PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Arimoclomol maleate is in a phase III clinical trials by Orphazyme for the treatment of Niemann-Pick disease type C (NP-C). It is also in phase II clinical studies for the treatment of amyotrophic lateral sclerosis (ALS).
Arimoclomol (INN; originally codenamed BRX-345, which is a citrate salt formulation of BRX-220) is an experimental drug developed by CytRx Corporation, a biopharmaceutical company based in Los Angeles, California. In 2011 the worldwide rights to arimoclomol were bought by Danish biotech company Orphazyme ApS.[1] The European Medicines Agency (EMA) and U.S. Food & Drug Administration (FDA) granted orphan drug designation to arimoclomol as a potential treatment for Niemann-Pick type C in 2014 and 2015 respectively.[2][3]
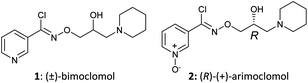
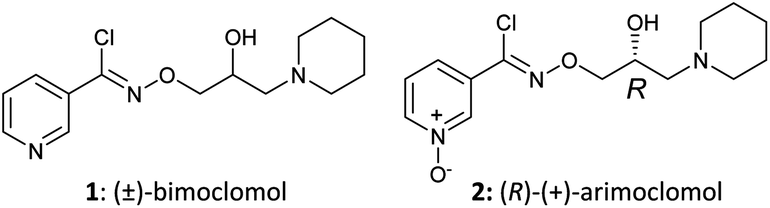
Fig. 1 Structures of (±)-bimoclomol (1) and (R)-(+)-arimoclomol (2).
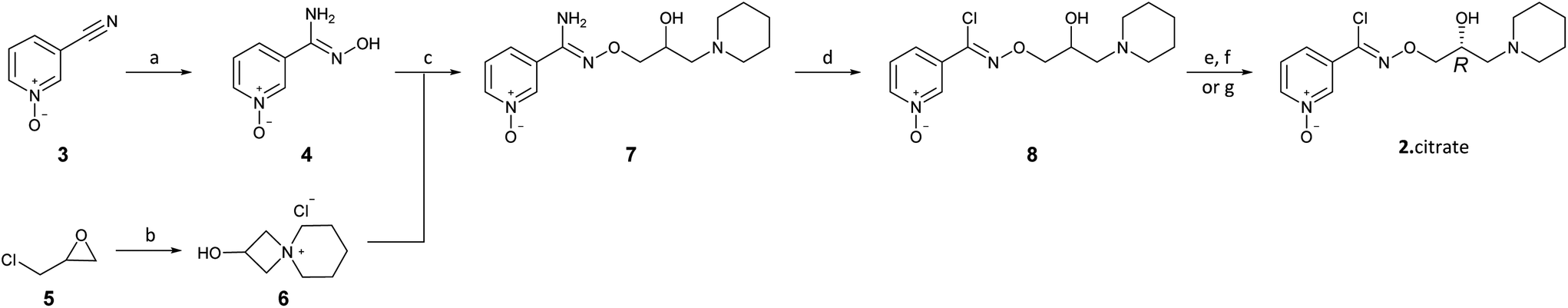
The present disclosure provides an optimized four-step process for preparing an ultra-pure composition comprising arimoclomol citrate, i.e. N-{[(2R)-2-hydroxy-3-piperidin-l-ylpropyl]oxy}pyridine-3-carboximidoyl chloride 1-oxide citrate. The optimized process comprises a plurality of optimized sub-steps, each contributing to an overall improved process, providing the ultra-pure composition comprising arimoclomol citrate. The ultra-pure composition comprising arimoclomol citrate meets the medicines agencies’ high regulatory requirements. An overview of the four-steps process is outlined below:
Step 1: Overview of process for preparing ORZY-01
Step 2: Overview of process for preparing ORZY-03
Step 4: Overview of process for preparing BRX-345 (ORZY-05)
The previously reported two-step synthesis of ORZY-01 as shown below includes a 2 hour reflux in step 1A, followed by purification of intermediate compound (V) to increase the batch quality.
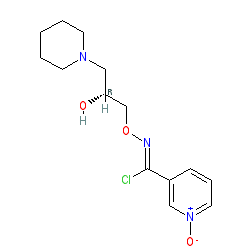
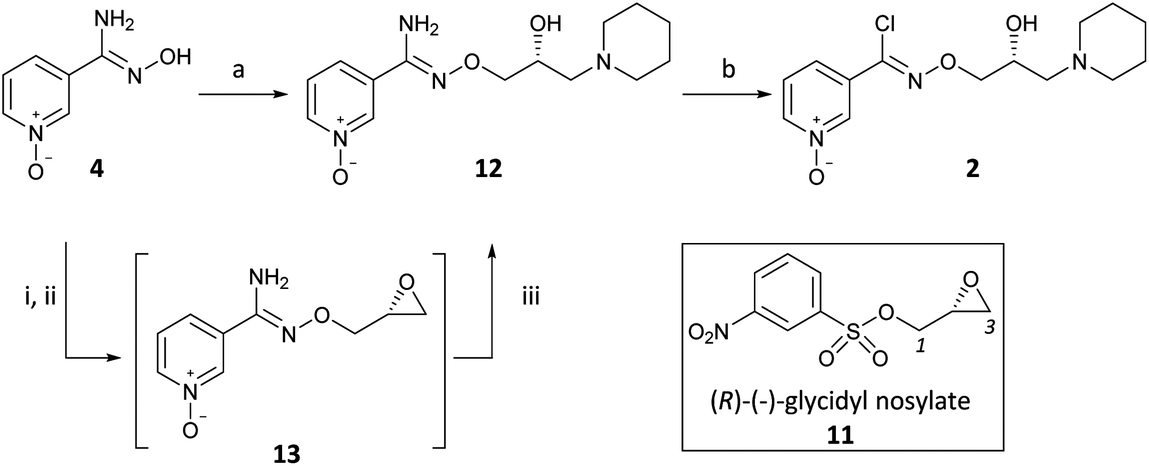
(R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide1 – (R)-(+)-Arimoclomol – 2 A solution of (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carbamimidoyl)pyridine-1-oxide 12 (205 mg, 0.70 mmol) in conc. hydrochloric acid (1.1 mL, 13.9 mmol) and water (3 mL) was cooled to -5 °C for 15 minutes. Sodium nitrite (63 mg, 0.91 mmol) in water (0.5 mL) was then added dropwise to the reaction mixture and the reaction was stirred at -5 °C for 2.5 hours. The reaction mixture was made alkaline with NaOH (7 M, 3 mL). An additional 10 mL of water was added followed by DCM (30 mL) containing EtOAc (5 mL) and the organics were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on Biotage Isolera using Biotage SNAP 10 g Si cartridge eluting with gradient elution 0-30% MeOH:DCM both containing 0.1% Et3N to afford the title compound (160 mg, 73% yield) as a colourless semi-solid. Analytical data was consistent with literature values. See ESI section SFC traces for specific enantiomeric ratios of 2 synthesised under the various methodologies quoted in the text. Optical rotation was not determined as it was determined in the ultimate product of this 2·citrate and comparative run times on SFC. 1H NMR (600 MHz, CDCl3) δ: 8.63 (t, J = 1.4 Hz, 1H), 8.16 (ddd, J = 6.4, 1.6, 0.9 Hz, 1H), 7.66 – 7.62 (m, 1H), 7.25 (dd, J = 8.0, 6.6 Hz, 1H), 4.26 (qd, J = 11.3, 5.2 Hz, 2H), 4.07 (dd, J = 9.2, 4.7 Hz, 1H), 2.62 (s, 2H), 2.47 – 2.31 (m, 4H), 1.65 – 1.51 (m, 4H), 1.42 (s, 2H); 13C NMR (151 MHz, CDCl3) δ: 140.3, 137.7, 133.1, 132.5, 125.7, 123.9, 78.7, 64.9, 60.9, 54.8, 25.8, 24.0.
