
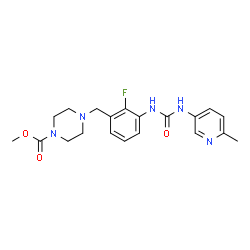
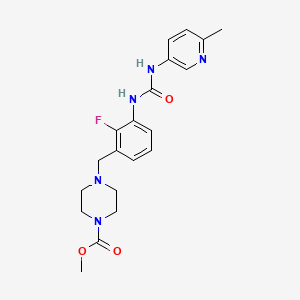
Omecamtiv mecarbil
- Molecular FormulaC20H24FN5O3
- Average mass401.435 Da
4-[2-fluoro-3-[(6-methyl-3-pyridyl)carbamoylamino]benzyl]piperazine-1-carboxylic acid methyl ester
AMG 423
AMG-423
CK1827452
CK-1827452; CK1827452
methyl 4-(2-fluoro-3-(3-(6-methylpyridin-3-yl)ureido)benzyl)piperazine-1-carboxylate
1-Piperazinecarboxylic acid, 4-[[2-fluoro-3-[[[(6-methyl-3-pyridinyl)amino]carbonyl]amino]phenyl]methyl]-, methyl ester
2M19539ERK
オメカムティブメカビル
9088
Methyl 4-(2-fluoro-3-{[(6-methyl-3-pyridinyl)carbamoyl]amino}benzyl)-1-piperazinecarboxylate
In January 2019, Cytokinetics and licensees Amgen and Servier are developing oral modified- and immediate-release formulations of the cardiac myosin activator omecamtiv mecarbil (phase III), the lead from a series of small-molecule, sarcomere-directed compounds, for the treatment of chronic heart diseases including high risk heart failure, stable heart failure and ischemic cardiomyopathy
Omecamtiv Mecarbil has been used in trials studying the treatment and basic science of Heart Failure, Echocardiogram, Pharmacokinetics, Chronic Heart Failure, and History of Chronic Heart Failure, among others.
Omecamtiv mecarbil, a small-molecule activator of cardiac myosin, is developed in phase III clinical trials by originator Cytokinetics and Amgen for the oral treatment of chronic heart failure.
WO2006009726 product patent of omecamtiv mecarbil expire in EU states until June 2025 and expire in the US in September 2027 with US154 extension.
- Originator Cytokinetics
- Developer Amgen; Cytokinetics; Servier
- Class Esters; Heart failure therapies; Organic chemicals; Piperazines; Pyridines; Small molecules
- Mechanism of Action Cardiac myosin stimulants
- Phase III Chronic heart failure
- Phase II Acute heart failure; Heart failure
- No development reported Angina pectoris; Cardiomyopathies
- 26 Apr 2018 Amgen and Cytokinetics plan the phase III METEORIC-HF trial in Heart failure by the end of 2018 (NCT03759392)
- 18 Sep 2017 Pharmacodynamics data from the phase III COSMIC-HF trial Chronic heart failure released by Cytokinetics
- 08 May 2017 Amgen completes the phase II trial in Heart failure in Japan (NCT02695420)
Omecamtiv mecarbil (INN), previously referred to as CK-1827452, is a cardiac-specific myosin activator. It is being studied for a potential role in the treatment of left ventricular systolic heart failure.[1]
Systolic heart failure involves a loss of effective actin-myosin cross bridges in the myocytes (heart muscle cells) of the left ventricle, which leads to a decreased ability of the heart to move blood through the body. This causes peripheral edema (blood pooling), which the sympathetic nervous system tries to correct[2] by overstimulating the cardiac myocytes, leading to left ventricular hypertrophy, another characteristic of chronic heart failure.
Current inotropic therapies work by increasing the force of cardiac contraction, such as through calcium conduction or modulating adrenoreceptors. But these are limited by adverse events, including arrhythmias related to increased myocardical oxygen consumption, desensitization of adrenergic receptors, and altering intracellular calcium levels.[3] Inotropes are also thought to be associated with worse prognosis.[4] Therefore, the novel mechanism of omecamtiv mecarbil may offer a useful new option for heart failure.
Mechanism of action
Cardiac myocytes contract through a cross-bridge cycle between the myofilaments, actin and myosin. Chemical energy in the form of ATP is converted into mechanical energy which allows myosin to strongly bind to actin and produce a power stroke resulting in sarcomere shortening/contraction.[5] Omecamtiv mecarbil specifically targets and activates myocardial ATPase and improves energy utilization. This enhances effective myosin cross-bridge formation and duration, while the velocity of contraction remains the same.[6]Specifically, it increases the rate of phosphate release from myosin, thereby accelerating the rate-determining step of the cross-bridge cycle, which is the transition of the actin-myosin complex from the weakly bound to the strongly bound state.[7][1] Furthermore, once myosin is bound to actin, it stays bound dramatically longer in the presence of omecamtiv mecarbil.[8][9] The combination of increased and prolonged cross-bridge formation prolongs myocardial contraction. Thus, the overall clinical result of omecamtiv mecarbil is an increase in left ventricular systolic ejection time and ejection fraction.[6][7]
There is a slight decrease in heart rate while myocardial oxygen consumption is unaffected. The increased cardiac output is independent of intracellular calcium and cAMP levels.[3][10] Thus omecamtiv mecarbil improves systolic function by increasing the systolic ejection duration and stroke volume, without consuming more ATP energy, oxygen or altering intracellular calcium levels causing an overall improvement in cardiac efficiency.[6]
Clinical trials
Experimental studies on rats and dogs, proved the efficacy and mechanism of action of omecamtiv mecarbil.[3] Current clinical studies on humans have shown there is a direct linear relationship between dose and systolic ejection time.[1][11][12] The dose-dependent effects persisted throughout the entire trial, suggesting that desensitization does not occur. The maximum tolerated dose was observed to be an infusion of 0.5 mg/kg/h. Adverse effects, such as ischemia, were only seen at doses beyond this level, due to extreme lengthening of systolic ejection time.[1] Thus due to the unique cardiac myosin activation mechanism, omecamtiv mecarbil could safely improve cardiac function within tolerated doses. Omecamtiv mecarbil effectively relieves symptoms and enhances the quality of life of systolic heart failure patients. It drastically improves cardiac performance in the short term; however, the hopeful long-term effects of reduced mortality have yet to be studied.[1][2]
PATENT
WO2006009726
PAPER
Synthesis of unsymmetrical diarylureas via pd-catalyzed C-N cross-coupling reactions
Org Lett 2011, 13(12): 3262
Synthesis of Unsymmetrical Diarylureas via Pd-Catalyzed C–N Cross-Coupling Reactions
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, United States
Org. Lett., 2011, 13 (12), pp 3262–3265
DOI: 10.1021/ol201210t
Abstract
A facile synthesis of unsymmetrical N,N′-diarylureas is described. The utilization of the Pd-catalyzed arylation of ureas enables the synthesis of an array of diarylureas in good to excellent yields from benzylurea via a one-pot arylation–deprotection protocol, followed by a second arylation.

Click to access ol201210t_si_001.pdf
Methyl 4-(2-fluoro-3-(3-(6-methylpyridin-3-yl)ureido)benzyl)piperazine-1- carboxylate (Omecamtiv Mecarbil).11 Following general procedure C, a mixture of methyl 4-(3-chloro-2-fluorobenzyl)piperazine-1-carboxylate (143.1 mg, 0.5 mmol), (2- Methylpyridin-5-yl)urea (90.6 mg, 0.6 mmol), Pd(OAc)2 (5 mol %), t-BuBrettPhos (15 mol %), Cs2CO3 (456.2 mg, 0.7 mmol), degassed water (4 mol %) and THF (1 mL) was heated to 65 °C for 6 h. The crude product was purified via flash chromatography (5-10% MeOH/DCM) to provide the title compound as a slightly brownish solid (164 mg, 82%),
mp = 180 °C.
1 H NMR (400 MHz, DMSO-d6 ) δ: 9.13 (s, 1H), 8.59 (d, J = 1.5 Hz, 1H), 8.47 (d, J = 2.3 Hz, 1H), 8.05 (t, J = 7.6 Hz, 1H), 7.83 (dd, J = 8.4, 2.4 Hz, 1H), 7.16 (d, J = 8.4 Hz, 1H), 7.09 (t, J = 7.9 Hz, 1H), 7.00 (t, J = 6.7 Hz, 1H), 3.57 (s, 3H), 3.55 (s, 2H), 3.35 (br, 4H), 2.40 (s, 3H), 2.36 (br, 4H) ppm.
13C NMR (101 MHz, DMSO-d6 ) δ: 155.0, 152.3, 151.1, 150.7 (d, J = 242.5 Hz), 139.2, 133.6, 127.3 (d, J = 10.9 Hz), 125.8, 124.1 (d, J = 13.3 Hz), 124.0 (d, J = 4.0 Hz), 123.8 (d, J = 3.8 Hz), 122.8, 119.5, 54.6, 52.2, 52.1, 43.4, 23.2 ppm (observed complexity is due to C–F splitting).
19F NMR (376 MHz, DMSO-d6 ) δ: -135.09.
IR (neat, cm-1 ): 3297, 2920, 2823, 1705, 1638, 1557, 1476, 1450, 1233, 1189, 1129, 779, 765.
Anal. Calcd. for C20H24FN5O3: C, 59.84; H, 6.03. Found: C, 59.64; H, 5.92.


