaHCO3 solution (5 vol) and the phases separated. The organic phase was washed with water (5 vol) and the organic and aqueous phases separated.
The product rich organic phase was polish filtered into a clean reaction vessel and then concentrated to ˜8 volumes (˜16 L) under vacuum (80 Torr, Tjacket=60° C.). Once at the prescribed volume, the reaction mixture was allowed to cool to 25° C. Once at the prescribed temperature the reaction mixture was seeded with 2-(2-(2-(2,6-dichlorophenyl)propan-2-yl)-1-(3,3′-difluoro-4′-(hydroxymethyl)-5′-(methylsulfonyl)biphenyl-4-yl)-1H-imidazol-4-yl)propan-2-ol (0.5%, 0.008 kg). The resulting slurry was stirred at 25° C. for about 18 h. At the conclusion of this period, the reaction mixture was concentrated to ˜8 L under vacuum (150 Torr, Tjacket=60° C.). Once at the prescribed volume, the reaction mixture was heated to 50° C. and isopropyl acetate (IPAc, 13.90 kg) was added to the reactor during a 90 min period. Upon completion of addition, the reaction mixture was cooled to 25° C. during a 3 h period. Once at the prescribed temperature the reaction mixture was stirred at room temperature for about 16 h. At the conclusion of this period, the reaction mixture was filtered, deliquored, and washed with additional IPAc (10.4 kg). The filter cake was dried via suction on the filter under a stream of dry nitrogen to yield a white solid. The white solid was transferred to a dryer and dried at 50° C. under full vacuum to afford 2.03 kg of product (81% yield, 99.40 AP, 98 wt %).
PAPER

Introducing a uniquely substituted phenyl sulfone into a series of biphenyl imidazole liver X receptor (LXR) agonists afforded a dramatic potency improvement for induction of ATP binding cassette transporters, ABCA1 and ABCG1, in human whole blood. The agonist series demonstrated robust LXRβ activity (>70%) with low partial LXRα agonist activity (<25%) in cell assays, providing a window between desired blood cell ABCG1 gene induction in cynomolgus monkeys and modest elevation of plasma triglycerides for agonist 15. The addition of polarity to the phenyl sulfone also reduced binding to the plasma protein, human α-1-acid glycoprotein. Agonist 15 was selected for clinical development based on the favorable combination of in vitroproperties, excellent pharmacokinetic parameters, and a favorable lipid profile.
Discovery of Highly Potent Liver X Receptor β Agonists
http://pubs.acs.org/doi/full/10.1021/acsmedchemlett.6b00234
| WO2007002563A1 | Jun 26, 2006 | Jan 4, 2007 | Exelixis, Inc. | Imidazole based lxr modulators |
| WO2008073825A1 | Dec 7, 2007 | Jun 19, 2008 | Exelixis, Inc. | Lxr and fxr modulators |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US8901106 | Mar 26, 2012 | Dec 2, 2014 | Bristol-Myers Squibb Company | Imidazole prodrug LXR modulators |
| US20140163081 * | Nov 21, 2013 | Jun 12, 2014 | Exelixis Patent Company Llc | Lxr modulators |
| US20150299136 * | May 4, 2015 | Oct 22, 2015 | Bristol-Myers Squibb Company | Lxr modulators |
///////////Discovery, Highly Potent, Liver X Receptor β Agonists, ABCA1, ABCG1, Liver X receptor, LXRα, LXRβ, α-1-acid glycoprotein, BMS-852927, BMS 852927
CS(=O)(=O)c1cc(cc(F)c1CO)c2cc(F)c(cc2)n3cc(nc3C(C)(C)c4c(Cl)cccc4Cl)C(C)(C)O
Merck’s Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP)
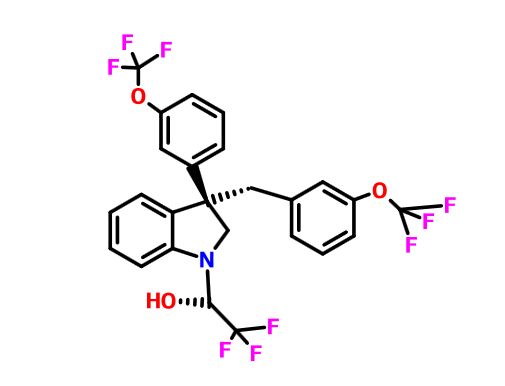
Indoline 7 as in ACS MEDCHEM LETTERS, DOI: 10.1021/acsmedchemlett.5b00404
and
eg 10 as in WO2015054088
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.
1H-Indole-1-ethanol, 2,3-dihydro-3-[3-(trifluoromethoxy)phenyl]-3-[[3-(trifluoromethoxy)phenyl]methyl]-α-(trifluoromethyl)-, (αR)-
cas 1699732-96-1 R ISOMER
Merck Sharp & Dohme Corp. INNOVATOR

