
Home » Posts tagged 'cyclin-dependent kinase (CDK) inhibitor'
Tag Archives: cyclin-dependent kinase (CDK) inhibitor
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Mocaciclib
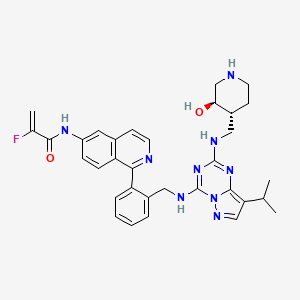
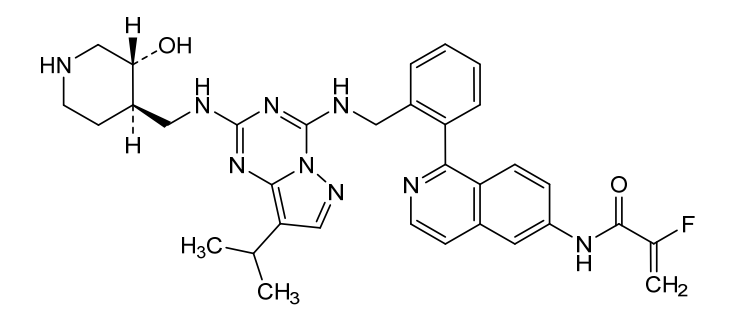
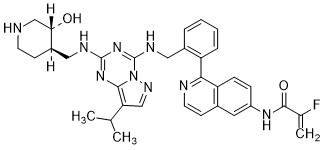
Mocaciclib
CAS 2766124-39-2
MF C33H36FN9O2 MW609.71
- 2-fluoro-N-[1-[2-[[[2-[[(3R,4R)-3-hydroxypiperidin-4-yl]methylamino]-8-propan-2-ylpyrazolo[1,5-a][1,3,5]triazin-4-yl]amino]methyl]phenyl]isoquinolin-6-yl]prop-2-enamide
- 2-Fluoro-N-[1-[2-[[[2-[[[(3R,4R)-3-hydroxy-4-piperidinyl]methyl]amino]-8-(1-methylethyl)pyrazolo[1,5-a]-1,3,5-triazin-4-yl]amino]methyl]phenyl]-6-isoquinolinyl]-2-propenamide
- 2-fluoro-N-[1-[2-[[[2-[[(3R,4R)-3-hydroxypiperidin-4-yl]methylamino]-8-propan-2-ylpyrazolo[1,5-a][1,3,5]triazin-4-yl]amino]methyl]phenyl]isoquinolin-6-yl]prop-2-enamide

cyclin-dependent kinase (CDK) inhibitor, antineoplastic, Q 901, CDK7-IN-21,
- OriginatorThe Lead Discovery Center; The Max Planck Institute of Biochemistry
- DeveloperQurient Co
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase I/IISolid tumours
- 31 May 2024Preliminary efficacy, pharmacodynamics, pharmacokinetics and adverse events data from a phase I/II trial in Solid tumours presented at the 60th Annual Meeting of the American Society of Clinical Oncology (ASCO-2024)
- 21 May 2024Qurient Therapeutics enters into an Cooperative Research and Development Agreement (CRADA) with the US National Cancer Institute (NCI) for phase I/II trial in Small cell lung cancer (SCLC) and Solid tumours
- 21 May 2024Qurient Therapeutics plans phase I/II trial in Small cell lung cancer (SCLC) and Solid tumours
Mocaciclib (Q-901) is an orally bioavailable, selective cyclin-dependent kinase (CDK) inhibitor with potent activity against CDK2, CDK4, and CDK6. Preclinical data show that Mocaciclib inhibits CDK2/cyclin E with an IC₅₀ of 1.1 nM, CDK4/cyclin D1 with an IC₅₀ of 2.5 nM, and CDK6/cyclin D3 with an IC₅₀ of 4.1 nM, demonstrating high potency in enzymatic assays. In cancer cell lines, Mocaciclib suppresses retinoblastoma protein (Rb) phosphorylation, leading to G1 cell cycle arrest and growth inhibition in Rb-positive tumor models. It has shown antiproliferative effects in various preclinical models, including breast and lung cancers.
Mocaciclib is a selective inhibitor of cyclin-dependent kinase 7 (CDK7), with potential antineoplastic activity. Upon administration, mocaciclib selectively targets, covalently binds to and inhibits the activity of CDK7, thereby inhibiting CDK7-mediated signaling. The inhibition of CDK7 prevents phosphorylation of the carboxy-terminal domain (CTD) of RNA polymerase II, thereby preventing transcription of important cancer-promoting genes. It prevents phosphorylation of the cell cycle kinases CDK1, 2, 4, and 6, thereby disrupting uncontrolled cell cycle progression. Altogether, this may induce apoptosis, cause cell cycle arrest, inhibit DNA damage repair and inhibit tumor cell proliferation in certain cancers that are dependent on CDK7-mediated transcriptional regulation and signaling. CDK7, a serine/threonine kinase, plays a role in controlling cell cycle progression and transcriptional regulation, and promotes the expression of key oncogenes through the phosphorylation of RNA polymerase II. It is overexpressed in multiple cancers.
