
Home » Posts tagged 'Borole'
Tag Archives: Borole
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
AN 2898

AN2898
(5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole)
1,2-Benzenedicarbonitrile, 4-((1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy)-,
AN-2898
cas: 906673-33-4
UNII: 6O60L94RMB,
MW 276.0581, MF C15 H9 B N2 O3
A PDE4 inhibitor potentially for the treatment of fungal infection.
AN-2898, a novel topical anti-inflammatory compound that inhibits phosphodiesterase 4 and 7 enzyme activit
PHASE 2 FUNGAL INFECTION, Anacor Pharmaceuticals for the treatment of atopic dermatitis
![]()
| Anacor Pharmaceuticals Inc. | |
| Description | Boron-containing small molecule phosphodiesterase-4 (PDE-4) inhibitor that reduces the production of tumor necrosis factor (TNF) alpha, IL-12 and IL-23 |
| Molecular Target | Phosphodiesterase-4 (PDE-4) |
| Mechanism of Action | Phosphodiesterase-4 (PDE-4) inhibitor |
| Therapeutic Modality | Small molecule |
AN2898 (5-(3,4-dicyanophenoxy)-1-hydroxy -1,3-dihydro-2,1-benzoxaborole) is a broad spectrum anti-inflammatory compound currently in development for the topical treatment of plaque and atopic psoriasis.
AN2898 inhibited phosphodiesterase 4 (PDE4) enzyme activity (IC50 0.060 μM) and the release of multiple cytokines including TNF-α (IC50 0.16 μM) from peripheral blood mononuclear cells (hPBMCs) stimulated by lipopolysaccharide (LPS) or phytohemag- glutinin.
Further, AN2898 was also found to inhibit IL-23 release (IC50 1.0 μM) from THP-1 cells stimulated by LPS and IFN-γ. Investigation of the structure-activity relation-ship around this compound was reported to identify a more potent dual TNF-α/IL-23 inhibitor
( ref………. Akama T, Antunes J, Freund Y, Kimura R, Dong C, Sanders V, et al. Structure-activity studies of novel oxaborole dual inhibitors of PDE4 and IL-23 release. 69th Annu Meet Soc Invest Dermatol (May 6-9, Montreal) 2009 Abst 282. ).
PATENT
WO 2007095638
https://www.google.co.in/patents/WO2007095638A2?cl=en
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en
US 7582621
http://www.google.co.in/patents/US7582621
WO 2009111676
http://www.google.im/patents/WO2009111676A2?cl=en
WO 2007078340
http://www.google.im/patents/WO2007078340A2?cl=en
US 20070286822
http://www.google.com/patents/US20070286822
REFERENCES
1 Structure-activity studies led to the discovery of AN2898 in development for topical treatment of psoriasis and atopic dermatitis, J Am Acad Dermatol 2009, 60(3, Suppl. 1): Abst P1317
2 FEBS Letters (2012), 586(19), 3410-3414
See all Bboroles at………http://apisynthesisint.blogspot.in/p/borole-compds.html
/////////AN2898, AN 2898, ANACOR, BOROLE
B1(c2ccc(cc2CO1)Oc3ccc(c(c3)C#N)C#N)O
AN-2718, a New Borole in the pipeline
AN-2718,
2,1-Benzoxaborole, 5-chloro-1,3-dihydro-1-hydroxy-
5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole
5-Chloro-2,1-benzoxaborol-1(3H)-ol
CAS 174672-06-1
UNII: 810U6C2DGG
MW 168.3864, MF C7 H6 B Cl O2
MP…150-154 °C [WO 9533754]
MP 142-144 °C [ jmc 2006, 49(15) 447]
M.p. 147- 149 °C. WO2013050591
Anacor Pharmaceuticals Inc, INNOVATOR
Onychomycosis is a disease of the nail caused by yeast, dermatophytes, or other molds, and represents approximately 50% of all nail disorders. Toenail infection accounts for approximately 80% of onychomycosis incidence, while fingernails are affected in about 20% of the cases. Dermatophytes are the most frequent cause of nail plate invasion, particularly in toenail onychomycosis. Onychomycosis caused by a dermatophyte is termed Tinea unguium. Trichophyton rubrum is by far the most frequently isolated dermatophyte, followed by T. mentagrophytes. Distal subungual onychomycosis is the most common presentation of tinea unguium, with the main site of entry through the hyponychium (the thickened epidermis underneath the free distal end of a nail) progressing in time to involve the nail bed and the nail plate. Discoloration, onycholysis, and accumulation of subungual debris and nail plate dystrophy characterize the disease. The disease adversely affects the quality of life of its victims, with subject complaints ranging from unsightly nails and discomfort with footwear, to more serious complications including secondary bacterial infections.