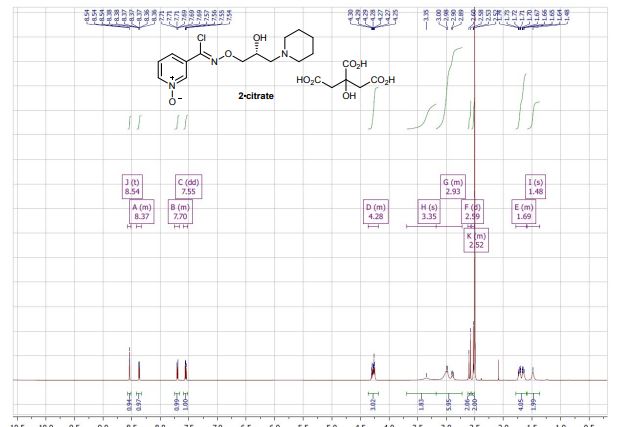
(R)-(+)- Arimoclomol citrate – 2·citrate (R,Z)-3-(N’-(2-hydroxy-3-(piperidin-1-yl)propoxy)carboximidoyl chloride)pyridine-1-oxide (159 mg, 0.51 mmol) was dissolved in acetone (3 mL) and citric acid (97 mg, 0.51 mmol) was added. The reaction mixture was left to stir at room temperature for 18 hours. After this time the mixture was sonicated and the precipitate was filtered, rinsed with cold acetone (1 mL) and dried under vacuum to afford the title compound (165 mg, 64% yield) as a white amorphous solid. Analytical data was consistent with literature values. m.p. 161-162 °C, Acetone (lit. 163-165 °C, EtOH); [α]D 20 +8.0 (c=1, H2O); IR νmax (neat): 3423, 3228, 2949, 2868, 1722, 1589, 1483, 1433, 1307, 1128, 972, 829 cm-1; 1H NMR (600 MHz, d6-DMSO) δ: 8.54 (t, J = 1.5 Hz, 1H), 8.39 – 8.35 (m, 1H), 7.72 – 7.68 (m, 1H), 7.55 (dd, J = 8.0, 6.5 Hz, 1H), 4.28 (ddd, J = 17.6, 13.3, 7.4 Hz, 3H), 3.35 (br. s, 2H), 3.13 – 2.74 (m, 6H), 2.59 (d, J = 15.2 Hz, 2H), 2.56 – 2.51 (m, 2H), 1.77 – 1.61 (m, 4H), 1.48 (s, 2H); 13C NMR (151 MHz, d6-DMSO) δ: 176.6, 171.3, 140.5, 136.4, 132.7, 131.5, 126.8, 123.3, 77.8, 71.4, 63.8, 58.7, 53.1, 44.0, 30.7, 23.0, 21.9; HRMS (m/z TOF MS ES+) for C14H20ClN3O3 [M+H]+ calc. 314.1271, observed 314.1263; SFC er purity R:S >99:1
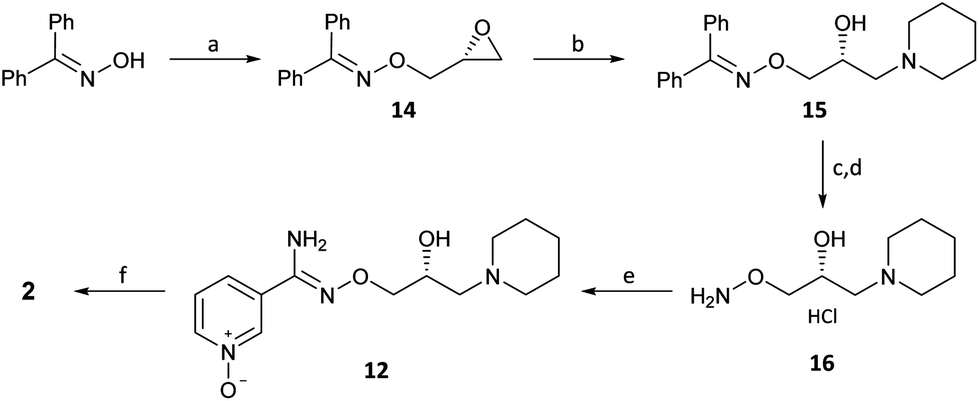
Procedure for the conversion of (R)-(+)-Bimoclomol 1 into (R)-(+)-Arimoclomol 2 To a solution of (R)-(+)-bimoclomol (61 mg, 0.21 mmol) in acetone (2 mL) was added benzenesulfonic acid (33 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 1.5 hours. The reaction mixture was concentrated in vacuo. Separately to a suspension of hydrogen peroxide-urea adduct (39 mg, 0.41 mmol) in acetonitrile (6 mL) at -5°C (ice-salt bath) was added trifluoroacetic anhydride (58 μL, 0.41 mmol) dropwise. A suspension of (R)-(+)-bimoclomol, 1, benzenesulfonic acid salt, as made above, in acetonitrile (3 mL) was then added dropwise to this solution. The reaction mixture was stirred for 18 hours, whilst slowly warming to room temperature. Aqueous Na2S2O5 solution (0.5 M, 1 mL) was added and the reaction mixture stirred for 1 hour. The reaction mixture was made alkaline with NaOH (7 M) and extracted with DCM (2 x 30 mL). The combined organics were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by FCC on a Biotage Isolera using Biotage SNAP 10g Si cartridge eluting with gradient elution 0-35% MeOH in DCM to afford the title compound (35 mg, 55% yield) as a colourless semi-solid. Analytical data of the products was consistent with literature and/or previous samples synthesised above.
Arimoclomol is believed to function by stimulating a normal cellular protein repair pathway through the activation of molecular chaperones. Since damaged proteins, called aggregates, are thought to play a role in many diseases, CytRx believes that arimoclomol could treat a broad range of diseases.
Arimoclomol has been shown to extend life in an animal model of ALS[11] and was well tolerated in healthy human volunteers in a Phase I study. CytRx is currently conducting a Phase II clinical trial.[12]
Arimoclomol also has been shown to be an effective treatment in an animal model of Spinal Bulbar Muscular Atrophy (SBMA, also known as Kennedy’s Disease).[13]
Arimoclomol was discovered by Hungarian researchers, as a drug candidate to treat insulin resistance[14][15] and diabetic complications such as retinopathy, neuropathy and nephropathy. Later, the compound, along with other small molecules, was screened for further development by Hungarian firm Biorex, which was sold to CytRx Corporation, who developed it toward a different direction from 2003.
^ Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (April 2004). “Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice”. Nat. Med. 10 (4): 402–5. doi:10.1038/nm1021. PMID15034571. S2CID2311751.
^ Kalmar B, Greensmith L, Malcangio M, McMahon SB, Csermely P, Burnstock G (December 2003). “The effect of treatment with BRX-220, a co-inducer of heat shock proteins, on sensory fibers of the rat following peripheral nerve injury”. Exp. Neurol. 184 (2): 636–47. doi:10.1016/S0014-4886(03)00343-1. PMID14769355. S2CID5316222.
^ Rakonczay Z, Iványi B, Varga I, et al. (June 2002). “Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats”. Free Radic. Biol. Med. 32 (12): 1283–92. doi:10.1016/S0891-5849(02)00833-X. PMID12057766.
^ Kalmar B, Burnstock G, Vrbová G, Urbanics R, Csermely P, Greensmith L (July 2002). “Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats”. Exp. Neurol. 176 (1): 87–97. doi:10.1006/exnr.2002.7945. PMID12093085. S2CID16071543.
Cancer; Febrile neutropenia; Non-small-cell lung cancer
Plinabulin (chemical structure, BPI-2358, formerly NPI-2358) is a small molecule under development by BeyondSpring Pharmaceuticals, and is in a world-wide Phase 3 clinical trial for non-small cell lung cancer. [1] Plinabulin blocks the polymerization of tubulin in a unique manner, resulting in multi-factorial effects including an enhanced immune-oncology response, [2] activation of the JNK pathway [3] and disruption of the tumor blood supply. Plinabulin is being investigated for the reduction of chemotherapy-induced neutropenia [4] and for anti-cancer effects in combination with immune checkpoint inhibitors [5][6] and in KRAS mutated tumors. [7]
Plinabulin is a synthetic analog of diketopiperazine phenylahistin (halimide) discovered from marine and terrestrial Aspergillus sp. Plinabulin is structurally different from colchicine and its combretastatin-like analogs (eg, fosbretabulin) and binds at or near the colchicine binding site on tubulin monomers. Previous studies showed that plinabulin induced vascular endothelial cell tubulin depolymerization and monolayer permeability at low concentrations compared with colchicine and that it induced apoptosis in Jurkat leukemia cells. Studies of plinabulin as a single agent in patients with advanced malignancies (lung, prostate, and colon cancers) showed a favorable pharmacokinetic, pharmacodynamics, and safety profile.
Beyondspring, under license from Nereus (now Triphase, which licensed the program from the Scripps Institute of Oceanography of the University of California San Diego), is developing plinabulin, the lead in the NPI-2350 halimide series of marine Aspergillus-derived, vascular-targeting antimicrotubule agents, for treating cancer, primarily non-small cell lung cancer.
It is thought that a single, universal cellular mechanism controls the regulation of the eukaryotic cell cycle process. See, e.g., Hartwpll, L.H. et al., Science (1989), 246: 629-34. It is also known that when an abnormality arises in the control mechanism of the cell cycle, cancer or an immune disorder may occur. Accordingly, as is also known, antitumor agents and immune suppressors may be among the substances that regulate the cell cycle. Thus, new methods for producing eukaryotic cell cycle inhibitors are needed as antitumor and immune-enhancing compounds, and should be useful in the treatment of human cancer as chemotherapeutic, anti-tumor agents. See, e.g., Roberge, M. et al., Cancer Res. (1994), 54, 6115-21.
Fungi, especially pathogenic fungi and related infections, represent an increasing clinical challenge. Existing antifungal agents are of limited efficacy and toxicity, and the development and/or discovery of strains of pathogenic fungi that are resistant to drags currently available or under development. By way of example, fungi that are pathogenic in humans include among others Candida spp. including C. albicans, C. tropicalis, C. keƒyr, C. krusei and C. galbrata; Aspergillus spp. including A. fumigatus and A. flavus; Cryptococcus neoƒormans; Blastomyces spp. including Blastomyces dermatitidis; Pneumocystis carinii; Coccidioides immitis; Basidiobolus ranarum; Conidiobolus spp.; Histoplasma capsulatum; Rhizopus spp. including R. oryzae and R. microsporus; Cunninghamella spp.; Rhizomucor spp.; Paracoccidioides brasiliensis; Pseudallescheria boydii; Rhinosporidium seeberi; and Sporothrix schenckii (Kwon-Chung, K.J. & Bennett, J.E. 1992 Medical Mycology, Lea and Febiger, Malvern, PA).
Recently, it has been reported that tryprostatins A and B (which are diketopiperazines consisting of proline and isoprenylated tryptophan residues), and five other structurally-related diketopiperazines, inhibited cell cycle progression in the M phase, see Cui, C. et al., 1996 J Antibiotics 49:527-33; Cui, C. et al. 1996 J Antibiotics 49:534-40, and that these compounds also affect the microtubule assembly, see Usui, T. et al. 1998 Biochem J 333:543-48; Kondon, M. et al. 1998 J Antibiotics 51:801-04. Furthermore, natural and synthetic compounds have been reported to inhibit mitosis, thus inhibit the eukaryotic cell cycle, by binding to the colchicine binding-site (CLC-site) on tubulin, which is a macromolecule that consists of two 50 kDa subunits (α- and β-tubulin) and is the major constituent of microtubules. See, e.g., Iwasaki, S., 1993 Med Res Rev 13:183-198; Hamel, E. 1996 Med Res Rev 16:207-31; Weisenberg, R.C. et al., 1969 Biochemistry 7:4466-79. Microtubules are thought to be involved in several essential cell functions, such as axonal transport, cell motility and determination of cell morphology. Therefore, inhibitors of microtubule function may have broad biological activity, and be applicable to medicinal and agrochemical purposes. It is also possible that colchicine (CLC)-site ligands such as CLC, steganacin, see Kupchan, S.M. et al., 1973 J Am Chem Soc 95:1335-36, podophyllotoxin, see Sackett, D.L., 1993 Pharmacol Ther 59:163-228, and combretastatins, see Pettit, G.R. et al., 1995 J Med Chem 38:166-67, may prove to be valuable as eukaryotic cell cycle inhibitors and, thus, may be useful as chemotherapeutic agents.
Although diketopiperazine-type metabolites have been isolated from various fungi as mycotoxins, see Horak R.M. et al., 1981 JCS Chem Comm 1265-67; Ali M. et al., 1898 Toxicology Letters 48:235-41, or as secondary metabolites, see Smedsgaard J. et al., 1996 J Microbiol Meth 25:5-17, little is known about the specific structure of the diketopiperazine-type metabolites or their derivatives and their antitumor activity, particularly in vivo. Not only have these compounds been isolated as mycotoxins, the chemical synthesis of one type of diketopiperazine-type metabolite, phenylahistin, has been described by Hayashi et al. in J. Org. Chem. (2000) 65, page 8402. In the art, one such diketopiperazine-type metabolite derivative, dehydrophenylahistin, has been prepared by enzymatic dehydrogenation of its parent phenylahistin. With the incidences of cancer on the rise, there exists a particular need for chemically producing a class of substantially purified diketopiperazine-type metabolite-derivatives having animal cell-specific proliferation-inhibiting activity and high antitumor activity and selectivity. There is therefore a particular need for an efficient method of synthetically producing substantially purified, and structurally and biologically characterized, diketopiperazine-type metabolite-derivatives.
