
Home » Posts tagged 'chronic lymphocytic leukemia'
Tag Archives: chronic lymphocytic leukemia
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
CC-90009

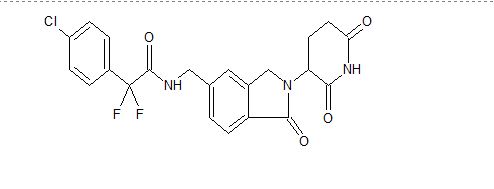
![2-(4-Chlorophenyl)-N-[[2-(2,6-dioxopiperidin-3-yl)-1-oxo-3H-isoindol-5-yl]methyl]-2,2-difluoroacetamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=118647211&t=l)
CC-90009
CC-90009-AML-001
CAS 1860875-51-9
461.8 g/mol, C22H18ClF2N3O4
2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide
- 4-Chloro-N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α,α-difluorobenzeneacetamide
- Benzeneacetamide, 4-chloro-N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α,α-difluoro-
Phase 1 Clinical, Acute myelogenous leukemia, Protein cereblon modulator
Useful for treating chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia or acute myeloid leukemia.
Celgene is developing CC-90009, a cereblon E3 ligase modulator, for treating AML; in January 2019, data from a phase I trial were expected later that year.
- 0iginator Celgene Corporation
- Class Antineoplastics
- Mechanism of Action CRBN protein modulators; Ubiquitin protein ligase complex modulators
- Phase I Acute myeloid leukaemia
- 28 Mar 2019 No recent reports of development identified for clinical-Phase-Unknown development in Acute-myeloid-leukaemia in USA (IV)
- 01 Sep 2016 Phase-I clinical trials in Acute myeloid leukaemia (Second-line therapy or greater) in Canada (IV) (NCT02848001)
- 04 Aug 2016 Celgene plans a phase I trial for Acute Myeloid Leukaemia in USA and Canada (NCT02848001)
In September 2016, Celgene initiated a phase I dose-finding trial of CC 90009 in patients with relapsed or refractory acute myeloid leukaemia (NCT02848001; CC-90009-AML-001). The open-label study intends to enrol 60 patients in the US and Canada
CC-90009 is a cereblon modulator. CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. .
Development Overview
cereblon modulator CC-90009A modulator of cereblon (CRBN), which is part of the cullin 4-RING E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase; CUL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and pro-apoptotic activities. Upon administration, CC-90009 specifically binds to CRBN, thereby affecting the activity of the ubiquitin E3 ligase complex. This leads to the ubiquitination of certain substrate proteins and induces the proteasome-mediated degradation of certain transcription factors, including Ikaros (IKZF1) and Aiolos (IKZF3), which are transcriptional repressors in T-cells. This reduces the levels of these transcription factors, and modulates the activity of the immune system, which may include the activation of T-lymphocytes. In addition, this downregulates the expression of other proteins, including interferon regulatory factor 4 (IRF4) and c-myc, which plays a key role in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins. Check for active clinical trials using this agent. (NCI Thesaurus)

WO 2017120446,
PATENT
WO2016007848
US 20170348298
WO 2017120415
WO 2017120446
WO 2017120437
PATENT
WO2017214014
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017214014&tab=PCTDESCRIPTION
Provided herein are methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy with 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of
stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof. Further provided is a compound for use in methods of treating, preventing, managing, and/or ameliorating a hematologic malignancy, wherein the compound is 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide or a stereoisomer or a mixture of stereoisomers, an isotopologue, pharmaceutically acceptable salt, tautomer, solvate, hydrate, co-crystal, clathrate, or polymorph thereof.
The term Compound 1 refers to”2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide” having the structure:
and its stereoisomers or mixture of stereoisomers, isotopologues, pharmaceutically acceptable salts, tautomers, solvates, hydrates, co-crystals, clathrates, or polymorphs thereof. In certain embodiments, Compound 1 refers to 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide and its tautomers. In certain embodiments, Compound 1 refers to a polymorph of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-
oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In certain embodiments, Compound 1 refers to polymorph Form C of 2-(4-chlorophenyl)-N-((2-(2,6-dioxopiperidin-3-yl)-l-oxoisoindolin-5-yl)methyl)-2,2-difluoroacetamide. In one embodiment, the stereoisomer is an enantiomer.
PATENT
WO-2019136016
Novel isotopologs of the compound presumed to be CC-90009 , processes for their preparation and compositions comprising them are claimed.



| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2017199193 | METHODS FOR TREATING CANCER AND THE USE OF BIOMARKERS AS A PREDICTOR OF CLINICAL SENSITIVITY TO THERAPIES | 2017-01-06 | |
| US2018224435 | METHODS FOR MEASURING SMALL MOLECULE AFFINITY TO CEREBLON | 2018-02-02 | |
| US2018353496 | FORMULATIONS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2018-07-19 | |
| US2017196847 | FORMULATIONS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2017-01-06 | |
| US2017348298 | TREATMENT OF A HEMATOLOGIC MALIGNANCY WITH 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE | 2017-06-05 |
| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US2018221361 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2018-04-09 | |
| US9968596 | Antiproliferative compounds and methods of use thereof | 2017-10-02 | 2018-05-15 |
| US2017197934 | SOLID FORMS OF 2-(4-CHLOROPHENYL)-N-((2-(2,6-DIOXOPIPERIDIN-3-YL)-1-OXOISOINDOLIN-5-YL)METHYL)-2,2-DIFLUOROACETAMIDE, AND THEIR PHARMACEUTICAL COMPOSITIONS AND USES | 2017-01-06 | |
| US9499514 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2015-07-09 | 2016-01-14 |
| US9808451 | ANTIPROLIFERATIVE COMPOUNDS AND METHODS OF USE THEREOF | 2016-09-23 |
////////CC-90009 , CC 90009 , CC90009, chronic lymphocytic leukemia, chronic myelocytic leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, phase I, CANCER, CC-90009-AML-001
Clc1ccc(cc1)C(F)(F)C(=O)NCc2ccc3C(=O)N(Cc3c2)C4CCC(=O)NC4=O
FDA approves new drug Venclexta (venetoclax) for chronic lymphocytic leukemia in patients with a specific chromosomal abnormality

