Cobicistat, GS-9350
1004316-88-4
N-[1(R)-Benzyl-4(R)-[2(S)-[3-(2-isopropylthiazol-4-ylmethyl)-3-methyl]ureido]-4-(4-morpholinyl)butyramido]-5-phenylpentyl]carbamic acid thiazol-5-ylmethyl ester
(1,3-thiazol-5-yl) methyl (5S, 8R, 11R) -8,11-dibenzyl-2-methyl-5-[2 – (morpholin-4-yl) ethyl] -1 – [2 – (propan-2-yl) -1,3-thiazol-4-yl] -3,6-dioxo-2 ,4,7,12-tetraazatridecan-13-oate
cytochrome P450 3A4 (CYP3A4) inhibitor
Gilead Sciences, Inc.
FDA Approves Tybost (cobicistat) for use in the treatment of HIV-1 Infection
September 24, 2014 — The U.S. Food and Drug Administration (FDA) has approved Tybost (cobicistat), a CYP3A inhibitor used in combination with atazanavir or darunavir for the treatment of human immunodeficiency virus type 1 (HIV-1) infection
Cobicistat is a pharmacokinetic enhancer that works by inhibiting the enzyme (CYP3A) that metabolizes atazanavir and darunavir. It increases the systemic exposure of these drugs and prolongs their effect. Cobicistat is also one of the ingredients in the combination HIV drug Stribild, which was approved by the FDA in August, 2012.
Tybost comes in 150 mg tablets and is administered once daily in combination with the protease inhibitors atazanavir (Reyataz), or darunavir (Prezista).
Because Tybost inhibits CYP3A, other medications metabolized by CYP3A may result in increased plasma concentrations and potentially severe side effects, which may be life-threatening or even fatal. Extra care should be exercised by healthcare professionals to ensure than other medications are reviewed and their concentrations monitored, especially when initiating new medicines or changing doses.
The approval of Tybost was based on the following clinical trials:
•The data to support the use of atazanavir and Tybost were from a phase 2 and 3 trial in treatment-naïve adults comparing atazanavir/cobicistat 300/150 mg and atazanavir/ritonavir 300/100 mg once daily each in combination with Truvada. The atazanavir/cobicistat based regimen was non-inferior to the atazanavir/ritonavir based regimen.
•The data to support the use of cobicistat with darunavir is from a multiple dose trial in healthy subjects comparing the relative bioavailability of darunavir/cobicistat 800/150 mg to darunavir/ritonavir 800/100 mg.

The most common adverse drug reactions observed with Tybost (in combination with atazanavir) in clinical trials were jaundice, ocular icterus, and nausea.
Tybost is a product of Gilead Sciences, Foster City, CA.

Cobicistat (formerly GS-9350) is a licensed drug for use in the treatment of infection with the human immunodeficiency virus (HIV).
Like ritonavir (Norvir), cobicistat is of interest not for its anti-HIV properties, but rather its ability to inhibit liver enzymes that metabolize other medications used to treat HIV, notablyelvitegravir, an HIV integrase inhibitor currently under investigation itself. By combining cobicistat with elvitegravir, higher concentrations of elvitgravir are achieved in the body with lower dosing, theoretically enhancing elvitgravir’s viral suppression while diminishing its adverse side-effects. In contrast with ritonavir, the only currently approved booster, cobicistat has no anti-HIV activity of its own.[1]
Cobicistat, a cytochrome P450 CYP3A4 inhibitor, was approved in the E.U. in 2013 as a pharmacokinetic enhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults. First launch took place in 2014 in United Kingdom. In 2012, Gilead filed a New Drug Application in the U.S. for the same indication. In April 2013, the FDA issued a Complete Response Letter from the FDA. In 2014 the FDA accepted Gilead’s resubmission.
Cobicistat is a component of the four-drug, fixed-dose combination HIV treatmentelvitegravir/cobicistat/emtricitabine/tenofovir (known as the “Quad Pill” or Stribild).[1][2] The Quad Pill/Stribild was approved by the FDA in August 2012 for use in the United States and is owned by Gilead Sciences.