Also, PCT Publication WO/0153290 (July 26, 2001) describes a non-synthetic method of producing dehydrophenylahistin by exposing phenylahistin or a particular phenylahistin analog to a dehydrogenase obtained from Streptomyces albulus.
The imidazolecarboxaldehyde may be prepared, for example, according the procedure disclosed in Hayashi et al., 2000 J Organic Chem 65: 8402 as depicted below:
EXAMPLE 2
Synthesis and Physical Characterization of tBu-dehydrophenylahistin Derivatives
[0207] Structural derivatives of dehydrophenylahistin were synthesized according to the following reaction schemes to produce tBu-dehydrophenylahistin. Synthesis by Route
A (see Figure 1) is similar in certain respects to the synthesis of the dehydrophenylahistin synthesized as in Example 1.
Route A:
[0208] N,N’-diacethyl-2,5-piperazinedione 1 was prepared as in Example 1.
. [0209] To a solution of 5-tert-butylimidazole-4-carboxaldehyde 15 (3.02 g, 19.8. mmol) in DMF (30 mL) was added compound 1 (5.89 g, 29.72 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (9.7 g, 29.72 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was stirred for 5 h at room temperature. After the solvent was removed by evaporation, the residue was dissolved in the mixture of EtOAc and 10% Na2CO3, and the organic phase was washed with 10% Na2CO3 again and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The residual oil was purified by column chromatography on silica using CHCl3-MeOH (100:0 to 50:1) as an eluant to give 1.90 g (33 %) of a pale yellow solid 16. 1H NMR (270 MHz, CDCl3) δ 12.14 (d, br-s, 1H), 9.22 (br-s, 1H), 7.57 (s, 1H), 7.18, (s, 1H), 4.47 (s, 2H), 2.65 (s, 3H), 1.47 (s, 9H).
2) t-Bu-dehydrophenylahistin
[0210] To a solution of 1-Acetyl-3-{(Z)-1-[5-tert-butyl-1H-4-imidazolyl]methylidene}]-2,5-piperazinedione (16) (11 mg, 0.038 mmol) in DMF (1.0 mL) was added benzaldehyde (19 μL, 0.19 mmol, 5 eq) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (43 mg, 0.132 mmol, 3.5 eq) and the evacuation-flushing process was repeated again. The resultant mixture was heated for 2.5 h at 80°C. After the solvent was removed by
evaporation, the residue was dissolved in EtOAc, washed with water for two times and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The resulting residue was dissolved in 90% MeOH aq and applied to reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 × 250 mm) and eluted using a linear gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min, and the desired fraction was collected and concentrated by evaporation to give a 6.4 mg (50%) of yellow colored tert-butyl-dehydrophenylahistin. 1H NMR (270 MHz, CDCl3) δ 12.34 br-s, 1H), 9.18 (br-s, 1H), 8.09 (s, 1H), 7.59 (s, 1H), 7.31 – 7.49 (m, 5H), 7.01 s, 2H), 1.46 (s, 9H).
[0211] The dehydrophenylahistin reaction to produce tBu-dehydrophenylahistin is identical to Example 1.
[0212] The total yield of the tBu-dehydrophenylahistin recovered was 16.5%. Route B:
[0213] N,N’-diacethyl-2,5-piperazinedione 1 was prepared as in Example 1.
[0214] To a solution of benzaldehyde 4 (0.54 g, 5.05. mmol) in DMF (5 mL) was added compound 1 (2.0 g, 10.1 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (1.65 g, 5.05 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was stirred for 3.5 h at room temperature. After the solvent was removed by evaporation, the residue was dissolved in the mixture of EtOAc and 10% Na2CO3, and the organic phase was washed with 10% Na2CO3 again and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The residual solid was recrystalized from MeOH-ether to obtain a off-white solid of 17; yield 1.95 g (79%).
2) t-Bu-dehydrophenylahistin
[0215] To a solution of 1-Acetyl-3-[(Z)-benzylidenel]-2,5-piperazinedione (17) (48 mg, 0.197 mmol) in DMF (1.0 mL) was added 5-tert-butylimidazole-4-carboxaldehyde 15 (30 mg, 0.197 mmol) and the solution was repeatedly evacuated in a short time to remove oxygen and flushed with Ar, followed by the addition of Cs2CO3 (96 mg, 0.296 mmol) and the evacuation-flushing process was repeated again. The resultant mixture was heated for 14 h at 80°C. After the solvent was removed by evaporation, the residue was dissolved in EtOAc, washed with water for two times and saturated NaCl for three times, dried over Na2SO4 and concentrated in vacuo. The resulting residue was dissolved in 90% MeOH aq and applied to reverse-phase HPLC column (YMC-Pack, ODS-AM, 20 x 250 mm) and eluted using a linear gradient from 70 to 74% MeOH in water over 16 min at a flow rate of 12 mL/min, and the desired fraction was collected and concentrated by evaporation to give a 0.8 mg (1.2%) of yellow colored tert-butyl-dehydrophenylahistin.
[0216] The total yield of the tBu-dehydrophenylahistin recovered was 0.9%.
[0217] The HPLC profile of the crude synthetic tBu-dehyrophenylahistin from Route A and from Route B is depicted in Figure 4.
[0218] Two other tBu-dehydrophenylahistin derivatives were synthesized according to the method of Route A. In the synthesis of the additional tBu-dehydrophenylahistin derivatives, modifications to the benzaldehyde compound 4 were made.
[0219] Figure 4 illustrates the similarities of the HPLC profiles (Column: YMC-Pack ODS-AM (20 × 250mm); Gradient: 65% to 75% in a methanol-water system for 20 min, then 10 min in a 100% methanol system; Flow rate: 12mL/min; O.D. 230 nm) from the synthesized dehydrophenylahistin of Example 1 (Fig 2) and the above exemplified tBu-dehydrophenylahistin compound produced by Route A.
[0220] The sequence of introduction of the aldehydes is a relevant to the yield and is therefore aspect of the synthesis. An analogue of dehydrophenylahistin was synthesized, as a confrol or model, wherein the dimethylallyl group was changed to the tert-butyl group with a similar steric hindrance at the 5-position of the imidazole ring.