April 11, 2016
Release
The U.S. Food and Drug Administration today approved Venclexta (venetoclax) for the treatment of patients with chronic lymphocytic leukemia (CLL) who have a chromosomal abnormality called 17p deletion and who have been treated with at least one prior therapy. Venclexta is the first FDA-approved treatment that targets the B-cell lymphoma 2 (BCL-2) protein, which supports cancer cell growth and is overexpressed in many patients with CLL.
According to the National Cancer Institute, CLL is one of the most common types of leukemia in adults, with approximately 15,000 new cases diagnosed each year. CLL is characterized by the progressive accumulation of abnormal lymphocytes, a type of white blood cell. Patients with CLL who have a 17p deletion lack a portion of the chromosome that acts to suppress cancer growth. This chromosomal abnormality occurs in approximately 10 percent of patients with untreated CLL and in approximately 20 percent of patients with relapsed CLL.
“These patients now have a new, targeted therapy that inhibits a protein involved in keeping tumor cells alive,” said Richard Pazdur, director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For certain patients with CLL who have not had favorable outcomes with other therapies, Venclexta may provide a new option for their specific condition.”
The efficacy of Venclexta was tested in a single-arm clinical trial of 106 patients with CLL who have a 17p deletion and who had received at least one prior therapy. Trial participants took Venclexta orally every day, beginning with 20 mg and increasing over a five-week period to 400 mg. Results showed that 80 percent of trial participants experienced a complete or partial remission of their cancer.
Venclexta is indicated for daily use after detection of 17p deletion is confirmed through the use of the FDA-approved companion diagnostic Vysis CLL FISH probe kit.
The most common side effects of Venclexta include low white blood cell count (neutropenia), diarrhea, nausea, anemia, upper respiratory tract infection, low platelet count (thrombocytopenia) and fatigue. Serious complications can include pneumonia, neutropenia with fever, fever, autoimmune hemolytic anemia, anemia and metabolic abnormalities known as tumor lysis syndrome. Live attenuated vaccines should not be given to patients taking Venclexta.
The FDA granted the Venclexta application breakthrough therapy designation, priority review status, and accelerated approval for this indication. These are distinct programs intended to facilitate and expedite the development and review of certain new drugs in light of their potential to benefit patients with serious or life-threatening conditions. Venclexta also received orphan drug designation, which provides incentives such as tax credits, user fee waivers and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Venclexta is manufactured by AbbVie Inc. of North Chicago, Illinois, and marketed by AbbVie and Genentech USA Inc. of South San Francisco, California. The Vysis CLL FISH probe kit is manufactured by Abbott Molecular of Des Plaines, Illinois.
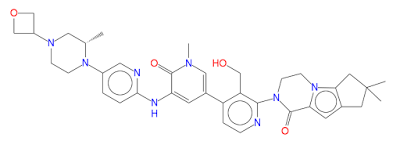
GDC 0853, Fenebrutinib
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
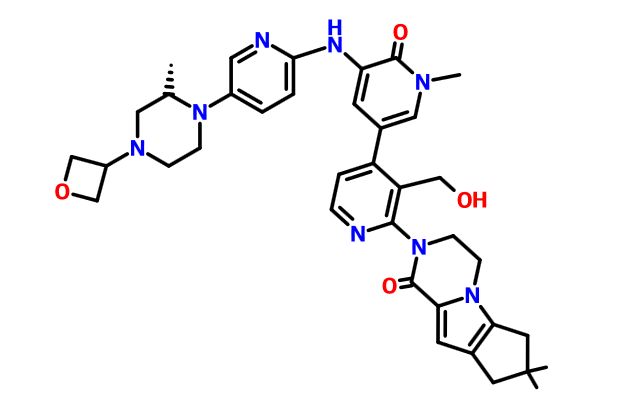
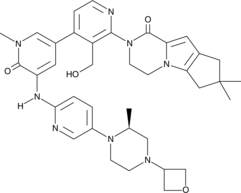
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
 , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† 
Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE



 Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
Infinity and AbbVie partner to develop and commercialise Duvelisib for cancer… for the treatment of chronic lymphocytic leukemia


Duvelisib
Infinity and AbbVie partner to develop and commercialise duvelisib for cancer
INK 1197; IPI 145; 8-Chloro-2-phenyl-3-[(1S)-1-(9H-purin-6-ylamino)ethyl]-1(2H)-isoquinolinone
1(2H)-Isoquinolinone, 8-chloro-2-phenyl-3-((1S)-1-(9H-purin-6-ylamino)ethyl)-
8-Chloro-2-phenyl-3-((1S)-1-(7H-purin-6-ylamino)ethyl)isoquinolin-1(2H)-one
(S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
UNII-610V23S0JI; IPI-145; INK-1197;
Originator…….. Millennium Pharmaceuticals
| Molecular Formula | C22H17ClN6O | |
| Molecular Weight | 416.86 | |
| CAS Registry Number | 1201438-56-3 |

Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma, to treat patients with cancer.
![]()
Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K gamma, to treat patients with cancer.
Duvelisib has shown clinical activity against different blood cancers, such as indolent non-Hodgkin’s lymphoma (iNHL) and chronic lymphocytic leukemia (CLL).
AbbVie executive vice-president and chief scientific officer Michael Severino said: “We believe that duvelisib is a very promising investigational treatment based on clinical data showing activity in a broad range of blood cancers.”
Duvelisib (IPI-145, INK-1197), an inhibitor of PI3K-delta and –gamma, originated at Takeda subsidiary Intellikine. It is now being developed by Infinity Pharmaceuticals, which began a phase III trial in November, following US and EU grant of orphan drug status for both CLL and small lymphocytic leukemia
INK-1197 is a dual phosphatidylinositol 3-Kinase delta (PI3Kdelta) and gamma (PI3Kgamma) inhibitor in phase III clinical development at Infinity Pharmaceuticals for the treatment of chronic lymphocytic leukemia and small lymphocytic lymphoma. The company is also carring phase II trials for the treatment of patients with mild asthma undergoing allergen challenge, for the treatment of rheumatoid arthritis and for the treatment of refractory indolent non-Hodgkin’s lymphoma. Phase I clinical trials for the treatment of advanced hematological malignancies (including T-cell lymphoma and mantle cell lymphoma) are currently under way.
IPI-145 is an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma. The PI3K-delta and PI3K-gamma isoforms are preferentially expressed in leukocytes (white blood cells), where they have distinct and non-overlapping roles in key cellular functions, including cell proliferation, cell differentiation, cell migration and immunity. Targeting PI3K-delta and PI3K-gamma may provide multiple opportunities to develop differentiated therapies for the treatment of blood cancers and inflammatory diseases.
Licensee Infinity Pharmaceuticals is developing INK-1197. In 2014, Infinity licensed Abbvie for joint commercialization in the U.S. and exclusive commercialization elsewhere. Originator Millennium Pharmaceuticals had also been developing the compound; however, no recent development has been reported for this research. In 2013, orphan drug designations were assigned by the FDA and the EMA for the treatment of chronic lymphocytic leukemia, for the treatment of small lymphocytic lymphoma and for the treatment of follicular lymphoma.
currently enrolling patients DYNAMO™, a Phase 2 study designed to evaluate the activity and safety of IPI-145 in approximately 120 people with refractory indolent non-Hodgkin lymphoma (iNHL) and DUO™, a Phase 3 clinical study of IPI-145 in approximately 300 people with relapsed/refractory chronic lymphocytic leukemia (CLL). These studies are supported by Phase 1 data reported at the 2013 American Society of Hematology (ASH) Annual Meeting which showed that IPI-145 was well tolerated and clinically active in a broad range of malignancies, including iNHL and CLL. These studies are part of DUETTS™, a worldwide investigation of IPI-145 in blood cancers.

WO 2011008302
http://www.google.com/patents/WO2011008302A1?cl=en
Reaction Scheme 1

Reaction Scheme 2:

201 202 203

204 205

Reaction Scheme 3:

Reaction Scheme 4A:

Reaction Scheme 4B:
2


Example 14b: Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (9)
(compound 4904)
Scheme 27b. The synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (9)
(compound 4904) is described.

[00493] The compound of Formula 4904 (compound 292 in Table 4) was synthesized using the synthetic transformations as described in Examples 12 and 14a, but 2-chloro-6-methyl benzoic acid (compound 4903) was used instead of 2, 6 ,dimethyl benzoic acid (compound 4403). By a similar method, compound 328 in Table 4 was synthesized using the synthetic transformations as described starting from the 2-chloro-6-methyl m-fluorobenzoic acid.
…………………………………….
http://www.google.com/patents/WO2012097000A1?cl=en OR http://www.google.com/patents/US8809349?cl=en
Formula (I):

(I),
or a pharmaceutically acceptable salt, solvate, or hydrate thereof. In one embodiment, the method comprises any one, two, three, four, five, six, seven, or eight, or more of the following steps:



“Formula (I)” includes (S)-3-(l -(9H-purin-6-ylamino)ethyl)-8-chloro-2- phenylisoquinolin-l(2H)-one in its imide tautomer shown below as (1-1) and in its lactim tautomer shown below as (1-2):

(1-1)………………………………………………………………………………… (1-2)
[0055] FIG. 27 shows an FT-IR spectra of Polymorph Form C.

[0056] FIG. 28 shows a ‘H-NMR spectra of Polymorph Form C.

[0057] FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.

Example 1
Synthesis of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
Example 1A

1 2
[00563] Compound 1 (6.00 kg) was treated with 1-hydroxybenzotriazole monohydrate (HOBt»H20), triethylamine, Ν,Ο-dimethylhydroxylamine hydrochloride, and EDCI in dimethylacetamide (DMA) at
10 °C. The reaction was monitored by proton NMR and deemed complete after 2.6 hours, affording Compound 2 as a white solid in 95% yield. The R-enantiomer was not detected by proton NMR using (R)-(- ) -alpha-ace tylmandelic acid as a chiral-shift reagent.

[00564] Compound 3 (4.60 kg) was treated with p-toluenesulfonic acid monohydrate and 3,4-dihydro-2H- pyran (DHP) in ethyl acetate at 75 °C for 2.6 hours. The reaction was monitored by HPLC. Upon completion of the reaction, Compound 4 was obtained as a yellow solid in 80% yield with >99% (AUC) purity by HPLC analysis.