Cobicistat is a potent inhibitor of cytochrome P450 3A enzymes, including the importantCYP3A4 subtype. It also inhibits intestinal transport proteins, increasing the overall absorption of several HIV medications, including atazanavir, darunavir and tenofovir alafenamide fumarate.[3]
The drug candidate acts as a pharmaco-enhancer to boost exposure of HIV protease inhibitors. In 2011, cobicistat was licensed to Japan Tobacco by Gilead for development and commercialization in Japan as a stand-alone product for the treatment of HIV infection. In 2012, orphan drug designation was assigned in Japan for the pharmacokinetic enhancement of anti-HIV agent.
Oxidative metabolism by cytochrome P450 enzymes is one of the primary mechanisms of drug metabolism.. It can be difficult to maintain therapeutically effective blood plasma levels of drugs which are rapidly metabolized by cytochrome P450 enzymes. Accordingly, the blood plasma levels of drugs which are susceptible to cytochrome P450 enzyme degradation can be maintained or enhanced by co-administration of cytochrome P450 inhibitors, thereby improving the pharmacokinetics of the drug.
While certain drugs are known to inhibit cytochrome P450 enzymes, more and/or improved inhibitors for cytochrome P450 monooxygenase are desirable. Particularly, it would be desirable to have cytochrome P450 monooxygenase inhibitors which do not have appreciable biological activity other than cytochrome P450 inhibition. Such inhibitors can be useful for minimizing undesirable biological activity, e.g., side effects. In addition, it would be desirable to have P450 monooxygenase inhibitors that lack significant or have a reduced level of protease inhibitor activity. Such inhibitors could be useful for enhancing the effectiveness of antiretroviral drugs, while minimizing the possibility of eliciting viral resistance, especially against protease inhibitors.
…………………………….
Cobicistat (GS-9350): A potent and selective inhibitor of human CYP3A as a novel pharmacoenhancer
ACS Med Chem Lett 2010, 1(5): 209
http://pubs.acs.org/doi/abs/10.1021/ml1000257
http://pubs.acs.org/doi/suppl/10.1021/ml1000257/suppl_file/ml1000257_si_001.pdf
Cobicistat (3, GS-9350) is a newly discovered, potent, and selective inhibitor of human cytochrome P450 3A (CYP3A) enzymes. In contrast to ritonavir, 3 is devoid of anti-HIV activity and is thus more suitable for use in boosting anti-HIV drugs without risking selection of potential drug-resistant HIV variants. Compound 3 shows reduced liability for drug interactions and may have potential improvements in tolerability over ritonavir. In addition, 3 has high aqueous solubility and can be readily coformulated with other agents.
1-Benzyl-4-{2-[3-(2-isopropyl-thiazol-4-ylmethyl)-3-methyl-ureido]-4-morpholin-4-yl-butyrylamino}-5-phenyl-pentyl)-carbamic acid thiazol-5-ylmethyl ester (GS-9350)
HPLC (Chiral CelROD-H, Chiral Technologies Inc;heptane/iPrOH = 70/30).
1H NMR (CD3OD)
δ8.98 (1 H, s), 7.82 (1 H, s), 7.25-7.05
(11 H, m), 5.25-5.10 (2 H, m), 4.60-4.50 (2 H, m), 4.21-4.03 (2 H, m), 3.82-3.72 (1
H, m), 3.65-3.65 (4 H, m), 3.35-3.25 (1 H, m), 2.98 (3 H, s), 2.8-2.6 (4 H, m), 2.4-2.2
(6 H, m), 1.95-1.8 (1 H, m), 1.8-1.6 (1 H, m), 1.6-1.4 (4 H, m), 1.42-1.32 (6 H, m).
MS (ESI) m/z: 776.2 (M+H)+.
HRMS calc. for C40H53N7O5S2: 775.355, found: 775.353.