PAPER
Morgan et al. ACS Med. Chem. Lett. 2010, 1, 472
Discovery of Omecamtiv Mecarbil the First, Selective, Small Molecule Activator of Cardiac Myosin
Bradley P. Morgan*, Alexander Muci, Pu-Ping Lu, Xiangping Qian, Todd Tochimoto, Whitney W. Smith, Marc Garard, Erica Kraynack, Scott Collibee, Ion Suehiro, Adam Tomasi, S. Corey Valdez, Wenyue Wang, Hong Jiang, James Hartman, Hector M. Rodriguez, Raja Kawas, Sheila Sylvester, Kathleen A. Elias, Guillermo Godinez, Kenneth Lee, Robert Anderson, Sandra Sueoka, Donghong Xu, Zhengping Wang, Nebojsa Djordjevic, Fady I. Malik, and David J. Morgans Jr.
Cytokinetics, Inc., 280 East Grand Avenue, South San Francisco, California 94080
ACS Med. Chem. Lett., 2010, 1 (9), pp 472–477
DOI: 10.1021/ml100138q
We report the design, synthesis, and optimization of the first, selective activators of cardiac myosin. Starting with a poorly soluble, nitro-aromatic hit compound (1), potent, selective, and soluble myosin activators were designed culminating in the discovery of omecamtiv mecarbil (24). Compound 24 is currently in clinical trials for the treatment of systolic heart failure.
omecamtiv mecarbil as a white powder (3.64 kg, 90% yield).
IR (KBR) 3292, 2950, 2866, 2833, 1720, 1640, 1550, 1600, 1490, 1455, 1406, 1378, 1352, 1274, 1244, 1191, 1125, 815, 769, 725, 668 cm-1 ;
1H NMR (400 MHz, DMSO-d6) δ 9.12 (s, 1 H, 2-pyridyl H), 8.59 (d, 1 H, J = 2.5 Hz, Urea N-H), 8.47 (d, 1 H, J = 2.6 Hz, Urea N-H), 8.04 (dt, 1 H, J = 1.5 Hz, 7.8 Hz, phenyl H), 7.83 (dd, 1 H, J = 2.6 Hz, 8.4 Hz, 4-pyridyl H), 7.18 (d, 1 H, J = 8.4 Hz, 5-pyridyl H), 7.10 (app t, 1 H, J = 7.8 Hz, phenyl H), 7.02 (app p, 1 H, J = 1.5 Hz, 6.3 Hz, 7.8 Hz, phenyl H), 3.58 (s, 3 H, OCH3), 3.56 (m, 4 H, piperazine Hs), 2.41 (s, 3 H, pyridineCH3), 2.37 (br m, 4 H, piperazine Hs); 13C NMR (100 MHz, DMSO-d6) δ 155.0,152.3, 151.1 150.7, 139.1, 133.6, 127.3, 127.2, 125.8, 124.1, 123.7, 122.8, 119.5, 54.5, 52.2, 52.0, 43.4, 23.2;
Exact mass calcd for C20H24FN5O3 requires m/z 402.1926. Found m/z 402.1940.
Anal. Calcd. For C20H24FN5O3: C, 59.84; H, 6.03; N, 17.45. Found: C, 59.99; H, 6.07; N, 17.41.
PATENT
WO2016210240
PATENT
WO-2019006231
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019006231&tab=PCTDESCRIPTION&maxRec=1000
Process for the preparation of omecamtiv mecarbil and its new intermediates. Useful for the treatment of heart failure..
Scheme 1 :



Scheme 2
I



Scheme 3
I

Piper 

Scheme 5
Aminopyridine
(APYR) Commercially Available

Scheme 6


IPAc Reaction

.
Scheme 7

Scheme 8
Pi 
(PIPA)
[0043] Thus, provided herein is a method of synthesizing PIPA comprising admixing PIPN (which can comprise PIPN hydrochloride salt), an aqueous solution of an inorganic base, and toluene to form a PIPN freebase solution. The inorganic base can be sodium bicarbonate or sodium hydroxide, for example. In some embodiments, the inorganic base comprises sodium hydroxide. The PIPN freebase solution is then hydrogenated in the presence of a palladium catalyst in toluene and an alcohol solvent to form crude PIPA. The alcohol solvent can comprise ethanol or isopropanol. PIPA is then crystallized from a heptane and toluene solvent mixture.
[0044] In some specific embodiments, to a mixture of 1 equiv. PIPN-HCI and toluene (4V) is added 1 M aq. NaOH (3.3V) at 20 °C. Stirring is continued for 1 hour before the phases are separated. The organic layer is washed twice with a mixture of water (2.4V) and saturated brine (0.6V), then the organic layer is distilled to 3.8V. The solution is filtered, the reactor rinsed with toluene (1V) and the rinse solution filtered before the organic layers are combined. To the toluene layer is added Pd/C (0.7 wt%) and the heterogeneous mixture is charged into a hydrogenation vessel. Ethanol (1V) is added to the mixture. Hydrogenation is performed at 20 °C under 60 psig of hydrogen. After the reaction is complete, the mixture is filtered and rinsed with toluene (1V). The mixture is distilled to 2.4V, seeded with 1 mol% PIPA in heptane (0.1V) at 35 °C and then cooled to 20 °C. The addition of heptane (5.6V) is completed in 3 hours. The mixture is filtered and dried under vacuum and nitrogen to afford PIPA (90% yield, > 97.0 wt%, > 98.0 LCAP).
[0045] In some other specific embodiments, 1 N aqueous sodium hydroxide (3.3 volumes) is added to 1 equiv. of PIPN (hydrochloride salt) suspended in toluene (4 volumes). The biphasic mixture is agitated at 20 °C for 1 hour and the phases are allowed to separate. The organic layer is washed twice with a 0.9 M aqueous sodium chloride solution (3 volumes). The reaction mixture is azeotropically dried by concentration to approximately 3.8 volumes and polish filtered. The transfer line is rinsed with toluene (1 volume) and the rinse solution is combined with the PIPN solution.
Ethanol (1 volume) is added to the PIPN solution and hydrogenation of the starting material is carried out in the presence of 5% Pd/C (on activated carbon sold by BASF as Escat 1421, 0.7 wt% catalyst loading) using a pressure of 4 bars of hydrogen at 15 °C. Upon reaction completion, the mixture is filtered. The hydrogenation autoclave and filtered catalyst are rinsed with toluene (1V) and the rinse solution is combined with the reaction mixture. The solution is concentrated to 2.4 volumes and seeded with 1 mol% PIPA in heptane (0.1 volume) at 38 °C. The mixture is agitated for 30 minutes at 38 °C, cooled to 20 °C over the course of 2 hours, and agitated at that temperature for 30 minutes. Heptane is added (5.6 volumes) over the course of 3 hours and the mixture is agitated for 30 minutes. The mixture is filtered and dried on filter/drier. The cake is washed once with
heptane:toluene (7:3, 2 total volumes) and once with heptane (2 volumes). PIPA is isolated in 88% yield with > 98.0 wt% assay and > 98.0 LC area%.
[0046] Preparation of omecamtiv mecarbil dihvdrochloride hydrate: The prior process to prepare omecamtiv mecarbil dihydrochloride hydrate involved a telescoped procedure by which the
omecamtiv mecarbil is prepared as a solution in THF, and the solvent is subsequently exchanged for isopropanol. However, considering that the solubility of omecamtiv mecarbil in isopropanol at 20°C is about 10 mg/mL and the total volume of isopropanol at the end of the solvent exchange, 95% of the material is out of solution at the end of the solvent exchange, leading to the formation of a slurry that is difficult or impossible to stir. Distillation can no longer be performed once this slurry is formed due to poor mass transfer, leaving behind THF levels in the slurry that are above the in-process control (IPC) specification, e.g., greater than or equal to 1 GC area%. In practice, this leads to delays in the manufacturing due to necessary recharging of isopropanol until the mixture can be stirred, followed by additional distillation and analysis of residual THF. In addition, the ratio of isopropanol and water has to be verified using an in-process control considering the variable amounts of isopropanol at the end of the distillation and the influence of the solvent ratio (isopropanol/water) on the mother liquor losses upon filtration.
Scheme 9

