
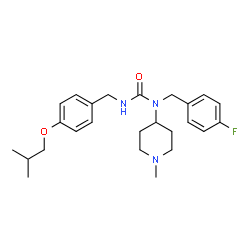
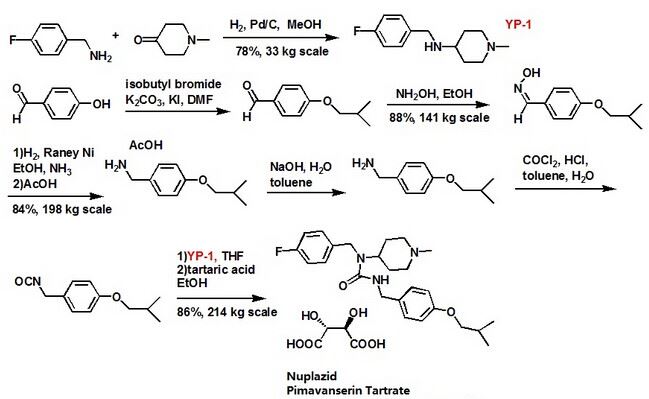
Pimavanserin
- MF C25H34FN3O2
- MW 427.555
Pimavanserin, ACP 103, ACP-103; BVF-048
N-(4-fluorophenylmethyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide,
706779-91-1 (Pimavanserin )
706782-28-7 (Pimavanserin Tartrate)
For treatment of psychotic symptoms in patients with Parkinson’s disease
WATCH OUT AS THIS POST IS UPDATED………..
Trade Name:Nuplazid®
MOA:5-HT2A inverse agonist
Indication:Hallucinations and delusions associated with Parkinson’s disease psychosis
Company:Acadia (Originator)
APPROVED US FDA 2016-04-29, ACADIA PHARMS INC, (NDA) 207318
To treat hallucinations and delusions associated with psychosis experienced by some people with Parkinson’s disease
|

706782-28-7 (tartrate) |
| Molecular Weight |
1005.2 |
| Formula |
(C25H34FN3O2)2 ● C4H6O6 |
Urea, N-[(4-fluorophenyl)methyl]-N-(1-methyl-4-piperidinyl)-N’-[[4-(2-methylpropoxy)phenyl]methyl]-, (2R,3R)-2,3-dihydroxybutanedioate (2:1)

Pimavanserin Tartrate was approved by the U.S. Food and Drug Administration (FDA) on Apr 29, 2016. It was developed by Acadia, then marketed as Nuplazid® by Acadia in US.
Pimavanserin Tartrate is a 5-HT2A receptor inverse agonists, used to treat hallucinations and delusions associated with psychosis experienced by some people with Parkinson’s disease.
Nuplazid® is available as tablet for oral use, containing 17 mg of pimavanserin. Recommended dose is 34 mg, taken orally as two tablets once daily.
Pimavanserin (INN), or pimavanserin tartate (USAN), marketed under the trade name Nuplazid, is a non-dopaminergic atypical antipsychotic[2] developed by Acadia Pharmaceuticals for the treatment of Parkinson’s disease psychosis and schizophrenia. Pimavanserin has a unique mechanism of action relative to other antipsychotics, behaving as a selective inverse agonist of theserotonin 5-HT2A receptor, with 40-fold selectivity for this site over the 5-HT2C receptor and no significant affinity or activity at the5-HT2B receptor or dopamine receptors.[1] The drug has met expectations for a Phase III clinical trial for the treatment ofParkinson’s disease psychosis,[3] and has completed Phase II trials for adjunctive treatment of schizophrenia alongside anantipsychotic medication.[4]
Pimavanserin is expected to improve the effectiveness and side effect profile of antipsychotics.[5][6][7] The results of a clinical trial examining the efficacy, tolerability and safety of adjunctive pimavanserin to risperidone and haloperidol were published in November 2012, and the results showed that pimavanserin potentiated the antipsychotic effects of subtherapeutic doses ofrisperidone and improved the tolerability of haloperidol treatment by reducing the incidence of extrapyramidal symptoms.[8]
On September 2, 2014, the United States Food and Drug Administration granted Breakthrough Therapy status to Acadia’s New Drug Application for pimavanserin.[9] It was approved by the FDA to treat hallucinations and delusions associated with psychosis experienced by some people with Parkinson’s disease on April 29, 2016.[10]

Clinical pharmacology
Pimavanserin acts as an inverse agonist and antagonist at serotonin 5-HT2A receptors with high binding affinity (Ki 0.087 nM) and at serotonin 5-HT2C receptors with lower binding affinity (Ki 0.44 nM). Pimavanserin shows low binding to σ1 receptors (Ki 120 nM) and has no appreciable affinity (Ki >300 nM) to serotonin 5-HT2B, dopaminergic (including D2), muscarinic, histaminergic, oradrenergic receptors, or to calcium channels.[2]

Pimavanserin tartrate, 1-(4-fluorobenzyl)-3-(4-isobutoxybenzyl)-1-(1-methylpiperidin-4-yl)urea L-hemi-tartrate, has the following chemical structure:

Pimavanserin tartrate was developed by Acadia Pharmaceuticals and was approved under the trade name NUPLAZID® for use in patients with Parkinson’s disease psychosis.
Pimavanserin free base and its synthesis are disclosed in US 7,601,740 (referred to herein as US ‘740 or the ‘740 patent) and US 7,790,899 (referred to herein as US ‘899 or the ‘899 patent). US ‘740 discloses the synthesis of Pimavanserin free base (also referred to herein as“Compound A”), which includes O-alkylation followed by ester hydrolysis, and then in situ azidation. This process suffers from low process safety, and utilizes the hazardous reagent diphenylphosphoryl azide. The process is illustrated by the following Scheme 1.
Scheme 1:

US ‘899 describes another process, which includes O-alkylation followed by aldehyde reductive amination to obtain an intermediate which is then reacted with the hazardous reagent phosgene. This process is illustrated by the following Scheme 2:
Scheme 2:

Both of the above processes for the preparation of Pimavanserin include a reaction between 1-isobutoxy-4-(isocyanatomethyl)benzene, a benzyl isocyanate intermediate, and N-(4-fluorobenzyl)-1-methylpiperidin-4-amine. Processes for preparing benzyl isocyanate derivatives are generally described in the literature, such as in US ‘740; US ‘899; Bioorganic & Medicinal Chemistry, 21(11), 2960-2967, 2013; JP 2013087107; Synthesis (12), 1955-1958, 2005; and Turkish Journal of Chemistry, 31(1), 35-43, 2007. These processes often use the hazardous reagents like phosgene derivatives or diphenylphosphoryl azide.


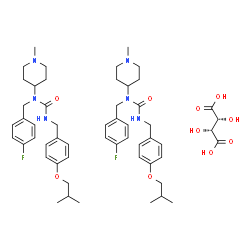
synthetic route:
First, reduction of the ketone and a secondary amine to amine condensation after S-3 . 4- hydroxybenzaldehyde etherification, followed by condensation with hydroxylamine to give the oxime S-. 7 , which is then reduced by hydrogenation to the amine S-. 8 , S.8- light gas reaction to give the isocyanate S-. 9 , S. 9- react with the primary amine can be obtained Nuplazid ( pimavanserin ).Kg product can be obtained by this route.

WO2006036874
https://www.google.com/patents/WO2006036874A1?cl=en
Example 1 : Preparation of N-(4-fluorobenzyl)-N-( 1 -methylpiperidin-4-yl)-N’ -( 4-(2- methylpropyloxy)phenylmethyl)carbamide a) Preparation of
Tπacetoxy borohydπde (6.5 kg) was added over 1.5 h to a solution of N- methylpiperid-4-one (3.17 kg) and 4-fluorobenzylamme (3.50 kg) in methanol (30 1), maintaining the temperature under 27 0C. The reaction mixture was stirred for 15 h at 22 0C. The residual amine was checked by gel chromatography (4-fluorobenzylamine: < 5%). A solution of 30% sodium hydroxide (12.1 kg) in water (13.6 kg) was added in 75 minutes (min) maintaining the temperature under 20 0C. Methanol was distilled off to a residual volume of 26 litters. Ethyl acetate was added (26 L), the solution was stirred for 15 min, the phases were decanted over 15 min and the lower aqueous phase was discarded. Ethyl acetate was distilled under reduced pressure from the organic phase at 73-127 0C. At this stage the residue was mixed with a second crude batch prepared according to this method. The combined products were then distilled at 139-140 0C / 20 mbar to yield 11.2 kg product (> 82%). b) Preparation of
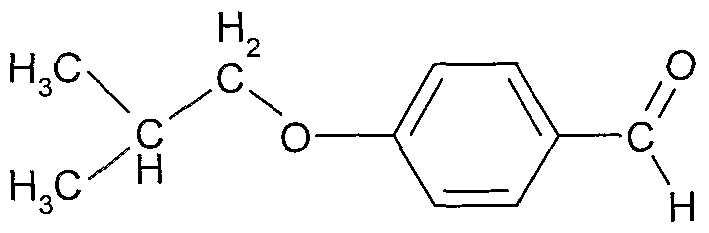
4-Hydroxybenzaldehyde (4.0 kg) and ethanol (20 1) were added to a solution of isobutyl bromide (9.0 kg) in ethanol (15 1). Potassium carbonate (13.6 kg) was added and the suspension was refluxed (74-78 0C) for 5 days. The residual 4- hydroxybenzaldehyde was checked by HPLC (< 10%). The suspension was cooled to 20 0C and used in the next step.
c) Preparation of