SYN
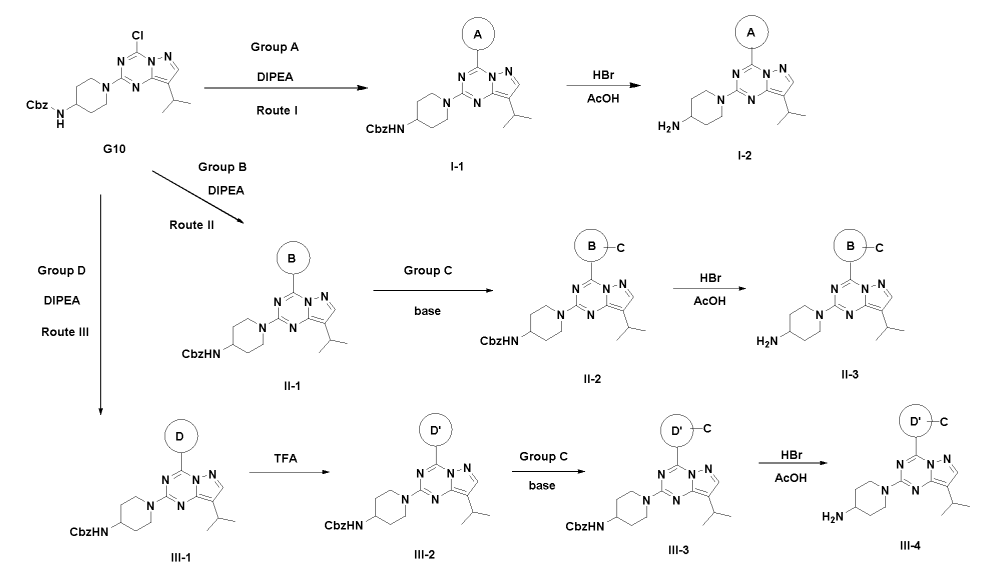
SYN
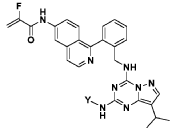
This is compound 64, as disclosed in WO2O19/197546.
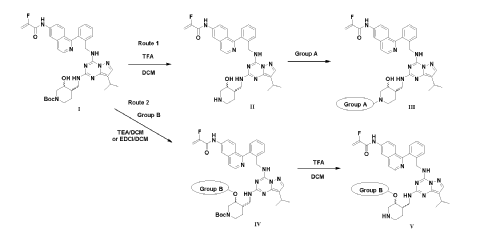
PAT
- Compounds having cyclin-dependent kinase(cdk)-inhibitory functionPublication Number: WO-2022117504-A1Priority Date: 2020-12-02
- Substituted pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a][1,3,5]triazines as CDK inhibitorsPublication Number: US-11858937-B2Priority Date: 2018-04-11Grant Date: 2024-01-02
- Pharmaceutically active pyrazolo-triazine and/or pyrazolo-pyrimidine derivativesPublication Number: US-2021139483-A1Priority Date: 2018-04-11



AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

//////////mocaciclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, Q 901, CDK7-IN-21,
Istisociclib
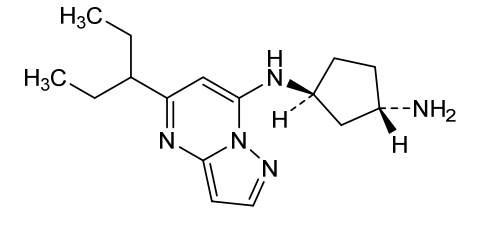
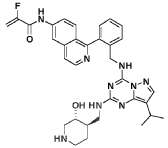
Istisociclib
KB 130742
CAS 2416873-83-9
MF C16H25N5, 287.40 g/mol
trans-(1S,3S)-3-N-(5-pentan-3-ylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine
- (1S,3S)-N1-[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-1,3-cyclopentanediamine
- 1,3-Cyclopentanediamine, N1-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-, (1S,3S)-
(1S,3S)-N1-[5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib is a small molecule drug. The usage of the INN stem ‘-ciclib’ in the name indicates that Istisociclib is a cyclin dependant kinase inhibitor. Istisociclib is under investigation in clinical trial NCT04718675 (A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)). Istisociclib has a monoisotopic molecular weight of 287.21 Da.
