Home » Posts tagged 'BMS-379224'
Tag Archives: BMS-379224
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fosravuconazole L-lysine ethanolate, ホスラブコナゾール L-リシンエタノール付加物

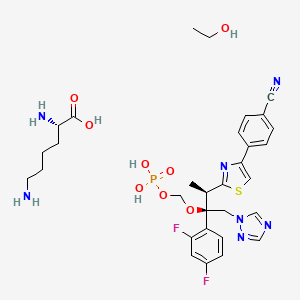
C23H20F2N5O5PS▪C6H14N2O2▪C2H6O : 739.73
[914361-45-8]
[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,4-difluorophenyl)-1-(1,2,4-triazol-1-yl)butan-2-yl]oxymethyl dihydrogen phosphate;(2S)-2,6-diaminohexanoic acid;ethanol
L-Lysine [[(2R,3R)-3-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butan-2-yl]oxy]methyl dihydrogen phosphate ethanol
BFE-1224
BMS-379224
E-1224
ravuconazole prodrugs, ravuconazole methyl phosphate
fosravuconazole bis(L-lysine)
ホスラブコナゾール L-リシンエタノール付加物
| Formula |
C23H20F2N5O5PS. C6H14N2O2. C2H6O
|
|---|---|
| CAS |
914361-45-8
|
| Mol weight |
739.727
|
Antifungal, Ergosterol biosynthesis inhibitor
Fungal infection; Onychomycosis; Trypanosoma cruzi infection
PMDA JAPAN APPROVED
| 2018/1/19 | PMDA | APPROVED | Fosravuconazole L-lysine ethanolate | Nailin | Sato Pharmaceutical
FOR Tinea, nail (onychomycosis) |
NOTE THIS STR
- 4-[2-[(1R,2R)-2-(2,4-Difluorophenyl)-1-methyl-2-[(phosphonooxy)methoxy]-3-(1H-1,2,4-triazol-1-yl)propyl]-4-thiazolyl]benzonitrile
- E 1224
- Fosravuconazole
- CAS 351227-64-0
Drugs for Neglected Diseases initiative (DNDi), under license from Eisai, is developing fosravuconazole for CD and eumycetoma
In February 2013, the drug was in phase II/III development by Seren Pharmaceuticals for onychomycosis in North America, Europe and Asia, including Japan,
In 2010, the product was licensed exclusively to Brain Factory (now Seren Pharma) for development, commercialization and sublicense in Japan for the treatment of fungal infections. In 2014, Seren Pharma signed an agreement with Sato Pharma, granting them the development and commercialization rights of the product in Japan
Sato Pharmaceutical Co., Ltd. has obtained marketing and manufacturing approval for the oral antifungal agent, Nailin capsules 100mg containing the active ingredient fosravuconazole L-lysine ethanolate (fosravuconazole) for the treatment of onychomycosis in Japan.
Sato Pharma conducted a phase III clinical study of the agent in patients with onychomycosis in Japan, and after confirming efficacy and safety of the agent in the study, the company applied for marketing and manufacturing authorization in January 2017.
Fosravuconazole, the active ingredient of Nailin capsules 100mg, is a new triazole class oral antifungal component discovered by Eisai.
Fosravuconazole, the active ingredient of Nailin capsules 100mg, is a new triazole class oral antifungal component discovered by Eisai. By providing Nailin capsules 100mg as a new option for the treatment of onychomycosis, Sato Pharma and Eisai will strive to fulfil the needs of onychomycosis patients and healthcare professionals.
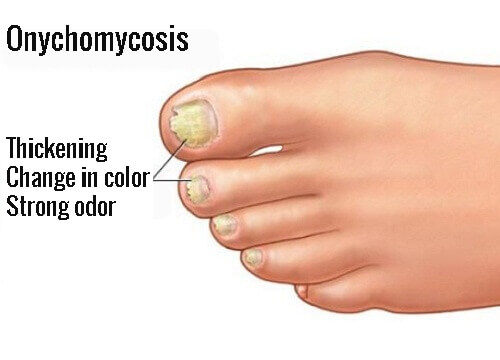
Onychomycosis is a fungal infection of the toenails or fingernails that may involve any component of the nail unit, including the matrix, bed, or plate. With Sato Pharma now having obtained marketing and manufacturing approval for Nailin capsules 100mg, as an oral treatment for onychomycosis, this is the first new treatment for the disease in approximately 20 years.
Fosravuconazole is a prodrug of ravuconazole originated by Eisai. In 2018, the product was approved in Japan for the treatment of onychomycosis. Fosravuconazole is being tested in phase II clinical studies at Eisai and Drugs for Neglected Diseases Initiative (DNDi) for the treatment of american trypanosomiasis (Chagas disease)