95% yield
[0048] Thus, provided herein is a method of preparing omecamtiv mecarbil dihydrochloride hydrate via admixing PIPA, PCAR, and a trialkylamine (e.g., triethylamine or diisopropylethylamine) in acetonitrile and THF to form omecamtiv mecarbil. The omecamtiv mecarbil is isolated as the free base and then admixed with 2 to 3 molar equivalents of hydrochloric acid in isopropanol and water to form omecamtiv mecarbil dihydrochloride hydrate, which can optionally be crystallized from isopropanol and water. Isolation of the omecamtiv mecarbil free base can be performed via crystallization by addition of water and filtration. PIPA and PCAR can be prepared as disclosed above.
[0049] In some embodiments, PIPA (2.1 kg, 1 equiv) is charged to a reactor, followed by PCAR (1.1 equiv), then THF (2.5 V), and finally acetonitrile (2.5 V). To the resulting slurry is added N,N-diisopropylethylamine (1.2 equiv) and the batch is heated to 55 °C for 16 h. Water (5 V) is then added over 15 minutes and omecamtiv mecarbil freebase seeds (0.05 equiv) are charged to the reactor. The batch is agitated for 15 minutes and water (10 V) is added over 3 h. The batch is cooled to 20 °C over 1 h and filtered. The cake is washed with 3:1 watenacetonitrile (3 V) and then acetonitrile (3 x 3 V). The cake is dried in a filter/drier. Omecamtiv mecarbil freebase is isolated as a solid in 80% yield, with 99.9 LC area%, and 99.3 wt% assay.
[0050] Omecamtiv mecarbil freebase (2.6 kg, 1 equiv) is charged to a reactor followed by 2-propanol (2.6 V) and water (1.53 V). The batch is then heated to 45 °C. 6 M aqueous HCI (2.2 equiv) is added at a rate to keep batch temperature below 60 °C. The batch is heated to 60 °C for 30 minutes and filtered into a clean reactor at 60 °C. The original vessel is rinsed with an
isopropanokwater mixture (1 :1 , 0.1 volume total) and the rinse volume is added to the reaction mixture. The solution is cooled to 45 °C and a slurry of omecamtiv mecarbil dihydrochloride hydrate seed (0.05 or 0.03 equiv) in isopropanol (0.14 or 0.1 V) is charged to the reactor. The suspension is agitated for 1 h. Isopropanol (3.68 V) is charged to the reactor over 2 h. The mixture is warmed to 55 °C over 1 h and held for 30 minutes at that temperature. The mixture is cooled to 45 °C over 1 h. The mixture is agitated for 2 h and then isopropanol (7.37 V) is added to the reactor over 3 h. The mixture is agitated for 1 h and then cooled to 20 °C over 2 h. The mixture is wet milled until d90 specifications are met (e.g., < 110 μιτι) and the suspension is filtered. The wet cake is washed twice with isopropanokwater (95:5, 2V) . The wet cake is dried under vacuum until isopropanol levels are below 1000 ppm. The cake is optionally re-hydrated if necessary using e.g., a stream of humidified nitrogen, until the water content of the solids are between 3.0 and 4.2 wt%. The material can be recrystallized if it doesn’t meet specification. Omecamtiv mecarbil dihydrochloride hydrate is isolated as a solid in 91.3% yield, with 99.96 LC area%, and 100.1 wt% assay.
[0051] Omecamtiv Mecarbil Dihydrochloride Hydrate Preparation using Continuous Manufacturing: Provided herein is a method of preparing omecamtiv mecarbil dihydrochloride hydrate using a continuous manufacturing process. The general synthetic procedure is outlined in Scheme 10 below.
Scheme 10
Conditions For 100 a Demo Run

CH3CN (6 V), 21 °C
Assay Yield = 95.2 %
Conversion = 98.2 %
L-Urea LCAP = 0 %
PIPA Methyl Carbamate LCAP = 1.49 %
Production Rate of Omecamtiv Mecarbil = 15.29 g/h
PATENT
WO2019006235
PATENT
https://patents.google.com/patent/WO2014152270A1
The cardiac sarcomere is the basic unit of muscle contraction in the heart. The cardiac sarcomere is a highly ordered cytoskeletal structure composed of cardiac muscle myosin, actin and a set of regulatory proteins. The discovery and development of small molecule cardiac muscle myosin activators would lead to promising treatments for acute and chronic heart failure. Cardiac muscle myosin is the cytoskeletal motor protein in the cardiac muscle cell. It is directly responsible for converting chemical energy into the mechanical force, resulting in cardiac muscle contraction.
[0004] Current positive inotropic agents, such as beta-adrenergic receptor agonists or inhibitors of phosphodiesterase activity, increase the concentration of intracellular calcium, thereby increasing cardiac sarcomere contractility. However, the increase in calcium levels increase the velocity of cardiac muscle contraction and shortens systolic ejection time, which has been linked to potentially life-threatening side effects. In contrast, cardiac muscle myosin activators work by a mechanism that directly stimulates the activity of the cardiac muscle myosin motor protein, without increasing the intracellular calcium concentration. They accelerate the rate-limiting step of the myosin enzymatic cycle and shift it in favor of the force-producing state. Rather than increasing the velocity of cardiac contraction, this mechanism instead lengthens the systolic ejection time, which results in increased cardiac muscle contractility and cardiac output in a potentially more oxygen-efficient manner. [0005] U.S. Patent No. 7,507,735, herein incorporated by reference, discloses a genus of com ounds, including omecamtiv mecarbil (AMG 423, CK- 1827452), having the structure:
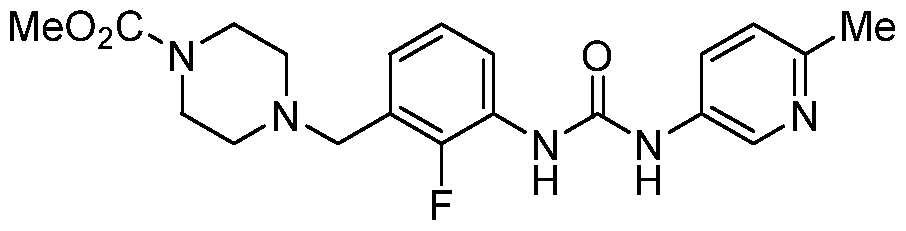
[0006] Omecamtiv mecarbil is a first in class direct activator of cardiac myosin, the motor protein that causes cardiac contraction. It is being evaluated as a potential treatment of heart failure in both intravenous and oral formulations with the goal of establishing a new continuum of care for patients in both the in-hospital and outpatient settings.
Manufacture of Omecamtiv Mecarbil dihydrochloride hydrate Synthetic Route to Omecamtiv Mecarbil
PiE§razine_Nitro^!C Piperazine Aniline
to IPA
omecamtiv mecarbil-2HCI-H20
Synthesis of the API SM Piperazine Nitro-HCl
Piperazine Carboxylate