Istisociclib is an orally bioavailable, selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon oral administration, istisociclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins and oncogenic transcription factors including MYC and androgen receptor (AR). This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII), and is an important cofactor for various oncogenic transcription factors. It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
ISTISOCICLIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)
CTID: NCT04718675
Phase: Phase 1/Phase 2
Status: Terminated
Date: 2025-02-17
REF
- Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-11-15PMCID: PMC10726352PMID: 37967851DOI: 10.1021/acs.jmedchem.3c01233
- CDK9 inhibitors in cancer researchPublication Name: RSC Medicinal ChemistryPublication Date: 2022-06-22PMCID: PMC9215160PMID: 35814933DOI: 10.1039/d2md00040g
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
PAT
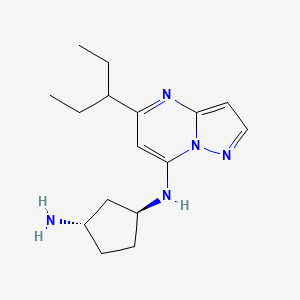
(lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine is a pharmaceutically active compound that has been studied for various uses, such as for the treatment of cancer. As used herein, the term “Compound A” is used to refer to both the free base and salt forms of (lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine. The free base of Compound A has the CAS number of 2416873-83-9 and structure of formula (I):
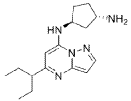
SYN
Example 35: (1S,3S)-N3-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (35)
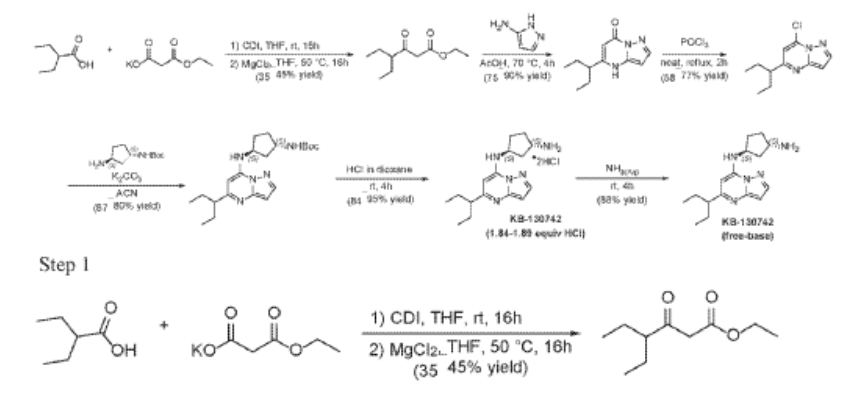
2-Ethylbutanoic acid (7.5 g, 64.57 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. Within 20 min CDI (16.23 g, 100.08 mmol) was added portion-wise. The reaction warmed to room temp (rt) and the mixture was stirred at rt overnight (Solution A). In another flask MgCl2 (6.14 g, 64.57 mmol) and potassium 3-ethoxy-3-oxo-propanoate (17 g, 100.1 mmol) were mixed with THF (150 mL) and stirred under argon overnight at 50 °C. The resultant white suspension was cooled to rt and solution A was added dropwise over 10 min and the reaction mixture (RM) was stirred for 16h at room temperature. After several minutes a sticky, amorphous solid appeared whereupon after several hours the reaction mixture became homogenous in appearance. The RM was concentrated to about a third, taken up in half sat. potassium bisulphate solution and extracted twice with ethyl acetate. The organic layers were subsequently washed with a sat. sodium bicarbonate solution, combined, dried over anhydrous sodium sulfate, filtered and evaporated. Purification by column chromatography gave ethyl 4-ethyl-3-oxo-hexanoate (4.3 g, 23.087 mmol, 35.8% yield) as a transparent liquid. The RM was monitored by TLC (10% EA in Hex, Product Rf=0.6, SM Rf=0.1).
Step 2
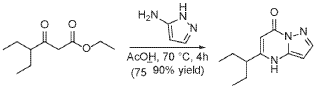
To a suspension of ethyl 4-ethyl-3-oxo-hexanoate (4.4 g, 23.62 mmol) in acetic acid (11 mL) at 70 °C was added 1H-pyrazol-5-amine (4.71 g, 56.7 mmol) in two portions (the second portion was added after 2 hours of stirring the first portion) over a 4 hour period. Upon consumption of SM as indicated by TLC, the reaction was cooled to rt and the solvent was evaporated in a rotary evaporator. The residue was treated with ethyl acetate and filtered to give 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 17.7 mmol, 74.9% yield) as an off-white solid. The reaction mixture was monitored by TLC (5% MeOH in DCM, Product Rf=0.3, SM Rf=0.8).
Step 3

A stirred solution of 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 18.03 mmol) in POCl3 (33.7 mL, 360.52 mmol) was heated to reflux for 4 hours. The reaction mixture was cooled to room temperature, excess reagent was evaporated in a rotary evaporator, and the residue was treated with ice-water. The chlorinated product was extracted from aqueous mixture by DCM. The organic layer was separated, dried over anhydrous Na2SO4, filtered and purified by column chromatography to give 7- chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (3.1 g, 13.9 mmol, 76.9% yield) as a light yellow liquid. The reaction mixture was monitored by TLC (20% EA in Hex, Product Rf=0.6, SM Rf=0.1).
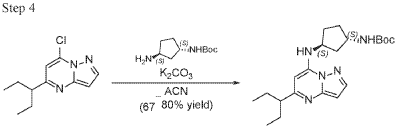
To a stirred solution 7-chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (2.3 g, 10.28 mmol), tert-Butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.27 g, 11.31 mmol) and K2CO3 (4.26 g, 30.84 mmol) in MeCN (20 mL) were heated to reflux for 16 hours. The reaction mixture was filtered, concentrated under reduced pressure and purified by column chromatography, eluent 30% EA in hexane to give tert-butyl N-[(1S,3S)-3-[[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (4.5 g, 11.6 mmol, 112.8% yield) as an off-white solid. The reaction mixture was monitored by TLC (40% EA in Hex, Product Rf=0.5, SM Rf=0.7).