Using the collective body of known (CETP) inhibitors as inspiration for design, a structurally novel series of tetrahydroquinoxaline CETP inhibitors were discovered. An exemplar from this series, compound 5, displayed potent in vitro CETP inhibition and was efficacious in a transgenic cynomologus-CETP mouse HDL PD (pharmacodynamic) assay. However, an undesirable metabolic profile and chemical instability hampered further development of the series. A three-dimensional structure of tetrahydroquinoxaline inhibitor 6 was proposed from 1H NMR structural studies, and this model was then used in silico for the design of a new class of compounds based upon an indoline scaffold. This work resulted in the discovery of compound 7, which displayed potent in vitro CETP inhibition, a favorable PK–PD profile relative to tetrahydroquinoxaline 5, and dose-dependent efficacy in the transgenic cynomologus-CETP mouse HDL PD assay.
chemical compounds that inhibit cholesterol ester transfer protein (CETP) and are expected to have utility in raising HDL-C, lowering LDL-C, and in the treatment and prevention of atherosclerosis.
see………….http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.5b00404
http://pubs.acs.org/doi/suppl/10.1021/acsmedchemlett.5b00404/suppl_file/ml5b00404_si_001.pdf
Discovery of Novel Indoline Cholesterol Ester Transfer Protein Inhibitors (CETP) through a Structure-Guided Approach
Atherosclerosis and its clinical consequences, including coronary heart disease
(CHD), stroke and peripheral vascular disease, represent a truly enormous burden to the health care systems of the industrialized world. In the United States alone, approximately 13 million patients have been diagnosed with CHD, and greater than one half million deaths are attributed to CHD each year. Further, this toll is expected to grow over the next quarter century as an epidemic in obesity and diabetes continues to grow.
It has long been recognized that in mammals, variations in circulating lipoprotein profiles correlate with the risk of atherosclerosis and CHD. The clinical success of HMG-CoA reductase inhibitors, especially the statins, in reducing coronary events is based on the reduction of circulating low density lipoprotein cholesterol (LDL-C), levels of which correlate directly with an increased risk for atherosclerosis. More recently, epidemiologic studies have
demonstrated an inverse relationship between high density lipoprotein cholesterol (HDL-C) levels and atherosclerosis, leading to the conclusion that low serum HDL-C levels are associated with an increased risk for CHD.
Metabolic control of lipoprotein levels is a complex and dynamic process involving many factors. One important metabolic control in man is the cholesteryl ester transfer protein (CETP), a plasma glycoprotein that catalyzes the movement of cholesteryl esters from HDL to the apoB containing lipoproteins, especially VLDL (see Hesler, C.B., et. al. (1987) Purification and characterization of human plasma cholesteryl ester transfer protein. J. Biol. Chem. 262(5), 2275-2282)). Under physiological conditions, the net reaction is a heteroexchange in which CETP carries triglyceride to HDL from the apoB lipoprotein and transports cholesterol ester from HDL to the apoB lipoprotein.
In humans, CETP plays a role in reverse cholesterol transport, the process whereby cholesterol is returned to the liver from peripheral tissues. Intriguingly, many animals do not possess CETP, including animals that have high HDL levels and are known to be resistant to coronary heart disease, such as rodents (see Guyard-Dangremont, V., et. al, (1998)
Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility, Comp. Biochem. Physiol. B Biochem. Mol. Biol. 120(3), 517-525). Numerous epidemiologic studies correlating the effects of natural variation in CETP activity with respect to coronary heart disease risk have been performed, including studies on a small number of known human null mutations (see Hirano, K.-L, Yamashita, S. and Matsuzawa, Y. (2000) Pros and cons of inhibiting cholesteryl ester transfer protein, Curr. Opin. Lipidol. 11(6), 589-596). These studies have clearly demonstrated an inverse correlation between plasma HDL-C concentration and CETP activity (see Inazu, A., et. al. (2000) Cholesteryl ester transfer protein and atherosclerosis, Curr. Opin. Lipidol. 11(4), 389-396), leading to the hypothesis that pharmacologic inhibition of CETP lipid transfer activity may be beneficial to humans by increasing levels of HDL-C while lowering LDL-C.
Despite the significant therapeutic advance that statins such as simvastatin and atorvastatin represent, statins only achieve a risk reduction of approximately one-third in the treatment and prevention of atherosclerosis and ensuing atherosclerotic disease events.
Currently, few pharmacologic therapies are available that favorably raise circulating levels of HDL-C. Certain statins and some fibrates offer modest HDL-C gains. Niacin provides an effective therapy for raising HDL-C but suffers from patient compliance issues, due in part to side effects such as flushing. Drugs that inhibit CETP (CETP inhibitors) have been under development with the expectation that they will effectively raise HDL cholesterol levels and also reduce the incidence of atherosclerosis in patients. Torcetrapib was the first drug that was tested in a long-term outcomes clinical trial. The clinical trial of torcetrapib was terminated early due to a higher incidence of mortality in patients to whom torcetrapib and atorvastatin were administered concomitantly compared with patients who were treated with atorvastatin alone. The cause of the increased mortality is not completely understood, but it is not believed to be associated with the CETP inhibiting effects of the drug.
Two other drug candidates, dalcetrapib and anacetrapib, are currently being tested in Phase III clinical trials, including large scale outcomes trials. Data from the recently completed DEFINE Phase III trial of anacetrapib are promising. Patients who were being treated with anacetrapib along with baseline statin therapy showed an increase of HDL-C of 138% and a decrease of LDL-C of 40%> compared with patients who were treated with just a statin. See: N. Engl. J. Med. 2010: 363: 2406-15. The data in the DEFINE trial were sufficient to indicate that an increase in mortality for patients treated with anacetrapib is unlikely. Additional drug candidates are still being sought that may have properties that are advantageous compared with the CETP inhibitors that have so far been studied or are currently being studied. Such properties may include, for example, higher potency, reduced off-target activity, better pharmacodynamics, higher bioavailability, or a reduced food effect compared with many of the highly lipophilic compounds that have so far been studied. “Food effect” refers to the variability in exposure to the active drug that occurs depending on when the patient had last eaten, whether or not the drug is administered with food, and the fat content of the food.
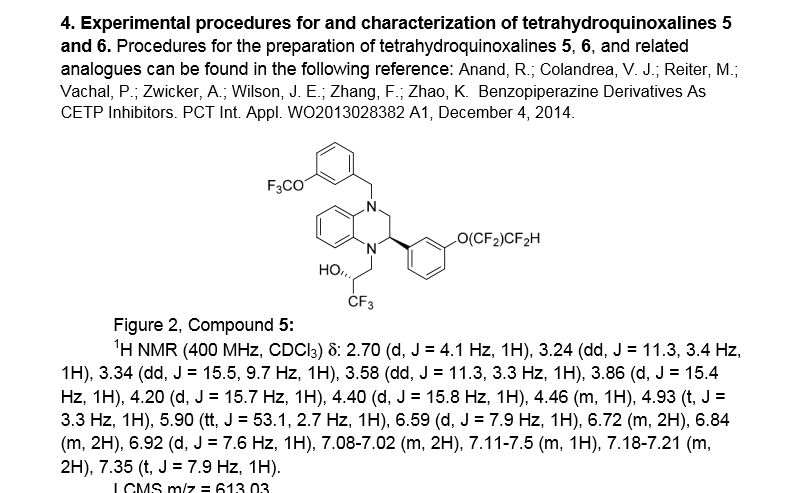
Example 18 as in patent