Onychomycosis due to Trichophyton rubrum, right and left great toe. Tinea unguium.
Image/CDC
Sato Pharmaceutical Co., Ltd. obtained marketing and manufacturing approval for the oral antifungal agent NAILIN Capsules 100mg containing the active ingredient fosravuconazole L-lysine ethanolate (fosravuconazole) for the treatment of onychomycosis in Japan on January 19, 2018.
Fosravuconazole, the active ingredient of NAILIN Capsules 100mg, is a new triazole class oral antifungal component discovered by Eisai. Sato Pharma conducted a Phase III clinical study of the agent in patients with onychomycosis in Japan, and after confirming efficacy and safety of the agent in the study, Sato Pharma applied for marketing and manufacturing authorization in January 2017.Sato Pharma and Eisai Co., Ltd. are jointly providing information on its proper use.
Onychomycosis is a fungal infection of the toenails or fingernails that may involve any component of the nail unit, including the matrix, bed, or plate.
Onychomycosis affects 1 in every 10 Japanese people, and there are an estimated approximately 11 million sufferers in Japan. With Sato Pharma now having obtained marketing and manufacturing approval for NAILIN Capsules 100mg, as an oral treatment for onychomycosis, this is the first new treatment for the disease in approximately 20 years.
Sato Pharmaceutical Co. Ltd., Eisai Co. Ltd., and Seren Pharmaceuticals Inc. announced that Sato Pharma and Eisai will co-promote a new triazole class oral antifungal agent (development code: BFE1224) containing the active ingredient fosravuconazole L-lysine ethanolate (fosravuconazole) in Japan, based on an agreement between the three companies. The agent is currently under regulatory review for the treatment of onychomycosis.
After receiving regulatory approval, Sato Pharma will begin distributing the agent, and Sato Pharma and Eisai will jointly provide information on its proper use.
Fosravuconazole is a new oral antifungal component developed by Eisai. In 2010, Eisai concluded a license agreement with Seren Pharma (formerly known as Brain Factory Co., Ltd.), granting them exclusive rights to develop, commercialize, and sublicense the agent in Japan.
In 2014, Seren Pharma concluded an agreement with Sato Pharma, granting them the development and commercialization rights, and both companies continued to develop the agent for treating onychomycosis. In January 2017, Sato Pharma applied for marketing authorization for the agent.
Sato Pharma, Eisai, and Serena Pharma will cooperate to maximize the value of fosravuconazole in order to fulfil the unmet medical needs of patients with fungal diseases.
PATENT
WO 2006118351

Journal of the American Chemical Society, 139(31), 10733-10741; 2017

PAPER
BMS-379224, a water-soluble prodrug of ravuconazole
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
PATENT
WO 2006118351
WO 2007072851
WO 2001052852
WO 2006026274
WO 2013082102
WO 2013157584
////////////ホスラブコナゾール L-リシンエタノール付加物, Fosravuconazole L-lysine ethanolate, Nailin, SATO, BFE-1224, BMS-379224, E-1224, JAPAN 2018, ravuconazole prodrugs, ravuconazole methyl phosphate, fosravuconazole bis(L-lysine), Drugs for Neglected Diseases initiative, DNDi
CCO.CC(C1=NC(=CS1)C2=CC=C(C=C2)C#N)C(CN3C=NC=N3)(C4=C(C=C(C=C4)F)F)OCOP(=O)(O)O.C(CCN)CC(C(=O)O)N
Fosravuconazole in phase 1 for the treatment of fungal infections.