Step 5
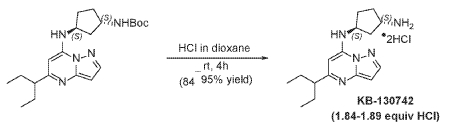
To tert-butyl N-[(1 S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (1.0 g, 2.58 mmol) in l,4-Dioxane (0.2 mL), 4 M HC1 in Dioxane (3.22 mL, 12.9 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give [(lS,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium dichloride (0.9 g, 2.5 mmol, 96.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC ( 100% EA, Product Rf=0.l, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 15.00 (s, 1H), 9.93-9.86 (m, 1H), 8.51 (s, 3H), 8.30 (s, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 4.95 (q, J = 7.8 Hz, 1H), 3.77- 3.66 (m, 1H), 2.84-2.71 (m, 1H), 2.29-2.05 (m, 4H), 1.94-1.63 (m, 6H), 0.81 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.0 [M+H+])
Step 6
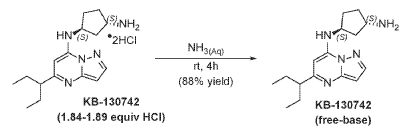
To [(1S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[I,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium-di chloride (0.2 g, 0.5600 mmol) in aq. NH3 (4.0 mL, 0.56 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give (lS,3S)-N3-[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]cyclopentane-l,3-diamine (140 mg, 0.49 mmol, 87.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC (100% EA, Product Rf=0.1, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 7.95 (d, J = 2.2 Hz, 1H), 6.86 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 5.95 (s, 1H), 4.31-4.19 (m, 1H), 3.57-3.44 (m, 1H), 2.52-2.44 (m, 1H), 2.36-2.22 (m, 1H),
2.09–1.79 (m, 3H), 1.80–1.59 (m, 5H), 1.58–1.24 (m, 3H), 0.83 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.5 [M+H+]).
PAT
- Compounds, compositions and methods for modulating CDK9 activityPublication Number: CN-112996790-BPriority Date: 2018-10-30Grant Date: 2023-11-03
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2024132506-A1Priority Date: 2018-10-30
- Compounds, Compositions, and Methods for Modulating CDK9 ActivityPublication Number: US-2020131189-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2022002305-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: EP-3873911-A1Priority Date: 2018-10-30
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11845754-B2Priority Date: 2018-10-30Grant Date: 2023-12-19
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11155560-B2Priority Date: 2018-10-30Grant Date: 2021-10-26
- Compounds and methods for modulating cdk9 activityPublication Number: EP-4240422-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: WO-2022098843-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: US-2025188084-A1Priority Date: 2020-11-05
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: WO-2021216828-A1Priority Date: 2020-04-24
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: US-2023158159-A1Priority Date: 2020-04-24
- Polymorphic and salt forms of (ls,3s)-n-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: EP-4436569-A1Priority Date: 2021-11-24
- Polymorphs and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-A]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: KR-20240110634-APriority Date: 2021-11-24
- Compositions and methods for enhanced protein productionPublication Number: EP-4412621-A2Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: US-2024252688-A1Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: WO-2023056202-A2Priority Date: 2021-09-22
- A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutationPublication Number: WO-2025217597-A1Priority Date: 2024-04-12
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3- yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: WO-2023096922-A8Priority Date: 2021-11-24
- Polymorphic and salt forms of (ls,3s)-n1-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: WO-2023096922-A1Priority Date: 2021-11-24
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl) cyclopentane-1,3-diaminePublication Number: US-2025059193-A1Priority Date: 2021-11-24
- Polymorphic forms and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: CN-118678952-APriority Date: 2021-11-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////istisociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib
Istisociclib
KB 130742
CAS 2416873-83-9
MF C16H25N5, 287.40 g/mol
trans-(1S,3S)-3-N-(5-pentan-3-ylpyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diamine
- (1S,3S)-N1-[5-(1-Ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-1,3-cyclopentanediamine
- 1,3-Cyclopentanediamine, N1-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]-, (1S,3S)-
(1S,3S)-N1-[5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Istisociclib is a small molecule drug. The usage of the INN stem ‘-ciclib’ in the name indicates that Istisociclib is a cyclin dependant kinase inhibitor. Istisociclib is under investigation in clinical trial NCT04718675 (A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)). Istisociclib has a monoisotopic molecular weight of 287.21 Da.
Istisociclib is an orally bioavailable, selective inhibitor of the serine/threonine cyclin-dependent kinase 9 (CDK9), the catalytic subunit of the RNA polymerase II (RNA Pol II) elongation factor positive transcription elongation factor b (PTEF-b; PTEFb), with potential antineoplastic activity. Upon oral administration, istisociclib targets, binds to and blocks the phosphorylation and kinase activity of CDK9, thereby preventing PTEFb-mediated activation of RNA Pol II, leading to the inhibition of gene transcription of various anti-apoptotic proteins and oncogenic transcription factors including MYC and androgen receptor (AR). This induces cell cycle arrest and apoptosis and prevents tumor cell proliferation. CDK9 regulates elongation of transcription through phosphorylation of RNA Pol II at serine 2 (p-Ser2-RNAPII), and is an important cofactor for various oncogenic transcription factors. It is upregulated in various tumor cell types and plays a key role in the regulation of Pol II-mediated transcription of anti-apoptotic proteins. Tumor cells are dependent on anti-apoptotic proteins for their survival.
ISTISOCICLIB is a small molecule drug with a maximum clinical trial phase of II and has 1 investigational indication.