[0221] The synthesis of this “tert-butyl (tBu)-dehydrophenylahistin” using “Route A” was as shown above: Particularly, the sequence of infroduction of the aldehyde exactly follows the dehydrophenylahistin synthesis, and exhibited a total yield of 16.5% tBu-dehydrophenylahistin. This yield was similar to that of dehydrophenylahistin (20%). Using “Route B”, where the sequence of introduction of the aldehydes is opposite that of Route “A” for the dehydrophenylahistin synthesis, only a trace amount of the desired tBu-dehydroPLH was obtained with a total yield of 0.9%, although in the introduction of first benzaldehyde 4 gave a 76% yield of the intermediate compound 17. This result indicated that it may be difficult to introduce the highly bulky imidazole-4-carboxaldehydes 15 with a substituting group having a quaternary-carbon on the adjacent 5-position at the imidazole ring into the intermediate compound 17, suggesting that the sequence for introduction of aldehydes is an important aspect for obtaining a high yield of dehydrophenylahistin or an analog of dehydrophenylahistin employing the synthesis disclosed herein:
[0222] From the HPLC analysis of the final crude products, as shown in Figure 4, a very high content of tBu-dehydrophenylahistin and small amount of by-product formations were observed in the crude sample of Route A (left). However, a relatively smaller amount of the desired tBu-dehydrophenylahistin and several other by-products were observed in the sample obtained using Route B (right).
[0280] Sulfuryl chloride (14.0 ml, 0.17 mol) was added to a cooled (0°) solution of ethyl pivaloylacetate (27.17 g, 0.16 mol) in chloroform (100 ml). The resulting mixture was allowed to warm to room temperature and was stirred for 30 min, after which it was heated under reflux for 2.5 h. After cooling to room temperature, the reaction mixture was diluted with chloroform, then washed with sodium bicarbonate, water then brine.
[0281] The organic phase was dried and evaporated to afford, as a clear oil, 2-chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester (33.1 g, 102%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB1341375 (Great Britain, 1973)).
[0285] A solution of 2-chloro-4,4-dimethyl-3-oxo-pentanoic acid ethyl ester (25.0 g, 0.12 mol) in formamide (47.5 ml) and water (2.5 ml) was shaken, then dispensed into 15 x 8 ml vials. All vials were sealed and then heated at 150° for 3.5 h. The vials were allowed to cool to room temperature, then water (20 ml) was added and the mixture was exhaustively extracted with chloroform. The chloroform was removed to give a concentrated formamide solution (22.2 g) which was added to a flash silica column (6 cm diameter, 12 cm height) packed in 1% MeOH/1% Et3N in chloroform. Elution of the column with 2.5 L of this mixture followed by 1 L of 2% MeOH/1% Et3N in chloroform gave, in the early fractions, a product suspected of being 5-tert-butyl-oxazole-4-carboxylic acid ethyl ester (6.3 g, 26%).
[0291] Recovered from later fractions was 5-tert-butyl-3H-imidazole-4-carboxylic acid ethyl ester (6.20 g, 26%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB 1341375 (Great Britain, 1973)).
[0296] Further elution of the column with 1L of 5% MeOh/1% Et3N gave a compound suspected of being 5-tert-butyl-3H-imidazole-4-carboxylic acid (0.50 g, 2%).
[0302] A solution of 5-tert-butyl-3-imidazole-4-carboxylic acid ethyl ester (3.30 g, 16.8 mmol) in THF (60 ml) was added dropwise to a suspension of lithium aluminium hydride (95% suspension, 0.89 g, 22.2 mmol) in THF (40 ml) and the mixture was stirred at room temperature for 3 h. Water was added until the evolution of gas ceased, the mixture was stirred for 10 min, then was filtered through a sintered funnel. The precipitate was washed with THF, then with methanol, the filtrate and washings were combined and evaporated. The residue was freeze-dried overnight to afford, as a white solid (5-tert-butyl- 3H-imidazol-4-yl)-methanol (2.71 g, 105%). (Durant et al., “Aminoalkylimidazoles and Process for their Production.” Patent No. GB1341375 (Great Britain, 1973)).
[0307] This material was used without further purification.
5-tert-Butyl-3H-imidazole-4-carbaldehyde
[0308] Manganese dioxide (30 g, 0.35 mol) was added to a heterogeneous solution of (5-tert-butyl-3H-imidazol-4-yl)-methanol (4.97 g, 0.03 mol) in acetone (700 ml) and the resulting mixture was stirred at room temperature for 4 h. The mixture was filtered through a pad of Celite and the pad was washed with acetone. The filfrate and washings were combined and evaporated. The residue was triturated with ether to afford, as a colorless solid, 5-tert-butyl-3H-imidazole-4-carbaldehyde (2.50 g, 51%). (Hayashi, Personal Communication (2000)).
[0313] To a solution of 5-tert-butyl-3H-imidazole-4-carbaldehyde (2.50 g, 164.4 mmol) in DMF (50 ml) was added 1,4-diacetyl-piperazine-2,5-dione (6.50 g, 32.8 mmol) and the solution was evacuated, then flushed with argon. The evacuation-flushing process was repeated a further two times, then cesium carbonate (5.35 g, 16.4 mmol) was added. The evacuation-flushing process was repeated a further three times, then the resultant mixture was stirred at room temperature for 5 h. The reaction mixture was partially evaporated (heat and high vacuum) until a small volume remained and the resultant solution was added dropwise to water (100 ml). The yellow precipitate was collected, then freeze-dried to afford 1-acetyl-3-(5′-tert-butyl-1Η-imidazol-4′-Z-ylmethylene)-piperazine-2,5-dione (2.24 g, 47%). (Hayashi, Personal Communication (2000)).
[0318] To a solution of 1-acetyl-3-(5′-tert-butyl-1H-imidazol-4′-Z-ylmethylene)-piperazine-2,5-dione (2.43 g, 8.37 mmol) in DMF (55 ml) was added benzaldehyde (4.26 ml, 41.9 mmol) and the solution was evacuated, then flushed with nitrogen. The evacuation-
flushing process was repeated a further two times, then cesium carbonate (4.09 g, 12.6 mmol) was added. The evacuation-flushing process was repeated a further three times, then the resultant mixture was heated under the temperature gradient as shown below. After a total time of 5 h the reaction was allowed to cool to room temperature and the mixture was added to ice-cold water (400 ml). The precipitate was collected, washed with water, then freeze-dried to afford a yellow solid (2.57 g, HPLC (214nm) tR = 6.83 (83.1%) min.). This material was dissolved in chloroform (100 ml) and evaporated to azeofrope remaining water, resulting in a brown oil. This was dissolved in chloroform (20 ml) and cooled in ice. After 90 min the yellow precipitate was collected and air-dried to afford 3-Z-benzylidene-6-(5″-tert-butyl-1H-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione (1.59 g, 56%). (Hayashi, Personal Communication (2000)).
[0319] HPLC (214nm) tR = 6.38 (2.1%), 6.80 (95.2) min.
[0321] LC/MS tR = 5.84 (337.4 [M+H]+, E isomer), 6.25 (337.4 [M+H]+, 673.4 [2M+H]+, Z isomer) min.
[0322] ESMS m/z 337.3 [M+H]+, 378.1 [M+OLGNT.
[0323] Evaporation of the chloroform solution gave additional 3-Z-benzylidene-6-(5″-tert-butyl-1H-imidazol-4″-Z-ylmethylene)-piperazine-2,5-dione (0.82 g, 29%). ΗPLC (214nm) tR = 6.82 (70.6%) min.