] Hydroxylamine (50% in water, 8.7 kg) was added to the product from previous step b)(174 1, 176 kg) and ethanol (54 1). The suspension was refluxed (77 0C) for 3 h. Unreacted residual amounts of the compound of step b was checked by HPLC (< 5%). The suspension was cooled to 30 0C, filtered and the filter was washed with ethanol (54 1). The solution was concentrated by distillation under reduced pressure at 30 0C to a residual volume of 67 litters. The solution was cooled to 25 0C and water (110 1) was added. The suspension was concentrated by distillation under reduced pressure at 30 0C to a residual volume of 102 litters. Petrol ether (60-90 fraction, 96 1) was added and the mixture was heated to reflux (70 0C). The solution Λvas cooled to 40 0C and crystallization was initiated by seeding. The suspension was cooled to 5 0C and stirred for 4h. The product was centrifuged and the cake was washed with petrol ether (60-90 fraction, 32 1). The wet cake was dried at about 40 0C to yield 16kg product (63%).
d) Preparation of
[0105] The product from previous step c) (15.7 kg) was dissolved in ethanol (123 1). Acetic acid (8.2 kg) and palladium on charcoal 5% wet (1.1 kg) were added. The oxime was hydxogenated at 22 0C and 1.5 bar for 4h. Consumption of oxime was checked by HPLC (for information). The catalyst was filtered and the solvent was distilled under reduced pressure at 36 0C to a final volume of 31 1. Ethyl acetate (63 1) was added and the mixture was heated to reflux (75 0C) until dissolution. The solution was cooled to 45 0C and the crystallization was initiated by seeding. The suspension was cooled to 6-10 0C and stirred for 2.5h. The product was centrifuged and the cake was washed with 2 portions of ethyl acetate (2 x 0.8 1). The wet cake was dried at a temperature of about 40 0C to yield 8 kg (41%).
e) Preparation of
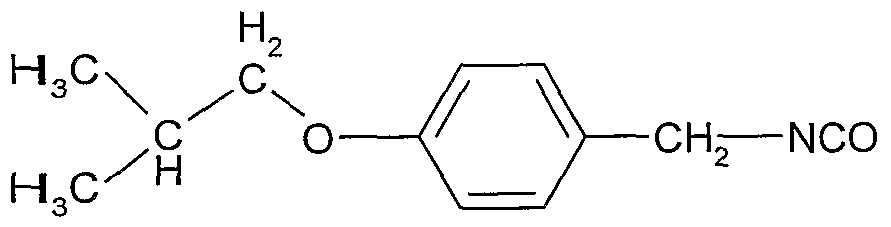
Aqueous sodium hydroxide (30%, 5.0 kg) was added to a suspension of the product from previous step d) (7.9 kg) in heptane (41 1). The solution was heated to 47 0C, stirred for 15 mm and decanted o~ver 15 mm. The pH was checked (pH>12) and the aqueous phase was separated. The solvent was removed by distillation under reduced pressure at 47-650C. Heptane was added (15 1) and it was removed by distillation under reduced pressure at 58-65 0C. Heptane was added (7 1), the solution was filtered and the filter was washed with heptane (7 1). The solvent was removed by distillation under reduced pressure at 28-60 0C. Tetrahydrofuran (THF, 107 1) and tπethylamme (TEA, 6.8 kg) were added and the temperature was fixed at 22 0C. In another reactor, phosgene (5.0 kg) was introduced in tetrahydrofuran (88 1) previously cooled to -3 0C. The THF and TEA s olution was added to the solution of phosgene in 3h 50 mm maintaining the temperature at -3 0C. The reactor was washed with tetrahydrofuran (22 1). The mixture was stirred for 45 min at 20 0C and then for 90 min at reflux (65 0C). The solvent was distilled under reduced pressure at 25-30 0C to a residual volume of 149 1. The absence of phosgene was controlled. At this stage, there still was phosgene and the suspension was degassed by bubbling nitrogen through it. After this operation the level of phosgene above the solution was below 0.075 ppm. The suspension was filtered and washed with tetrahydrofuran (30 1). The solvent was distilled under reduced pressure at 20-25 0C to a residual volume of 40 1. Tetrahydrofuran (51 1) was added and the solvent was distilled under reduced pressure at 20-25 0C to a residual volume of 40 1. The final volume was adjusted to about 52 litters by addition of tetrahydrofuran (11 1). The solution was analysed and used in the next step. f) Preparation of the title compound of formula I
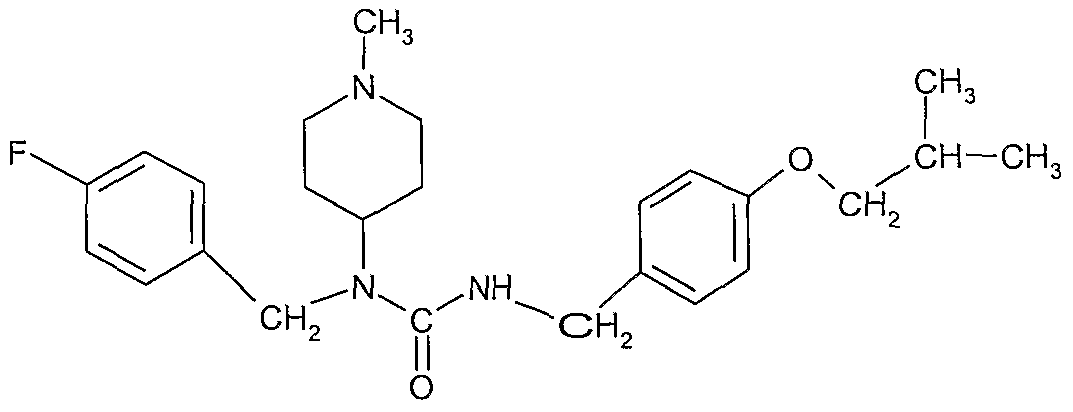
The product from previous step e) (51 1) was added in 1 h to a solution of the product from step a) (7.3 kg) in tetrahydrofuian (132 1) at 17 0C. The line was washed with tetrahydrofuran (12 1) and the mixture was stirred for 15h. Residual product from the first step was checked by HPLC The solvent was removed “by distillation under reduced pressure at 20-38 0C to a residual volume of 165 1. Charcoal (Noπt SXl-G, 0 7 kg) was added, the mixture was stirred for 15 mm and filtered. The lme was washed with tetrahydrofuran (7 1) and the solvent was removed by distillation under reduced pressure at 20-25 0C to a residual volume of 30 1. Isopropyl acetate (96 1) was added to obtain a solution of the title compound of formula I, which contains a small amount of impurities, which were mainly side products from the previous reactions. Removal of the solvent from a sample yields a substantially amorphous solid
g) Preparation of N-(4-fluorobenzyl)-N-(l-methylpipeπdm-4-yl)-N’-(4-(2-methylpropyloxy)phe- nylmethyl)carbamide hemi-tartrate
To the solution of the compound of Formula I in isopropyl acetate (96 1) from step f was added at 23 0C a previously prepared solution of tartaric acid (1 7 kg) in water (1.7 1) and tetrahydrofuran (23 1) The residual suspension was stirred for 2.5 days at 22 0C The tartrate crude product was centrifuged and the cake was washed with 4 portions of isopropyl acetate (4 x 23 1). A total of 107 kg of mother liquors was saved for later use in obtaining the tartrate salt The wet cake was dπed at about 40 0C to yield 8.3 kg (50%) product.
h) First Purification
The tartrate crude product of step g) (8.1 kg) was dissolved m demmeralized water (41 1) at 22 0C. Isopropyl acetate (40 L), 30% aqueous sodium hydroxide (4.3 kg) and sodium chloride (2 kg) were added. The pH was checked (>12) and the solution was stirred for 15 mm. The solution was decanted over 15 mm and the aqueous phase was separated. The aqueous phase was re-extracted with isopropyl acetate (12 1) Demmeralized water (20 1) and sodium chloride (2 0 kg) were added to the combined organic phases, the solution was stirred for 15 mm, decanted over 15 mm and the aqueous phase was discarded. Charcoal (0.4 kg) was added, the mixture was stirred for 20 mm and filtered. After a line wash with isopropyl acetate (12 1), the solvent was removed under reduced pressure at 20-25 0C Heptane (49 1) was added and the suspension was stirred for 15 mm at 40 °C. Then, 8 1 of solvent was removed by distillation under reduced pressure at 38-41 0C The slurry was cooled to 20 0C and stirred for 1 h. The product was centrifuged and the cake was washed with heptane (5 1) The wet compound of Forrnu-la I (5.5 kg) was dissolved m ethanol (28 1) at 45 0C. A solution of tartaric acid (0.72 kg) m ethanol (11 1) was added at 45 0C and the line was washed with ethanol (91). The solution was cooled to 43 0C, seeded with the tartrate salt of the compound o f Formula I, then the slurry was cooled to 350C m 30 mm, stirred at this temperature for 1 h and cooled to -5 0C After 14 h at this temperature the product was centrifuged and washed with two portions of ethanol (2×6 1) The wet cake was dried at about 45 0C for 76 h to yield 4 kg of the herm-tartrate
i) Re -crystallization
150 O g of herm-tartrate obtained m h) was dissolved under stirring at 65 0C m 112 ml absolute ethanol and then cooled under stirring to 48 0C at a cooling rate of 1 °C/mm Crystallization started after a few minutes at this temperature and the suspension turned to a thick paste withm 1 h. The suspension was heated again to 60 0C and then cooled to 480C at a rate of 1 °C/mm The obtained suspension was stirred and was cooled to 15 0C at a cooling rate of 3 °C/h. The crystalline precipitate was separated by filtration and the bottle was washed with 10 ml absolute ethanol cooled to 5 0C. The crystalline residue was dried under vacuum and 40 0C for 50 hours to yield 146 g crystalline pure herm-tartrate.
j) Second purification
15 78 g of the tartrate salt prepared from step i) was dissolved 121 130 ml water 500 ml TBME was added and the pH -was adjusted to 9 8 by addition of 2 ISf NaOH solution. After precipitation of a white solid, the aqueous phase was extracted 5 times by 500 ml TBME The organic phases were concentrated until a volume of about 400 ml remained. The solution was stored at 60C. The precipitate was filtered, washed with TBME and finally dried m vacuum for 5 hours. Yield: 8.24 g of a white poΛvder. The mother liquor was concentrated to a fourth and stored at 60C. The precipitate was filtered and dried m vacuum for 18 hours. Yield: 1.6 g of a white powder.
PXRD revealed a crystalline compound of formula I. No Raman peaks from tartaric acid were found. The first scan of DSC (-500C to 2100C5 10°K/mm) revealed a melting point at 123.6°C. Above about 19O0C, the sample started to decompose. Example 2. Preparation of N-(4-fluoroben2yl)-N-(l-methylpiperidin-4-yl)-N’-(4-(2- methylpropγloxy)phenylmethyl)carbamide citrate of formula FV
a) 90 mg of the product from Example 1 and 40 mg citnc acid were suspended m 5.0 ml ethylacetate. The suspension was stirred at 60 0C for 15 minutes (mm), cooled to 23±2 0C, and then stored for 30 mm at 23±2 0C. The precipitate was filtered off and dried in air for 30 mm to yield 52 mg of a crystalline white powder. Optical microscopy shows that the obtained solid was crystalline
b) 182 mg of the product from Example 2 and 78.4 mg citric acid were suspended m 10.0 ml ethyl acetate The suspension was stirred at 60 0C for 30 mm, then stirred at 40 0C for 90 mm, and finally stirred for 60 mm at 23 0C The suspension was filtered and washed with heptane, yielding 237 mg of a white crystalline powder -with an endothermic peak near 153 0C (enthalpy of fusion of about 87 J/g), determined by differential scanning caloπmetry at a rate of 10K/mm (DSC). Thermogravimetry (TG-FTIR) showed a mass loss of about 0.7% between 60 and 160 0C, which was attributed to absorbed water Decomposition started at about 170 0C Solubility m water was about 14 mg/ml The crystalline powder remained substantially unchanged when stored for 1 week at 60 0C and about 75% r_h. m an open container (HPLC area was 99.4% compared to reference value of 99.9%). Elemental analysis and 1H-NMR complies with an 1 : 1 stoichiometry.
PATENT
http://www.google.im/patents/WO2008144326A2?cl=en