Many methods are known for the treatment of fungal infections, including the oral and topical use of antibiotics (e.g., nystatin and amphotericin B), imidazole anti-fungal agents such as miconazole, clotrimazole, fluconazole, econazole and sulconazole, and non-imidazole fungal agents such as the allylamine derivatives terbinafme and naftifϊne, and the benzylamine butenafine. However, onychomycosis has proven to be resistant to most treatments. Nail fungal infections reside in an area difficult to access by conventional topical treatment and anti-fungal drugs cannot readily penetrate the nail plate to reach the infection sites under the nail. Therefore, onychomycosis has traditionally been treated by oral administration of anti-fungal drugs; however, clearly this is undesirable due to the potential for side effects of such drugs, in particular those caused by the more potent anti-fungal drugs such as itraconazole and ketoconazole. An alternative method of treatment of onychomycosis is by removal of the nail before treating with a topically active anti-fungal agent; such a method of treatment is equally undesirable. Systemic antimycotic agents require prolonged use and have the potential for significant side effects. Topical agents have usually been of little benefit, primarily because of poor penetration of the anti-fungal agents into and through the nail mass.
- 51. Hui X, Baker SJ, Wester RC, In Vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. J Pharm Sci 2007;96(10):2622-31 [CrossRef], [PubMed], [Web of Science ®]
- 55. Mao W, Seiradake E, Crepin T, AN2718 has Broad Spectrum Antifungal Activity Necessary for the Topical Treatment of Skin and Nail Fungal Infections. J Am Acad Dermatol 2007:56(2 Suppl):AB124
- 56. ClinicalTrials.gov. Cumulative Irritation Test. Available from: http://clinicaltrials.gov/ct2/show/NCT00781664 [Cited 11 December 2011]
SYNTHESIS

Reduction of 2-bromo-5-chlorobenzoic acid with BH3/THF in THF gives 2-bromo-5-chlorobenzyl alcohol , which is protected as the methoxymethyl derivative by treatment with MOM-Cl in the presence of DIEA in CH2Cl2. Metalation and boronylation of aryl bromide either with t-BuLi or BuLi (1,2,3,4) and (i-PrO)3B (4) or B(OMe)3 in THF affords the title compound .
Alternatively, reaction of 3-chlorobenzaldehyde with p-toluenesulfonylhydrazide in MeOH provides tosyl hydrazone which undergoes thermal decomposition in the presence of BBr3 and catalytic FeCl3 in refluxing CH2Cl2, followed by heating with aqueous NaOH to produce the title oxaborole compound .
You can construct from this…………….
 OH A
OH A
(2-bromo-5-chloro-phenyl)methanol (A)
 B
B
l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
 1
1
5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
PATENT
WO 9533754
https://www.google.com/patents/WO1995033754A1?cl=en
Example 1 Preparation of 5-chloro-1,3-dihydro-l-hydroxy-2,1- benzoxaborole (Method B).
a)
Preparation of 3-chlorobenzaldehyde tosyl hydrazide
A solution of 3-chlorobenzaldehyde (15.56 parts; 0.109M;
Aldrich) in methylated spirits (40 ml) was added slowly at below 10°C to a stirred suspension of p-toluene-sulphonylhydrazide (20.7 parts;
0.108M) in methylated spirits (150 ml). The reaction mass was then stirred at 20 to 25°C for 1 hour and then heated at 60-70°C for 1M hours when the reactants and products dissolved. The solvent was then removed by rotary evaporation and the product was obtained as a solid which was slurried with ether and washed with n-hexane. Yield = 27.2 parts (81.5% theory) mpt 122-3°C. Elemental analysis
Theory 54.5%C; 4.2%H; 9.1%N
Found 54.5%C; 4.3%H; 9.1%N
Proton NMR (CDCl3:ppm)
8.5, s, 1H(-NH-); 7.9, d, 2H(Tosyl aromatic); 7.7,s, 1H(CH=N); 7.5, s, 1H (aromatic); 7.2-7.4, m, 5H(Tosyl aromatic); 2.3, s, 3H(-CH3) b) Preparation of title compound
A suspension of anhydrous ferric chloride catalyst (0.75 parts, Fisons) in dry dichloromethane (20 ml) was added at 20 to 25°C simultaneously with boron tribromide (25 parts, 0.1M, Aldrich) in dry dichloromethane (100 mis) to a stirred suspension of the hydrazide from a) above (10.18 part, 0.033M) in dry dichloromethane (160 mis) under a nitrogen blanket. The reactants were then stirred under reflux and the evolved hydrogen bromide trapped under aqueous sodium hydroxide. After 3 hours stirring at reflux, the reactants were allowed to stand at 20- 25°C for 48 hours and then stirred under reflux for a further 4 hours. The reaction mass was then cooled and the solvent removed by rotary evaporation. The solid obtained was then stirred under reflux with 2N sodium hydroxide solution (160 ml) for 3 hours. The brown aqueous suspension was extracted with dichloromethane (50 ml), screened and then acidified to about pH 2 by addition of 2N hydrochloric acid. The solid was filtered, slurried with dichloromethane (400 ml) and then washed with a saturated solution of sodium bicarbonate followed by water.