Plinabulin (11, NPI-2358) is a potent microtubule-targeting agent derived from the natural diketopiperazine “phenylahistin” (1) with a colchicine-like tubulin depolymerization activity. Compound 11 was recently developed as VDA and is now under phase II clinical trials as an anticancer drug. To develop more potent antimicrotubule and cytotoxic derivatives based on the didehydro-DKP skeleton, we performed further modification on the tert-butyl or phenyl groups of 11, and evaluated their cytotoxic and tubulin-binding activities. In the SAR study, we developed more potent derivatives 33 with 2,5-difluorophenyl and 50 with a benzophenone in place of the phenyl group. The anti-HuVEC activity of 33 and 50 exhibited a lowest effective concentration of 2 and 1 nM for microtubule depolymerization, respectively. The values of 33 and 50 were 5 and 10 times more potent than that of CA-4, respectively. These derivatives could be a valuable second-generation derivative with both vascular disrupting and cytotoxic activities.
Synthesis and Structure–Activity Relationship Study of Antimicrotubule Agents Phenylahistin Derivatives with a Didehydropiperazine-2,5-dione Structure
† Department of Medicinal Chemistry, Tokyo University of Pharmacy and Life Sciences, Hachioji, Tokyo 192-0392, Japan
‡ Department of Medicinal Chemistry, Center for Frontier Research in Medicinal Science, Kyoto Pharmaceutical University, Kyoto 607-8412, Japan
§Nereus Pharmaceuticals, San Diego, California 92121, United States
∥ Department of Analytical and Bioinorganic Chemistry, Kyoto Pharmaceutical University, Kyoto 607-8414, Japan
⊥ Laboratory of Comparative Agricultural Science, Division of Environmental Science and Technology, Graduate School of Agriculture, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
# Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Ibaraki 305-8572, Japan
∇Marine Biotechnology Institute Co., Ltd., Kamaishi, Iwate 026-0001, Japan
Click for improved solubility: A water-soluble prodrug of plinabulin was designed and synthesized efficiently by using click chemistry in three steps (see scheme). The product was highly water-soluble, and the parent compound could be regenerated by esterase hydrolysis.
Lloyd, G.K.; Muller, Ph.; Kashyap, A.; Zippelius, A.; Huang, L. (January 7–9, 2016), Plinabulin: Evidence for an Immune Mediated Mechanism of Action (Philadelphia (PA) AACR 2016 Abstract nr A07), San Diego CA
Lloyd, G.K.; Du, L.; Lee, G.; Dalsing-Hernandez, J.; Kotlarczyk, K.; Gonzalez, K.; Nawrocki, S.; Carew, J.; Huang, L. (October 5–9, 2015), Activity of Plinabulin in Tumor Models with Kras Mutations (Philadelphia (PA) AACR 2015 Abstract nr. 184), Boston MA
A thyrotropin-releasing hormone potentially for the treatment of spinocerebellar ataxia.
CAS No.204386-76-5(Rovatirelin)
879122-87-9(Rovatirelin Hydrate)
C17H24N4O4S
Exact Mass: 380.1518
Rovatirelin is a novel synthetic agent that mimics the actions of thyrotropin-releasing hormone (TRH). Rovatirelin binds to the human TRH receptor with higher affinity (Ki=702nM) than taltirelin (Ki=3877nM). Rovatirelin increased the spontaneous firing of action potentials in the acutely isolated noradrenergic neurons of rat locus coeruleus (LC). Rovatirelin increased locomotor activity. Rovatirelin may have an orally effective therapeutic potential in patients with SCD.
Rovatirelin ([1-[-[(4S,5S)-(5-methyl-2-oxo oxazolidin-4-yl) carbonyl]-3-(thiazol-4-yl)-l-alanyl]-(2R)-2-methylpyrrolidine) is a novel synthetic agent that mimics the actions of thyrotropin-releasing hormone (TRH). The aim of this study was to investigate the electrophysiological and pharmacological effects of rovatirelin on the central noradrenergic system and to compare the results with those of another TRH mimetic agent, taltirelin, which is approved for the treatment of spinocerebellar degeneration (SCD) in Japan. Rovatirelin binds to the human TRH receptor with higher affinity (Ki=702nM) than taltirelin (Ki=3877nM). Rovatirelin increased the spontaneous firing of action potentials in the acutely isolated noradrenergic neurons of rat locus coeruleus (LC). The facilitatory action of rovatirelin on the firing rate in the LC neurons was inhibited by the TRH receptor antagonist, chlordiazepoxide. Reduction of the extracellular pH increased the spontaneous firing of LC neurons and rovatirelin failed to increase the firing frequency further, indicating an involvement of acid-sensitive K+ channels in the rovatirelin action. In in vivo studies, oral administration of rovatirelin increased both c-Fos expression in the LC and extracellular levels of noradrenaline (NA) in the medial prefrontal cortex (mPFC) of rats. Furthermore, rovatirelin increased locomotor activity. The increase in NA level and locomotor activity by rovatirelin was more potent and longer acting than those by taltirelin. These results indicate that rovatirelin exerts a central nervous system (CNS)-mediated action through the central noradrenergic system, which is more potent than taltirelin. Thus, rovatirelin may have an orally effective therapeutic potential in patients with SCD.
Preparation of the compound represented by Example 1 set (IX)
The second step
Two
(First step)
Method described in the literature (Synth. Commun., 20, 3507 (1990)) synthesized N- in (tert- butoxide deer Lupo sulfonyl) one 3- (4 one-thiazolyl) one L Aranin (1, 21.79 g, 80 mmol) in Torifuruoro and the mixture was stirred acetic acid (80 ml) were added under ice-cooling for 2 hours and a half. Then stirred for 30 minutes at room temperature was added to the reaction mixture p- toluenesulfonic acid hydrate (15.22 g, 80 mmol). The reaction mixture was concentrated to dryness under reduced pressure. To remove excess Torifuruoro acetic acid by the obtained residue concentrated to dryness under reduced pressure by addition of water and methanol.Obtained obtained residue was collected by filtration crystals ether was added to precipitate the compound (2) 29.8 g (quantitative).
I 匕合 product (2) 38.85 g E evening Nord (200 ml) of (112.8 mmol) – in THF (600 ml) solution, diphenyl di § zone methane while 攪袢 at room temperature (39 g, 201 mmol) in small portions over 30 minutes were added. The reaction mixture was stirred for 1 hour at room temperature, Ziv E sulfonyl di § zone methane (10 g, 51.5 mmol) was added and stirred for one hour. To the reaction mixture
After decomposing the excess reagent by the addition of acetic acid (0.1 ml), it was concentrated to dryness under reduced pressure and distilled off the solvent. The resulting residue (92 g) with ether (1 L) was crystallized to give compound (3) 49.05 g (96.1%).
Measured value: C, 61.14; H, 5.32; N, 5.41; S, 12.46.