Example 1 : Preparation of N-(4-fluorobenzyl)-N-Cl-methylpiperidin-4-yl)-N’-(‘4-f2- methylpropyloxy)phenylmethγl)carbamide a) Preparation of

Triacetoxy borohydride (6.5 kg) was added over 1.5 h to a solution of N- methylpiperid-4-one (3.17 kg) and 4-fluorobenzylamine (3.50 kg) in methanol (30 L) maintaining the temperature under 27 0C. The reaction mixture was stirred for 15 h at 22 0C. The residual amine was checked by gel chromatography (4-fluorobenzylamine: < 5%). A solution of 30% sodium hydroxide (12.1 kg) in water (13.6 kg) was added in 75 minutes (min) maintaining the temperature under 20 0C. Methanol was distilled off to a residual volume of 26 litres. Ethyl acetate was added (26 L), the solution was stirred for 15 min, the phases were decanted over 15 min and the lower aqueous phase was discarded. Ethyl acetate was distilled under reduced pressure from the organic phase at 73-127 0C. At this stage the residue was mixed with a second crude batch prepared according to this method. The combined products were then distilled at 139-140 0C / 20 mbar to yield 11.2 kg product (> 82%). b) Preparation of

4-Hydroxybenzaldehyde (4.0 kg) and ethanol (20 L) were added to a solution of isobutyl bromide (9.0 kg) in ethanol (15 L). Potassium carbonate (13.6 kg) was added and the suspension was refluxed (74-78 0C) for 5 days. The residual 4- hydroxybenzaldehyde was checked by HPLC (< 10%). The suspension was cooled to 20 °C and used in the next step.
c) Preparation of

[0117] Hydroxylamine (50% in water, 8.7 kg) was added to the product from previous step b) (174 L5 176 kg) and ethanol (54 L). The suspension was refluxed (77 0C) for 3 h. Unreacted residual was checked by HPLC (< 5%). The suspension was cooled to 30 °C, filtered and the filter was washed with ethanol (54 L). The solution was concentrated by distillation under reduced pressure at 30 0C to a residual volume of 67 litters. The solution was cooled to 25 0C and water (1 10 L) was added. The suspension was concentrated by distillation under reduced pressure at 30 °C to a residual volume of 102 litters. Petrol ether (60-90 fraction, 96 L) was added and the mixture was heated to reflux (70 °C). The solution was cooled to 40 0C and crystallization was initiated by seeding. The suspension was cooled to 5 0C and stirred for 4h. The product was centrifuged and the cake was washed with petrol ether (60-90 fraction, 32 L). The wet cake was dried at about 40 °C to yield 16kg product (63%). d) Preparation of

The product from previous step c) (15.7 kg) was dissolved in ethanol (123 L). Acetic acid (8.2 kg) and palladium on charcoal 5% wet (1.1 kg) were added. The oxime was hydrogenated at 22 0C and 1.5 bar for 4h. Consumption of oxime was checked by HPLC. The catalyst was filtered and the solvent was distilled under reduced pressure at 36 °C to a final volume of 31 L. Ethyl acetate (63 L) was added and the mixture was heated to reflux (75 0C) until dissolution. The solution was cooled to 45 0C and the crystallization was initiated by seeding. The suspension was cooled to 6-10 °C and stirred for 2.5h. The product was centrifuged and the cake was washed with 2 portions of ethyl acetate (2 x 0.8 L). The wet cake was dried at a temperature of about 40 0C to yield 8 kg (41%).
e) Preparation of