Fosravuconazole
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane
BEF-1224
BMS-379224
E-1224
Phosphoric acid 2(R)-[4-(4-cyanophenyl)thiazol-2-yl]-1(R)-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-ylmethyl)propyoxymethyl monoester bis(L-lysine) salt is used as drug
The azole antifungal agent E-1224 is a prodrug of ravuconazole. In 2009, originator Eisai licensed E-1224 to Drugs for Neglected Diseases Initiative for the treatment of American trypanosomiasis (Chagas disease) in Latin America and the Caribbean. DNDi was conducting phase II clinical trials with the prodrug for this indication, however, development of the compound has been discontinued due to lack of sustained efficacy. Ravuconazole was originally licensed by Eisai to Bristol-Myers Squibb (BMS). BMS developed the drug’s prodrug, referred to by BMS as BMS-379224. For strategic reasons, BMS did not pursue development of the compound. In 2010, E-1224 was licensed exclusively to Brain Factory for development, commercialization and sublicense in Japan for the treatment of fungal infections.
About Ravuconazole and Ravuconazole Prodrug
The compound on the left is ravuconazole; the compound on the right is the dihydrogen phosphonoxy methoxy derived ravuconazole prodrug which has improved solubility and bioavailability.

……………………………………………………………
WO 2001052852
http://www.google.com/patents/WO2001052852A1?cl=en
Triazole antifungal compounds are well known in the prior art. Of the several classes known, one particularly potent class contains a tertiary hydroxyl group. For example, U. S. Patent 5,648,372 discloses that (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)- butan-2-ol has anti-fungal activity.

The utility of this class of compounds is limited by their low water solubility. For example, the solubility of the above triazole compound in water at pH 6.8 is 0.0006 mg/mL. This greatly impedes developing suitable parenteral dosage forms.
One method of addressing this problem was disclosed in European Patent Application 829478, where the water solubility of an azole antifungal agent was increased by attaching a linked amino-acid to the azole portion of the molecule

Alternatively, WO 97/28169 discloses that a phosphate moiety can be attached directly to the tertiary hydroxyl portion of the anti-fungal compound, e.g. the compound having the formula

U.S. Patent 5,707,977 and WO 95/19983 disclose water soluble prodrugs having the general formula

wherein X is OP(O)(OH)2 or an easily hydrolyzable ester OC(O)RNR l’rR>2.
WO 95/17407 discloses water-soluble azole prodrugs of the general formula

wherein X is P(O)(OH)2, C(O)-(CHR’)n-OP(O)(OH)2 or C(O)-(CHR’)π
-(OCHR,CHR1)mOR2.
WO 96/38443 discloses water-soluble azole prodrugs of the general formula

U.S. Patent 5,883,097 discloses water-soluble amino acid azole prodrugs such as the glycine ester

The introduction of the phosphonooxymethyl moiety into hydroxyl containing drugs has been disclosed as a method to prepare water-soluble prodrugs of hydroxyl containing drugs.
European Patent Application 604910 discloses phosphonooxymethyl taxane derivatives of the general formula

wherein at least one of R1 ‘, R2″, R3′, R6′ or R7′ is OCH2OP(O)(OH)2.
European Patent Application 639577 discloses phosphonooxymethyl taxane derivatives of the formula T-[OCH2(OCH2)mOP(O)(OH)2]n wherein T is a taxane moiety bearing on the C13 carbon atom a substituted 3-amino-2- hydroxypropanoyloxy group; n is 1, 2 or 3; m is 0 or an integer from 1 to 6 inclusive, and pharmaceutically acceptable salts thereof. WO 99/38873 discloses O-phosphonooxymethyl ether prodrugs of a diaryl 1,3,4-oxadiazolone potassium channel opener.
Golik, J. et al, Bioorganic & Medicinal Chemistry Letters, 1996, 6:1837- 1842 discloses novel water soluble prodrugs of paclitaxel such as


EXAMPLE 1
(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)- 1 -(1 H- 1 ,2,4- triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxylbutane, sodium salt

(2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol- 1 -yl)-2-[(di-tert-butyl phosphonoxy)methoxy1butane