Yield = 24 parts (43% theory). The solid was slurried in hot
dichloromethane and filtered to give 0.36 parts oxaborole mp 140-45°C. The dichloromethane solution was cooled and the solid filtered giving a further 0.35 parts oxaborole mp 146-8°C. The solids were combined and recrystallised from methylated spirits.
Yield = 0.51 parts (9.2% theory) mp 150-4°C.
Elemental Analysis
Theory 49.8%C, 3.5%H, 21.06%C1
Found 49.5%C, 3.5%H, 21.0%C1
Proton NMR (CDCl3) ppm
9.3, s, 1H(-OH); 7.5, d, s, d, 3H(aromatic);
5.0, s, 2H(-CH2-O).
PATENT
WO 2006089067
http://www.google.co.in/patents/WO2006089067A2?cl=en

Analytical data for exemplary compounds of structure I are provided below.
4.2. a 5-Chloro-1.3-dihydro-l -hvdroxy-2J-benzoxaborole (Cl) M.ρ. 142-15O0C. MS (ESI): m/z = 169 (M+l, positive) and 167 (M-I, negative). HPLC (220 nm): 99% purity. 1H NMR (300 MHz, DMSO-d6): δ 9.30 (s, IH), 7.71 (d, J = 7.8 Hz, IH), 7.49 (s, IH), 7.38 (d, J = 7.8 Hz, IH) and 4.96 (s, 2H) ppm.
PAPER
http://pubs.acs.org/doi/abs/10.1021/jm0603724?source=chemport
COMPD IS 19d
5-Chloro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (19d) This compound was made from 18d in the same manner as compound 19b (triturated with hexane, 75% yield):: mp 142-144°C. 1 H NMR (300MHz, DMSO-d6) δ (ppm) 4.96 (s, 2H), 7.38 (d, J = 7.8 Hz, 1H), 7.49 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 9.30 (s, 1H); ESI-MS m/z 167 (M-H)- ; HPLC purity 99.0%; Anal (C7H6BClO2 ⋅ 0.1H2O) C, H
precursor 18d
2-Bromo-5-chloro-1-(methoxymethoxymethyl)benzene (18d) To a solution of 2-bromo-5-chlorobenzoic acid (5.49 g, 23.3 mmol) in anhydrous THF (70 mL) under nitrogen was added dropwise a BH3 THF solution (1.0 M, 55 mL) at 0o C and the reaction mixture was stirred overnight at room temperature. Then the mixture was cooled on an ice bath and MeOH (20 mL) was added dropwise to decompose excess BH3. The resulting mixture was stirred until no bubble was released and then 10% NaOH (10 mL) was added. The mixture was concentrated and the residue was S6 mixed with water (200 mL) and extracted with EtOAc. The residue from rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 2-bromo-5-chlorobenzyl alcohol as a white solid (4.58 g, 88%): 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 7.57 (d, J = 8.7 Hz, 1H), 7.50-7.49 (m, 1H), 7.28-7.24 (m, 1H), 5.59 (t, J = 6.0 Hz, 1H), 4.46 (d, J = 6.0 Hz, 2H). 2-Bromo-5-chlorobenzyl alcohol obtained above was dissolved in CH2Cl2 (150 mL) and cooled to 0o C on an ice bath. To this solution under nitrogen were added in sequence i-Pr2NEt (5.4 mL, 31 mmol) and chloromethyl methyl ether (2.0 mL, 26 mmol). The reaction mixture was stirred overnight at room temperature and washed with NaHCO3-saturated water and then brine. The residue after rotary evaporation was purified by silica gel column chromatography (5:1 hexane/EtOAc) to give 18d (4.67 g, 85%) as a colorless oil: 1 H NMR (300 MHz, DMSO-d6): δ (ppm) 3.30 (s, 3H), 4.53 (s, 2H), 4.71 (s, 2H), 7.32 (dd, J = 8.4, 2.4 Hz, 1H), 7.50 (dd, J = 2.4, 0.6 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H).