Aqueous sodium hydroxide (30%, 5.0 kg) was added to a suspension of the product from previous step d) (7.9 kg) in heptane (41 L). The solution was heated to 47 °C, stirred for 15 min and decanted over 15 min. The pH was checked (pH>12) and the aqueous phase was separated. The solvent was removed by distillation under reduced pressure at 47-65 °C. Heptane was added (15 L) and then removed by distillation under reduced pressure at 58-65 0C. Heptane was added (7 L), the solution was filtered, and the filter was washed with heptane (7 L). The solvent was removed by distillation under reduced pressure at 28-60 0C. Tetrahydrofuran (THF, 107 L) and triethylamine (TEA, 6.8 kg) were added and the temperature was fixed at 22 0C. In another reactor, phosgene (5.0 kg) was introduced in tetrahydrofuran (88 L) previously cooled to -30C. The THF and TEA solution was added to the solution of phosgene in 3h 50 min, maintaining the temperature at – 3 0C. The reactor was washed with tetrahydrofuran (22 L). The mixture was stirred for 45 min at 20 0C and then for 90 min at reflux (65 0C). The solvent was distilled under reduced pressure at 25-30 0C to a residual volume of 149 L. The absence of phosgene was controlled. At this stage, phosgene was still present and the suspension was degassed by bubbling nitrogen through it. After this operation, the level of phosgene above the solution was below 0,075 ppm. The suspension was filtered and washed with tetrahydrofuran (30 L). The solvent was distilled under reduced pressure at 20-25 0C to a residual volume of 40 L. Tetrahydrofuran (51 L) was added and the solvent was distilled under reduced pressure at 20- 25 0C to a residual volume of 40 L. The final volume was adjusted to about 52 litters by addition of tetrahydrofuran (1 1 L). The solution was analysed and used in the next step.
f) Preparation of the title compound of formula I
The product from previous step e) (51 L) was added in 1 h to a solution of the product from step a) (7.3 kg) in tetrahydrofuran (132 L) at 17 0C. The line was washed with tetrahydrofuran (12 L) and the mixture was stirred for 15h. Residual product from the first step was checked by HPLC. The solvent was removed by distillation under reduced pressure at 20-38 0C to a residual volume of 165 L. Charcoal (Norit SXl-G5 0.7 kg) was added, the mixture was stirred for 15 min and filtered. The line was washed with tetrahydrofuran (7 L) and the solvent was removed by distillation under reduced pressure at 20-25 0C to a residual volume of 30 L. Isopropyl acetate (96 L) was added to obtain a solution of the title compound of formula I, which contains a small amount of impurities (mainly side products from the previous reactions.) Removal of the solvent from a sample yields a substantially amorphous solid.
The solution with the crude product was used for the direct preparation of the hemi-tartrate and simultaneously for the purification of the free base via the hemi-tartrate through crystallization from suitable solvents.
Example 5: Preparation of the hemi-tartrate of formula IV from crude free base of formula I
Crude product according to Example l(f) (4.3 kg) was dissolved at 45 0C in ethanol (23 L). A solution of (+)-L-tartaric acid (0.58 kg) in ethanol was added at 45 0C and the line was washed with 6 L of ethanol. The solution was stirred for 20 min (formation of solid precipitate) and the slurry was cooled to 35 0C over 30 min. The slurry was stirred at this temperature for 1 hour and then cooled to -5 0C. After 14 hours stirring at this temperature, the product was centrifuged and washed with 2 portions of ethanol (2 x 4 L). The wet cake was dried at 45 0C for 80 hours yielding 3.3 kg of product (85%, based on tartaric acid). PXRD of the product revealed that polymorph A was formed.
PATENT
WO2014085362A1.
CN101031548A
CN101035759A
CN102153505A
CN1816524A
US2008280886A1.
WO0144191
PATENT
WO-2016141003
Scheme 4:

The reaction depicted in Scheme 4 can be carried out in a suitable organic solvent such as acetone at rather mild conditions (e.g.40-50°C). If necessary, the R1 substituent may subsequently be converted to an isobutoxy group to obtain Pimavanserin or a salt thereof.
An overview about certain processes for preparation of Pimavanserin is shown in Scheme 5 below.
Scheme 5:

Compound A L-Tartaric acid
Hemi-tartrate salt *Compound A is Pimavanserin



Scheme 10:

Compound 1 Compound 2 Pimavanserin
Scheme 13:

An overview about synthetic routes to Pimavanserin via Compound XVI is shown in the following Scheme 14:
Scheme 14:


Example 16: Preparation of hemi-tartrate salt of Pimavanserin
To a 25 mL seal tube, equipped with a stir bar, was charged 344.4 mg of the above crude PMV (1.0 mmol in theory), 75 mg of L-tartaric acid (FW: 150.09, 0.5 mmol, 0.5 equiv.), and 7 mL (16.4 vol.) of absolute ethanol. The tube was sealed and heated to 70°C to afford a clear solution, then cooled down gradually to room temperature. The product precipitated, and the batch was further cooled down to 0-5°C and stirred at this temperature for 0.5 hour. The product was collected by vacuum filtration, and the filter cake was washed with 2 × 1 mL (2.3 vol.) of EtOH. The product was dried in the Buchner funnel under vacuum overnight, affording 177.6 mg of salt, representing a 35.4% yield in 99.6 A% purity. 1H NMR (CDCl3, 400 MHz): δ = 1.01 (d, J = 6.4 Hz, 6 H), 1.79-1.82 (m, 2H), 2.02-2.19 (m, 3H), 2.63 (brs, 5H), 3.38-3.47 (m, 2H), 3.67 (d, J = 6.4 Hz, 2H), 4.25 (d, J = 4.8 Hz, 2H), 4.32 (s, 1H), 4.38 (s, 2H), 4.58 (brs, 2H), 6.77 (d, J = 8.0 Hz, 2H), 6.95-6.99 (m, 4H), 7.17 (d, J = 7.2 Hz, 2H).
Example 21: Preparation of Pimavanserin via compound V as dihydrochloride salt
Step 1: Preparation of N-(4-fluorobenzyl)-1-methylpiperidin-4-amine dihydrochloride (Compound V x 2HCl)
The reaction was performed in 300 mL reactor. The reactor was purged with N2, then Argon. 4-Fluorobenzylamine (10 g; 80 mmol, 1.0 eq) was dissolved in dry MeCN (100 mL), then 1-methylpiperidin-4-one (10.9 g; 96 mmol, 1.2 eq) was added and the reaction mixture was stirred at ambient temperature for 18h. Then, the reaction mixture was cooled to 0°C and 25.4 g of NaBH(OAc)3 (25.4 g; 120 mmol, 1.5 eq) was added in portions over 20 min and the reaction was allowed to stir to room temperature. After 1h, the reaction was quenched by the addition of 200 ml of water, pH was adjusted to 2 with 5M HCl and then extracted using 3 x 250 mL of DCM. Basification of the aqueous layer to pH 9.5 with 30% sol. NaOH and extraction 3 x 300 ml of DCM followed. The organic layers were collected and dried over anh. Na2SO4, filtered and evaporated to dryness yielding 17.24 g (92%) of oily product, N-(4-fluorobenzyl)-1-methylpiperidin-4-amine (Compound V).
To a 250 mL, three necked, round bottom flask, equipped with a stir bar and thermometer, N-(4-fluorobenzyl)-1-methylpiperidin-4-amine (10 g; 0.045 mol) and DCM (50 mL) were charged and cooled to 10-15 °C. To the resulting solution, 5-6 N HCl in 2-PrOH (3 equiv., 0.135 mmol) was added dropwise over 25 min., white crystals formed, and the solution then cooled to 0-5 °C for 2 hours. Crystals were filtered off, washed with 50 mL of DCM, dried at 50°C/10 mbar for 10 hours yielding 12.8 g (96.4%) of N-(4-fluorobenzyl)-1-methylpiperidin-4-amine dihydrochloride (Compound V x 2HCl).
Step 2: Preparation of 4-isobutoxybenzaldehyde (Compound XIII)
4-Hydroxybenzaldehyde (10 g; 0.082 mol), potassium carbonate (33.95 g; 0.246 mol) and potassium iodide (1.36 g; 0.008 mol) were suspended in N,N-dimethylformamide (50 mL). Isobutyl bromide (26.7 mL; 0.246 mol) was added and the reaction was heated at 70°C under nitrogen for 3 hours. The reaction was cooled down, diluted by using 150 mL of water and extracted by using 300 mL of ethyl acetate. The organic layer was extracted five times by using 150 mL of 10% NaCl solution, dried under Na2SO4, filtered and concentrated which resulted in 14.3 g (98%) of yellow oily product of 4-isobutoxybenzaldehyde
(Compound XIII).
Step 3: Preparation of (4-isobutoxyphenyl)methanamine hydrochloride (Compound XI x HCl)
[0142] To a solution of 4-isobutoxybenzaldehyde (Compound XIII) (19.9 g; 0.112 mol) in methanol (90 mL), Raney nickel (6 g) and 7N methanol ammonia solution (90 mL) were added. The reaction mixture was stirred under hydrogen atmosphere (0.5 bar) at 10-15°C for 24 hours. The reaction solution was filtered through Celite to remove the catalyst. Methanol was distilled off and toluene (500 mL) was added. The solution was concentrated to 250 mL and 5-6 N HCl in 2-PrOH (30 mL; 0.15 mol) was added dropwise at ambient temperature. The resulting suspension was then cooled to 5 °C and stirred for additional 2 hours. Crystals were filtered off, washed with 60 mL of toluene, dried at 50°C/10 mbar for 10 hours yielding 20.88 g (86.7%) of (4-isobutoxyphenyl)methanamine hydrochloride (Compound XI x HCl). The product was analyzed by PXRD– form I was obtained, the PXRD pattern is shown in Figure 3.
Step 4: Option 1: Preparation of 1-(4-fluorobenzyl)-3-(4-isobutoxybenzyl)-1-(1-methylpiperidin-4-yl)urea (Pimavanserin
Part a: Preparation of Compound VI-a:
To a 250 mL, three necked, round bottom flask, equipped with a stir bar, condenser and thermometer, (4-isobutoxyphenyl)methanamine hydrochloride (Compound XI x HCl) (5 g, 0.023 mol), CDI (6.01 g; 0.037 mol) and acetonitrile (40 mL) were charged. The resulting solution was stirred for 1 h at 65-70 °C and monitored by HPLC until full conversion to Compound VI-a.
Part b: Preparation of Pimavanserin:
N-(4-fluorobenzyl)-1-methylpiperidin-4-amine (Compound V) (7.73 g; 0.035 mol) was added to Compound VI-a obtained above. After 2h, complete conversion was observed. Upon completion, the reaction solution was cooled to 50 °C and water was added dropwise in a 1:3 ratio (120 mL). After addition of a whole amount of water, crystals were formed and suspension was allowed to cool to ambient temperature. The crystals were filtered off, washed with 2 x 40 mL solution of CH3CN:H2O 1:3, then 40 mL of water, dried at 45°C/10 mbar for 10 hours yielding 9.35 g (94.4%) of 1-(4-fluorobenzyl)-3-(4-isobutoxybenzyl)-1-(1-methylpiperidin-4-yl)urea (Pimavanserin).
Step 4– option 2: Preparation of 1-(4-fluorobenzyl)-3-(4-isobutoxybenzyl)-1-(1-methylpiperidin-4-yl)urea (Pimavanserin)
Part a: Preparation of Compound VI-a:
To a 500 mL, three necked, round bottom flask, equipped with a stir bar, condenser and thermometer, (4-isobutoxyphenyl)methanamine hydrochloride (Compound XI x HCl) (10 g; 0.046 mol), CDI (11.28 g; 0.07 mol) and acetonitrile (100 mL) were charged. The resulting solution was stirred for 1 h at 65-70 °C and monitored by HPLC until full conversion to Compound VI-a.
Part b: Preparation of Pimavanserin:
[0146] The reaction solution containing Compound VI-a obtained above was cooled to 30°C and N-(4-fluorobenzyl)-1-methylpiperidin-4-amine dihydrochloride (Compound V x 2HCl) (20.53 g; 0.07 mol) and K2CO3 (9.61 g; 0.07 mol) were added. The reaction mixture was heated to 65-70 °C and stirred for next 18 hours. Upon completion, the reaction solution was cooled to 50 °C, pH of solution was adjusted to 10.5 with 6N NaOH solution, and water was added dropwise in ratio 1:3 (300 mL). After addition of a whole amount of water, crystals were formed, and suspension was allowed to cool to ambient temperature, and then cooled on ice-bath (0-5°C) for 1.5 hour. The crystals were filtered off, washed with 2 x 100 mL solution of CH3CN:H2O 1:3, then 100 mL of water, dried at 45°C/10 mbar for 10 hours yielding 18.797 g (95.6%) of 1-(4-fluorobenzyl)-3-(4-isobutoxybenzyl)-1-(1-methylpiperidin-4-yl)urea (Pimavanserin).
Example 26: One pot preparation of Pimavanserin (without isolation of Compound 1)
Step 1: Preparation of 2-(4-isobutoxyphenyl)acetic acid
To a 250 mL, 3 neck, round bottom flask, equipped with thermocouple and nitrogen sweep, was charged 10 g of 4-hydroxy phenyl acetic acid (Molecular weight (FW): 152.15, 65.7 mmol, 1.0 equiv.), 30 g of potassium carbonate (FW: 138.21, 216.8 mmol, 3.3 equiv.), 1.1 g of potassium iodide (KI, FW: 166, 6.57 mmol, 0.1 equiv.), followed by 100 mL (10 vol.) of DMF. After stirring for 5 minutes at room temperature, 15.7 mL of isobutyl bromide (FW: 137.02, 144.6 mmol, 2.2 equiv.) was charged into the batch. The mixture was then heated to 75°C and kept stirring at the same temperature for 2 days until no limited starting material remaining as determined by HPLC. The reaction was cooled down to room temperature, and quenched by charging with 100 mL of deionized (DI) water. The pH of the reaction mixture was adjusted to less than 1 by charging 100 mL of 2N HCl. The product was extracted with 150 mL of ethyl acetate. After partitioning, the upper organic layer was washed with additional 100 mL of DI water, concentrated to dryness on the rotary evaporator under vacuum. The residue was dissolved in 100 mL each of THF (10 vol) and DI water (10 vol). After charging 20 g of lithium hydroxide, the mixture was heated to reflux for 3 hours until complete reaction. The batch was cooled to room temperature, concentrated on rotary
evaporator to remove THF. The residue was acidified with 300 mL of 2N HCl and 45 mL of 6N HCl aqueous solution until pH <1. The product was extracted with 2×250 mL of methylene chloride, dried over sodium sulfate, and filtered on Buchner funnel. The filtrate was concentrated to dryness on rotary evaporator under vacuum to afford 10.18 g of 2-(4-isobutoxyphenyl)acetic acid, representing a 74.4% yield in 98.5 A% purity. 1H NMR (d6-DMSO, 400 MHz): δ = 0.97 (d, J = 6.8 Hz, 6 H), 1.96-2.02 (m, 1H), 3.47 (s, 2H), 3.71 (d, J = 6.4 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H).