To a solution of (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)-l-(lH-l,2,4-triazol-l-yl)butan-2-ol, II, (8.74 g, 20 mmol) in THF (40 mL) under a nitrogen atmosphere was added sodium hydride (0.80 g, 60% in oil, 20 mmol) at rt. The resulting mixture was stirred at rt for 0.25 h and then di- tert-butyl chloromethyl phosphate, III (10.3 g, 40 mmol) was added. The reaction mixture was heated at 50 °C for 16 h. The reaction mixture was then allowed to cool to rt and was concentrated under reduced pressure. The residue was dissolved in Et2O and was washed with H2O and brine. The organic layer was dried over MgSO4 and was concentrated under reduced pressure to obtain 17.0 g of crude subtitled compound. IV, as a gum. A small portion of this crude compound was purified by reverse phase chromatography on C- 18. The column was eluted with 30% CH3CN/H2O, 38% CH3CN/H2O, 45% CH3CN/H2O and then 50% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure in order to remove CH3CN. The resulting aqueous layer was then extracted with Et2O. The Et O layers were washed with brine, dried and concentrated under reduced pressure to afford purified subtitled compound, IV, as a white solid. 1H NMR (300 MHz, CDC13): δ 8.35 (s, 1H), 7.98 (d, 2H, J=9), 7.76 (s, 1H), 7.71 (d, 2H, J=9), 7.63 (s, 1H), 7.36-7.27 (m, 1H), 6.86-6.78 (m, 2H), 5.53 (dd, 1H, J=28,6), 5.53 (dd, 1H, J=9,6), 5.17 (d, 1H, J=15), 5.03 (d, 1H, J=15), 4.01 (q, 1H, J=7), 1.47 (s, 9H), 1.45 (s, 9H), 1.37 (d, 3H, J=7). MS [ESI+ (M+H)+] 660.2 obs. B. (2R,3R)-3-r4-(4-cyanoρhenyl)thiazol-2-yll-2-(2,4-difluorophenyl)-l-(lH- 1 ,2,4-triazol-l-yl)-2-[(dihydrogen phosphonoxy)methoxy]butane, sodium saltdeprotection