88% overall [0081] In a 60 L reactor (containing no exposed Stainless steel, Hastelloy®, or other metal parts) equipped with a reflux/return condenser and scrubber charged with a 5N NaOH solution, a mechanically stirred mixture of FN-Toluene (2.0 kg, 12.89 mol, 1.0 equiv.), N- Bromosuccinimide (3.9 kg, 21.92 mol, 1.70 equiv.), benzoyl peroxide (125.0 g, 0.03 equiv., 0.39 mol, containing 25 wt% water), and acetic acid (7.0 L, 3.5 volumes) was heated to 85 °C under an atmosphere of nitrogen for 7 hours. A solution of H3PO3 (106.0 g, 1.29 mol, 0.1 equiv.) and acetic acid (200 mL, 0.1 volume), prepared in separate vessel, was added. The reaction mixture was agitated for 0.5 h and analysis of an aliquot confirmed complete decomposition of benzoyl peroxide (not detected, HPLC254 nm)- The reaction mixture was cooled to 22 °C. DI Water (8.0 L, 4 volumes) and toluene (16.0 L, 8 volumes) were charged, the biphasic mixture was agitated (20 min), and the layers were separated. Aqueous 1.6N NaOH (14.0 L, 7.0 volumes) was added to the organic layer at a rate allowing the batch temperature to stay under 25 °C and the pH of the resultant aqueous phase was measured (> 11). The biphasic mixture was filtered through a 5 μιη Teflon® cartridge line and the layers were separated. The filter line was washed with another 2L of toluene.
[0082] The assay yields were 2.5 % of FN-Toluene, 62.3 % of FN-Bromide and 30.0 % of Di-Bromide. The toluene solution contained no benzoyl peroxide, succinimide, or cc- bromoacetic acid and water content by KF titration was 1030 ppm (This solution could be held under nitrogen at room temperature for > 12 h without any change in the assay yield).
[0083] To this solution at room temperature was added diisopropylethylamine (880.0 g, 6.63 mol, 0.53 equiv.) followed by methanol (460 mL, 11.28 mol, 0.88 equiv.) and heated to 40 °C. A solution of diethylphosphite (820.0 g, 5.63 mol, 0.46 equiv.) in methanol (460 mL, 11.28 mol, 0.88 equiv.) was prepared and added to the reaction mixture at 40 °C through an addition funnel over a period of 1 hour at such a rate that the batch temperature was within 40 + 5 °C. The contents were stirred for a period of 3h at 40 °C from the start of addition and cooled to room temperature and held under nitrogen atmosphere for 12 hours. The assay yield of the reaction mixture was 2.5 % FN-Toluene 92.0% FN-Bromide and 0.2% Di-Bromide. This solution is used as such for the alkylation step.
[0084] Characterization for components of final product mixture (collected for pure compounds).
[0085] 2-Fluoro-3-Nitrotoluene (FN-Toluene): 1H NMR (400 MHz, CHLOROFORM- J) δ ppm 2.37 (s, 1 H), 7.13-7.20 (m, 1 H), 7.45-7.51 (m, 1 H), 7.79-7.85 (m, 1 H). 13C NMR (100 MHz, CHLOROFORM- d) δ ppm 14.3 (d, J = 5 Hz), 123.3 (d, J = 3 Hz), 123.6 (d, J = 5 Hz), 128.2 (d, J = 16 Hz), 136.7 (d, J = 5 Hz), 137.5 (broad), 153.7 (d, J = 261 Hz); 1- (bromomethyl)-2-fluoro-3-nitrobenzene (FN-Bromide): 1H NMR (400 MHz,
CHLOROFORM-J) δ ppm 4.56 (s, 1 H), 7.28-7.34 (m, 1 H), 7.69-7.76 (m, 1 H), 7.98-8.05 (m, 1 H). 13C NMR (100 MHz, CHLOROFORM- J) δ ppm 23.6 (d, / = 5 Hz), 124.5 (d, / = 5 Hz), 126.1 (d, / = 3 Hz), 128.5 (d, / = 14 Hz), 136.5 (d, / = 4 Hz), 137.7 (broad), 153.3 (d, / = 265 Hz). DSC: single melt at 53.59 °C. Exact Mass [C7H5BrFN02 + H]+: calc. = 233.9566, measured = 233.9561; l-(dibromomethyl)-2-fluoro-3-nitrobenzene (Dibromide): 1H NMR (400 MHz, CHLOROFORM- d) δ ppm 6.97 (s, 1 H), 7.39-7.45 (m, 1 H), 8.03-8.10 (m, 1 H), 8.16-8.21 (m, 1 H). 13C NMR (100 MHz, CHLOROFORM-J) δ ppm 29.2 (d, / = 7 Hz), 124.9 (d, / = 5 Hz), 127.1 (d, / = 2 Hz), 132.1 (d, / = 11 Hz), 135.7 (d, / = 2 Hz), 137.2 (broad), 149.8 (d, / = 266 Hz). DSC: single melt at 49.03 °C. Exact Mass [C7H4Br2FN02 + H]+: calc. = 311.8671, measured = 311.8666.
Piperazine Nitro-HCl:
[0086] To a mechanically stirred toluene solution (9 volumes) of FN-Bromide (prepared from previous step) in a 60 L reactor at 22 °C under an atmosphere of nitrogen,
diisopropylethylamine was charged (1.90 kg, 14.69 mol, 1.14 equiv.). To this mixture a solution of piperazine carboxylate methylester (Piperazine Carboxylate) (2.03 kg, 14.05 mol, 1.09 equiv.) in toluene (1.0 L, 0.5 volumes) was added at a rate allowing the batch temperature to stay under 30.0 °C (Exothermic. During the addition, jacket temperature was adjusted to 5 °C in order to maintain batch temperature below 30 °C. The mixture was agitated at 22 °C for 3 hours and analysis of an aliquot confirmed completion of the alkylation reaction (<1.0 LCAP FN-Bromide, HPLC254 nm). The reaction mixture was treated with aqueous NH4C1 (20 wt%, 10.0 L, 5 volumes; prepared from 2.0 kg of NH4C1 and 10.0 L of DI water), the biphasic mixture was agitated (30 min), and the layers were separated. The organic layer was sequentially washed with aqueous NaHC03 (9 wt%, 10.0 L, 5 volumes; prepared from 0.90 kg of NaHC03 and 10.0 L of DI water). The organic layer was filtered through a 5 μιη Teflon® cartridge line and transferred in a drum, washed the filter line with another 1.0 L toluene and the combined toluene solution (10.0 volumes) weighed, and assayed (HPLC) to quantify Piperazine Nitro free base. The assay yield for the Piperazine Nitro-freebase is 89.0%, FN-Toluene 2.5% and FN-Bromide 0.2% with FN-Bromide undetected. The total loss of product to the aqueous washes is < 1.0 %. This solution under nitrogen atmosphere is stable for more than 12h.
[0087] To a mechanically stirred toluene solution of Piperazine Nitro free base, prepared as described above, at 22 °C in a 60 L reactor under an atmosphere of nitrogen, IPA (19.4 L, 9.7 volumes) and DI water (1.0 L, 0.5 volume) were charged. The mixture was heated to 55 °C and 20% of the 1.4 equiv. of cone. HCl (Titrated prior to use and charge based on titer value; 276.0 mL, 3.21 mol) was charged. The contents were agitated for 15 min and
Piperazine Nitro-HCl seed (130.0 g, 0.39 mol, 0.03 equiv.) was charged as slurry in IPA (400 mL, 0.2 volume). The mixture was agitated for 30 min and the remaining cone. HCl (80% of the charge, 1.10 L, 12.82 mol) was added over a period of 4 hours. The mixture was stirred at 55 °C for 1 h, cooled to 20 °C in a linear manner over 1.5 hours, and agitated at this temperature for 12 hours. The supernatant concentration of Piperazine Nitro-HCl was measured (2.8 mg/g). The mixture was filtered through an aurora filter equipped with a 5 μιη Teflon® cloth. The mother liquor were transferred to a clean drum and assayed. The filter cake was washed twice with IPA (11.2 L, 5.6 volumes) and dried to constant weight (defined as < 1.0% weight loss for 2 consecutive TGA measurements over a period of 2 hours) on filter with vacuum and a nitrogen sweep (14 h). The combined losses of Piperazine Nitro- HCl in the mother liquors and the washes were 2.5 %. Piperazine Nitro-HCl was isolated 3.59 kg in 87.6% corrected yield with >99.5 wt% and 99.0% LCAP purity.