(R)- 1,1, 1 -trifluoro-3-((R)-4-(3-trifluoromethoxy)benzyl)-2-(3-(l, 1 ,2,2,-tetrafluoroethoxy)phenyl)-3,4- dihydroquinoxalin- 1 (2H)-yl)propan-2-ol
SPA: 15 nM
Example 18 was prepared from 2-bromo-l-(3-(l , 1 ,2,2,-tetrafluoroethoxy)phenyl)ethanone in three steps, using the reactions detailed in Schemes A6, A2 and Al . Spectral data are as follows: 1H NMR (400 MHz, CDC13) £2.70 (bd, J=4.1 Hz, IH), 3.24 (dd, J=l 1.3, 3.4 Hz, IH), 3.34 (dd, J=15.5, 9.7 Hz, IH), 3.58 (dd, J=l 1.3, 3.3 Hz, IH), 3.86 (d, J=15.4 Hz, IH), 4.20 (d, J=15.7 Hz, IH), 4.40 (d, J=15.8 Hz, IH), 4.46 (m, IH), 4.927 (t, J=3.3 Hz, IH), 5.90 (tt, J=53.1 , 2.7 Hz, IH), 6.59 (d, J= 7.9 Hz, IH), 6.72 (m, 2H), 6.84 (m, 2H), 6.92 (d, J=7.6 Hz, IH), 7.20 (m, 2H), 7.35 (t, J=7.9 Hz, IH), MS m/z = 613.03.
Scheme A12