…………………………………
http://www.google.com/patents/CN103694196A?cl=en
CN 103694196
oxidative metabolism by cytochrome P450 enzymes is one of the main mechanisms of drug metabolism, generally by administration of cytochrome P450 inhibitors to maintain or increase the degradation of cytochrome P450 enzymes are sensitive to the drug plasma levels, in order to improve the pharmacokinetics of drugs dynamics, can be used to enhance the effectiveness of anti-retroviral drugs. For example W02008010921 discloses compounds of formula I as a cytochrome P450 monooxygenase specific compounds (Cobicistat):
W02008010921 discloses the synthesis of compounds of formula I with a variety of, as one of the methods of the following routes
Shows:
The reagents used in the method is expensive, and more difficult to remove by-products, long reaction time, high cost, is not conducive to industrial
Production.
W02010115000 on these routes has been improved:
The first step in the route used for the ring-opening reaction reagent trimethylsilyl iodide, trimethylsilyl iodide expensive. W02010115000 reports this step and the subsequent ring-opening reaction of morpholine substitution reaction yield of two steps is not high, only 71%, so that only iodotrimethylsilane a high cost of raw material is not suitable for industrial production.



Preparation of compounds of formula I
Example [0126] Implementation
[0127] I1-a (20g) was dissolved in dichloromethane, was added 50% K0H (5.5g) solution, control the internal temperature does not exceed 25 ° C, TLC analysis ΙΙ-a disappears. Was cooled to O ~ 10 ° C, was added (2R, 5R) -5 – amino-1 ,6 – diphenyl-2 – hexyl-carbamic acid 5 – methyl-thiazole ester hydrochloride (14.8g), stirred for I ~ 2 h, 1 – hydroxybenzotriazole triazole (5.5g), stirred for I h, 1 – ethyl – (3 – dimethylaminopropyl) carbodiimide hydrochloride (15g), and incubated for 5 ~ 10 hours, TLC analysis of the starting material disappeared, the reaction was completed. The reaction was quenched with aqueous acetic acid, methylene chloride layer was separated, washed with saturated aqueous NaHCO3, washed with water, dried and concentrated. By HPLC purity of 99.1%. Adding ethanol, the ethanol was evaporated to give the product compound of part I of a solution in ethanol. Molar yield 88%, LC-MS: M +1 = 777.1 [0128] All publications mentioned in the present invention are incorporated by reference as if each reference was individually incorporated by reference, as cited in the present application. It should also be understood that, after reading the foregoing teachings of the present invention, those skilled in the art that various modifications of the present invention or modifications, and these equivalents falling as defined by the appended claims scope of claims of the present application.
…………………………
US 2014088304
http://www.google.com/patents/US20140088304
International Patent Application Publication Number WO 2008/010921 and International Patent Application Publication Number WO 2008/103949 disclose certain compounds that are reported to be useful to modify the pharmacokinetics of a co-administered drug, e.g. by inhibiting cytochrome P450 monooxygenase. One specific compound identified therein is a compound of the following formula I:
There is currently a need for improved synthetic methods and intermediates that can be used to prepare the compound of formula I and its salts
Schemes 1-4 below.