[00565] Compound 5 (3.30 kg) was treated with thionyl chloride and a catalytic amount of DMF in methylene chloride at 25 °C for five hours. The reaction was monitored by HPLC which indicated a 97.5% (AUC) conversion to compound 6. Compound 6 was treated in situ with aniline in methylene chloride at 25 °C for 15 hours. The reaction was monitored by HPLC and afforded Compound 7 as a brown solid in 81% yield with >99% (AUC) purity by HPLC analysis. [00566] Compound 2 was treated with 2.0 M isopropyl Grignard in THF at -20 °C. The resulting solution was added to Compound 7 (3.30 kg) pre -treated with 2.3 M n-hexyl lithium in tetrahydrofuran at -15 °C. The reaction was monitored by HPLC until a 99% (AUC) conversion to Compound 8 was observed.
Compound 8 was treated in situ with concentrated HC1 in isopropyl alcohol at 70 °C for eight hours. The reaction was monitored by HPLC and afforded Compound 9 as a brown solid in 85% yield with 98% (AUC) purity and 84% (AUC) ee by HPLC analysis.
Example ID
[00567] Compound 9 (3.40 kg) was treated with D-tartaric acid in methanol at 55 °C for 1-2 hours. The batch was filtered and treated with ammonium hydroxide in deionized (DI) water to afford enantiomerically enriched Compound 9 as a tan solid in 71% yield with >99% (AUC) purity and 91% (AUC) ee by HPLC analysis.
Example 2
Synthesis of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one

Example 2A
[00568] To Compound 7 (20.1 g) was charged 100 mL of anhydrous THF. The resulting solution was cooled to about -10 °C and 80 mL of n-hexyl lithium (2.3 M in hexanes, 2.26 equiv.) was slowly added (e.g. , over about 20 min). The resulting solution was stirred at about -10 °C for about 20 min.
[00569] To Compound 2 (26.5 g; 1.39 equiv.) was charged 120 mL of anhydrous THF. The resulting mixture was cooled to about -10 °C and 60 mL of isopropyl magnesium chloride (2.0 M in THF, 1.47 equiv.) was slowly added (e.g. , over about 15-20 min). The resulting mixture was then stirred at about -10 °C for about 20 min. The mixture prepared from Compound 2 was added to the solution prepared from Compound 7 while maintaining the internal temperature between about -10 and about 0 °C. After the addition was complete (about 5 min), the cold bath was removed, and the resulting mixture was stirred at ambient temperature for about 1 h, then cooled. [00570] A solution of 100 mL of anisole and 33 mL of isobutyric acid (4.37 equiv.) was prepared. The anisole solution was cooled to an internal temperature of about -3 °C. The above reaction mixture was added to the anisole solution such that the internal temperature of the anisole solution was maintained at below about 5 °C. The ice bath was then removed (after about 15 min, the internal temperature was about 7 °C). To the mixture, 100 mL of 10 wt aqueous NaCl solution was rapidly added (the internal temperature increased from about 7 °C to about 15 °C). After stirring for about 30 min, the two phases were separated. The organic phase was washed with another 100 mL of 10 wt aqueous NaCl. The organic phase was transferred to a flask using 25 mL of anisole to facilitate the transfer. The anisole solution was then concentrated to 109 g. Then, 100 mL of anisole was added.
[00571] To the approximately 200 mL of anisole solution was added 50 mL of TFA (8 equiv.) while maintaining the internal temperature below about 45-50 °C. The resulting solution warmed to about 45-50 °C and stirred for about 15 hrs, then cooled to 20-25 °C. To this solution was added 300 mL of MTBE dropwise and then the resulting mixture was held at 20-25 °C for 1 h. The mixture was filtered, and the wet cake washed with approximately 50 mL of MTBE. The wet cake was conditioned on the filter for about 1 h under nitrogen. The wet cake was periodically mixed and re-smoothed during conditioning. The wet cake was then washed with 200 mL of MTBE. The wet cake was further conditioned for about 2 h (the wet cake was mixed and resmoothed after about 1.5 h). The wet cake was dried in a vacuum oven at about 40 °C for about 18 h to afford Compound 9»TFA salt in about 97.3% purity (AUC), which had about 99.1 % S- enantiomer (e.g. , chiral purity of about 99.1 %).
[00572] Compound 9»TFA salt (3 g) was suspended in 30 mL of EtOAc at about 20 °C. To the EtOAc suspension was added 4.5 mL (2.2 eq.) of a 14% aqueous ammonium hydroxide solution and the internal temperature decreased to about 17 °C. Water (5 mL) was added to the biphasic mixture. The biphasic mixture was stirred for 30 min. The mixing was stopped and the phases were allowed to separate. The aqueous phase was removed. To the organic phase (combined with 5 mL of EtOAc) was added 10 mL of 10% aqueous NaCl. The biphasic mixture was stirred for about 30 min. The aqueous phase was removed. The organic layer was concentrated to 9 g. To this EtOAc mixture was added 20 mL of i-PrOAc. The resulting mixture was concentrated to 14.8 g. With stirring, 10 mL of n-heptane was added dropwise. The suspension was stirred for about 30 min, then an additional 10 mL of n-heptane was added. The resulting suspension was stirred for 1 h. The suspension was filtered and the wet cake was washed with additional heptane. The wet cake was conditioned for 20 min under nitrogen, then dried in a vacuum oven at about 40 °C to afford Compound 9 free base in about 99.3% purity (AUC), which had about 99.2% S-enantiomer (e.g., chiral purity of about 99.2%).
Example 2B [00573] A mixture of Compound 7 (100 g, 0.407 mol, 1 wt) and THF (500 mL, 5 vol) was prepared and cooled to about 3 °C. n-Hexyllithium (2.3 M in hexanes, 400 mL, 0.920 mol, 2.26 equiv) was charged over about 110 minutes while maintaining the temperature below about 6 °C. The resulting solution was stirred at 0 ± 5 °C for about 30 minutes. Concurrently, a mixture of Compound 2 (126 g, 0.541 mol, 1.33 equiv) and THF (575 mL, 5.8 vol) was prepared. The resulting slurry was charged with isopropylmagnesium chloride (2.0 M in THF, 290 mL, 0.574 mol, 1.41 equiv) over about 85 minutes while maintaining the temperature below about 5 °C. The resulting mixture was stirred for about 35 minutes at 0 ± 5 °C. The Compound 2 magnesium salt mixture was transferred to the Compound 7 lithium salt mixture over about 1 hour while maintaining a temperature of 0 ± 5 °C. The solution was stirred for about 6 minutes upon completion of the transfer.
[00574] The solution was added to an about -5 °C stirring solution of isobutyric acid (165 mL, 1.78 mol, 4.37 equiv) in anisole (500 mL, 5 vol) over about 20 minutes during which time the temperature did not exceed about 6 °C. The resulting solution was stirred for about 40 minutes while being warmed to about 14 °C. Then, a 10% sodium chloride solution (500 mL, 5 vol) was rapidly added to the reaction. The temperature rose to about 21 °C. After agitating the mixture for about 6 minutes, the stirring was ceased and the lower aqueous layer was removed (about 700 mL). A second portion of 10% sodium chloride solution (500 mL, 5 vol) was added and the mixture was stirred for 5 minutes. Then, the stirring was ceased and the lower aqueous layer was removed. The volume of the organic layer was reduced by vacuum distillation to about 750 mL (7.5 vol).
[00575] Trifluoroacetic acid (250 mL, 3.26 mol, 8.0 equiv) was added and the resulting mixture was agitated at about 45 °C for about 15 hours. The mixture was cooled to about 35 °C and MTBE (1.5 L, 15 vol) was added over about 70 minutes. Upon completion of the addition, the mixture was agitated for about 45 minutes at about 25-30 °C. The solids were collected by vacuum filtration and conditioned under N2 for about 20 hours to afford Compound 9*TFA salt in about 97.5% purity (AUC), which had a chiral purity of about 99.3%.
[00576] Compound 9»TFA salt (100 g) was suspended EtOAc (1 L,10 vol) and 14% aqueous ammonia (250 mL, 2.5 vol). The mixture was agitated for about 30 minutes, then the lower aqueous layer was removed. A second portion of 14% aqueous ammonia (250 mL, 2.5 vol) was added to the organic layer. The mixture was stirred for 30 minutes, then the lower aqueous layer was removed. Isopropyl acetate (300 mL, 3 vol) was added, and the mixture was distilled under vacuum to 500 mL (5 vol) while periodically adding in additional isopropyl acetate (1 L, 10 vol).
[00577] Then, after vacuum-distilling to a volume of 600 mL (6 vol), heptanes (1.5 L, 15 vol) were added over about 110 minutes while maintaining a temperature between about 20 °C and about 30 °C. The resulting slurry was stirred for about 1 hour, then the solid was collected by vacuum filtration. The cake was washed with heptanes (330 mL, 3.3 vol) and conditioned for about 1 hour. The solid was dried in an about 45 °C vacuum oven for about 20 hours to afford Compound 9 free base in about 99.23% purity (AUC), which has a chiral purity of about 99.4%.
Example 3
Chiral Resolution of (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9)
[00578] In some instances, (S)-3-(l-aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) obtained by synthesis contained a minor amount of the corresponding (R)-isomer. Chiral resolution procedures were utilized to improve the enantiomeric purity of certain samples of (S)-3-(l-aminoethyl)-8- chloro-2-phenylisoquinolin- 1 (2H)-one.
[00579] In one experiment, Compound 9 (3.40 kg) was treated with D-tartaric acid in methanol at about 55 °C for about 1 to about 2 hours. The mixture was filtered and treated with ammonium hydroxide in deionized (DI) water to afford Compound 9 in greater than about 99% (AUC) purity, which had a chiral purity of about 91% (AUC).
[00580] In another procedure, MeOH (10 vol.) and Compound 9 (1 equiv.) were stirred at 55 ± 5 °C. D- Tartaric acid (0.95 equiv.) was charged. The mixture was held at 55 ± 5 °C for about 30 min and then cooled to about 20 to about 25 °C over about 3 h. The mixture was held for about 30 min and then filtered. The filter cake was washed with MeOH (2.5 vol.) and then conditioned. The cake was returned to the reactor and water (16 vol.) was charged. The mixture was stirred at 25 ± 5 °C. NH4OH was then charged over about 1 h adjusting the pH to about 8 to about 9. The mixture was then filtered and the cake was washed with water (4 vol.) and then heptanes (4 vol.). The cake was conditioned and then vacuum dried at 45-50 °C to afford Compound 9 free base with a chiral purity of about 99.0%.
Example 4
Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one