[0088] Methyl 4-(2-fluoro-3-nitrobenzyl)piperazine-l-carboxylate hydrochloride
(Piperazine Nitro-HCl): 1H NMR (300 MHz, DMSO-J) δ ppm 3.25 (br. s, 3 H), 3.52-3.66 (m, 8 H), 4.47 (s, 2 H), 7.44-7.63 (t, 1 H, J = 8 Hz), 7.98-8.15 (m, 1 H), 8.17-8.34 (m, 1 H). 13C NMR (75 MHz, DMSO-J) 5 ppm 50.3, 51.4, 52.8, 119.6 (d, J = 14 Hz), 125.1 (d, J = 5 Hz), 127.9, 137.4 (d, J = 8 Hz), 139.8 (d, J = 3 Hz), 152.2, 154.7, 155.7. DSC: melt onset at 248.4 °C. Exact Mass [Q3H16FN3O4 + H]+: calculated = 298.1203, measured = 298.1198. lternative processes for the synthesis of Piperazine Nitro:
2-fluoro-3-nitrobenzoic acid (2-fluoro-3-nitrophenyl)metlianol 2-fluoro-3-nitrobenzy? methanesulfonate
methyl 4-(2-fluoro-3-nitrobenzyl)piperazine-l -carboxylate hydrochloride
[0089] A mixture of NaBH4 ( 1.7 g, 44 mmol) in THF (68 mL) was treated 2-fluoro-3- nitrobenzoic acid (3.4 g, 18.4 mmol) and cooled to 0-5 °C. A solution of iodine (4.7 g, 18.4 mmol) in THF (12 mL) was then added drop wise at a rate to control off-gassing. The progress of the reaction was assessed by HPLC. After 2 hours HPLC assay indicated 4% AUC of 2-fluoro-3-nitrobenzoic acid remained. The mixture was quenched into 1 M HCl (30 mL) and extracted with MTBE (5 mL). The organics were then washed with 20% aqueous KOH solution and 10% sodium thiosulfate. The organics were dried with Na2S04, filtered over Celite and concentrated to afford (2-fluoro-3-nitrophenyl)methanol (2.8 g, 88%, 89% AUC by HPLC).
[0090] A solution of (2-fluoro-3-nitrophenyl)methanol (2.8 g, 16 mmol) in 2-MeTHF (26 mL) was treated with triethylamine (4.5 mL, 32 mmol) and cooled to 0-5 °C. The solution was then treated with methanesulfonyl chloride (1.6 mL, 21 mmol). The progress of the reaction was assessed by HPLC. After 30 minutes at 0-5 °C, the reaction was deemed complete. The mixture was quenched with water (14 mL) and the phases were separated. The organics were washed with brine, dried with Na2S04, filtered over Celite and
concentrated to afford 2-fluoro-3-nitrobenzyl methanesulfonate (3.3 g, 83.1%, 81% AUC by HPLC) as a yellow oil.
[0091] A solution of 2-fluoro-3-nitrobenzyl methanesulfonate (3.3 g, 13 mmol, AMRI lot # 46DAT067B) in toluene (33 mL), was treated with diisopropylethylamine (2.7 mL, 15 mmol) in one portion. A solution of methylpiperazine- 1 -carboxylate (2.1 g, 15 mmol) in toluene (1.1 mL) was added slowly via syringe to maintain between 23-29 °C. The reaction was stirred for 16 hours following the addition. An HPLC assay after this time showed that the reaction was complete. 20% Aqueous NH4C1 (11 mL) was added at 20-25 °C. The biphasic mixture was stirred for 15 minutes, and the phases were separated. This process was repeated using 9% aqueous sodium bicarbonate (11 mL). The toluene layer was then filtered over Celite at 20-25 °C. 2-propanol (50 mL) and water (1.1 mL) were added to the toluene solution and the mixture heated to 55-60 °C. The mixture was then treated with 37wt% HC1 (1.6 mL, 18.7 mmol) over 20 minutes. A precipitate was noted following the addition. When the addition was complete, the mixture was allowed to cool gradually to 20-25 °C and was stirred for hours before filtering and washing with IPA (2 bed volumes).
[0092] The cake was then dried at under vacuum to afford 4-(2-fluoro-3- nitrobenzyl)piperazine-l-carboxylate hydrochloride (2.41 g, 54%, 90% AUC by HPLC, 88 wt% by HPLC).
Piperazine Nitro Freebase:
[0093] In a 60 L reactor equipped with a reflux/return condenser, a mixture of Piperazine Nitro-HCl (2.0 kg, 5.99 mol, 1.0 equiv.) and isopropyl acetate (6.0 L, 3.0 volumes) was mechanically agitated at ambient temperature under an atmosphere of nitrogen. A solution of sodium bicarbonate (629 g, 7.49 mol, 1.25 equiv.) and water (7.5 L, 3.75 volume), prepared in separate vessel, was added. The biphasic mixture was agitated (15 min), and the layers were separated. The upper organic layer (containing product) was transferred to a separate vessel while the reactor was rinsed with water and isopropanol. The organic layer was then transferred through an inline 5 μιη Teflon® cartridge back into the clean 60 L reactor. The filter line was washed with 4.0 L (2.0 volumes) of isopropanol into the 60 L reactor. An additional 12.0 L (6.0 volumes) of isoproponal was added to the 60 L reactor and heated to 40 °C. Under reduced pressure (50 torr) the batch was concentrated down to approximately 6 L (3.0 volumes). The solution was cooled from 27 °C to 20 °C in a linear manner over 10 minutes. Water (4.0 L, 2.0 volumes) was added at 20 °C over 30 minutes followed by Piperazine Nitro Freebase seed (18 g, 0.06 mol, 0.01 equiv). The mixture was aged for 5 minutes and the remaining water (24.0 L, 12.0 volumes) was added over 90 minutes. After holding overnight at 20 °C, the supernatant concentration of Piperazine Nitro Freebase was measured (< 10 mg/mL). The mixture was filtered through an aurora filter equipped with a 12 μιη Teflon® cloth. The filter cake was washed with a mixture of water (3.3 L, 1.65 volumes) and isopropanol (700 mL, 0.35 volumes) and dried to constant weight (defined as < 1.0% weight loss for 2 consecutive TGA measurements over a period of 2 hours) on filter with vacuum and a nitrogen sweep (48 h). The combined losses of Piperazine Nitro Freebase in the mother liquors and the wash were aproximately 7.5 %. Piperazine Nitro Freebase was isolated 1.67 kg in 92.5% corrected yield with 100.0 wt% and 99.4% LCAP purity.
Synthesis of the API SM Phenyl Carbamate-HCl
Amino Pyridine Phenyl Carbamate-HCl
[0094] A 60 L, glass-lined, jacketed reactor set at 20 °C under nitrogen atmosphere and vented through a scrubber (containing 5N NaOH) was charged with 2.5 kg of Amino