PATENT
http://www.google.com/patents/WO2013050591A2?cl=en
EXAMPLES Example 1 : Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
Step 1 : Preparation of (2-bromo-5-chloro-phenyl)methanol (A)
A solution of borane-tetrahydrofuran complex in THF (0.15 L, 1.5 eq) was added dropwise to a solution of 2-bromo-5-chlorobenzoic acid (24 g) in anhydrous tetrahydrofuran (0.24 L) at 0°C and under argon atmosphere. The reaction mixture was stirred at room temperature for 16 h, before being slowly poured onto 0.10 L of a 2N aqueous solution of hydrogen chloride at 0°C. The mixture was stirred for 15 minutes and the volatiles were removed under reduced pressure. The aqueous layer was extracted with ethyl acetate and the combined organic layers were washed with a IN aqueous solution of sodium hydroxide and then water. After drying over sodium sulfate, filtration and concentration under reduced pressure, the crude product was purified by column chromatography; 23.2 g; M.p. 79-80 °C.
Step 2: Preparation of l-bromo-4-chloro-2-(methoxymethoxymethyl)benzene (B)
(2-bromo-5-chloro-phenyl)methanol (A, 12 g) was dissolved in dichloromethane (0.35 mL) and cooled to 0 °C. Under argon atmosphere, diisopropylethylamine (14 mL, 1.5 eq) and chloromethyl methyl ether (5.0 mL, 1.2 eq) were added. After 1 night of stirring at room temperature, the crude reaction mixture was washed with a saturated solution of sodium hydrogen carbonate, dried over sodium sulfate and evaporated under reduced pressure. Purification by column chromatography afforded 10.5 g of l-bromo-4-chloro-2- (methoxymethoxymethyl)benzene (B) as an oil.
Step 3: Preparation of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1)
To a solution of (B) (6.0 g) in anhydrous tetrahydrofuran (120 mL) at -78°C was added dropwise a solution of butyllithium in hexane (15.6 mL, 1.1 eq). To the resulting yellow- brown solution trimethyl borate (2.5 mL, 1.0 eq) was injected in one portion and the cooling bath was removed. The mixture was warmed gradually for 30 minutes. After stirring at room temperature for 2 hours, 8.0 ml of a 6N aqueous solution of hydrogen chloride were added and the reaction mixture was left stirring overnight at room temperature. Evaporation of the volatiles gave a residue which was taken up in ethyl acetate, washed with water, brine, dried over sodium sulfate and then evaporated. The crude product was crystallized from ethyl acetate to give 1.4 g of 5-chloro-l-hydroxy-3H-2,l-benzoxaborole (1) as a white powder. Purification of the filtrate by column chromatography afforded 1.2 g more of 1. M.p. 147- 149 °C.
PATENT
http://www.google.fr/patents/WO2010110400A1?cl=en&hl=fr
Reference Example 187
5-chloro-2,1-benzoxazine ball roll -1 (3H) – All

5-chloro-2- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) benzaldehyde 8.50g synthesized in Reference Example 185 was dissolved in methanol 100ml, borohydride Sodium 2.40g was added, and after stirring for 30 minutes at room temperature and stirred for 2 hours at 60 ℃. The reaction solution was concentrated, and the organic layer after the layers were separated with ethyl acetate and saturated aqueous ammonium chloride and then concentrated under reduced pressure. The residue was added 100ml of tetrahydrofuran, and 6N hydrochloric acid 60ml, and stirred for 8 hours at room temperature.After the reaction mixture was dried and the organic layer was extracted with ethyl acetate, and concentrated. The residue was purified by silica gel column chromatography to give the title compound 9.6g. 1 H-NMR (400MHz, DMSO-d 6) δ: 4.95 (2H, s), 7.36 (1H, dd, J = 8.0,1.6Hz), 7.47 (1H, s) , 7.70 (1H, d, J = 8.0Hz), 9.28 (1H, s).
PATENT
http://www.google.com/patents/WO2008070257A2?cl=en
5-Fluoro-l,3-dihvdro-l-hvdroxy-2, 1-benzoxaborole (Cl)
1-Hydroxy-dihydrobenzoxaboroles, such as Cl, were synthesized as shown in Scheme 1. The protected o-bromobenzyl alcohol derivative (18), prepared from 16 or 17, was converted into the corresponding phenyl boronic acid. Deprotection of the methoxymethyl ether using hydrochloric acid followed by spontaneous cyclization gave the target compounds.