[00581] A mixture of Compound 7 (1 equiv.) and anhydrous THF (5 vol.) was prepared. Separately, a mixture of Compound 2 (1.3 equiv.) and anhydrous THF (5 vol.) was prepared. Both mixtures were stirred for about 15 min at about 20 to about 25 °C and then cooled to -25 ± 15 °C. n-Hexyl lithium (2.05 equiv.) was added to the Compound 7 mixture, maintaining the temperature at > 5 °C. i-PrMgCl (1.33 equiv.) was added to the Compound 2 mixture, maintaining the temperature at > 5 °C. The Compound 2 mixture was transferred to the Compound 7 mixture under anhydrous conditions at 0 ± 5 °C. The resulting mixture was warmed to 20 ± 2 °C and held for about 1 h. Then, the reaction was cooled to -5 ± 5 °C, and 6 N HC1 (3.5 equiv.) was added to quench the reaction, maintaining temperature at below about 25 °C. The aqueous layer was drained, and the organic layer was distilled under reduced pressure until the volume was 2-3 volumes. IPA (3 vol.) was added and vacuum distillation was continued until the volume was 2-3 volumes. IPA (8 vol.) was added and the mixture temperature was adjusted to about 60 °C to about 75 °C. Cone. HC1 (1.5 vol.) was added and the mixture was subsequently held for 4 hours. The mixture was distilled under reduced pressure until the volume was 2.5-3.5 volumes. The mixture temperature was adjusted to 30 ± 10 °C. DI water (3 vol.) and DCM (7 vol.) were respectively added to the mixture. Then, NH4OH was added to the mixture, adjusting the pH to about 7.5 to about 9. The temperature was adjusted to about 20 to about 25 °C. The layers were separated and the aqueous layer was washed with DCM (0.3 vol.). The combined DCM layers were distilled until the volume was 2 volumes. i-PrOAc (3 vol.) was added and vacuum distillation was continued until the volume was 3 volumes. The temperature was adjusted to about 15 to about 30 °C. Heptane (12 vol.) was charged to the organic layer, and the mixture was held for 30 min. The mixture was filtered and filter cake was washed with heptane (3 vol.). The cake was vacuum dried at about 45 °C afford Compound 9.
[00582] Then, MeOH (10 vol.) and Compound 9 (1 equiv.) were combined and stirred while the temperature was adjusted to 55 ± 5 °C. D-Tartaric acid (0.95 equiv.) was charged. The mixture was held at 55 ± 5 °C for about 30 min and then cooled to about 20 to about 25 °C over about 3 h. The mixture was held for 30 min and then filtered. The filter cake was washed with MeOH (2.5 vol.) and then conditioned. Water (16 vol.) was added to the cake and the mixture was stirred at 25 ± 5 °C. NH4OH was charged over 1 h adjusting the pH to about 8 to about 9. The mixture was then filtered and the resulting cake washed with water (4 vol.) and then heptanes (4 vol.). The cake was conditioned and then vacuum dried at 45-50 °C to afford Compound 9.
[00583] To a mixture of i-PrOH (4 vol.) and Compound 9 (1 equiv.) was added Compound 4 (1.8 equiv.), Et3N (2.5 equiv.) and i-PrOH (4 vol.). The mixture was agitated and the temperature was adjusted to 82 ± 5 °C. The mixture was held for 24 h. Then the mixture was cooled to about 20 to about 25 °C over about 2 h. The mixture was filtered and the cake was washed with i-PrOH (2 vol.), DI water (25 vol.) and n-heptane (2 vol.) respectively. The cake was conditioned and then vacuum dried at 50 ± 5 °C to afford Compound 10.
To a mixture of EtOH (2.5 vol.) and Compound 10 (1 equiv.) was added EtOH (2.5 vol.) and DI water (2 vol.). The mixture was agitated at about 20 to about 25 °C. Cone. HC1 (3.5 equiv.) was added and the temperature was adjusted to 35 ± 5 °C. The mixture was held for about 1.5 h. The mixture was cooled to 25 ± 5 °C and then polish filtered to a particulate free vessel. NH4OH was added, adjusting the pH to about 8 to about 9. Crystal seeds of Form C of a compound of Formula (I) (0.3 wt ) were added to the mixture which was held for 30 minutes. DI water (13 vol.) was added over about 2 h. The mixture was held for 1 h and then filtered. The resulting cake was washed with DI water (4 vol.) and n-heptane (2 vol.) respectively. The cake was conditioned for about 24 h and then DCM (5 vol.) was added. This mixture was agitated for about 12 h at about 20 to about 25 °C. The mixture was filtered and the cake washed with DCM (1 vol.). The cake was conditioned for about 6 h. The cake was then vacuum-dried at 50 ± 5 °C. To the cake was added DI water (10 vol.), and i-PrOH (0.8 vol.) and the mixture was agitated at 25 ± 5 °C for about 6 h. An XRPD sample confirmed the compound of Formula (I) was Form C. The mixture was filtered and the cake was washed with DI water (5 vol.) followed by n-heptane (3 vol.). The cake was conditioned and then vacuum dried at 50 ± 5 °C to afford a compound of Formula (I) as polymorph Form C. Example 5
Synthesis of (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one