A Study of KB-0742 in Participants With Relapsed or Refractory Solid Tumors Including Platinum Resistant High Grade Serous Ovarian Cancer (HGSOC)
CTID: NCT04718675
Phase: Phase 1/Phase 2
Status: Terminated
Date: 2025-02-17
REF
- Discovery of KB-0742, a Potent, Selective, Orally Bioavailable Small Molecule Inhibitor of CDK9 for MYC-Dependent CancersPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-11-15PMCID: PMC10726352PMID: 37967851DOI: 10.1021/acs.jmedchem.3c01233
- CDK9 inhibitors in cancer researchPublication Name: RSC Medicinal ChemistryPublication Date: 2022-06-22PMCID: PMC9215160PMID: 35814933DOI: 10.1039/d2md00040g
- From Structure Modification to Drug Launch: A Systematic Review of the Ongoing Development of Cyclin-Dependent Kinase Inhibitors for Multiple Cancer TherapyPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-04-29PMID: 35485642DOI: 10.1021/acs.jmedchem.1c02064
- Lessons Learned from Past Cyclin-Dependent Kinase Drug Discovery EffortsPublication Name: Journal of Medicinal ChemistryPublication Date: 2022-03-02PMID: 35235745DOI: 10.1021/acs.jmedchem.1c02190
PAT
(lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine is a pharmaceutically active compound that has been studied for various uses, such as for the treatment of cancer. As used herein, the term “Compound A” is used to refer to both the free base and salt forms of (lS,3S)-N1-(5-(pentan-3-yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diamine. The free base of Compound A has the CAS number of 2416873-83-9 and structure of formula (I):
SYN
Example 35: (1S,3S)-N3-[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]cyclopentane-1,3-diamine (35)
2-Ethylbutanoic acid (7.5 g, 64.57 mmol) was dissolved in THF (150 mL) and cooled to 0 °C. Within 20 min CDI (16.23 g, 100.08 mmol) was added portion-wise. The reaction warmed to room temp (rt) and the mixture was stirred at rt overnight (Solution A). In another flask MgCl2 (6.14 g, 64.57 mmol) and potassium 3-ethoxy-3-oxo-propanoate (17 g, 100.1 mmol) were mixed with THF (150 mL) and stirred under argon overnight at 50 °C. The resultant white suspension was cooled to rt and solution A was added dropwise over 10 min and the reaction mixture (RM) was stirred for 16h at room temperature. After several minutes a sticky, amorphous solid appeared whereupon after several hours the reaction mixture became homogenous in appearance. The RM was concentrated to about a third, taken up in half sat. potassium bisulphate solution and extracted twice with ethyl acetate. The organic layers were subsequently washed with a sat. sodium bicarbonate solution, combined, dried over anhydrous sodium sulfate, filtered and evaporated. Purification by column chromatography gave ethyl 4-ethyl-3-oxo-hexanoate (4.3 g, 23.087 mmol, 35.8% yield) as a transparent liquid. The RM was monitored by TLC (10% EA in Hex, Product Rf=0.6, SM Rf=0.1).
Step 2
To a suspension of ethyl 4-ethyl-3-oxo-hexanoate (4.4 g, 23.62 mmol) in acetic acid (11 mL) at 70 °C was added 1H-pyrazol-5-amine (4.71 g, 56.7 mmol) in two portions (the second portion was added after 2 hours of stirring the first portion) over a 4 hour period. Upon consumption of SM as indicated by TLC, the reaction was cooled to rt and the solvent was evaporated in a rotary evaporator. The residue was treated with ethyl acetate and filtered to give 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 17.7 mmol, 74.9% yield) as an off-white solid. The reaction mixture was monitored by TLC (5% MeOH in DCM, Product Rf=0.3, SM Rf=0.8).
Step 3
A stirred solution of 5-(1-ethylpropyl)-4H-pyrazolo[1,5-a]pyrimidin-7-one (3.7 g, 18.03 mmol) in POCl3 (33.7 mL, 360.52 mmol) was heated to reflux for 4 hours. The reaction mixture was cooled to room temperature, excess reagent was evaporated in a rotary evaporator, and the residue was treated with ice-water. The chlorinated product was extracted from aqueous mixture by DCM. The organic layer was separated, dried over anhydrous Na2SO4, filtered and purified by column chromatography to give 7- chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (3.1 g, 13.9 mmol, 76.9% yield) as a light yellow liquid. The reaction mixture was monitored by TLC (20% EA in Hex, Product Rf=0.6, SM Rf=0.1).
To a stirred solution 7-chloro-5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidine (2.3 g, 10.28 mmol), tert-Butyl ((1S,3S)-3-aminocyclopentyl)carbamate (2.27 g, 11.31 mmol) and K2CO3 (4.26 g, 30.84 mmol) in MeCN (20 mL) were heated to reflux for 16 hours. The reaction mixture was filtered, concentrated under reduced pressure and purified by column chromatography, eluent 30% EA in hexane to give tert-butyl N-[(1S,3S)-3-[[5-(1-ethylpropyl)pyrazolo[1,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (4.5 g, 11.6 mmol, 112.8% yield) as an off-white solid. The reaction mixture was monitored by TLC (40% EA in Hex, Product Rf=0.5, SM Rf=0.7).