Scheme 7

Conditions (a) NaBH4, MeOH, rt (when X = H ), or BH3-THF, THF, rt (when X = OH), (b) MeOCH2CI, /-Pr2NEt, CH2CI2, rt, (c) MeMgBr, THF, -78 0C to rt , (d) NBS, AIBN, CCI4, reflux, (e) NaOAc, DM F, 70 0C, (f) NaOH, MeOH, reflux, (g) n-BuLι, (/-PrO)3B, THF, -780C to rt, (h) 6N HCI, THF, rt
References
- Hui, Xiaoying; Journal of Pharmaceutical Sciences 2007, 96(10), Pg2622-2631
- Baker, Stephen J.; Journal of Medicinal Chemistry 2006, 49(15), Pg4447-4450
- Austin, Peter William; WO 9533754 A1 1995 CAPLUS
| US5880188 * | 26 May 1995 | 9 Mar 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US6083903 * | 16 May 1995 | 4 Jul 2000 | Leukosite, Inc. | Boronic ester and acid compounds, synthesis and uses |
| WO2005013892A2 | 15 Jun 2004 | 17 Feb 2005 | Tsutomu Akama | Hydrolytically-resistant boron-containing therapeutics and methods of use |
| Reference | ||
|---|---|---|
| 1 | * | Austin et al., 1996, CAS: 124:234024. |
| 2 | * | fungicide: definition from Answre.com, 1998. |
| 3 | S. J. Baker, et al., “Progress on New Therapeutics for Fungal Nail Infections,“Annual Reports in Medicinal Chemistry, 40:323-335 (2005). | |
| 4 | Sudaxshina Murdan, “Drug Delivery to the Nail Following Topical Application,” International Journal of Pharmaceutics, 236:1-26 (2002). | |
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
//////////AN-2718, Borole, PHASE 2
B1(c2ccc(cc2CO1)Cl)O
крисаборол , كريسابورول , Crisaborole, AN 2728
Crisaborole
Treatment for Inflammatory Skin Diseases, including Atopic Dermatitis and Psoriasis
C14H10BNO3, Average mass251.045 Da
4-[(1-Hydroxy-1,3-dihydro-2,1-benzoxaborol-5-yl)oxy]benzonitrile ,
4-((1-Hydroxy-1,3-dihydrobenzo(c)(1,2)oxaborol-6-yl)oxy)benzonitrile
CAS 906673-24-3, AN-2728
Benzonitrile, 4-[(1,3-dihydro-1-hydroxy-2,1-benzoxaborol-5-yl)oxy]-
1,3-Dihydro-1-hydroxy-5-(4-cyanophenoxy)-2,1-benzoxaborole
5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole
crisaborol, crisaborole, Crisaborole, crisaborolum
UNII-Q2R47HGR7P
крисаборол
كريسابورول
In phase 3 for treatment of mild to moderate atopic dermatitis……Anacor Pharmaceuticals, Inc.
Psoriasis is a chronic skin disorder caused by inflammatory cell infiltration into the dermis and epidermis, and is accompanied by keratinocyte hyperproliferation. Once triggered, a strong T-cell response is mounted, and a cascade of cytokine and chemokine production is induced.
Down-regulation of certain cytokines and chemokines is considered to be a good approach to treatment, and indeed, the biologics targeting TNF-α demonstrate the effectiveness of this approach.However, biologics have intrinsic challenges, such as limited administration route, side effects, quality control and production cost.
Small molecule approaches to treat psoriasis include systemic or topical steroids, cyclosporine, psoralen plus UVA (PUVA), retinoids, methotrexete, and vitamin D3 analogs.Atopic dermatitis is an allergic skin disorder, which is typically treated with topical steroids, antihistamines, and calcineurin inhibitors.
However, there is still a need for new treatment with improved safety profile. Recently phosphodiesterase 4 (PDE4) inhibitors have been in development for such skin diseases. CC-10004 is in development as an oral treatment for psoriasis and atopic dermatitis. AWD-12-281 was, until recently, in development for the topical treatment of atopic dermatitis. In addition, roflumilast is under Phase 1 development for both diseases.

Figure 1.
PDE4 inhibitors aiming at skin inflammatory diseases.

Anacor’s lead product candidate is crisaborole, an investigational non-steroidal topical PDE-4 inhibitor in development for the potential treatment of mild-to-moderate atopic dermatitis and psoriasis
crisaborole is an investigational topical antiinflammatory drug in phase III clinical development by Anacor Pharmaceuticals for the treatment of mild to moderate atopic dermatitis and in phase II clinical trials in mild to moderate psoriasis
A novel boron-containing small molecule, Crisaborole inhibits the release of pro-inflammatory cytokines including TNF-alpha, IL-12, and IL-23, known mediators of the inflammation associated with psoriasis.