(Third step)
Cis-one L one 5-methyl-2-one O Kiso O Kisa ethylbenzthiazoline one 4-carboxylic acid 13.95 g (96.14 mmol), compound (3) 49.09 g (96.14 mmol ), N-hydroxybenzotriazole To Riazoru 2.6 g (19.23 mmol) and under ice-cooling in THF (1L) solution of Toryechiruamin 14.1 ml (lOlmmol), was added to the DCC (20.83g, 101 mmol). The cooling bath was removed after stirring for 10 minutes at the same temperature, and stirred for an additional 2 0 hours at room temperature. After removing the precipitated precipitate and the filtrate concentrated to dryness under reduced pressure an oily residue (82.7 g was obtained). The residue was filtered off and dissolved by heating to insoluble matter in acetic acid Echiru (700 ml). The filtrate was successively washed with sodium carbonate aqueous solution and water.After the addition of methanol (20 ml) the organic layer was dried with sulfuric acid mug Neshiumu, was concentrated to a small volume under reduced pressure.Precipitated collected by filtration and acetic acid E Ji Le crystals – ether (2: 3) washing to compound with a mixture (4) 35.69 g (79.8% ) was obtained. After addition was concentrated to dryness under reduced pressure of the mother liquor, and crystallized from acetic acid E Chiru ether mixture compound (4) 2.62 g (5.9% ) was obtained.
Measured value: C ! 61.95; H, 5.01; N, 8.94; S ) 6.62.
(Fourth step)
Compound (4) 41.24 under ice-cooling to g (88.59 mmol), and the mixture was stirred Anisoru (240ml) and To Rifuruoro acetic acid (120 ml) and the mixture for 15 minutes. And the mixture was stirred for 2 hours 3 0 minutes further room temperature after removal of the cooling bath. The reaction mixture was added to the E one ether (500 ml) to the oily residue obtained by concentrated to dryness under reduced pressure was collected by filtration and pulverized. The resulting powder is water (50 ml) – was removed by filtration methanol (300 ml) warming dissolved insoluble matter in a mixture. The filtrate was concentrated to small volume under reduced pressure, and allowed to stand at room temperature for 3 days adding a seed crystal and methanol. The precipitated crystals were obtained Shi preparative filtration compound (5) 14.89 g (56.1%). The mother liquor was concentrated to dryness under reduced pressure, to give again further compound was crystallized from methanol one ether mixture of the (5) 10.3 g (38%). mp: 214-215 ° C
Calculated: C ; 44.14; H, 4.38; N, 14.04; S ) 10.71.
Measured value: C, 43.94; H, 4.478; N, 14.09; S, 10.58.
(Fifth step)
Compound (5) 12.1 g, (40.48 mmol) and N- hydroxysuccinimide (4.66 g, 40,48 mM) under ice-cooling to THF (242 ml) suspension of,: DCC (8.35 g, 40.48 mmol) was added to 3 and the mixture was stirred for 10 minutes. The cooling bath was removed, and the mixture was further stirred at room temperature for 2 hours. The resulting compound N- hydroxysuccinimide ester solution of (5) was synthesized in a way described in the literature (Tetrahedron, 27, 2599 (1971 )) (R) – (+) – 2- Mechirupiro lysine hydrochloride (5.42 g) and Toryechiruamin (8.46 ml, was added at room temperature to THF (121 ml) suspension of 60.72 mmol). The reaction mixture was stirred for an additional 1 5 hrs. The filtrate after removal of the insoluble matter that has issued analysis was concentrated to dryness under reduced pressure. Residue (24.6 Ga) the insoluble material was removed by filtration was dissolved in water (150 ml). The filtrate was purified by gel filtration column chromatography one (MCI Gel CHP-20P, 600 ml). 4 0% aqueous methanol solution compound of the collected crude eluted cut off fractionated (IX) was obtained 8.87 g. Then after purification by silica gel column chromatography (black port Holm one methanol mixture), to give the compound was freeze-dried (IX) 5.37 g (35.7% ).
N.N-dicyclohexylcarbodiimide (10.83 g, 52.5 mmol), N-hydroxybenzotriazole (2.03 g, 15 mmol) and triethylamine (7.7 ml, 55.2 mmol) were added to a solution (130 ml) of N-(tert-butoxycarbonyl)-3-(thiazol-4-yl)-L-alanine (1) (13.62 g, 50 mmol) obtained by the method described in literatures (J. Am. Chem. Soc. 73, 2935 (1951) and Chem. Pharm. Bull. 38, 103 (1950)) and 2(R)-2-methylpyrrolidine p-toluenesulfonic acid (2) (12.79 g, 50 mmol) obtained by the method described in a literature (HeIv. Chim. Acta, 34, 2202 (1951)) in tetrahydrofuran. The mixture was stirred for 20 hours at room temperature. After the precipitates are filtered off, the obtained filtrate was concentrated under reduced pressure. Thus-obtained residue was dissolved in ethyl acetate (200 ml) and the solution were washed with an aqueous solution of sodium hydrogencarbonate and water, successively. The organic layers were dried over magnesium sulfate and concentrated under reduced pressure to give a title compound (3) (16.45 g, 100%) as oil.
NMR (CDCl3): OH 8.76 and 8.75 (1 H, each d, J=2.1Hz, Thia-H-2), 7.08 (1 H, d, J=2.fflz, thia-H-5), 5.45 (1 H, m, NH), 3.45-3.64 (1 H, m, AIa-CoH), 4.14 and 3.81 (1 H, each m, Pyr-CαH), 3.51 (1 H, m, PVr-NCH2), 3.1-3.4 (3 H, m, Pyr-CH2and AIa-CH2), 1.39 (9 H, s, BOC), 1.3-2.0 (4 H, m, PyT-CH2), 1.06 (3 H, d, J=6Hz, Pyr-Me)
Compound (3) (33.77 g, 99.48 mmol) and p-toluenesulfonic acid hydrate (37.85 g, 199 mmol) were dissolved in ethyl acetate (101 ml) and the solution was cooled with ice. To the mixture, 4 mol/L solution of hydrogen chloride-ethyl acetate (125 ml) was added, and the mixture was stirred for 2 hours 45 minutes. After the mixture was concentrated under reduced pressure, methanol was added to the residue. The mixture was concentrated. Methanol-toluene (1: 1) was added to the residue and concentrated under reduced pressure to give crystalline residue. The residue was washed with acetone and filtered to give compound (4) as crystals (36 g, 62%). After the mother liquor was concentrated under reduced pressure, methanol and toluene were added to the residue and concentrated. Obtained crystalline residue was washed with acetone to give compound (4) (10.67 g, 18.4%). mp 188-189 0C [α]D24 +2.2 (c, 1.0, MeOH) IR(KBr)Cm“1: 3431, 3125, 3080, 2963, 1667, 1598, 1537, 1497, 1451, 1364, 1229, 1198, 1170, 1123, 1035, 1011.
NMR (CD3OD): δH 9.04 and 9.03 (1 H, each d, J=2.1Hz, Thia-H-2), 7.70 (2 H, m, aromaticH), 7.46 (1H, d, J=2.1Hz, thia-H-5), 7.23 (2H, m, aromaticH), 4.49and4.46 (1 H, each d, J=6.9Hz, Ala-CαH), 4.14 and 3.75 (1 H, each m, Pyr-CαH), 3.51 (1 H, m, pyr-NCH2), 3.2-3.4 (3 H, m, PyT-CH2 and AIa-CH2), 2.36 (3 H, s, aromatic Me), 1.3-2.0 (4 H, m, pyr-CH2), 1.19 and 1.07 (3 H, each d, J=6.3Hz, Pyr-Me) Anal Calcd For C11H17N3OS 2C7H8O3S Calculated: C, 51.44%; H1 5.70%; N, 7.20%; S, 16.48%. Found: C, 51.36%; H, 5.69%; N, 7.23%; S, 16.31%.