Step 5
To tert-butyl N-[(1 S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]amino]cyclopentyl]carbamate (1.0 g, 2.58 mmol) in l,4-Dioxane (0.2 mL), 4 M HC1 in Dioxane (3.22 mL, 12.9 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give [(lS,3S)-3-[[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium dichloride (0.9 g, 2.5 mmol, 96.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC ( 100% EA, Product Rf=0.l, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 15.00 (s, 1H), 9.93-9.86 (m, 1H), 8.51 (s, 3H), 8.30 (s, 1H), 6.84 (s, 1H), 6.58 (s, 1H), 4.95 (q, J = 7.8 Hz, 1H), 3.77- 3.66 (m, 1H), 2.84-2.71 (m, 1H), 2.29-2.05 (m, 4H), 1.94-1.63 (m, 6H), 0.81 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.0 [M+H+])
Step 6
To [(1S,3S)-3-[[5-(l-ethylpropyl)pyrazolo[I,5-a]pyrimidin-4-ium-7-yl]amino]cyclopentyl]ammonium-di chloride (0.2 g, 0.5600 mmol) in aq. NH3 (4.0 mL, 0.56 mmol) was added and stirred at room temperature for 4 hours. The reaction mixture was evaporated in vacuo, triturated with pentane and lyophilized from MeCN:H20 to give (lS,3S)-N3-[5-(l-ethylpropyl)pyrazolo[l,5-a]pyrimidin-7-yl]cyclopentane-l,3-diamine (140 mg, 0.49 mmol, 87.8% yield) as a pale-yellow sticky solid. The reaction mixture was monitored by TLC (100% EA, Product Rf=0.1, SM Rf=0.8). 1H NMR (400 MHz, DMSO-d6) d 7.95 (d, J = 2.2 Hz, 1H), 6.86 (s, 1H), 6.29 (d, J = 2.2 Hz, 1H), 5.95 (s, 1H), 4.31-4.19 (m, 1H), 3.57-3.44 (m, 1H), 2.52-2.44 (m, 1H), 2.36-2.22 (m, 1H),
2.09–1.79 (m, 3H), 1.80–1.59 (m, 5H), 1.58–1.24 (m, 3H), 0.83 (t, J = 7.4 Hz, 6H). LC-MS (m/z 287.21, found 288.5 [M+H+]).
PAT
- Compounds, compositions and methods for modulating CDK9 activityPublication Number: CN-112996790-BPriority Date: 2018-10-30Grant Date: 2023-11-03
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2024132506-A1Priority Date: 2018-10-30
- Compounds, Compositions, and Methods for Modulating CDK9 ActivityPublication Number: US-2020131189-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: US-2022002305-A1Priority Date: 2018-10-30
- Compounds, compositions, and methods for modulating cdk9 activityPublication Number: EP-3873911-A1Priority Date: 2018-10-30
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11845754-B2Priority Date: 2018-10-30Grant Date: 2023-12-19
- Substituted pyrazolo[1,5-a]pyrimidines for modulating CDK9 activityPublication Number: US-11155560-B2Priority Date: 2018-10-30Grant Date: 2021-10-26
- Compounds and methods for modulating cdk9 activityPublication Number: EP-4240422-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: WO-2022098843-A1Priority Date: 2020-11-05
- Compounds and methods for modulating cdk9 activityPublication Number: US-2025188084-A1Priority Date: 2020-11-05
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: WO-2021216828-A1Priority Date: 2020-04-24
- Chimeric degraders of cyclin-dependent kinase 9 and uses thereofPublication Number: US-2023158159-A1Priority Date: 2020-04-24
- Polymorphic and salt forms of (ls,3s)-n-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: EP-4436569-A1Priority Date: 2021-11-24
- Polymorphs and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-A]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: KR-20240110634-APriority Date: 2021-11-24
- Compositions and methods for enhanced protein productionPublication Number: EP-4412621-A2Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: US-2024252688-A1Priority Date: 2021-09-22
- Compositions and methods for enhanced protein productionPublication Number: WO-2023056202-A2Priority Date: 2021-09-22
- A cdk9 inhibitor for use in the treatment of cancer in a subject having an asxl1 mutationPublication Number: WO-2025217597-A1Priority Date: 2024-04-12
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3- yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: WO-2023096922-A8Priority Date: 2021-11-24
- Polymorphic and salt forms of (ls,3s)-n1-(5-(pentan-3- yl)pyrazolo[l,5-a]pyrimidin-7-yl)cyclopentane-l,3-diaminePublication Number: WO-2023096922-A1Priority Date: 2021-11-24
- Polymorphic and salt forms of (1s,3s)-n1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl) cyclopentane-1,3-diaminePublication Number: US-2025059193-A1Priority Date: 2021-11-24
- Polymorphic forms and salt forms of (1S,3S)-N1-(5-(pentan-3-yl)pyrazolo[1,5-a]pyrimidin-7-yl)cyclopentane-1,3-diaminePublication Number: CN-118678952-APriority Date: 2021-11-24
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
//////istisociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, KB-0742, 2416873-83-9, KB 0742, F7J6KSY5I8, UB-18422, KB-130742, KB 00130742
Atirmociclib



Atirmociclib
CAS 2380321-51-5
MF C22H27ClFN5O3,
463.9 g/mol
(3S,4R)-4-[[5-chloro-4-[7-fluoro-2-(2-hydroxypropan-2-yl)-3-propan-2-ylbenzimidazol-5-yl]pyrimidin-2-yl]amino]oxan-3-ol
(3S,4R)-4-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan2-yl)-1H-1,3-benzimidazol-6-yl]pyrimidin-2-yl}amino)oxan-3-ol
1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxpropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol
D-threo-Pentitol, 1,5-anhydro-3-[[5-chloro-4-[4-fluoro-2-(1-hydroxy-1-methylethyl)-1-(1-methylethyl)-1H-benzimidazol-6-yl]-2-pyrimidinyl]amino]-2,3-dideoxy-
cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6
Atirmociclib is an orally bioavailable inhibitor of cyclin-dependent kinase 4 (CDK4), with potential antineoplastic activity. Upon administration, atirmociclib selectively inhibits CDK4, which inhibits the phosphorylation of retinoblastoma protein (Rb) early in the G1 phase, prevents CDK-mediated G1-S-phase transition and leads to cell cycle arrest. This suppresses DNA replication and inhibits tumor cell proliferation. CDK4, a serine/threonine kinase, is upregulated in many tumor cell types and plays a key role in the regulation of both cell cycle progression from the G1-phase into the S-phase and tumor cell proliferation.