Synthesis
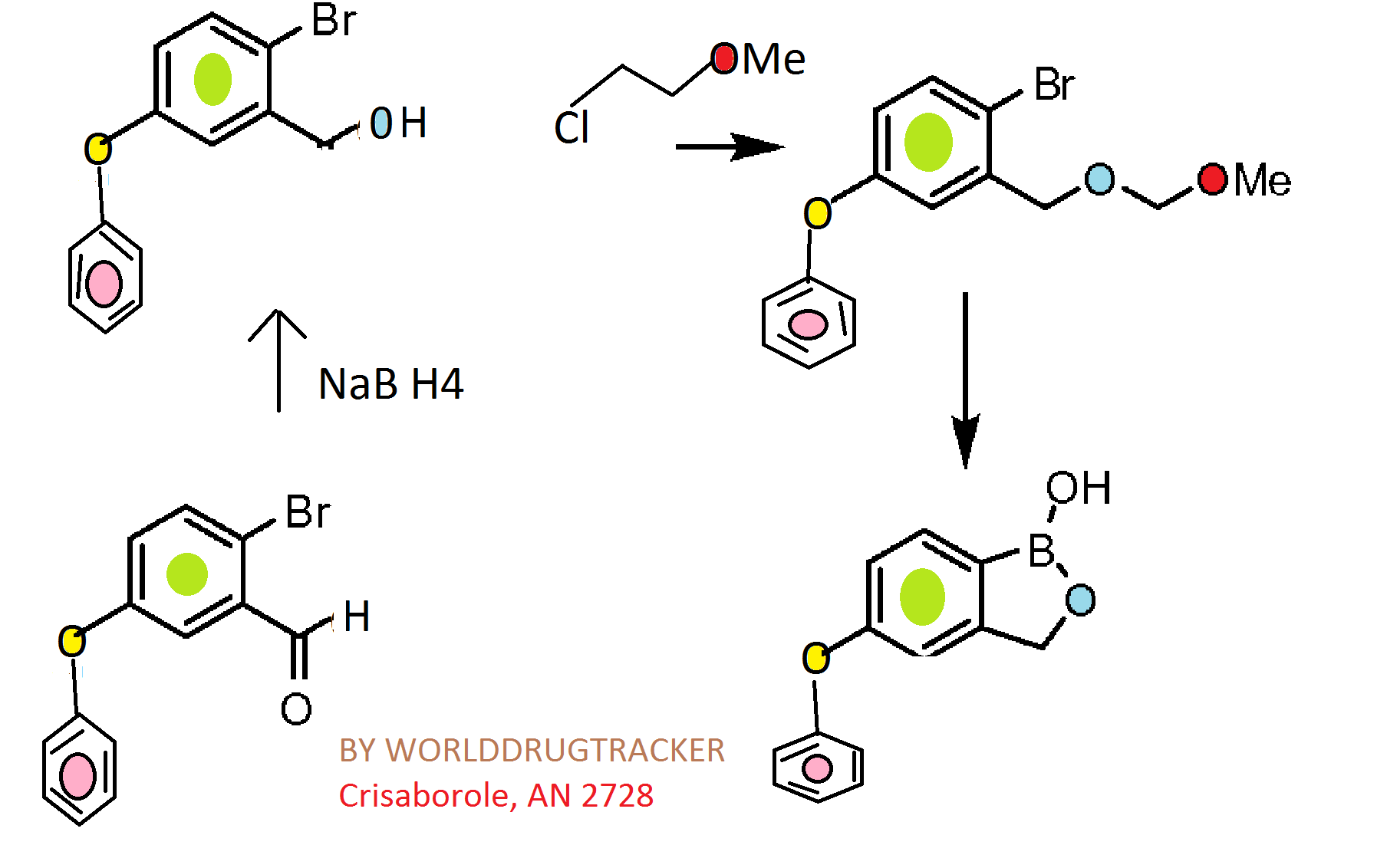
CKICK ON IMAGE FOR CLEAR VIEW
| Originator | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Therapeutic Claim | |||||||||||||||||||||
| Class | |||||||||||||||||||||
| Mechanism of action | |||||||||||||||||||||
| WHO ATC code(s) | |||||||||||||||||||||
| EPhMRA code(s) | |||||||||||||||||||||
| Clinical trial(s) |
|
PAPER
Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis
Bioorg Med Chem Lett 2009, 19(8): 2129
http://www.sciencedirect.com/science/article/pii/S0960894X09002996
- Anacor Pharmaceuticals, Inc., 1020 E. Meadow Circle, Palo Alto, CA 94303, USA
A series of phenoxy benzoxaboroles were synthesized and screened for their inhibitory activity against PDE4 and cytokine release. 5-(4-Cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1-benzoxaborole (AN2728) showed potent activity both in vitro and in vivo. This compound is now in clinical development for the topical treatment of psoriasis and being pursued for the topical treatment of atopic dermatitis


Scheme 1.
Reagents and conditions: (a) ethylene glycol, p-TsOH, toluene, reflux, 6 h (quant.); (b) K2CO3, DMF, 100 °C, overnight (82–96%); (c) 3 M HCl, THF, reflux, 2 h (80–100%); (d) NaBH4, MeOH, rt, 1 h (quant.); (e) 3,4-dihydro-2H-pyran, camphorsulfonic acid, CH2Cl2, rt, 2 h (quant.); (f) (i-PrO)3B, n-BuLi, THF, −78 °C to rt, 3 h; (g) 6 M HCl, THF, rt, 3 h (37–44%); (h) 6 M NaOH, MeOH, 1,4-dioxane, reflux, 6 days (79%); (i) diethylamine (for 5f) or morpholine (for 5g), EDCI, HOBt, DMAP, DMF, rt, overnight (41–70%).