The crude (2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluoropheny 1)- 1 -( 1 H- 1 ,2 ,4-triazol- 1 -y l)-2- [(di-tert-buty 1 phosphonoxy)methoxy]butane, IV, (17 g) was dissolved in CH C1 (100 mL). To this solution was added TFA (50 mL) and the reaction mixture was stirred at rt for 0.25 h. The reaction mixture was then concentrated under reduced pressure. To the residue was added H2O (200 mL), Et2O (100 mL) and EtOAc (100 mL). The pH of the aqueous layer was adjusted to 7.6 by addition of solid Na2CO3 and then the organic and aqueous layers were separated. The aqueous layer was then subjected to reverse phase chromatography on 400 g of C-18 eluted with H2O to 5% CH3CN/Η2O. The product containing fractions were concentrated under reduced pressure, frozen and lyophilized to afford 1.5 g of the subtitled compound, I, as a white solid. (1.5 g, 12% over two steps). Η NMR (500 MHz, D2O) δ 8.91 (s, IH), 7.92 (s, IH), 7.81 (d, 2H, J=8), 7.80 (s, IH), 7.77 (d, 2H, J=8), 7.21 (dd, IH, J=15,9), 6.99 (ddd, IH, J=9,9,2), 6.91 (ddd, IH, J=9,9,2), 5.35 (dd, IH, J=6,6), 5.29 (d, IH, J=15), 5.21 (dd, IH, J=6,6), 5.19 (d, IH, J=15), 3.86 (q, IH, J=7), and 1.35 (d, 3H, J=7); MS [(ESI“ (M-HV 546.1]; Anal. Calcd for C23Hi8F2N5θ5SιPι Na2/3.5 H2O: C, 42.21 : H, 3.85: N, 10.70: Na, 7.03. Found: C, 42.32: H, 3.83: N, 10.60: Na, 7.04.
Di-tert-butyl chloromethyl phosphate, III:
Di-tert-butyl chloromethyl phosphate, III, may be made by any of the following methods.
Method 1
Silver di-t-butyl phosphate (6.34 g, 20 mmol), which was prepared by mixing di- t-butyl phosphate (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971 , 27, 3163) with one equivalent of silver carbonate in 50% aqueous acetonitrile and by lyophilizing to dryness, was placed together with chloroiodomethane (35 g, 200 mmol) in benzene and stirred at room temperature for 18 hrs. The reaction mixture was filtered and the filtrate concentrated under reduced pressure. The residue was chromatographed on silica and eluted with 2:1 hexanes-ethyl acetate. Appropriate fractions were concentrated to dryness to obtain the subtitled compound III (3.7 g, 71% yield): H NMR (CDCI3) δ 5.63 (d, 2H, J=17), 1.51 (s, 18H); MS (MH+ = 259).
Method 2
Tetrabutylammonium di-t-butyl phosphate was prepared by dissolving di-t-butyl phosphate [ 20g, 94 mmol (obtained from di-t-butyl phosphite by the method of Zwierzak and Kluba, Tetrahedron, 1971, 27, 3163)] in methanolic tetrabutylammonium hydroxide (47 mL of 1M solution, 47 mmol). The reaction mixture had a temperature of 23 °C and pH of 4.33. The pH of the reaction mixture was adjusted to 6.5-7.0 by addition of methanolic tetrabutylammonium hydroxide (48 mL of 1M solution, 48 mmol) over 0.2 h. The reaction mixture was stirred for 0.5 h at approximately 26 °C and then was concentrated under reduced pressure at a bath temperature below 40 °C. The crude residue was azeotroped three times by adding toluene (3×100 mL) and then the mixture was concentrated under reduced pressure. The crude residue was then triturated in cold hexanes (0°C) for 1 h and then the solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a first crop of tetrabutylammonium di-t-butyl phosphate as a white solid. (24. Og). The mother liquor was concentrated under reduced pressure and then triturated in cold hexanes (20 mL) for 1 h. The solid was collected by filtration, washed with a minimum amount of cold hexanes and dried to give a second crop of tetrabutylammonium di-t-butyl phosphate as a white solid. [(8.5g), 32.5g total (77%)]. A solution of tetrabutylammonium di-t-butyl phosphate (218 g, 480 mmol) in benzene (200 mL) was added dropwise to stirred chloroiodomethane (800g, 4535 mmol) over 1.5 h at rt. The reaction mixture was stirred an additional 1.5 h at rt and then was concentrated under reduced pressure. The oily residue was dissolved in Et2O and filtered to remove white solids which had precipitated. The organic layer was washed with saturated NaHCO3 and H O/brine (1/1). The organic layer was then dried over magnesium sulfate, filtered and concentrated under reduced pressure to yield a red brown oil (320 g). The red brown oil was subjected to chromatography on silica gel (800g) eluted with 20% EtOAc/Hexanes, 25% EtOAc/Hexanes then 30% EtOAc/Hexanes. The product containing fractions were concentrated under reduced pressure to yield a golden oil. The oil was diluted with CH2C12 (30 mL) , concentrated under reduced pressure and then dried under vacuum to yield the subtitled compound III (61.3g, 49% yield). 1H NMR (Benzene-d6) δ 5.20 (2H, d, J=15), 1.22 (18H, s).
Method 3
Iodochloromethane (974 g, 402 mL, 5.53 mol) at 25°C was treated with tetrabutylammonium di-t-butylphosphate (250 g, 0.553 mol). The phosphate was added portion wise over 10 minutes. The heterogeneous mixture became a clear pink solution after approximately 15 minutes. The mixture was stirred for three hours, and the iodochloromethane was then removed by rotary evaporation with a bath temperature of <30°C. The residue was taken up in 1 L t-butyl methyl ether and stirred for 15 minutes to precipitate tetrabutylammonium iodide by-product. Tetrabutylammonium iodide was removed by vacuum filtration through a sintered glass funnel. The filtrate was concentrated by rotary evaporation to an oil which contained a 5:1 mixture of III and undesired dimer impurity