Methyl 3 – { 1 – [(R)-3 ,3 ,3 -trifluoro-2-hy droxypropyl] -4- [3 -(trifluoromethoxy) benzyl]-l,2,3,4-tetrahydroquinoxalin-2-yl}benzoate (700 mg, 1.262 mmol) is made as described in Example 16 but with one stereochemical center unresolved. The compound was dissolved in MeOH (12.6mL), lithium hydroxide monohydrate (530 mg, 12.62 mmol) was added, and the reaction mixture was heated to 60°C for 4 hours. The crude mixture was dissolved in saturated ammonium chloride solution and extracted into EtOAc, the organic phase was dried with anhydrous magnesium sulfate, filtered, concentrated, and purified on a silica gel column with a 0-100% Hex/EtOAc gradient. The major peak was concentrated to afford 3-{l-[(R)-3,3,3-trifluoro-2-hydroxypropyl]-4-[3-(trifluoromethoxy)benzyl]-l,2,3,4-tetra-hydroquinoxalin-2-yl} benzoic acid. MS m/z = 541.09.

Patent
WO2015054088
http://google.com/patents/WO2015054088A1?cl=en
Scheme Al

Scheme A2

Scheme A3

R = Ar, NR2l C02R, CN, S02Me
es
es
SEE EXAMPLE ………SIMILAR BUT NOT SAME

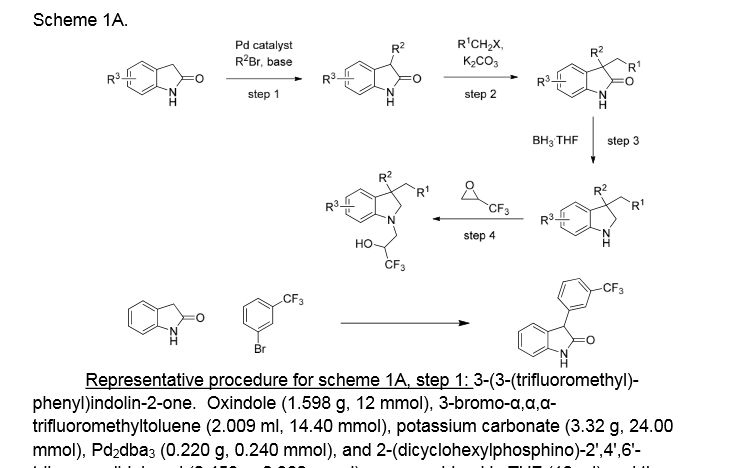
Example 1. (2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. This material was prepared according to Scheme Al, as described below.