Pyridine (1.0 equiv, 23.1 moles), followed by 25 L (19.6 kg, 10 vol) acetonitrile. After initiating agitation and (the endothermic) dissolution of the Amino Pyridine, the vessel was charged with 12.5 L of N-methyl-2-pyrolidinone (12.8 kg, 5 vol). An addition funnel was charged with 1.8 L (0.6 equiv, 13.9 moles) phenyl chloroformate which was then added over 68 minutes to the solution of the Amino Pyridine keeping the internal temperature < 30°C. The reaction was agitated for > 30 minutes at an internal temperature of 20 ± 5 °C. The vessel was then charged with 61 ± 1 g of seed as a slurry in 200 mL acetonitrile and aged for > 30 min. The addition funnel was charged with 1.25 L (0.45 equiv, 9.7 moles) of phenyl chloroformate which was then added over 53 minutes to the reaction suspension while again keeping the temperature < 30°C. The contents of the reactor were aged > 30 hours at 20 ± 5°C. After assaying the supernatant (< 15mg/g for both product and starting material), the solids were filtered using an Aurora filter equipped with a 12μιη Teflon cloth. The mother liquor was forwarded to a 2nd 60 L, glass-lined, jacketed reactor. The reactor and cake were rinsed with l x lO L of 5: 10 NMP/ ACN and 1 x 10 L ACN. The washes were forwarded to the 2nd reactor as well. The cake was dried under vacuum with a nitrogen bleed for > 24 hours to afford 5.65 kg (90.2% yield) of the product, Phenyl Carbamate-HCl as an off-white solid in 98.8 wt% with 99.2% LCAP purity.
[0095] Phenyl (6-methylpyridin-3-yl)carbamate hydrochloride (Phenyl Carbamate-HCl) 1H NMR (400 MHz, DMSO-J6) 5 ppm 11.24 (s, 1 H), 8.81 (s, 1 H), 8.41 (d, 1 Η, / = 8.8 Hz), 7.85 (d, l H, / = 8.8 Hz), 7.48 – 7.44 (m, 2 H), 7.32 – 7.26 (m, 3 H), 2.69 (s, 3 H); 13C NMR (100 MHz, DMSO- ) δ ppm 151.66, 150.01, 147.51, 136.14, 133.79, 129.99, 129.49, 127.75, 125.87, 121.70, 18.55: HR-MS : Calculated for Cuii W . 228.0899, M + H+ = 229.0972; Observed mass: 229.0961
Alternative Synthesis of Phenyl Carbamate HC1
[0096] 5-Amino-2-methylpyridine (53.2 kg, 1.0 equiv) and acetonitrile (334 kg, 8.0 mL/g) were charged to a nitrogen flushed glass-lined reactor. The contents of the reactor were stirred while warming to 25-30 °C. The mixture was then recirculated through a filter packed with activated carbon (11 kg, 20 wt ) for 3 h intervals while maintaining 25-30 °C.
Following each 3 h interval, a sample of the mixture was analyzed for color by comparison to a color standard and UV Absorbance at 440nm. Once a satisfactory result was achieved, the filter was blown out into the reactor and the filter was rinsed with acetonitrile (85 kg, 2.0 mL/g). The acetonitrile rinse was transferred into the reaction mixture. l-Methyl-2- pyrrolidinone (274 kg, 5.0 mL/g) was charged to the reaction mixture in the glass-lined reactor. Phenyl chloroformate (46.6 kg, 0.6 equiv) was slowly added to the mixture while maintaining 15-30 °C (typically 60-70 min). The reaction mixture was stirred for approximatly 60 minutes while maintaining 20-25 °C. Phenyl(6-methylpyridin-3- yl)carbamate hydrochloride (0.58 kg, 0.010 equiv) seed crystals were charged to the stirring mixture. The slurry was then stirred for approximatly 4 h at 20+ 5°C. Phenyl chloroformate (33.4 kg, 0.45 equiv) was slowly added to the slurry while maintaining 15-30 °C. The mixture was then allowed to age while stirring for 8+1 h whereupon concentration of 5- amino-2-methylpyridine (target <15 mg/mL) and phenyl (6-methylpyridin-3-yl)carbamate hydrochloride (target <15 mg/mL) were checked by HPLC. The batch was then filtered under vacuum and washed with a mixture of acetonitrile (112 kg, 2.68 mL/g) and l-methyl-2- pyrrolidinone (72 kg, 1.32 mL/g) followed by washing thrise with acetonitrile (167 kg, 4.0 mL/g). The solids were deliquored followed by transfering to a tray dryer maintained between 20-40°C and 1.3-0.65 psia until an LOD of <lwt was achieved, whereupon phenyl(6-methylpyridin-3-yl)carbamate hydrochloride 106.3 kg (81.6% yield) was isolated from the dryer. Methyl 4-(3-amino-2-fluorobenzyl)piperazine-l-carboxylate (Piperazine Aniline)
Neutralization
Piperazine NitrcHCI
+ NaCI (1 equiv)
+ C02 (1 equiv)
+ H20 (1 equiv)
+ NaHC03 (0.25 equiv)
[0097] To a 100-L jacketed glass-lined reactor were added methyl 4-(2-fluoro-3- nitrobenzyl)piperazine-l-carboxylate hydrochloride (2.00 kg, 1.00 equiv) and isopropyl acetate (6.00 L, 3.00 Vol with-respect to starting material). The resulting slurry was agitated under a nitrogen sweep. To the mixture was added dropwise over 45 + 30 min: 7.7 % w/w aqueous sodium bicarbonate solution (629 g, 1.25 equiv of sodium bicarbonate dissolved in 7.50 L water), maintaining an internal temperature of 20 + 5 °C by jacket control (NOTE: addition is endo thermic, and may evolve up to 1 equiv of carbon dioxide gas). The mixture was stirred for > 15 min, resulting in a clear biphasic mixture. Agitation was stopped and the layers were allowed to settle.
[0098] The bottom (aqueous) layer was drained and analyzed by pH paper to ensure that the layer is pH > 6. Quantititative HPLC analysis of the upper (organic) layer revealed 97- 100% assay yield of the methyl 4-(2-fluoro-3-nitrobenzyl)piperazine-l-carboxylate freebase (1.73 – 1.78 kg). The upper (organic) layer was transferred through an in-line filter into a 20- L Hastelloy® hydro genator, and the 100-L reactor and lines were rinsed with an additional aliquot of isopropyl acetate (2.00 L, 1.00 Vol). The hydrogenator was purged with nitrogen and vented to atmospheric pressure. To the reaction mixture was added a slurry of 5.0 wt% palladium on carbon (20.0 g, Strem/BASF Escat™ 1421, approx 50% water) in isopropyl acetate (400 mL), followed by a 400 mL rinse. The resulting reaction mixture was diluted with an additional aliquot of isopropyl acetate (1.2 L; total isopropyl acetate amount is 10.0 L, 5.00 Vol). The hydrogenator was purged three times with nitrogen (pressurized to 60 + 10 psig, then vented to atmospheric pressure), then pressurized to 60 + 5 psig with hydrogen. The reaction mixture was stirred at < 100 rpm at 30 + 5 °C while maintaining 60 + 5 psig hydrogen, for >2 hours until reaction was deemed complete. This temperature and pressure correspond to a measured kLa value of approx 0.40 in a 20-L Hydrogenator. End of reaction is determined by dramatic decrease in hydrogen consumption accompanied by a relief in the heat evolution of the reaction. To control potential dimeric impurities, the reaction is continued for at least 30 minutes after this change in reaction profile, and HPLC analysis is performed to confirm that >99.5% conversion of the hydroxyl-amine to the aniline is achieved.
[0099] At the end of reaction, the hydrogenator was purged with nitrogen twice