Step 2: Preparation of Pimavanserin
To a 50 mL, single neck, round bottom flask, equipped with thermocouple and nitrogen sweep, was charged 333.2 mg of 2-(4-isobutoxyphenyl)acetic acid (FW: 208.25, 1.6 mmol, 1.0 equiv.), 311.3 mg of CDI (FW: 162.15, 1.92 mmol, 1.2 equiv.), and 3.3 mL of CH3CN (10 vol.). After stirring at room temperature for 1 hour, this was charged 139 mg (FW: 69.5, 2.0 mmol, 1.25 equiv.) of NH2OH.HCl and stirred for additional 15-18 hours at room temperature. Additional 518.9 mg of CDI (FW: 162.15, 3.2 mmol, 2.0 equiv.) was charged and the batch turned from a slurry to a clear solution again. This was followed by charging a solution of 334 mg of Compound V (FW: 222.3, 1.5 mmol, 0.94 equiv.), and heating up to 60 oC. The reaction was stirred at this temperature for approximately 5 hour before cooling back to room temperature. The reaction was quenched with 20 mL of DI water, and concentrated on rotary evaporator to remove acetonitrile. The aqueous residue was diluted with 40 mL of ethyl acetate, and washed with 2×20 mL of brine. The organic phase was concentrated to dryness on rotary evaporator under vacuum. The residue was purified by chromatography (160 g RediSep Alumina column), eluting with 0-5% of methanol in dichloromethane to afford 305 mg of Pimavanserin, representing a 47.6% yield in 99.3 A% purity.1H NMR (CDCl3, 400 MHz): δ = 1.01 (d, J = 6.8 Hz, 6 H), 1.62-1.73 (m, 4H), 2.03-2.09 (m, 3H), 2.25 (s, 3H), 2.84-2.87 (m, 2H), 3.68 (d, J = 6.4 Hz, 2H), 4.27-4.34 (m, 5H), 4.45-4.48 (m, 1H), 6.67-6.79 (m, 2H), 6.99-7.02 (m, 4H), 7.16-7.27 (m, 2H). HRMS-ESI (m/z): [M+1]+ Calcd for C25H35F1N3O2: 428.2708; found 428.2723.
Example 27: Preparation of Pimavanserin (with isolation of Compound 1)
Step 1: Preparation of Compound 1
To a 100 mL, single neck, round bottom flask, equipped with thermocouple and nitrogen sweep, was charged 1 g of Compound XV (FW: 208.25, 4.8 mmol, 1.0 equiv.), 934.0 mg of CDI (FW: 162.15, 5.76 mmol, 1.2 equiv.), followed by 10 mL (10 vol.) of acetonitrile. After stirring for 45 minutes at room temperature, 417 mg of NH2OH.HCl (FW: 69.5, 6.0 mmol, 1.25 equiv.) was charged into the batch. The mixture was kept stirring at the ambient temperature overnight and turned into a thick slurry. HPLC determined 1.6 A% of starting material remaining. The batch was diluted with 6 mL of acetonitrile (6 vol.) and 16 mL (16 vol.) of DI water, and cooled down to 0-5 ºC. After stirring at the same temperature for additional 1 hour, the batch was filtered on the Buchner funnel. The filter cake was washed with 2×10 mL (10 vol.) of DI water, and dried in the funnel under vacuum overnight to afford 774.1 mg of hydroxamic acid Compound 1, representing a 72% yield in 99.6 A% purity. 1H NMR (CDCl3, 400 MHz): δ = 0.96 (d, J = 6.8 Hz, 6 H), 1.95-2.02 (m, 1H), 3.19 (s, 2H), 3.70 (d, J = 6.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 8.80 (s, 1H), 10.61 (s, 1H).
Step 2: Synthesis of Pimavanserin
To a 50 mL sealed tube, equipped with nitrogen sweep, was charged 250 mg of compound 1 (FW: 223.27, 1.12 mmol, 1.0 equiv.), 217.9 mg of CDI (FW: 162.15, 1.34 mmol, 1.2 equiv.), and 1.7 mL of acetonitrile (6.8 vol.). After stirring at room temperature for 40 minutes, the batch was heated to 60 oC and kept stirring at the same temperature for additional 10 minutes. This was followed by charging 373.5 mg of Compound 3 (FW: 222.3, 1.68 mmol, 1.5 equiv.). The container of Compound V was rinsed with 0.5 mL (2 vol.) of acetonitrile, and the wash was combined with the batch. The reaction was monitored by HPLC and complete in 2 hours. The batch was cooled down to room temperature, diluted with 5 mL (20 vol.) of ethyl acetate, which was washed with 3×5 mL (20 vol.) of DI water. After partitioning, the upper organic layer was concentrated to dryness on rotary evaporator. The residue was re-dissolved into 3 mL (12 vol.) of ethyl acetate after heating up to reflux to afford a slightly milky solution. This was charged with 12 mL (48 vol.) of heptane, and cooled down to 0-5oC. The batch was kept stirring at the same temperature for 1 hour and filtered on a Buchner funnel. The filter cake was washed with 2×5 mL (20 vol.) of heptane, and dried in the funnel with a nitrogen sweep for 1 hour to afford 270.8 mg of Pimavanserin as a white solid, representing a 56.6% yield in 98.8 A% purity. 1H NMR (CDCl3, 400 MHz): δ = 1.01 (d, J = 6.8 Hz, 6 H), 1.62-1.73 (m, 4H), 2.03-2.09 (m, 3H), 2.25 (s, 3H), 2.84-2.87 (m, 2H), 3.68 (d, J = 6.4 Hz, 2H), 4.27-4.34 (m, 5H), 4.45-4.48 (m, 1H), 6.67-6.79 (m, 2H), 6.99-7.02 (m, 4H), 7.16-7.27 (m, 2H). HRMS-ESI (m/z): [M+1]+ Calcd for C25H35F1N3O2: 428.2708; found 428.2723.
Example 34: Preparation of Pimavanserin from Compound 2
To a 25 mL, three neck, round bottom flask, equipped with a stir bar, condenser and thermocouple, Compound 2, 0.210 g, was charged (FW: 249.26, 0.84 mmol, 1.0 equiv.). This was followed 3 mL of acetonitrile, anhydrous, 99.8%. The mixture was stirred at 60°C for 4 h. Then, to the reaction mixture, Compound V, 0.375 g (FW: 222.30, 1.69 mmol, 2.0 equiv.), was added. After 1h, complete conversion was observed. The reaction was diluted with EtOAc (20 mL) and washed twice with a saturated solution of NH4Cl (2 x 15 mL), then H2O (10 mL) and finally with a saturated NaCl solution (10 mL). The organic layer was dried over anh. sodium sulfate, filtered and concentrated under partial vacuum to about 5 mL of EtOAc. To this solution, n-heptane (10 ml) was added with vigorous stirring, in a dropwise manner, over half an hour. A white precipitate was formed, followed by filtration and drying in vacuum at 45°C for 3h, affording 0.188 g of Pimavanserin. HPLC-MS (m/z) [M+1]+ 428.2; 1H NMR (CDCl3, 400 MHz): δ = 1.01 (d, J = 6.7 Hz, 6 H), 1.68-1.77 (m, 4H), 2.03-2.10 (m, 3H), 2.30 (s, 3H), 2.91-2.97 (m, 2H), 3.67(d, J = 6.7 Hz, 2H), 4.27 (d, J = 5.4 Hz, 2H), 4.31-4.43 (m, 3H), 4.50 (brt, J = 5.5 Hz, 1H), 6.74-6-79 (m, 2H), 6.95-7.05 (m, 4H), 7.14-7.22 (m, 2H).
Example 38: Preparation of Pimavanserin from Compound and Compound V x 2HCl
250 mL reactor was charged with N-hydroxy-2-(4-isobutoxyphenyl)acetamide (Compound 1) (10 g, 0.045 mol), CDI (10.53 g, 0.076 mol) and 100 mL of MeCN, p.a. The resulting solution was stirred for 1.5 h at 60-65 °C and monitored by HPLC. Upon full conversion to the corresponding isocyanate, reaction solution was cooled to 35 °C and N-(4- fluorobenzyl)-1-methylpiperidin-4-amine dihydrochloride (Compound V x 2HCl) (22.48 g, 0.065 mol) and K2CO3 (6.19 g, 0.045 mol) were added. Reaction mixture was heated up to 60-65 °C and stirred for 6 hours and followed by 17 h at ambient temperature.
Upon completion, the reaction solution was cooled to 20 °C and water was added dropwise in ratio 1:3 (300 mL) with adjustment of pH to 11 with 6N NaOH solution. After addition of whole amount of water, crystals were formed and suspension was stirred at 20 °C for 2 h and 0-5°C for next 2 hour. Crystals were filtered off, washed with 2 x 100 mL solution of MeCN:H2O 1:3, then 100 mL of H2O, dried at 30°C/10 mbar for 24 hours yielding 17.56 g (91.7%) of Pimavanserin.
PAPER
Bioorg. Med. Chem. Lett. 2015, 25, 1053–1056.
11C-labeling and preliminary evaluation of pimavanserin as a 5-HT2A receptor PET-radioligand
- Valdemar L. Andersena, b, c,
- Hanne D. Hansena, b,
- Matthias M. Hertha, b,
- Agnete Dyssegaarda, b,
- Gitte M. Knudsena, b,
- Jesper L. Kristensenb, c, ,
- a Neurobiology Research Unit, Rigshospitalet and University of Copenhagen, Blegdamsvej 9, 2100 Copenhagen, Denmark
- b Center for Integrated Molecular Brain Imaging, University of Copenhagen Rigshospitalet, Blegdamsvej 9, 2100 Copenhagen, Denmark
- c Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark
Pimavanserin is a selective serotonin 2A receptor (5-HT2AR) inverse agonist that has shown promise for treatment of psychotic symptoms in patients with Parkinson’s disease. Here, we detail the 11C-labeling and subsequently evaluate pimavanserin as a PET-radioligand in pigs. [11C]Pimavanserin was obtained by N-methylation of an appropriate precursor using [11C]MeOTf in acetone at 60 °C giving radiochemical yields in the range of 1–1.7 GBq (n = 4). In Danish Landrace pigs the radio ligand readily entered the brain and displayed binding in the cortex in accordance with the distribution of 5-HT2ARs. However, this binding could not be blocked by either ketanserin or pimavanserin itself, indicating high nonspecific binding. The lack of displacement by the 5-HT2R antagonist and binding in the thalamus suggests that [11C]pimavanserin is not selective for the 5-HT2AR in pigs.
Clip
THURSDAY Oct. 31, 2013 — Many people living with Parkinson’s disease suffer from hallucinations and delusions, but an experimental drug might offer some relief without debilitating side effects.
READ ALL AT
http://www.drugs.com/news/new-shows-early-promise-treating-parkinson-s-psychosis-48630.html