PATENT
http://www.google.co.in/patents/WO2006089067A2?cl=en
4.2. q 5-(4-Cyanophenoxy)-l, 3-dihydro-l-hydroxy-2, 1-benzoxaborole (C17) [0264] 1H-NMR (300 MHz,
δ ppm 4.95 (s, 2H), 7.08 (dd, J= 7.9, 2.1 Hz, IH), 7.14 (d, J= 8.8 Hz, IH), 7.15 (d, J= 2.1 Hz, IH), 7.78 (d, J= 7.9 Hz, IH), 7.85 (d, J= 9.1 Hz, 2H), 9.22 (s, IH).
PATENT
EXAMPLE 15
http://www.google.com/patents/WO2007095638A2?cl=en
4-(4-Cvanophenoxy)phenylboronic acid (C97)

(a) (4-cyanophenyl) (4-bromophenyl) ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 1000C for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with IN NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4- bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-de): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p.l94-198°C. MS: m/z = 239 (M+), 240 (M+ 1) (ESI+) and m/z = 238 (M-I) (ESI-). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6 + D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.
FURTHER METHOD

2-Bromo-5-(4-cvanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J= 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
PATENT
http://www.google.im/patents/EP1976536A2?cl=en
2.2.a 2-Bromo-5-(4-cyanophenoxy)benzyl Alcohol
1H-NMR (300 MHz, CDCl3) δ (ppm) 2.00 (br s, IH), 4.75 (s, 2H), 6.88 (dd, J= 8.5, 2.9 Hz, IH), 7.02 (d, J= 8.8 Hz, IH), 7.26 (d, J- 2.6 Hz, IH), 7.56 (d, J = 8.5 Hz, IH), 7.62 (d, J= 8.8 Hz, 2H).
2.2.b 2-Bromo-4-(4-cyanophenoxγ)benzyl Alcohol
1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.58 (d, IH), 7.39 (d, IH), 7.18 (dd, IH), 7.11- (d, 2H), 5.48 (t, IH) and 4.50 (d, 2H) ppm.
2.2.c 5- (4-Cyanophenoxy) -1 -Indanol
M.p.50-53°C. MS (ESI+): m/z = 252 (M+l). HPLC: 99.7% purity at 254 nm and 99.0% at 220 nm. 1H NMR (300 MHz, DMSOd6): δ 7.80 (d, 2H), 7.37 (d, IH), 7.04 (d, 2H), 6.98-6.93 (m, 2H), 5.27 (d, IH)5 5.03 (q, IH), 2.95-2.85 (m, IH), 2.75-2.64 (m, IH), 2.39-2.29 (m, IH) and 1.85-1.74 (m, IH) ppm.
2.2. d 2-Bromo-5-(tert-butyldimethylsiloxy)benzyl Alcohol [0429] 1H-NMR (300 MHz, CDCl3) δ (ppm) 0.20 (s, 6H), 0.98 (s, 9H), 4.67 (br s,lH), 6.65 (dd, J= 8.2, 2.6 Hz, IH), 6.98 (d, J= 2.9 Hz, IH), 7.36 (d, J= 8.8 Hz, IH).
3.2.k 2-Bromo-5-(2-cyanophenoχy)-l-(methoxymethoxymethyl)benzene [0443] 1H-NMR (300 MHz, CDCl3) δ (ppm) 3.41 (s, 3H), 4.64 (s, 2H), 4.76 (s, 2H), 6.8-6.9 (m, 2H), 7.16 (td, J= 7.6, 0.9 Hz, IH), 7.28 (d, J= 2.9 Hz, IH), 7.49 (ddd, J= 8.8, 7.6, 1.8 Hz, IH)5 7.56 (d, J= 8.5 Hz, IH), 7.67 (dd, J= 7.9, 1.8 Hz, IH).
EXAMPLE 32
Alternative Preparation of C17 -Intermediate

The procedure described in Example II I was followed for 1H NMR characterization of the current alcohol-borate intermediate. 1H NMR determination indicated there were 72.7 mol% of the desired alcohol-borate intermediate [2-bromo- 5-(4-cyanophenoxy)benzyl] diisopropyl borate, 20.7 mol% of an unknown intermediate and 6.5 mol% of unreacted alcohol. 1H NMR (CDCl3, 300 MHz) of [2- bromo-5-(4-cyanophenoxy)benzyl] diisopropyl borate: δ= 7.61 (d, J= 9.0 Hz, 2H), 7.52 (d, J= 8.4 Hz, IH), 7.15 (d, J= 3.0 Hz, IH), 7.03 (d, J= 8.7 Hz, 2H), 6.84 (dd, J= 8.7 Hz, J= 3.0 Hz, IH), 4.85 (s, 2H), 4.35 (septet, J= 6.1 Hz, 2H), 1.11 (d, J= 6.1 Hz, 12H) ppm.