(pressurized to 60 + 10 psig, then vented to atmospheric pressure). The crude reaction mixture was filtered through a 5 μιη filter followed by a 0.45 μιη filter in series, into a 40-L glass-lined reactor. The hydrogenator and lines were washed with an additional aliquot of isopropyl acetate (2.00 L). Quantitative HPLC analysis of the crude reaction mixture revealed 95-100% assay yield (1.52 – 1.60 kg aniline product). The reaction mixture was distilled under reduced pressure (typically 250 – 300 mbar) at a batch temperature of 50 + 5 °C until the total reaction volume was approximately 8.00 L (4.00 Vol). The batch was subjected to a constant-volume distillation at 50 + 5 °C, 250 – 300 mbar, by adding heptane to control the total batch volume. After approximately 8.00 L (4.00 Vol) of heptane were added, GC analysis indicated that the solvent composition was approximately 50 % isopropyl acetate, 50% heptane. Vacuum was broken, and the internal batch temperature was maintained at 50 + 5 °C. To the reaction mixture was added a slurry of seed (20.0 grams of product methyl 4-(3-amino-2-fluorobenzyl)piperazine-l-carboxylate, in a solvent mixture of 80 mL heptane and 20 mL isopropyl acetate). The resulting slurry was allowed to stir at 50 + 5 °C for 2 + 1 hours, then cooled to 20 + 5 °C over 2.5 + 1.0 h. Additional heptane (24.0 L, 12.0 Vol) was added dropwise over 2 hours, and the batch was allowed to stir at 20 + 5 °C for > 1 hours (typically overnight). Quantitative HPLC analysis of this filtered supernatant revealed < 5 mg/mL product in solution, and the product crystals were 50 – 400 μιη birefringent rods. The reaction slurry was filtered at 20 °C onto a filter cloth, and the cake was displacement-washed with heptane (6.00 L, 2.00 Vol). The cake was dried on the filter under nitrogen sweep at ambient temperature for > 4 hours, until sample dryness was confirmed by LOD analysis (indicated <1.0 wt% loss). The product methyl 4-(3-amino-2- fluorobenzyl)piperazine-l-carboxylate (1.56 kg) was isolated as a pale-yellow powder in 86% yield at 99.8 wt% by HPLC with 100.0 LCAP2i0. [Analysis of the combined filtrates and washes revealed 108 grams (7.0%) of product lost to the mother liquors. The remaining mass balance is comprised of product hold-up in the reactor (fouling).] 1H NMR (DMSO-Jg, 400 MHz) δ: 6.81 (dd, J = 7.53, 7.82 Hz, 1H), 6.67 (m, 1H), 6.49 (m, 1H), 5.04 (s, 2H), 3.58 (s, 3H), 3.45 (m, 2H), 3.34 (m, 4H), 2.33 (m, 4H). 19F NMR (d6-DMSO, 376 MHz) δ: – 140.2. 13C NMR (d6-DMSO, 125 MHz) δ: 155.0, 150.5, 148.2, 136.2 (m), 123.7 (m), 117.6, 115.1, 73.7, 54.9 (m), 52.1 (m), 43.4. mp = 89.2 °C.
Alternate route to Piperazine Aniline
[00100] To a jacketed glass-lined reactor were added methyl 4-(2-fluoro-3- nitrobenzyl)piperazine-l-carboxylate hydrochloride (46.00 kg, 1.00 equiv) and isopropyl acetate (200 kg, 5.0 mL/g). The resulting slurry was agitated under a nitrogen sweep. To the mixture was added 7.4 % w/w aqueous sodium bicarbonate solution (1.25 equiv) while maintaining an internal temperature of 25 + 5 °C. The mixture was agitated for > 30 min, resulting in a clear biphasic mixture. Agitation was stopped and the bottom (aqueous) layer was discharged. Analysis of aqueous layer indicates pH >6. Water (92 kg, 2.0 mL/g) was charged the organic layer and agitated for >15 min. Agitation was then stopped and the bottom (water wash) layer was discharged. Water (92 kg, 2.0 mL/g) was charged the organic layer and agitated for > 15 min. Agitation was then stopped and the bottom (water wash) layer was discharged. The batch was distilled under reduced pressure while maintaining the batch temperature between 40-50 °C. The batch volume was held constant throughout the distillation by the continuous addition of isopropyl acetate. Once the water content of the batch was < 1,500 ppm, the solution was passed through an inline filter into a Hastelloy reactor containing 5.0 wt% palladium on carbon (BASF Escat 1421, 0.69 kg, 1.5 wt%). The jacketed glass-lined reactor was rinsed with isopropyl acetate (100 kg, 2.5 mL/g) and added to the Hastelloy reactor though the inline filter.
[00101] The batch was adjusted to approximately 25-35 °C (preferably 30 °C) and hydrogen gas was added to maintain about 4 barg with vigorous agitation. Hydrogenation was continued for 1 h after hydrogen uptake has ceased, and >99.0% conversion by HPLC were achieved. The palladium on carbon catalyst was collected by filtration and the supernatant was collected in a reactor. Isopropyl acetate (40 kg, 1.0 mL/g) was charged to the Hastelloy reactor and transferred through the filter and collected in the jacketed glass-lined reactor.
[00102] The batch was concentrated under reduced pressure while maintaining the batch temperature between 35-55 °C until the final volume was approximately 4.0 mL/g. Heptane (219 kg, 7.0 mL/g) was added to the jacketed glass-lined reactor while maintaining the batch between 50-60 °C, until 20-25% isopropyl acetate in heptane was achieved as measured by GC. The solution was cooled to between 40-50 °C and seeded with methyl 4-(3-amino-2- fluorobenzyl)piperazine-l-carboxylate (0.46 kg, 1.0 wt%) as a slurry in heptane (6.4 kg, 0.20 mL/g). The slurry was aged for approximately 2 h, whereupon, the batch was distilled under reduced pressure while maintaining the batch temperature between 35-45 °C. The batch volume was held constant throughout the distillation by the continuous addition of heptane (219 kg, 7.0 mL/g). The batch was then cooled to between 15-25 °C over approximately 3 h. Concentration of the supernatant was measured to be <5 mg/mL methyl 4-(3-amino-2- fluorobenzyl)piperazine-l-carboxylate by HPLC.
[00103] The batch was filtered and the resulting solids were successively washed with heptane (63 kg, 2.0 mL/g) then heptane (94 kg, 3.0 mL/g). The solids were dried on the filter with a stream of dry nitrogen with vacuum until an LOD of <_lwt% was achieved whereupon 33.88 kg (90.7% yield) was isolated from the filter dryer.
Omecamtiv Mecarbil Dihydrochloride Hydrate procedure
f lu
1) 2-PrOH (11 V)
2) Distill to 4V
3) Water (2.30 V)
4) 6N HCI (2.4 equiv)
5) 2-PrOH (16.5V)
6) Wet Mill
[00104] To a 15L glass lined reactor were charged methyl 4-(3-amino-2-fluoro- benzyl)piperazine-l-carboxylate (1,202 g, 4.50 mol), phenyl (6-methylpyridin-3- yl)carbamate hydrochloride (1,444 g, 5.40 mol), and tetrahydrofuran (4.81 L). The resulting slurry was agitated under a nitrogen sweep and N,N-diisopropylethylamine (1,019 L, 5.85 mol) was then charged to the slurry which resulted in a brown solution. The temperature of the solution was increased to 65 °C and agitated for 22 h, until <1% AUC piperazine aniline remained by HPLC analysis.