The drug — pimavanserin — appears to significantly relieve these troubling symptoms, according to the results of a phase 3 trial to test its effectiveness.
Pimavanserin (ACP-103) is a drug developed by Acadia Pharmaceuticals which acts as an inverse agonist on the serotonin receptor subtype 5-HT2A, with 40x selectivity over 5-HT2C, and no significant affinity or activity at 5-HT2B or dopamine receptors.[1] As of September 3 2009, pimavanserin has not met expectations for Phase III clinical trials for the treatment of Parkinson’s disease psychosis,[2] and is in Phase II trials for adjunctive treatment of schizophrenia alongside an antipsychotic medication.[3] It is expected to improve the effectiveness and side effect profile of antipsychotics.[4][5][6]
3-D MODEL OF DRUG PIMAVANSERIN, THE DEVELOPMENT OF WHICH HAS BEEN EXPEDITED BY THE FDA

Psychiatrist Herb Meltzer sadly watched the agitated woman accuse her son of trying to poison her. Although not her physician, Dr. Meltzer certainly recognized the devastating effects of his mother-in-law’s Parkinson’s disease psychosis (PDP). Occurring in up to half of all patients with Parkinson’s, symptoms of the psychotic disorder may include hallucinations and delusions. The development of PDP often leads to institutionalization and increased mortality.
“I was on the sidelines,” explains Dr. Meltzer, professor of psychiatry and physiology and director of the Translational Neuropharmacology Program at Northwestern University Feinberg School of Medicine. “I told my brother-in-law it was the disease talking, not his mother.”
Ironically, Dr. Meltzer has been far from the sidelines and right on the PDP playing field for quite a while. In fact, he may soon see a drug he helped develop become the first approved treatment for the disorder. In early April, Dr. Meltzer celebrated, along with colleagues at ACADIA Pharmaceuticals in San Diego for which he has been a clinical advisor, the stunning announcement: the Food and Drug Administration (FDA) had expedited the company’s path to filing a new drug application (NDA) for pimavanserin, a selective serotonin 5-HT2Areceptor blocker. Typically, the FDA requires data from two successful pivotal Phase III clinical studies affirming a drug candidate’s safety and efficacy before the agency will even consider an NDA. Just as ACADIA was planning to launch another Phase III study this spring to fulfill this requirement, the FDA decided the company had amassed enough data to support an NDA filing.