Preparation of a Compound of Formula IV
Example 14Preparation of Compound I

To the solution of L-thiazole morpholine ethyl ester oxalate salt XIVa (35.6 kg) in water (66.0 kg) was charged dichloromethane (264 kg), followed by a slow addition of 15 wt % KHCO3 solution (184.8 kg). The resulting mixture was agitated for about 1 hour. The layers were separated and the organic layer was washed with water (132 kg). The organic layer was concentrated under vacuum to dryness. Water (26.5 kg) was charged and the content temperature was adjusted to about 10° C., followed by slow addition of 45% KOH solution (9.8 kg) while maintaining the content temperature at less than or equal to 20° C. The mixture was agitated at less than or equal to 20° C. until the reaction was judged complete by HPLC. The reaction mixture was concentrated under vacuum to dryness and co-evaporated five times with dichloromethane (132 kg each time) under reduced pressure to dryness. Co-evaporation with dichloromethane (132 kg) was continued until the water content was <4% by Karl Fischer titration. Additional dichloromethane (264 kg) was charged and the content temperature was adjusted to −18° C. to −20° C., followed by addition of monocarbamate.HCl salt IXa (26.4 kg). The resulting mixture was agitated at −18° C. to −20° C. for about 1 hour. HOBt (11.4 kg) was charged and the reaction mixture was again agitated at −18° C. to −20° C. for about 1 hour. A pre-cooled solution (−20° C.) of EDC.HCl (21.4 kg) in dichloromethane (396 kg) was added to the reaction mixture while the content temperature was maintained at less than or equal to −20° C. The reaction mixture was agitated at −18° C. to −20° C. until the reaction was judged complete. The content temperature was adjusted to about 3° C. and the reaction mixture quenched with a 10 wt % aqueous citric acid solution (290 kg). The layers were separated and the organic layer was washed once with 15 wt % potassium bicarbonate solution (467 kg) and water (132 kg). The organic layer was concentrated under reduced pressure and then co-evaporated with absolute ethanol.
The product I was isolated as the stock solution in ethanol (35.0 kg product, 76.1% yield).