(4S, 5S)-5-methyl-2-oxooxazolidin-4-yl carboxylic acid (5) (1.368 g, 9.43 mmol) obtained by the method described in literatures (J. Chem. Soc. 1950, 62; Tetrahedron 48; 2507 (1992) and Angew. Chem. 101, 1392 (1989)), Compound (4) (5 g, 8.56 mmol) and N-hydiOxysuccinimide (217 mg, 1.89 mmol) were dissolved in N, N-dimethylformamide (10 ml), and tetrahydrofuran (65 ml) was added. After the mixture was cooled with ice in a cool bath, triethylamine (2.63 ml, 18.86 mmol) and N, N-dicyclohexylcarbodiimide (2.04 g, 9.89 mmol) were added with stirred and the mixture was stirred for additional 30 minutes. The cooling bath was removed and the mixture was stirred for 15 hours at room temperature. The precipitated were filtered off and the filtrate was concentrated under reduced pressure. Water (100 ml) was added to thus-obtained residue (9.95 g) and the mixture was stirred for 1.5 hours at room temperature. After insoluble substance was filtered off, the filtrate was concentrated until it was reduced to about half volume under reduced pressure. The small amount of insoluble substance was filtered off and the filtrate was concentrated until it was reduced to about 2O g under reduced pressure. After the mixture was allowed to stand in a refrigerator for 3 days, the precipitated crystals (2.98 g) were collected by filtration and washed with cold water. The filtrate was extracted twice with chloroform, dried over magnesium sulfate and concentrated under reduced pressure. Ethyl acetate (5 ml) was added to oil residue (1.05 g) and the mixture was stirred to give crystals (136 mg). The obtained crystals were combined and dissolved in purified water (45 ml) with heating. After the solution was allowed to cool to room temperature, the precipitated insoluble substance was filtered off The filtrate was concentrated under reduced pressure and allowed to stand at room temperature overnight. The mixture was cooled with ice, and the crystals were collected by filtration to give Compound (1-1, 2.89 g, 80.3%). mp 194-196 0C
1379, 1235, 1089. NMR(CD3OD): δH 8.97 and 8.96 (total 1 H, d, J=2.1Hz, Thia-H-2), 7.34 and 7.33 (total 1
H, d, J=2.1Hz, Thia-H-5), 5.18 and 5.04 (total 1 H, each t, J=7.5Hz, Ala-CαH), 4.92 (1
H, dq, J=6.6 and 8.7Hz, Oxa-H-5), 4.36 and 4.35 (total 1 H, d, J=8.7Hz, Oxa-H-4), 4.07 and 3.92 (total 1 H, each m, Pyr-Cα-H), 3.78 (1 H, m, Pyr-NCH2), 3.42 (1 H, m, Pyr- 5 NCH2), 3.22 (2 H, m, AIa-CH2), 1.5-2.0 (4 H, m, Pyr-CH2), 1.28 and 1.22 (total 3 H, each d, J=6.6Hz, Oxa-5-Me), 1.21 and 1.02 (total 3 H, each d, J=6.6Hz, Pyr-2-Me)
Anal. Calcd For C16H22N4O4S 3H2O
Calculated: C, 45.00%; H, 6.71%; N, 13.33%; S, 7.63%.
Found: C, 45.49%; H, 6.60%; N, 13.58%, S, 7.88%. 10
Step 3 (2)
Method B
After Compound (1-2) (410 g, 1.119 mmol) was dissolved in purified water (6.3 L) with heating, the solution was concentrated until the total weight of the mixture was 15 reduced to 1370 g under reduced pressure. The concentrated solution was allowed to stand at room temperature overnight. The solution was cooled with ice for 1 hour and filtered to give the precipitated crystals. The obtained crystals were washed with cold water to give
Compound (T- 1) (448 g, 95.2%) as colorless crystals. Mother liquor was mixed with purified water (300 mL) with heating and the solution was concentrated to 55 g under reduced pressure. 20 After the concentrated solution was allowed to stand at room temperature overnight, the solution was filtered to give the precipitated crystals (T-1, 16.3 g, 3.5%, total amount 464.3 g, 98.7%). mp 194-196 0C
After l-[N-[(4S,5S)-(5-methyl-2-oxooxazolidin-4-yl)carbonyl]-3-(thiazol-4-yl)-L- 35 alanyl-(2R)-2-methylpyrrolidine monohydrate (4.77 g) obtained by the method described in Patent Literature 8 was crushed in a mortar, it was dried under reduced pressure (66.5 Pa) at 100 0C for 15 hours to give 4.54 g of Compound (1-2). mp 194.5-196.5 0C [α]D25 -2.1 +. 0.4 ° (c, 1.004, H2O), [α]365 +36.8 ± 0.8 ° (c, 1.004, H2O) Water measurement (Karl Fischer method): 0.27%
IR(NuJOr)Cm”1: 3276, 3180, 3104, 1766, 1654, 1626, 1548, 1517, 1457, 1380, 1235, 1102, 979. NMR(CD3OD):δH 8.97 and 8.96 (total 1 H, d, J 2.1 Hz, Thia-H-2), 7.34 and 7.33 (total 1 H, d, J 2.1 Hz, Thia-H-5), 5.19 and 5.04 (total 1 H, each t, J 7.5 Hz, Ala- CaH), 4.92 (1 H, dq, J 6.6 and 8.7 Hz, Oxa-H-5), 4.36 and 4.35 (total 1 H, d, J 8.7 Hz, Oxa-H-4), 4.07 and 3.92 (total 1 H, each m, Pyr-Cα-H), 3.78 (1 H, m, Pyr-NCH2), 3.42 (1 H, m, Pyr-NCH2), 3.22 (2 H, m, AIa-CH2), 1.5-2.0 (4 H, m, Pyr-CH2), 1.28 and 1.22 (total 3 H, each d, J 6.6 Hz, Oxa-5-Me), 1.21 and 1.02 (total 3 H, each d, J 6.6 Hz, Pyr-2-Me). Anal Calcd For: C16H22N4O4S
Calculated: C, 52.44%; H, 6.05%; N, 15.29%; S, 8.75%. Found: C, 52.24%; H, 5.98%; N, 15.27%, S, 8.57%.
Method B
After Compound (1-1) (17.89 g, 47.3 mmol) was crushed in a mortar, it was dried under reduced pressure (66.5 Pa) at 100 °C for 14 hours to give Compound (1-2, 17.31 g). mp 193-194 0C [α]D25 -1.9 ± 0.4 ° (c, 1.002, H2O), [α]365 +37.2 ± 0.8 ° (c, 1.002, H2O)

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader














![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)









































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}