Atirmociclib (development code PF-07220060) is an investigational orally bioavailable and CDK4-specific inhibitor being developed by Pfizer for the treatment of various solid tumors, particularly hormone receptor-positive, HER2-negative breast cancer.[1][2] The safety and efficacy of atirmociclib have not been established, as it remains in clinical development as of September 2025.[3][4][5]
SYN
https://pubs.acs.org/doi/10.1021/acs.jmedchem.5c02137


PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0K3I-13424-1

Example A94 (Scheme A-15): Preparation of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol

Step 8: Synthesis of 1,5-anhydro-3-({5-chloro-4-[4-fluoro-2-(2-hydroxypropan-2-yl)-1-(propan-2-yl)-1H-benzimidazol-6-yl]pyrimidin-2-yl}amino)-2,3-dideoxy-D-threo-pentitol (Example A94)
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US275481329&_cid=P22-MK0KHW-23947-1

PAT
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-102661053-B1Priority Date: 2018-04-26Grant Date: 2024-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: KR-20230152182-APriority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-11220494-B2Priority Date: 2018-04-26Grant Date: 2022-01-11
- CYCLINE-DEPENDENT KINASE INHIBITORSPublication Number: PE-20201202-A1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-2022089580-A1Priority Date: 2018-04-26
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: HR-P20250254-T1Priority Date: 2018-04-26
- Cyclin dependent kinase inhibitorsPublication Number: US-12378232-B2Priority Date: 2018-04-26Grant Date: 2025-08-05
- 2-amino-pyridine or 2-amino-pyrimidine derivatives as cyclin dependent kinase inhibitorsPublication Number: EP-3784664-B1Priority Date: 2018-04-26Grant Date: 2025-02-19
- 2-Amino-pyridine or 2-amino-pyrimidine derivatives as cyclin-dependent kinase inhibitorsPublication Number: CN-112313219-BPriority Date: 2018-04-26Grant Date: 2024-04-26
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Mechanism of action
Atirmociclib is designed as a CDK4-specific inhibitor, distinguishing it from dual CDK4/6 inhibitors currently approved for cancer treatment.[6] The drug targets cyclin-dependent kinase 4, which plays a role in cell cycle regulation.[1][7][8]
Atirmociclib functions as a selective inhibitor of the CDK4/cyclin D complex, which plays a crucial role in cell cycle regulation.[4] The drug works by targeting the CDK4 kinase, rendering the retinoblastoma (Rb)/E2F transcription system inactive, which ultimately leads to cell cycle arrest in the G1 phase.[4] This mechanism is particularly effective in tumors that have lost Rb cell cycle-suppressive function, a common feature in various solid tumors.[5]
The selective nature of atirmociclib represents a significant advancement over existing dual CDK4/6 inhibitors.[6] By specifically targeting CDK4 while limiting CDK6 inhibition, atirmociclib is designed to maintain antitumor efficacy while potentially reducing dose-limiting hematologic toxicities, particularly neutropenia, which is believed to be primarily driven by CDK6 inhibition.[9]
Clinical development
Atirmociclib is currently being evaluated in clinical trials for the treatment of advanced solid tumors.[1] Clinical studies are ongoing with estimated completion dates extending to 2027–2028, reflecting the early stage of development for this investigational compound.[1]
Preclinical research published in Cancer Cell in March 2025 reported atirmociclib as a next-generation CDK4-selective inhibitor with enhanced anti-tumor activity and reduced predicted toxicity compared to FDA-approved dual CDK4/6 inhibitors, though these findings require validation in clinical studies.[6]
Preclinical studies
Preclinical research has demonstrated that atirmociclib exhibits enhanced anti-tumor activity compared to FDA-approved dual CDK4/6 inhibitors while showing reduced predicted toxicity.[6] Studies have shown that CDK4-selective inhibition can provide improved preclinical anti-tumor efficacy and safety profiles compared to dual CDK4/6 inhibition strategies.[10]
The preclinical development program has explored combination approaches with various therapeutic modalities, including endocrine therapy, CDK2 inhibition, HER2 antibodies, and immune checkpoint inhibitors.[6] These combination strategies are designed to counter resistance mechanisms to CDK4 inhibition and expand the potential therapeutic applications of cell cycle targeting therapy.[6]
Clinical trials
Atirmociclib has entered clinical development as part of Pfizer’s extensive oncology pipeline.[11] The clinical program is evaluating atirmociclib both as a single agent and in combination with other therapeutic approaches, particularly focusing on patients with hormone receptor-positive, HER2-negative breast cancer.[9][12][13][14][15][16][17]
Early clinical studies have included heavily pretreated patient populations, including those who have previously received CDK4/6 inhibitor therapy.[9] This approach allows for the evaluation of atirmociclib’s potential to overcome resistance to existing CDK4/6 inhibitors and provide therapeutic benefit in patients with limited treatment options.[9]
Safety profile and toxicity
One of the key differentiating features of atirmociclib is its potential for improved safety profile compared to existing dual CDK4/6 inhibitors.[6] The selective targeting of CDK4 while limiting CDK6 inhibition is specifically designed to reduce neutropenia, the most common dose-limiting toxicity associated with current CDK4/6 inhibitors.[18]
The rationale for this approach is based on preclinical evidence suggesting that neutropenia is primarily driven by CDK6 inhibition rather than CDK4 inhibition.[18] By selectively targeting CDK4, atirmociclib aims to maintain therapeutic efficacy while potentially allowing for higher or more sustained dosing without the dose-limiting hematologic toxicities that can compromise treatment outcomes with existing agents.[18]
Regulatory status
As of September 2025, atirmociclib remains an investigational drug that has not received approval from the FDA or other regulatory agencies.[5] The compound is part of Pfizer’s oncology development pipeline.[5]
References
- Pfizer (2 February 2025). A Phase 1/2A Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Anti-Tumor Activity of Pf-07220060 as a Single Agent and as Part of Combination Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Shapiro GI (March 2017). “The evolving role of cyclin-dependent kinase inhibitors in cancer management”. Clinical Advances in Hematology & Oncology. 15 (3): 174–177. PMID 28398270.