PATENT
http://www.google.com/patents/US20090291917
- Example 154-(4-Cyanophenoxy)phenylboronic acid (C97)
-

-
(a) (4-cyanophenyl)(4-bromophenyl)ether. Under nitrogen, the mixture of 4-fluorobenzonitrile (7.35 g, 60.68 mmol), 4-bromophenol (10 g, 57.8 mmol) and potassium carbonate (12 g, 1.5 eq) in DMF (100 mL) was stirred at 100° C. for 16 h and then filtered. After rotary evaporation, the residue was dissolved in ethyl acetate and washed with 1N NaOH solution to remove unreacted phenol. The organic solution was dried and passed through a short silica gel column to remove the color and minor phenol impurity. Evaporation of the solution gave (4-cyanophenyl)(4-bromophenyl)ether (13.82 g, yield 87.2%) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 7.83 (d, 2H), 7.63 (d, 2H), 7.13 (d, 2H) and 7.10 (d, 2H) ppm.
-
(b) 4-(4-cyanophenoxy)phenylboronic acid. The procedure described in Example 2d was used for the synthesis of 4-(4-cyanophenoxy)phenylboronic acid using (4-cyanophenyl)(4-bromophenyl)ether as starting material. The title compound was obtained as a white solid. M.p. 194-198° C. MS: m/z=239 (M+), 240 (M+1) (ESI+) and m/z=238 (M−1) (ESI−). HPLC: 95.3% purity at 254 nm and 92.1% at 220 nm. 1H NMR (300 MHz, DMSO-d6+D2O): δ 7.83-7.76 (m, 4H), 7.07 (d, 2H) and 7.04 (d, 2H) ppm.

see
http://www.google.co.in/patents/WO2006089067A2?cl=en
see
http://www.google.com/patents/US20090291917
| US5688928 * | Jun 7, 1995 | Nov 18, 1997 | Prolinx, Inc. | Phenylboronic acid complexing reagents derived from aminosalicylic acid |
| US5880188 * | May 26, 1995 | Mar 9, 1999 | Zeneca Limited | Oxaboroles and salts thereof, and their use as biocides |
| US5962498 * | Dec 2, 1994 | Oct 5, 1999 | Procyon Pharmaceuticals, Inc. | Protein kinase C modulators. C. indolactam structural-types with anti-inflammatory activity |
| US6369098 * | Oct 4, 2000 | Apr 9, 2002 | Bethesda Pharmaceuticals, Inc. | Dithiolane derivatives |
| US20030032673 * | Jul 19, 2002 | Feb 13, 2003 | Isis Innovation Limited | Therapeutic strategies for prevention and treatment of alzheimer’s disease |
| US20050239170 * | Jul 16, 2001 | Oct 27, 2005 | Hedley Mary L | Alpha-MSH related compounds and methods of use |
| US20060009386 * | May 12, 2005 | Jan 12, 2006 | The Brigham And Women’s Hospital, Inc. | Use of gelsolin to treat infections |
|
Methods of treating anti-inflammatory conditions through the use of boron- containing small molecules are disclosed.
|
|
… Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … Francisco, CA Mar. 6-10, 2009. 7 , “AN2728 … Kyoto, Japan, May 14-18, 2008. 10, “AN2728 …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3- dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
… Dermatology Annual Meeting, San Francisco, CA Mar. 6-10, 2009. 6, “AN2728 … 7, “AN2728 … Francisco, CA May 6-10, 2009. 10, “AN2728 …
|
|
… from the group consisting of AN-2728, AN-2898, CBS- 3595, apremilast, ELB- 353, KF-66490, K-34, LAS-37779, IBFB-211913, AWD-12-281, …
|
|
“AN2728” is the compound 4-(l-hydroxy-l,3-dihydro-2 … GSK256066, oglemilast, tetomilast, apremilast, AN2728, Compound A, Compound B, …
|
|
AN2728, 5-(4-cyanophenoxy)-2,3-dihydro-1-hydroxy-2,1- …. UK-500,001, AN2728, DE-103, Tofisopam, Dextofisopam, Levotofisopam (USAN).
|
|
85.用于治疗疼痛的UK-500,001。 85. for the treatment of pain UK-500,001. 86.用 于治疗疼痛的AN2728。 86. for the treatment of pain AN2728.
|
see full series on boroles
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
http://apisynthesisint.blogspot.in/p/borole-compds.html
do not miss out
///////////crisaborole, AN 2728, PHASE 3, Anti-inflammatory, Phosphodiesterase, Oxaborole, Psoriasis, Atopic dermatitis, borole