3-(3-(trifluoromethyl)phenyl)indolin-2-one. Oxindole (1.598 g, 12 mmol), 3-bromo-a,a,a-trifluoromethyltoluene (2.009 ml, 14.40 mmol), potassium carbonate (3.32 g, 24.00 mmol), Pd2dba3 (0.220 g, 0.240 mmol), and 2-(dicyclohexylphosphino)-2′,4′,6′-triisopropylbiphenyl (0.458 g, 0.960 mmol) were combined in THF (12 ml) and the mixture was degassed with nitrogen. The solution was then heated to 80 °C for 18h. The mixture was cooled to room temperature, filtered through silica eluting with ethyl acetate, and concentrated. The material was then purified by silica gel chromatography (Biotage lOOg SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethyl)phenyl)indolin-2-one as a white solid.
1H NMR (500 MHz) δ 8.58 (s, 1H), 7.61 (d, J=7 Hz, 1H), 7.53-7.45 (m, 3H), 7.33-7.29 (m, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (m, 1H), 7.01-6.90 (m, 1H), 4.73 (s, 1H).

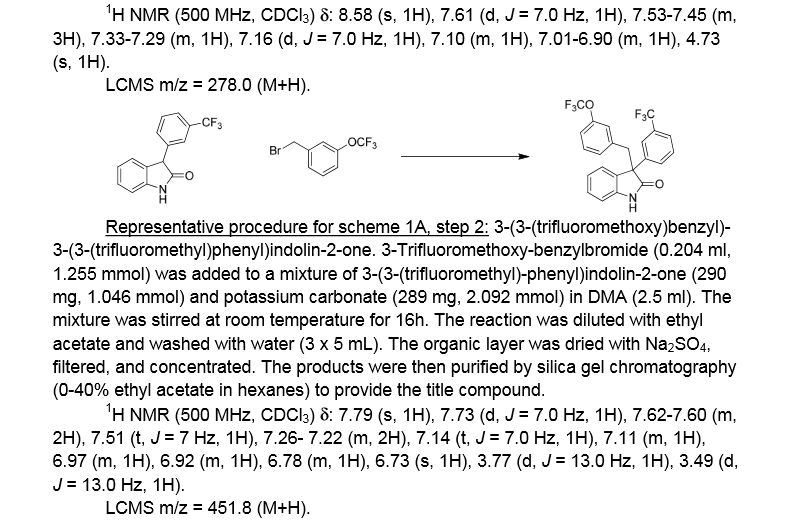
3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin-2-one . 3 -Trifluoromethoxy-benzylbromide (0.204 ml, 1.255 mmol) was added to a mixture of 3-(3-(trifluoromethyl)-phenyl)indolin-2-one (290 mg, 1.046 mmol) and potassium carbonate (289 mg, 2.092 mmol) (sodium carbonate may be used in place of potassium carbonate) in DMA (2.5 ml). The mixture was stirred at r.t. for 16h. The reaction was diluted with ethyl acetate and washed with water (3×5 mL). The organic layer was dried with Na2S04, filtered, and concentrated. The products were then purified by silica gel chromatography (Biotage 50g SNAP cartridge; 0-40%> ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-2-one .
1H NMR (500 MHz) δ 7.79 (s, 1H), 7.73 (d, J=7 Hz, 1H), 7.62-7.60 (m, 2H), 7.51 (t, J=7 Hz, 1H), 7.26- 7.22 (m, 2H), 7.14 (t, J=7.0 Hz, 1H), 7.11 (m, 1H), 6.97 (m, 1H), 6.92 (m, 1H), 6.78 (m, 1H), 6.73 (s, 1H), 3.77 (d, J=13 Hz, 1H), 3.49 (d, J=13 Hz, 1H).
LCMS m/z = 451.8 (M+H)