1H NMR (dDMSO) δ□ 9.05 (s, 1H), 7.85 (s, 1H), 7.52 (d, 1H), 7.25-7.02 (m, 12H), 6.60 (d, 1H), 5.16 (s, 2H), 4.45 (s, 2H), 4.12-4.05 (m, 1H), 3.97-3.85 (m, 1H), 3.68-3.59 (m, 1H), 3.57-3.45 (m, 4H), 3.22 (septets, 1H), 2.88 (s, 3H), 2.70-2.55 (m, 4H), 2.35-2.10 (m, 6H), 1.75 (m, 1H), 1.62 (m, 1H), 1.50-1.30 (m, 4H), 1.32 (d, 6H).
13C NMR (CD3OD) δ 180.54, 174., 160.1, 157.7, 156.9, 153.8, 143.8, 140.1, 140.0, 136.0, 130.53, 130.49, 129.4, 127.4, 127.3, 115.5, 67.7, 58.8, 56.9, 55.9, 54.9, 53.9, 51.6, 49.8, 42.7, 42.0, 35.4, 34.5, 32.4, 32.1, 29.1, 23.7.
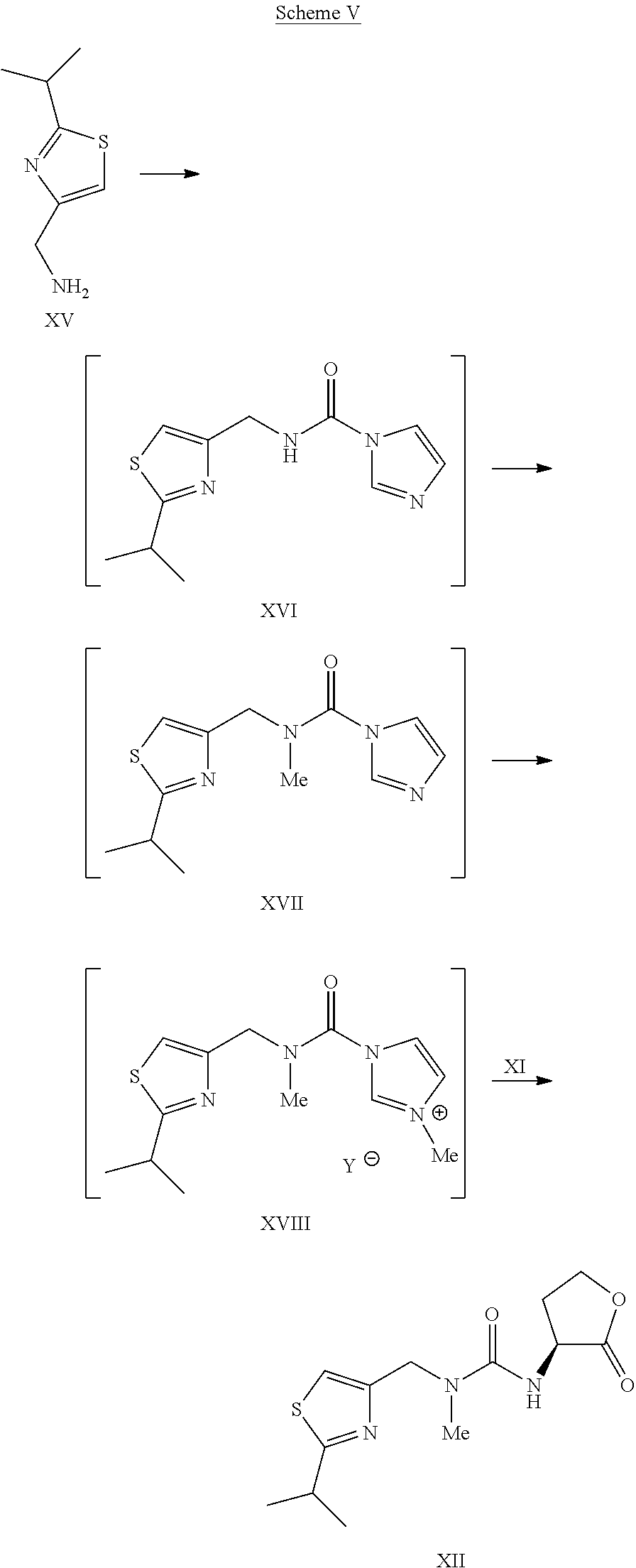
Example 13Preparation of L-Thiazole Morpholine Ethyl Ester Oxalate Salt XIVa

To a solution of (L)-thiazole amino lactone XII (33.4 kg) in dichloromethane (89.5 kg) was charged dichloromethane (150 kg) and absolute ethanol (33.4 kg). The content temperature was then adjusted to about 10° C., followed by slow addition of TMSI (78.8 kg) while the content temperature was maintained at less than or equal to 22° C. and agitated until the reaction was judged complete. The content temperature was adjusted to about 10° C., followed by a slow addition of morpholine (49.1 kg) while the content temperature was maintained at less than or equal to 22° C. Once complete, the reaction mixture was filtered to remove morpholine.HI salt and the filter cake was rinsed with two portions of dichloromethane (33.4 kg). The filtrate was washed twice with water (100 kg). The organic layer was concentrated under vacuum to dryness. Acetone (100 kg) was then charged to the concentrate and the solution was concentrated under reduced pressure to dryness. Acetone (233.8 kg) was charged to the concentrate, followed by a slow addition of the solution of oxalic acid (10 kg) in acetone (100 kg). The resulting slurry was refluxed for about 1 hour before cooling down to about 3° C. for isolation. The product XIVa was filtered and rinsed with acetone (66.8 kg) and dried under vacuum at 40° C. to afford a white to off-white solid (40 kg, 71% yield). 1H NMR (CDCl3) δ □7.00 (s, 1H), 6.35 (broad s, 1H), 4.60-4.40 (m, 3H), 4.19 (quartets, 2H), 4.00-3.90 (m, 4H), 3.35-3.10 (m, 7H), 3.00 (s, 3H), 2.40-2.30 (m, 1H), 2.15-2.05 (m, 1H), 1.38 (d, 6H), 1.25 (triplets, 3H).
……………………………………..