[0100] The batch was cooled to 50 °C and distilled under reduced pressure while maintaining the internal temperature of the vessel below 50 °C by adjusting vacuum pressure. 2-Propanol was added with residual vacuum at a rate to maintain a constant volume in the 15 L reactor. A total of 10.5 kg of 2-propanol was required to achieve <5% THF by GC. Water (2.77 kg) was then charged to the reactor followed by the addition of 6N HC1 (1.98 kg) at a rate to maintain the internal temperature below 60 °C. The reactor was brought to ambient pressure under a nitrogen sweep. The solution was then heated to 60 °C, and transferred to a 60L glass lined reactor through an inline filter. The 15L reactor was then rinsed with 1: 1 water/2-propanol (1.2L) which was sent through the inline filter to the 60L reactor.
[0101] The 60L reactor was adjusted to 45 °C and a slurry of seed (114 g, 0.23 mol) in 2- propanol (0.35 L) was added to the reactor resulting in a slurry. The batch was aged at 45 °C for 1 h, followed by the addition of 2-propanol (3.97 kg) through an inline filter over 2 h. The batch was heated to 55°C over 1 h and held for 0.25 h, then cooled back to 45°C over 1 h and held overnight at 45 °C. 2-propanol (11.71 kg) was then added through an inline filter to the batch over 3 h. The batch was aged for 1 h and then cooled to 20°C over 2 h and held at 20 °C for 0.5 h. The batch was then recirculated though a wet mill affixed with 1-medium and 2- fine rotor-stators operating at 56 Hz for 2.15 h, until no further particle size reduction was observed by microscopy.
[0102] The batch was then filtered through a 20″ Hastelloy® filter fitted with a 12 urn filter cloth under 500 torr vacuum. A wash solution of 95:5 2-propanol:water (1.82 L) was charged through an inline filter to the 60L reactor, then onto the filter. A second wash of 2- propanol (2.85L) was charged through an inline filter to the 60L reactor, then onto the filter. The batch was then dried under 5 psi humidified nitrogen pressure until <5,000 ppm 2- propanol, and 2.5-5% water remained. The final solid was discharged from the filter to afford 2.09 kg of methyl 4-(2-fluoro-3-(3-(6-methylpyridin-3-yl)ureido)benzyl)piperazine-l- carboxylate as an off-white crystalline solid in 89% yield at 99.88 wt% by HPLC, 100.0% AUC. Total losses to liquors was 0.10 kg (4.7%).
[0103] DSC: Tonset = 61.7 °C, Tmax = 95.0 °C; TGA = 2.2%, degradation onset = 222 °C; 1H HMR (D20, 500 MHz) δ 8.87 (s, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.83 (t, J = 1.5 Hz, 1H), 7.71 (d, J = 8.8 Hz, 1H), 7.35-7.29 (m, 2H), 4.48 (s, 2H), 4.24 (br s, 2H), 3.73 (s, 3H), 3.31 (br s, 6H), 2.68 (s, 3H); 13C HMR (D20, 150 MHz) δ 156.8, 154.2, 153.9 (J = 249 Hz), 147.8, 136.3, 136.1, 130.1, 129.4, 128.0, 127.2, 125.5 (J = 11.8 Hz), 125.1 (J = 4.2 Hz), 116.1 (J = 13.5 Hz), 53.54, 53.52, 53.49, 50.9, 40.5, 18.2.
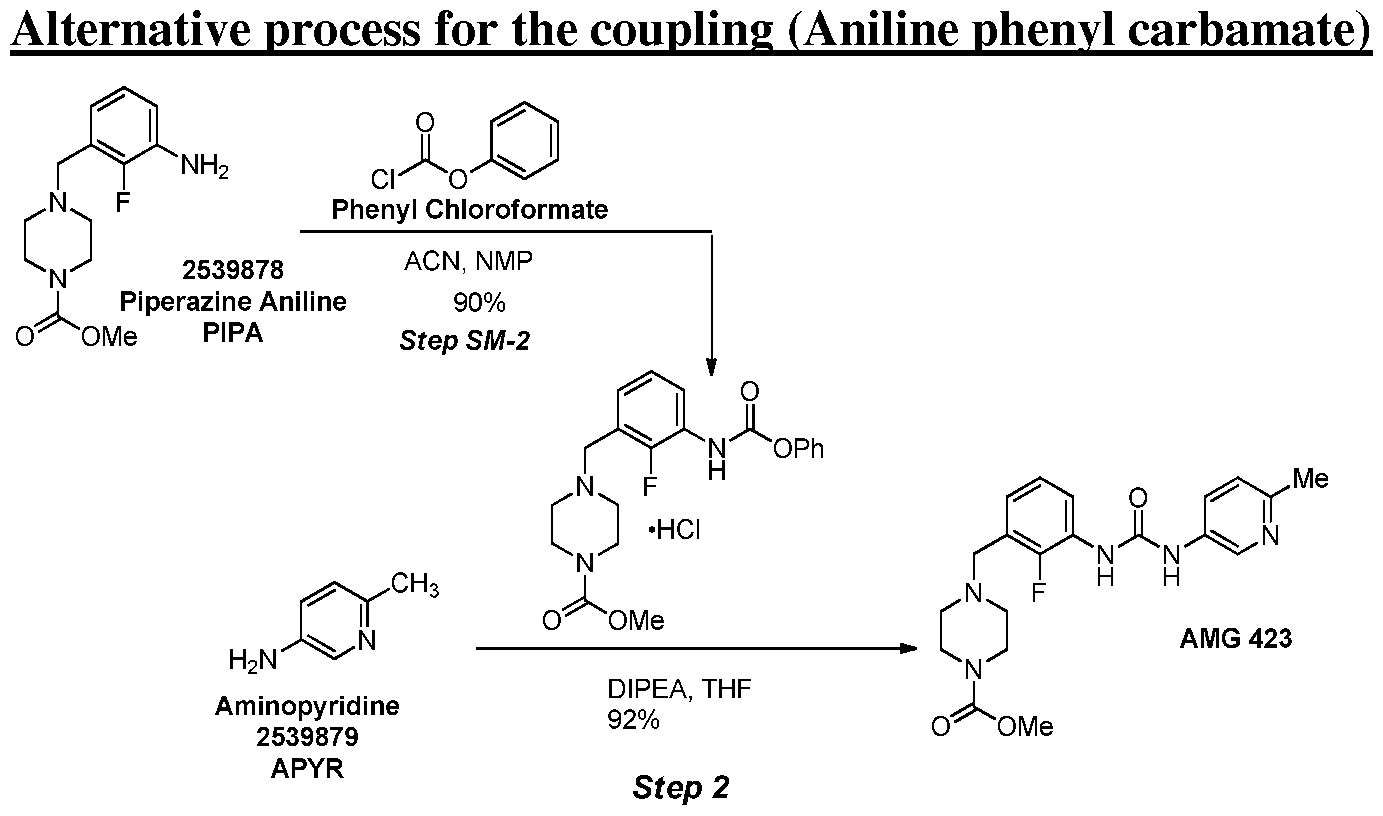
[0104] A reaction vessel was charged methyl 4-(3-amino-2-fluorobenzyl)piperazine-l- carboxylate (2.5 g, 1.0 equiv), acetonitrile (25.0 mL, 10.0 mL/g) and l-methyl-2- pyrrolidinone (12.5 mL, 5.0 mL/g). The batch was cooled to 0 °C whereupon phenyl chloroformate (1.20 mL, 1.02 equiv) was added over approximately 5 min. After 45 minutes the resulting slurry resulted was allowed to warm to 20 °C. The solids were collected by filtration and rinsed twice with acetonitrile (10.0 mL, 4.0 mL/g). The solids were dried under a stream of dry nitrogen to afford methyl 4-(2-fluoro-3-
((phenoxycarbonyl)amino)benzyl)piperazine- l -carboxylate hydrochloride 2.8 g (71 % yield) as a white solid.
[0105] 4-(2-fluoro-3-((phenoxycarbonyl)amino)benzyl)piperazine-l-carboxylate hydrochloride: 1H NMR (400 MHz, DMSO-J6) δ ppm 3.08 (br. s., 2 H), 3.24 – 3.52 (m, 4 H), 3.62 (s, 3 H), 4.03 (d, J=11.25 Hz, 2 H), 4.38 (br. s., 2 H), 7.11 – 7.35 (m, 4 H), 7.35 – 7.49 (m, 2 H), 7.49 – 7.66 (m, 1 H), 7.80 (s, 1 H), 10.12 (br. s, 1 H), 11.79 (br. s, 1 H); HRMS = 388.1676 found, 388.1667 calculated. [0106] A reaction vessel was charged methyl 4-(2-fluoro-3-
((phenoxycarbonyl)amino)benzyl)piperazine-l-carboxylate hydrochloride (0.50 g, 1.0 equiv), 6-methylpyridin-3-amine (0.15 g, 1.2 equiv), tetrahydrofuran (2.0 mL, 4.0 mL/g) and
N,N-diisopropylethylamine (0.23 mL, 1.1 equiv). The batch was heated to 65 °C for 22 h, whereupon quantitative HPLC analysis indicated 0.438 g (92% assay yield) of omecamtiv mecarbil.
Alternative Omecamtiv Mecarbil Dihydrochloride Hydrate procedure
[0107] Omecamtiv Mecarbil, free base (3.0 kg, 1.0 equiv) was charged to a nitrogen purged jacketed vessel followed by water (4.6 L, 1.5 mL/g) and 2-propanol (6.1 L, 2.60 mL/g). The slurry was agitated and heated to approximately 40 °C, whereupon 6N HC1 (2.6 L, 2.10 equiv) was charged to the slurry resulting in a colorless homogenous solution. The solution was heated to between 60-65 °C and transferred through an inline filter to a 60L reactor pre -heated to 60 °C. The batch was cooled to 45 °C whereupon Omecamtiv Mecarbil dihydrochloride hydrate (150 g, 5.0 wt%) was charged to the vessel as a slurry in 95:5 (v/v) 2-Propanol/Water (600 mL, 0.20 mL/g). The resulting slurry was maintained at 45 °C for 0.5 h followed by cooling to approximately 20 °C then held for 3-16 h. 2-Propanol (33.0 L, 11.0 mL/g) was added over >2h followed by a >1 h isothermal hold at approximately 20 °C.
(Supernatant pH <7).
[0108] The batch was recirculated through a wet mill for 5-10 batch turnovers until sufficient particle reduction was achieve as compared to offline calibrated visual microscopy reference. The slurry was filtered by vacuum and the resulting solids were washed with two washes of 95:5 (v/v) 2-Propanol/Water (3.0 L, 1.0 mL/g) and a final cake wash with 2- Propanol (6.0 L, 2.0 mL/g). The cake was dried on the filter by pushing humidified nitrogen through the cake until <5,000 ppm 2-propanol and 2.5-5% water were measured by GC and KF analysis, respectively. Omecamtiv Mecarbil dihydrochloride hydrate was isolated as a colorless crystalline solid (3.40 kg, 93% yield). pH dependent release profiles
CLIP
J Am Chem Soc. 2012 July 11; 134(27): 11132–11135. doi:10.1021/ja305212v.

CLIP
/////////////Omecamtiv mecarbil, オメカムティブメカビル , AMG 423, AMG-423, CK1827452, CK-1827452, K1827452, Cladribine, PHASE 3

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



