Example 5A
[00584] Compound 9 (2.39 kg) was treated with Compound 4 and triethylamine in isopropyl alcohol at 80 °C for 24 hours. The reaction was monitored by HPLC until completion, affording 8-chloro-2-phenyl-3- ((lS)-l-(9-(tetrahydro-2H^yran-2-yl)-9H^urin-6-ylamino)ethyl)isoquinolin-l(2H)-one (compound 10) as a tan solid in 94% yield with 98% (AUC) purity by HPLC analysis.
[00585] 8-Chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-ylamino)ethyl)- isoquinolin-l(2H)-one (compound 10) (3.63 kg) was treated with HC1 in ethanol at 30 °C for 2.3 hours. The reaction was monitored by HPLC until completion, and afforded a compound of Formula (I) as a tan solid in 92% yield with >99% (AUC) purity and 90.9% (AUC) ee by HPLC analysis.
Example 5B
[00586] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) (0.72 mmol), 6-chloro- 9-(tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) (344 mg, 1.44 mmol) and DIPEA
(279 mg, 2.16 mmol) were dissolved in «-BuOH (20 mL), and the resulting mixture was stirred at reflux for 16 h. The reaction mixture was concentrated in vacuo and purified by flash column chromatography on silica gel (eluting with 30% to 50% Hex/EA) to afford the product, 8-chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H- pyran-2-yl)-9H-purin-6-ylamino)ethyl)isoquinolin-l(2H)-one (Compound 10), as a white solid (60% yield). [00587] 8-Chloro-2-phenyl-3-((lS)-l-(9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-ylamino)ethyl)- isoquinolin-l(2H)-one (Compound 10) (0.42 mmol) was dissolved in HCl/EtOH (3 M, 5 mL), and the resulting mixture was stirred at room temperature for 1 h. The reaction mixture was quenched with saturated NaHC03 aqueous solution and the pH was adjusted to about 7-8. The mixture was extracted with CH2C12 (50 mL x 3), dried over anhydrous Na2S04, and filtered. The filtrate was concentrated in vacuo, and the residue was recrystallized from ethyl acetate and hexanes (1 : 1). The solid was collected by filtration and dried in vacuo to afford the product (S)-3-(l-(9H-purin-6-ylamino) ethyl)-8-chloro-2-phenylisoquinolin- l(2H)-one (Formula (I)) (90% yield) as a white solid as polymorph Form A.
Example 5C
[00588] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) and 6-chloro-9- (tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) are combined in the presence of triethylamine and isopropyl alcohol. The reaction solution is heated at 82 °C for 24 hours to afford Compound 10. The intermediate compound 10 is treated with concentrated HCl and ethanol under aqueous conditions at 35 °C to remove the tetrahydropyranyl group to yield (S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2- phenylisoquinolin-l(2H)-one. Isolation/purification under aqueous conditions affords polymorph Form C.
Example 6
Synthesis of (S)-3-(l-(9H^urin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one