W02008010921
http://www.google.co.in/patents/WO2008010921A2?cl=en
Preparation of Example A
Scheme 1
Example A Compound 2
To a solution of Compound 1 (ritonavir) (1.8 g, 2.5 mmol) in 1,2- dichloroethane (15 mL) was added l,l’-thiocarbonyldiimidazole (890 mg, 5.0 mmol). The mixture was heated at 75 SC for 6 hours and cooled to 25 SC. Evaporation under reduced pressure gave a white solid. Purification by flash column chromatography (stationary phase: silica gel; eluent: EtOAc) gave Compound 2 (1.6 g). m/z: 831.1 (M+H)+. Example A
To the refluxing solution of tributyltin hydride (0.78 mL, 2.9 mmol) in toluene (130 mL) was added a solution of Compound 2 (1.6 g, 1.9 mmol) and 2,2′- azobisisobutyronitrile (31 mg, 0.19 mmol) in toluene (30 mL) over 30 minutes. The mixture was heated at 1152C for 6 hours and cooled to 25 BC. Toluene was removed under reduced pressure. Purification by flash column chromatography (stationary phase: silica gel; eluent: hexane/EtOAc = 1/10) gave Example A (560 mg). m/z: 705.2 (M+H)+. 1H-NMR (CDCl3) δ 8.79 (1 H, s), 7.82 (1 H, s), 7.26-7.05 (10 H, m), 6.98 (1 H, s), 6.28 (1 H, m), 6.03 (1 H, m), 5.27 (1 H7 m), 5.23 (2 H, s), 4.45-4.22 (2 H, m), 4.17 (1 H, m), 3.98 (1 H, m), 3.75 (1 H, m), 3.25 (1 H7 m), 2.91 (3 H, s), 2.67 (4 H, m), 2.36 (1 H, m), 1.6-1.2 (10 H, m), 0.85 (6 H, m).
| EP1183026A2 * |
25 May 2000 |
6 Mar 2002 |
Abbott Laboratories |
Improved pharmaceutical formulations |
| US20060199851 * |
2 Mar 2006 |
7 Sep 2006 |
Kempf Dale J |
Novel compounds that are useful for improving pharmacokinetics |
| US7939553 * |
Jul 6, 2007 |
May 10, 2011 |
Gilead Sciences, Inc. |
co-administered drug (as HIV protease inhibiting compound, an HIV (non)nucleoside/nucleotide inhibitor of reverse transcriptase, capsid polymerization inhibitor, interferon, ribavirin analog) by inhibiting cytochrome P450 monooxygenase; ureido- or amido-amine derivatives; side effect reduction |
Highleyman, L.
Elvitegravir “Quad” Single-tablet Regimen Shows Continued HIV Suppression at 48 Weeks
- R Elion, J Gathe, B Rashbaum, and others. The Single-Tablet Regimen of Elvitegravir/Cobicistat/Emtricitabine/Tenofovir Disoproxil Fumarate (EVG/COBI/FTC/TDF; Quad) Maintains a High Rate of Virologic Suppression, and Cobicistat (COBI) is an Effective Pharmacoenhancer Through 48 Weeks. 50th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2010). Boston, September 12–15, 2010.
- Lepist, E. -I.; Phan, T. K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A. S. (2012). “Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and GS-7340, in Vitro”. Antimicrobial Agents and Chemotherapy 56 (10): 5409–5413. doi:10.1128/AAC.01089-12. PMC 3457391. PMID 22850510.
-
| Patent No
all US |
Expiry |
|
| 5814639 |
Sep 29, 2015 |
|
| 5814639*PED |
Mar 29, 2016 |
|
| 5914331 |
Jul 2, 2017 |
|
| 5914331*PED |
Jan 2, 2018 |
|
| 5922695 |
Jul 25, 2017 |
|
| 5922695*PED |
Jan 25, 2018 |
|
| 5935946 |
Jul 25, 2017 |
|
| 5935946*PED |
Jan 25, 2018 |
|
| 5977089 |
Jul 25, 2017 |
|
| 5977089*PED |
Jan 25, 2018 |
|
| 6043230 |
Jul 25, 2017 |
|
| 6043230*PED |
Jan 25, 2018 |
|
| 6642245 |
Nov 4, 2020 |
|
| 6642245*PED |
May 4, 2021 |
|
| 6703396 |
Mar 9, 2021 |
|
| 6703396*PED |
Sep 9, 2021 |
|
| 7176220 |
Nov 20, 2023 |
|
| 7635704 |
Oct 26, 2026 |
|
| 8148374 |
Sep 3, 2029 |
|
|

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....