HERBERT MELTZER, MD, DESIGNED ACADIA PHARMACEUTICAL’S INITIAL PROOF OF CONCEPT TRIAL OF THE DRUG PIMAVANSERIN TO TREAT PARKINSON’S DISEASE PSYCHOSIS.
“This action on the part of the FDA is extremely unusual,” says Dr. Meltzer, who designed ACADIA’s initial proof-of-concept trial of pimavanserin, a drug he had initially suggested ACADIA develop to treat schizophrenia, with PDP as a secondary indication. “The FDA staff decided that results from my small clinical study and the first successful Phase III study were sufficient to establish efficacy and safety.”
Bringing a safe and effective drug to market is a monumental achievement. Pimavanserin is not yet there but has significantly moved within striking distance with this recent nod from the regulatory agency.
24 YEARS IN THE MAKING
The neuropharmacologist’s collaboration with ACADIA began in 2000. The company wanted to develop a drug targeting the serotonin 5-HT 2A receptor, a neurotransmitter ACADIA believed played a key role in schizophrenia based upon basic research from Meltzer and their own studies. A distinguished schizophrenia investigator, then at Case Western Reserve University, he welcomed ACADIA’s offer to translate his ideas about developing safer and more effective drug treatments for psychosis. Through his provocative and groundbreaking research, Dr. Meltzer originally championed the idea that blocking the 5-HT2A receptor would lead to better antipsychotic drugs with fewer side effects. Existing drugs often impaired motor function because they targeted the dopamine D2 receptor. Of the 14 different types of serotonin receptors in this complex area of study, Dr. Meltzer zeroed in on the 5-HT2A type—the same receptor that leads to hallucinogenic properties of LSD and mescaline. It was an ideal target to complement weak D2 receptor blockade in schizophrenia and as a standalone treatment for PD psychosis.
External links
References
- Friedman, JH (October 2013). “Pimavanserin for the treatment of Parkinson’s disease psychosis”. Expert Opinion on Pharmacotherapy. 14 (14): 1969–1975.doi:10.1517/14656566.2013.819345. PMID 24016069.
- ^ Jump up to:a b c “Nuplazid (pimavanserin) Tablets, for Oral Use. U.S. Full Prescribing Information” (PDF). ACADIA Pharmaceuticals Inc. Retrieved 1 May 2016.
- Jump up^ ACADIA Pharmaceuticals. “Treating Parkinson’s Disease – Clinical Trial Pimavanserin – ACADIA”. Archived from the original on February 25, 2009. Retrieved 2009-04-11.
- Jump up^ “ACADIA Announces Positive Results From ACP-103 Phase II Schizophrenia Co-Therapy Trial” (Press release). ACADIA Pharmaceuticals. 2007-03-19. Retrieved 2009-04-11.
- Jump up^ Gardell LR, Vanover KE, Pounds L, Johnson RW, Barido R, Anderson GT, Veinbergs I, Dyssegaard A, Brunmark P, Tabatabaei A, Davis RE, Brann MR, Hacksell U, Bonhaus DW (Aug 2007). “ACP-103, a 5-hydroxytryptamine 2A receptor inverse agonist, improves the antipsychotic efficacy and side-effect profile of haloperidol and risperidone in experimental models”. The Journal of Pharmacology and Experimental Therapeutics. 322 (2): 862–70. doi:10.1124/jpet.107.121715.PMID 17519387.
- Jump up^ Vanover KE, Betz AJ, Weber SM, Bibbiani F, Kielaite A, Weiner DM, Davis RE, Chase TN, Salamone JD (Oct 2008). “A 5-HT2A receptor inverse agonist, ACP-103, reduces tremor in a rat model and levodopa-induced dyskinesias in a monkey model”. Pharmacology, Biochemistry, and Behavior. 90 (4): 540–4. doi:10.1016/j.pbb.2008.04.010. PMC 2806670
 .PMID 18534670.
.PMID 18534670.
- Jump up^ Abbas A, Roth BL (Dec 2008). “Pimavanserin tartrate: a 5-HT2A inverse agonist with potential for treating various neuropsychiatric disorders”. Expert Opinion on Pharmacotherapy. 9 (18): 3251–9.doi:10.1517/14656560802532707. PMID 19040345.
- Jump up^ Meltzer HY, Elkis H, Vanover K, Weiner DM, van Kammen DP, Peters P, Hacksell U (Nov 2012). “Pimavanserin, a selective serotonin (5-HT)2A-inverse agonist, enhances the efficacy and safety of risperidone, 2mg/day, but does not enhance efficacy of haloperidol, 2mg/day: comparison with reference dose risperidone, 6mg/day”. Schizophrenia Research. 141 (2-3): 144–152. doi:10.1016/j.schres.2012.07.029. PMID 22954754.
- Jump up^ “ACADIA Pharmaceuticals Receives FDA Breakthrough Therapy Designation for NUPLAZID™ (Pimavanserin) for Parkinson’s Disease Psychosis”. Press Releases. Acadia. 2014-09-02.
- Jump up^ “Press Announcements — FDA approves first drug to treat hallucinations and delusions associated with Parkinson’s disease”. U.S. Food and Drug Administration. Retrieved1 May 2016.
NUPLAZID contains pimavanserin, an atypical antipsychotic, which is present as pimavanserin tartrate salt with the chemical name, urea, N-[(4-fluorophenyl)methyl]-N-(1-methyl-4-piperidinyl)-N’-[[4-(2- methylpropoxy)phenyl]methyl]-,(2R,3R)-2,3-dihydroxybutanedioate (2:1). Pimavanserin tartrate is freely soluble in water. Its molecular formula is (C25H34FN3O2)2•C4H6O6 and its molecular weight is 1005.20 (tartrate salt). The chemical structure is:
The molecular formula of pimavanserin free base is C25H34FN3O2 and its molecular weight is 427.55.
NUPLAZID tablets are intended for oral administration only. Each round, white to off-white, immediaterelease, film-coated tablet contains 20 mg of pimavanserin tartrate, which is equivalent to 17 mg of pimavanserin free base. Inactive ingredients include pregelatinized starch, magnesium stearate, and microcrystalline cellulose. Additionally, the following inactive ingredients are present as components of the film coat: hypromellose, talc, titanium dioxide, polyethylene glycol, and saccharin sodium.
| WO2006036874A1 * |
26 Sep 2005 |
6 Apr 2006 |
Acadia Pharmaceuticals Inc. |
Salts of n-(4-fluorobenzyl)-n-(1-methylpiperidin-4-yl)-n’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and their preparation |
| WO2006037043A1 * |
26 Sep 2005 |
6 Apr 2006 |
Acadia Pharmaceuticals Inc. |
Synthesis of n-(4-fluorobenzyl)-n-(1-methylpiperidin-4-yl)-n’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms |
| WO2007133802A2 * |
15 May 2007 |
22 Nov 2007 |
Acadia Pharmaceuticals Inc. |
Pharmaceutical formulations of pimavanserin |
| US20060205780 * |
3 May 2006 |
14 Sep 2006 |
Thygesen Mikkel B |
Synthesis of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms |
| US20060205781 * |
3 May 2006 |
14 Sep 2006 |
Thygesen Mikkel B |
Synthesis of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms |
| US20070260064 * |
15 May 2007 |
8 Nov 2007 |
Bo-Ragnar Tolf |
Synthesis of n-(4-fluorobenzyl)-n-(1-methylpiperidin-4-yl)-n’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms |
Jeffrey Cummings, Stuart Isaacson, Roger Mills, Hilde Williams, Kathy Chi-Burris, Anne Corbett, Rohit Dhall, Clive Ballard.
Pimavanserin for patients with Parkinson’s disease psychosis: a randomised, placebo-controlled phase 3 trial.
The Lancet, Volume 383, Issue 9916, Pages 533 – 540, 8 February 2014.
Findings: Between Aug 11, 2010, and Aug 29, 2012, we randomly allocated 199 patients to treatment groups. For 90 recipients of placebo and 95 recipients of pimavanserin included in the primary analysis, pimavanserin was associated with a −5·79 decrease in SAPS-PD scores compared with −2·73 for placebo (difference −3·06, 95% CI −4·91 to −1·20; p=0·001; Cohen’s d 0·50). Ten patients in the pimavanserin group discontinued because of an adverse event (four due to psychotic disorder or hallucination within 10 days of start of the study drug) compared with two in the placebo group. Overall, pimavanserin was well tolerated with no significant safety concerns or worsening of motor function.This study is registered with ClinicalTrials.gov, number NCT01174004.Bo-Ragnar Tolf, Nathalie Schlienger, Mikkel Boas Thygesen.
Synthesis of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N′-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms.
US patent number:US7790899 B2
Also published as:CA2692001A1, CN101778821A, EP2146960A2, US20070260064, WO2008144326A2, WO2008144326A3.
Publication date:Sep 7, 2010.
Original Assignee:Acadia Pharmaceuticals, Inc.Tolf, Bo-Ragmar; Schlienger, Nathalie; Thygesen, Mikkel Boas.
Preparation of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N’-[4-(2-methylpropyloxy)phenylmethyl]carbamide and its tartrate salt and crystalline forms.
PCT Int. Appl. (2008), WO2008144326 A2 20081127.Tolf, Bo-Ragnar; Schlienger, Nathalie; Thygesen, Mikkel Boas.
Synthesis of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy)phenylmethyl)carbamide and its tartrate salt and crystalline forms.
U.S. Pat. Appl. Publ. (2007), US20070260064 A1 20071108.Pyke, Robert; Ceci, Angelo.
Pharmaceutical compositions for the treatment and/or prevention of schizophrenia and related diseases.
PCT Int. Appl. (2006), WO2006096439 A2 20060914.Wang, Y.; Bolos, J.; Serradell, N.ACP-103:
5-HT2A receptor inverse agonist treatment of psychosis treatment of sleep disorders.
Drugs of the Future (2006), 31(11), 939-943.Roberts, Claire.
Drug evaluation: ACP-103, a 5-HT2A receptor inverse agonist.
Current Opinion in Investigational Drugs (Thomson Scientific) (2006), 7(7), 653-660.hygesen, Mikkel; Schlienger, Nathalie; Tolf, Bo-Ragnar; Blatter, Fritz; Berghausen, Jorg.
Process for preparation of salts of N-(4-fluorobenzyl)-N-(1-methylpiperidin-4-yl)-N’-(4-(2-methylpropyloxy) phenylmethyl)carbamide.
PCT Int. Appl. (2006), WO2006036874 A1 20060406.Clip
FDA approves first drug to treat hallucinations and delusions associated with Parkinson’s disease
The U.S. Food and Drug Administration today approved Nuplazid (pimavanserin) tablets, the first drug approved to treat hallucinations and delusions associated with psychosis experienced by some people with Parkinson’s disease.
Hallucinations or delusions can occur in as many as 50 percent of patients with Parkinson’s disease at some time during the course of their illness. People who experience them see or hear things that are not there (hallucinations) and/or have false beliefs (delusions). The hallucinations and delusions experienced with Parkinson’s disease are serious symptoms, and can lead to thinking and emotions that are so impaired that the people experiencing them may not relate to loved ones well or take appropriate care of themselves.
“Hallucinations and delusions can be profoundly disturbing and disabling,” said Mitchell Mathis, M.D., director of the Division of Psychiatry Products in the FDA’s Center for Drug Evaluation and Research. “Nuplazid represents an important treatment for people with Parkinson’s disease who experience these symptoms.”
An estimated 50,000 Americans are diagnosed with Parkinson’s disease each year, according to the National Institutes of Health, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, when cells in the brain that produce a chemical called dopamine become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movement — like eating, writing and shaving. Early symptoms of the disease are subtle and occur gradually. In some people Parkinson’s disease progresses more quickly than in others. As the disease progresses, the shaking, or tremor, which affects the majority of people with Parkinson’s disease, may begin to interfere with daily activities. Other symptoms may include depression and other emotional changes; hallucinations and delusions; difficulty in swallowing, chewing, and speaking; urinary problems or constipation; skin problems; and sleep disruptions.
The effectiveness of Nuplazid was shown in a six-week clinical trial of 199 participants. Nuplazid was shown to be superior to placebo in decreasing the frequency and/or severity of hallucinations and delusions without worsening the primary motor symptoms of Parkinson’s disease.
As with other atypical antipsychotic drugs, Nuplazid has a Boxed Warning alerting health care professionals about an increased risk of death associated with the use of these drugs to treat older people with dementia-related psychosis. No drug in this class is approved to treat patients with dementia-related psychosis.
In clinical trials, the most common side effects reported by participants taking Nuplazid were: swelling, usually of the ankles, legs, and feet due to the accumulation of excessive fluid in the tissue (peripheral edema); nausea; and abnormal state of mind (confused state).
Nuplazid was granted breakthrough therapy designation for the treatment of hallucinations and delusions associated with Parkinson’s disease. Breakthrough therapy designation is a program designed to expedite the development and review of drugs that are intended to treat a serious condition and where preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint. The drug was also granted a priority review. The FDA’s priority review program provides for an expedited review of drugs that offer a significant improvement in the safety or effectiveness for the treatment, prevention, or diagnosis of a serious condition.
Nuplazid is marketed by Acadia Pharmaceuticals Inc. of San Diego, California.
//////////Pimavanserin, FDA 2016, Nuplazid®, Acadia , Breakthrough Therapy, PRIORITY REVIEW,

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



















 ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)
ONPATTRO™ (patisiran) packaging and product vial (Photo: Business Wire)
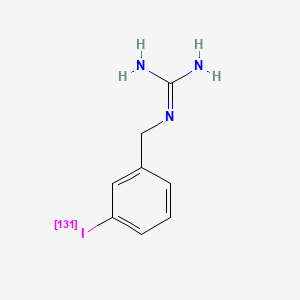

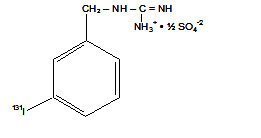













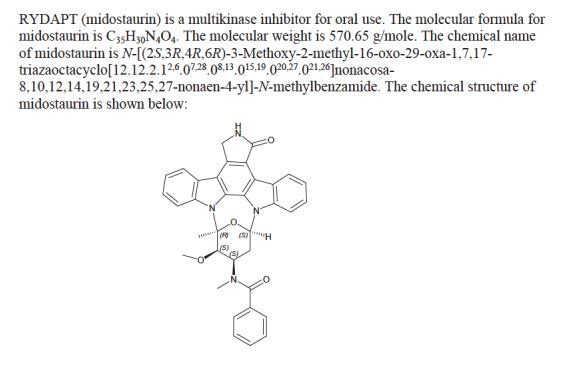
 MIDOSTAURIN
MIDOSTAURIN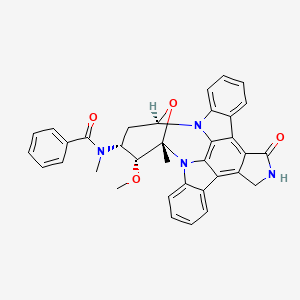 MIDOSTAURIN
MIDOSTAURIN























{kind=link}