3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. Borane tetrahydrofuran complex (1.673 ml, 1.673 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indolin-2-one (302 mg, 0.669 mmol) in THF (1.5 ml). The mixture was heated to 70 °C for 20h. The reaction was cooled to room temperature and quenched with saturated NH4C1 solution, and this mixture was stirred vigorously for 20 minutes. The product was extracted with ethyl acetate. The extracts were dried over Na2S04, filtered, and concentrated. The product was purified by silica gel chromatography (Biotage 25g SNAP cartridge, 0-50% ethyl acetate in hexanes) to provide 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline. This material may also be used without purification in the final step of the sequence, epoxide opening.
1H NMR (500 MHz) δ 7.66 (s, IH), 7.59 (d, J=7 Hz, IH), 7.53 (d, J=7 Hz, IH), 7.45 (t, J=8 Hz, IH), 7.18-7.13 (m, 2H), 7.04 (d, J=8 Hz, IH), 6.98 (d, J=7 Hz, IH), 6.81 (t, J=7.5 Hz, IH), 6.71 (m, 2H), 6.60 (s, IH), 3.83 (m, IH), 3.75-3.73 (m, 2H), 3.46 (d, J=13 Hz, IH), 3.41 (d, J=13 Hz, IH).
= 437.9 (M+H)

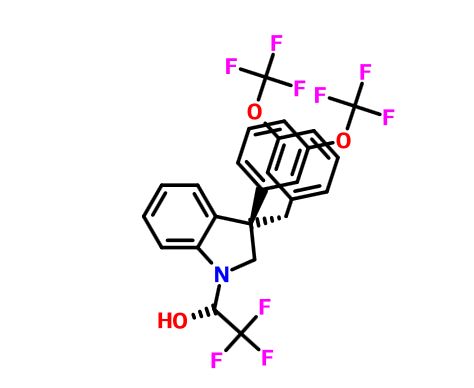
(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)-phenyl)indolin-l-yl)propan-2-ol. (S)-2-(trifluoromethyl)oxirane (81 μΐ, 0.933 mmol) was added to a solution of 3-(3-(trifluoromethoxy)benzyl)-3-(3-(trifluoromethyl)phenyl)indoline (136 mg, 0.311 mmol) in l,l,l,3,3,3-hexafluoro-2-propanol (412 μΐ, 3.91 mmol). The reaction was stirred at room temperature overnight. The solvent was removed and the product was purified by silica gel chromatography (Biotage 25 g SNAP cartridge; 0-25% ethyl acetate in hexanes) to provide (2R)- 1 ,1,1 -trifluoro-3 -(3 -(3 -(trifluoromethoxy)benzyl)-3 -(3 -(trifluoromethyl)phenyl)indolin- 1 -yl)propan-2-ol.
1H NMR (500 MHz) (mixture of diastereomers) δ 7.72 (s, 0.5 H), 7.69 (s, 0.5 H), 7.65 (d, J=6.5 Hz, 0.5 H), 7.61 (d, J=7.5 Hz, 0.5 H), 7.56 (s, 1H), 7.50 (m, 1H), 7.25-7.17 (m, 2H), 7.07 (broad s, 2H), 6.91-6.89 (m, 1H), 6.79-6.75 (m, 1H), 6.53 (m, 2H), 4.00 (broad s, 1H), 3.83 (d, J= 9 Hz, 0.5H), 3.77 (d, J=9 Hz, 0.5H), 3.59-3.55 (m, 1H), 3.45-3.43 (m, 1H), 3.39-3.29 (m, 2H), 3.21-3.15 (m, 1H), 2.32 (m, 0.5H), 2.15 (m, 0.5H).
LCMS m/z = 549.8 (M+H)
Examples 1-25, in the table below, were prepared according to Scheme Al in a
SEE EG 10…….(2R)- 1,1,1 -trifluoro-3-(3-(3-(trifluoromethoxy)benzyl)-3-(3- (trifluoromethoxy)-phenyl)indolin-l-yl)propan-2-ol.