- “CDK4 inhibitor PF-07220060”. http://www.cancer.gov. 2 February 2011. Retrieved 3 September 2025.
- “Pfizer Pipeline”. Pfizer.
- “Atirmociclib PF-07220060”. Pfizer Oncology Development. Retrieved 3 September 2025.
- Chang J, Lu J, Liu Q, Xiang T, Zhang S, Yi Y, et al. (March 2025). “Single-cell multi-stage spatial evolutional map of esophageal carcinogenesis”. Cancer Cell. 43 (3): 380–397.e7. doi:10.1016/j.ccell.2025.02.009. PMID 40068596.
- Topacio BR, Zatulovskiy E, Cristea S, Xie S, Tambo CS, Rubin SM, et al. (May 2019). “Cyclin D-Cdk4,6 Drives Cell-Cycle Progression via the Retinoblastoma Protein’s C-Terminal Helix”. Molecular Cell. 74 (4): 758–770.e4. doi:10.1016/j.molcel.2019.03.020. PMC 6800134. PMID 30982746.
- Helsten T, Kato S, Schwaederle M, Tomson BN, Buys TP, Elkin SK, et al. (July 2016). “Cell-Cycle Gene Alterations in 4,864 Tumors Analyzed by Next-Generation Sequencing: Implications for Targeted Therapeutics”. Molecular Cancer Therapeutics. 15 (7): 1682–1690. doi:10.1158/1535-7163.MCT-16-0071. PMID 27196769.
- “ESMO 2024 – combos could be the way forward for CDK2”. ApexOnco. 15 September 2024.
- Palmer CL, Boras B, Pascual B, Li N, Li D, Garza S, et al. (March 2025). “CDK4 selective inhibition improves preclinical anti-tumor efficacy and safety”. Cancer Cell. 43 (3): 464–481.e14. doi:10.1016/j.ccell.2025.02.006. PMID 40068598.
- “Pfizer Highlights Diverse Oncology Portfolio and Combination Approaches at ESMO 2024”. Pfizer. 2024.
- Pfizer (12 August 2025). A Phase 1/2a Dose Escalation and Expansion Study to Evaluate Safety, Tolerability, Pharmacokinetic, Pharmacodynamic, and Anti-Tumor Activity of Pf-07248144 in Participants With Advanced or Metastatic Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (2 July 2025). An Interventional Safety and Efficacy Phase 1/2, Open-Label Study to Investigate Tolerability, Pk, and Antitumor Activity of Vepdegestrant (Arv-47/Pf-07850327), an Oral Proteolysis Targeting Chimera, in Combination With Pf-07220060 in Participants Aged 18 Years and Older With Er+/her2- Advanced or Metastatic Breast Cancer (Report). clinicaltrials.gov.
- Pfizer (14 November 2024). A Phase 1/2, Open-Label, Multicenter, Dose Escalation and Dose Expansion Study to Evaluate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Antitumor Activity of PF-07220060 in Combination With Pf-07104091 Plus Endocrine Therapy in Participants With Advanced Solid Tumors (Report). clinicaltrials.gov.
- Pfizer (17 June 2025). (FOURLIGHT-3) (Report). clinicaltrials.gov.
- Pfizer (13 March 2025). An Interventional, Open-Label, Randomized, Multicenter Phase 3 Study of PF-07220060 Plus Letrozole Compared to cdk4/6 Inhibitor Plus Letrozole in Participants Over 18 Years of Age With Hormone Receptor (Hr)-Positive, her2-Negative Advanced/Metastatic Breast Cancer Who Have Not Received Any Prior Systemic Anticancer Treatment for Advanced/Metastatic Disease (FOURLIGHT-1) (Report). clinicaltrials.gov.
- Pfizer (15 November 2024). An Interventional, Open-Label, Randomized, Multicenter, Phase 2 Study of Pf-07220060 Plus Letrozole Compared to Letrozole Alone in Postmenopausal Women 18 Years or Older With Hormone Receptor-Positive, her2-Negative Breast Cancer in the Neoadjuvant Setting (Report). clinicaltrials.gov.
- “Pfizer dials down its atirmociclib ambitions”. ApexOnco. 1 May 2025.
| Identifiers | |
|---|---|
| IUPAC name | |
| CAS Number | 2380321-51-5 |
| PubChem CID | 146219790 |
| ChemSpider | 115009592 |
| UNII | S743GOJ5LJ |
| KEGG | D12834 |
| ChEMBL | ChEMBL5187755 |
| Chemical and physical data | |
| Formula | C22H27ClFN5O3 |
| Molar mass | 463.94 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Atirmociclib, cyclin-dependent kinase (CDK) inhibitor, antineoplastic, PF 07220060, S743GOJ5LJ, CDK4/6-IN-6

{kind=link}