[00589] 3-(l-Aminoethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one (Compound 9) (150 g; 90% ee) and 6- chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (Compound 4) (216 g, 1.8 equiv) were charged to a round bottom flask followed by addition of IPA (1.2 L; 8 vol) and triethylamine (175 mL; 2.5 equiv). The resultant slurry was stirred at reflux for one day. Heptane (1.5 L; 10 vol) was added dropwise over two hours. The batch was then cooled to 0-5 °C, held for one hour and filtered. The cake was washed with heptane (450 mL; 3 vol) and returned to the reactor. IPA (300 mL; 2 vol) and water (2.25 L; 15 vol) were added and the resultant slurry stirred at 20-25 °C for three and half hours then filtered. The cake was washed with water (1.5 L; 10 vol) and heptane (450 mL; 3 vol) and then vacuum dried at 48 °C for two and half days to give 227 g (90.1 %) of the intermediate (Compound 10) as an off-white solid with >99% (AUC) purity and >94 ee (chiral HPLC). The ee was determined by converting a sample of the cake to the final product and analyzing it with chiral HPLC.
[00590] The intermediate (Compound 10) (200 g) was slurried in an ethanol (900 mL; 4.5 vol) / water (300 mL; 1.5 vol) mixture at 22 °C followed by addition of cone. HC1 (300 mL; 1.5 vol) and holding for one and half hours at 25-35 °C. Addition of HC1 resulted in complete dissolution of all solids producing a dark brown solution. Ammonium hydroxide (260 mL) was added adjusting the pH to 8-9. Product seeds of polymorph Form C (0.5 g) (Form A seeds can also be used) were then added and the batch which was held for ten minutes followed by addition of water (3 L; 15 vol) over two hours resulting in crystallization of the product. The batch was held for 3.5 hours at 20-25 °C and then filtered. The cake was washed with water (1 L; 5 vol) followed by heptane (800 mL; 4 vol) and vacuum dried at 52 °C for 23 hours to give 155.5 g (93.5%) of product with 99.6% (AUC) purity and 93.8% ee (chiral HPLC).
Example 7
-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one

[00591] A mixtue of isopropanol (20.20 kg, 8 vol.), Compound 9 (3.17 kg, 9.04 mol, 1 eq.), Compound 4 (4.61 kg, 16.27 mol, 1.8 eq.) and triethylamine (2.62 kg, 20.02 mol, 2.4 eq.) was prepared and heated to an internal temperature of 82 ± 5 °C. The mixture was stirred at that temperature for an additional about 24 h. The temperature was adjusted to 20 ± 5 °C slowly over a period of about 2 h and the solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a Sharkskin paper. The filter cake was rinsed sequentially with IPA (5.15 kg, 3 vol.), purified water (80.80 kg, 25 vol.) and n-heptane (4.30 kg, 2 vol.). The cake was further dried for about 4 days in vacuo at 50 ± 5 °C to afford Compound 10.
[00592] To a mixture of ethanol (17.7 kg, 5 vol.) and Compound 10 (4.45 kg, 8.88 mol. 1.0 eq.) was added purified water (8.94 kg, 2 vol.). To this mixture was slowly added concentrated HC1 (3.10 kg, 3.5 eq.) while maintaining the temperature below about 35 °C. The mixture was stirred at 30 ± 5 °C for about 1.5 h and HPLC analysis indicated the presence the compound of Formula (I) in 99.8% (AUC) purity with respect to compound 10.
[00593] Then, the compound of Formula (I) mixture was cooled to 25 ± 5 °C. The pH of the mixture was adjusted to about 8 using pre filtered ammonium hydroxide (1.90 kg). After stirring for about 15 min, Form C crystal seeds (13.88 g) were added. After stirring for about 15 min, purified water (58.0 kg, 13 vol.) was charged over a period of about 2 h. After stirring the mixture for 15 h at 25 ± 5 °C, the solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper. The filter cake was rinsed with purified water (18.55 kg, 4 vol.) followed by pre -filtered n-heptane (6.10 kg, 2 vol.). After conditioning the filter cake for about 24 h, HPLC analysis of the filter cake indicated the presence the compound of Formula (I) in about 99.2% (AUC) purity.
[00594] To the filter cake was added dichloromethane (29.9 kg, 5 vol.) and the slurry was stirred at 25 ± 5 °C for about 24 h. The solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper, and the filter cake was rinsed with DCM (6.10 kg, 1 vol.). After conditioning the filter cake for about 22 h, the filter cake was dried for about 2 days in vacuo at 50 ± 5 °C to afford the compound of Formula (I) in 99.6% (AUC) purity. The compound of Formula (I) was consistent with a Form A reference by XRPD.
[00595] To this solid was added purified water (44.6 kg, 10 vol.) and pre filtered 2-propanol (3.0 kg, 0.8 vol.). After stirring for about 6 h, a sample of the solids in the slurry was analyzed by XRPD and was consistent with a Form C reference. The solids were isolated via vacuum filtration through a 24″ polypropylene table top filter equipped with a PTFE cloth over Sharkskin paper, and the filter cake was rinsed with purified water (22.35 kg, 5 vol.) followed by pre filtered n-heptane (9.15 kg, 3 vol.). After conditioning the filter cake for about 18 h, the filter cake was dried in vacuo for about 5 days at 50 ± 5 °C.
[00596] This process afforded a compound of Formula (I) in about 99.6% (AUC) purity, and a chiral purity of greater than about 99% (AUC). An XRPD of the solid was consistent with a Form C reference standard. :H NMR (DMSO-<i6) and IR of the product conformed with reference standard.
…………………………..
http://www.google.com/patents/US20140120083
In some embodiments, the compound has the following structure:

which is also referred to herein as Compound 292.
In some embodiments, a polymorph of a compound disclosed herein is used. Exemplary polymorphs are disclosed in U.S. Patent Publication No. 2012-0184568 (“the ‘568 publication”), which is hereby incorporated by reference in its entirety.
In one embodiment, the compound is Form A of Compound 292, as described in the ‘568 publication. In another embodiment, the compound is Form B of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form C of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form D of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form E of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form F of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form G of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form H of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form I of Compound 292, as described in the ‘568 publication. In yet another embodiment, the compound is Form J of Compound 292, as described in the ‘568 publication.
In specific embodiments, provided herein is a crystalline monohydrate of the free base of Compound 292, as described, for example, in the ‘568 application. In specific embodiments, provided herein is a pharmaceutically acceptable form of Compound 292, which is a crystalline monohydrate of the free base of Compound 292, as described, for example, in the ‘568 application.
Any of the compounds (PI3K modulators) disclosed herein can be in the form of pharmaceutically acceptable salts, hydrates, solvates, chelates, non-covalent complexes, isomers, prodrugs, isotopically labeled derivatives, or mixtures thereof.
Chemical entities described herein can be synthesized according to exemplary methods disclosed in U.S. Patent Publication No. US 2009/0312319, International Patent Publication No. WO 2011/008302A1, and U.S. Patent Publication No. 2012-0184568, each of which is hereby incorporated by reference in its entirety, and/or according to methods known in the art.
……………………………………………

KEY Duvelisib, IPI-145, INK-1197, AbbVie, INFINITY, chronic lymphocytic leukemia, phase 3, orphan drug
| WO2013088404A1 | Dec 14, 2012 | Jun 20, 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2014004470A1 * | Jun 25, 2013 | Jan 3, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using pi3 kinase inhibitors |
| WO2014072937A1 | Nov 7, 2013 | May 15, 2014 | Rhizen Pharmaceuticals Sa | Pharmaceutical compositions containing a pde4 inhibitor and a pi3 delta or dual pi3 delta-gamma kinase inhibitor |
| US7449477 * | Nov 22, 2004 | Nov 11, 2008 | Eli Lilly And Company | 7-phenyl-isoquinoline-5-sulfonylamino derivatives as inhibitors of akt (protein kinase B) |
| US20090312319 * | Jul 15, 2009 | Dec 17, 2009 | Intellikine | Certain chemical entities, compositions and methods |
| US20100168153 * | Nov 16, 2007 | Jul 1, 2010 | Novartis Ag | Salts and crystall forms of 2-methyl-2-[4-(3-methyl-2-oxo-8-quinolin-3-yl-2,3-dihydro-imidazo[4,5-c]quinolin-1-yl)-phenyl]-propionitrile |
| WO2013012915A1 | Jul 18, 2012 | Jan 24, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013012918A1 | Jul 18, 2012 | Jan 24, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013032591A1 | Jul 18, 2012 | Mar 7, 2013 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| WO2013049332A1 | Sep 27, 2012 | Apr 4, 2013 | Infinity Pharmaceuticals, Inc. | Inhibitors of monoacylglycerol lipase and methods of their use |
| WO2013088404A1 | Dec 14, 2012 | Jun 20, 2013 | Novartis Ag | Use of inhibitors of the activity or function of PI3K |
| WO2013154878A1 | Apr 3, 2013 | Oct 17, 2013 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2014004470A1 * | Jun 25, 2013 | Jan 3, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using pi3 kinase inhibitors |
| WO2014071105A1 | Nov 1, 2013 | May 8, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of rheumatoid arthritis and asthma using p13 kinase inhibitors |
| WO2014071109A1 | Nov 1, 2013 | May 8, 2014 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| WO2014072937A1 | Nov 7, 2013 | May 15, 2014 | Rhizen Pharmaceuticals Sa | Pharmaceutical compositions containing a pde4 inhibitor and a pi3 delta or dual pi3 delta-gamma kinase inhibitor |
| WO2001081346A2 | Apr 24, 2001 | Nov 1, 2001 | Icos Corp | Inhibitors of human phosphatidyl-inositol 3-kinase delta |
| US6800620 | Jan 6, 2003 | Oct 5, 2004 | Icos | Contacting leukocytes, osteoclasts with an enzyme inhibitors, a 9h-purin-3h-quinazolin-4-one derivatives, treating bone-resorption disorder, antiproliferative agents treating leukemia cells |
| US20060276470 * | Aug 18, 2003 | Dec 7, 2006 | Jackson Shaun P | (+-)-7-Methyl-2-morpholin-4-yl-9-(1-phenylaminoethyl)-pyrido[1,2-a]pyrimidin-4-one, for example; selective inhibitors of phosphoinositide (PI) 3-kinase beta for use in anti-thrombotic therapy |
| US20080032960 * | Apr 4, 2007 | Feb 7, 2008 | Regents Of The University Of California | PI3 kinase antagonists |