ABOUT AUTHOR
Jonathan Wilson
Associate Principal Scientist at Merck

https://www.linkedin.com/in/jonathan-wilson-23206523
Experience
Associate Principal Scientist
Merck
October 2013 – Present (2 years 4 months)
Senior scientist
Merck
May 2009 – October 2013 (4 years 6 months)

Postdoctoral researcher
Princeton University
October 2007 – May 2009 (1 year 8 months)
Associate Medicinal Chemist
Merck
2000 – 2002 (2 years)
Education


///////CETP inhibition, cholesterol ester transfer protein, HDL, indoline, tetrahydroquinoxaline, merck, discovery
c21ccccc1N(C[C@@]2(c3cccc(c3)OC(F)(F)F)Cc4cc(ccc4)OC(F)(F)F)C(C(F)(F)F)O
FC(F)(F)Oc1cccc(c1)C3(CN(C[C@@H](O)C(F)(F)F)c2ccccc23)Cc4cccc(OC(F)(F)F)c4
see…………http://worlddrugtracker.blogspot.in/2016/01/mercks-novel-indoline-cholesterol-ester.html
WCK 5222, Wockhardt receives QIDP status for its new drug WCK 5222 from USFDA
WCK 5222
Watch this post as I get to the structure…………..
DEC2015
Wockhardt has received Qualified Infectious Disease Product (QIDP) status for its new drug WCK 5222, a product from its breakthrough New Drug Discovery program in Anti Infectives from the US Food and Drug Administration (FDA).
This is the fourth product from the company to receive this coveted status. During last year, the company has received approval for WCK 771 & WCK 2349 and in early this year approval was received for WCK 4873. The only company globally to receive QIDP status for 4 drugs from US FDA.
Wockhardt is one of the few companies with end to end integrated capabilities for its products, starting with the manufacture of the oral and sterile API’s, the dose forms and marketing through wholly owned subsidiary in the US, enabling the company to capture maximum value.
Ten compounds generally represented by a general Formula (I) were used and are as follows:
(a) Sodium salt of ir ns-7-oxo-6-sulphooxy-l ,6-diazabicyclo[3.2.1]-octane-2-carbonitrile (Compound A);

(b) trans-sulphuric acid mono-[2-(5-carboxamido)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound B);

(c) trans-sulphuric acid mono-[2-(5-(piperidin-4-yl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound C);

(d) trans-sulphuric acid mono-[2-(5-azetidin-3-ylmethyl-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound D);

(e) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-piperidine-3-carbonyl)-hydrazinocarbonyl] -1,6-diaza-bicyclo[3.2.1]octane (Compound E);

(f) (25, 5i?)-7-Oxo-N-[(25)-pyrrolidin-2-ylmethoxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1] octane-2-carboxamide (Compound F);

(g) (25,5i?)-7-Oxo-6-sulphooxy-2-[N’-((i?)-pyrrolidine-3-carbonyl)-hydrazinocarbonyl]-l ,6-diaza -bicyclo[3.2.1]octane (Compound G);

(h) (25,5i?)-7-Oxo-N-[(25)-piperidine-2-ylmethyloxy]-6-(sulfooxy)-l ,6-diazabicyclo
octane-2-carboxamide (Compound H);

(i) trans-sulphuric acid mono-[2-(5-((5)-l-amino-ethyl)-[l ,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound I); and

j) trans-sulphuric acid mono-[2-(5-((5)-pyrrolidin-2-yl)-[l,3,4]-oxadiazol-2-yl)-7-oxo-l,6-diazabicyclo[3.2.1]-octan-6-yl] ester (Compound J).

////