III”
The mixture can be purified by a silica gel chromatography to obtain III as pure compound in ~60% yield as an oil.
EXAMPLE 2
(2R,3R)-3-[4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)-l-(lH-l,2,4- triazol- 1 -yl)-2- (dihydrogen phosphonoxy)methoxy]butane
A. An oven dried, 1L round-bottom flask equipped with a mechanical stirrer, nitrogen inlet adapter, pressure-equalizing addition funnel fitted with a rubber septum and temperature probe was charged with sodium hydride (2.89 g, 0.069 mol, 60%) and THF (50 mL). To this stirred suspension, (2R,3R)-3-[4-(4- cyanophenyl)thiazol-2-yl]-2-(2,4-difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)butan- 2-ol, II, (10 g, 0.023 mol) in 30 mL of THF was added dropwise over 20 minutes at room temperature. After stirring for 45 minutes, a solution of iodine (2.99 g, 0.0115 mol) in THF (30 mL)) was added dropwise over 10 minutes followed by dropwise addition of compound di tert butylchloromethyl phosphate, III (13.29 g, 0.035 mol, -68% purity) over 15 minutes. The reaction mixture was stirred for 4 hours at about 41 °C to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into ice cold water (100 mL). The aqueous phase was separated and extracted with ethyl acetate (3 x 50 mL) and the combined organic extract was washed with 10% sodium thiosulfite (50 mL), water (50 mL), brine (50 mL), dried over magnesium sulfate and concentrated under reduced pressure to give pale yellow oil (22.8 g, In-process HPLC: ~ 97% pure). The crude product was used “as is” in step B.
B. To a round-bottom flask equipped with magnetic stirrer, cooling bath, pH probe and N2 inlet-outlet was charged the product of Step A above (7.5 g) in CH2C12 (23 mL) and cooled to 0 °C. To this stirred solution, trifluoroacetic acid (8.8 mL) was added slowly and stirred for 3 h to complete the reaction. The completion of the reaction was judged by in-process HPLC. The reaction mixture was poured into a cold solution of 2N NaOH (64 mL). The reaction mixture was extracted with t-butyl acetate (2 x 65 mL) to remove all the organic impurities. The aqueous layer containing the title product as bis sodium salt was treated with activated charcoal (10 g) and filtered through a bed of Celite. The clear filtrate was acidified with IN HC1 to pH 2.5. The free acid, the title product, was extracted into ethyl acetate (2 x 50 mL). The combined organic layer was washed with water, dried over MgSO4) filtered, and the filtrate concentrated under reduced pressure to afford 3.39 g of crude title product.
EXAMPLE 3
Bis lysine salt of (2R,3R)-3-r4-(4-cyanophenyl)thiazol-2-yl]-2-(2,4- difluorophenyl)- 1 -( 1 H- 1 ,2,4-triazol- 1 -yl)-2-[(dihydrogen phosphonoxy)methoxy]butane
The above obtained title product from Example 2 was dissolved in methanol (75 mL) and to this L-lysine (1.8 g) was added and heated at 60 °C for 4.5 h. The hot reaction mixture was filtered through a bed of Celite. The filtrate was concentrated to about 5 mL, mixed with ethanol (100 mL) and heated to 65 °C to crystallize the bis lysine salt. The salt was collected on a Buchner funnel and dried under vacuum to afford 3.71 g of the title compound as an off white crystalline solid.
About Eisai Co., Ltd.
Eisai Co., Ltd. is a research-based human health care (hhc) company that discovers, develops, and markets products throughout the world. Eisai focuses its efforts in three therapeutic areas: integrative neuroscience, including neurology and psychiatric medicines; integrative oncology, which encompasses oncotherapy and supportive-care treatments; and vascular and immunological reactions. Eisai contributes to the well-being of people around the world through a global network of research facilities, manufacturing sites and marketing subsidiaries. For more information about Eisai Co., Ltd., please visit http://www.eisai.co.jp/index-e.html.
ref
BMS-379224, a water-soluble prodrug of ravuconazole
42nd Intersci Conf Antimicrob Agents Chemother (ICAAC) (September 27-30, San Diego) 2002, Abst F-817
| WO2000030655A1 * | Nov 17, 1999 | Jun 2, 2000 | Squibb Bristol Myers Co | Water soluble prodrugs of azole compounds |
| WO2006118351A1 | May 1, 2006 | Nov 9, 2006 | Eisai Co Ltd | Mono-lysine salts of azole compounds |
| WO2012060448A1 | Nov 4, 2011 | May 10, 2012 | Eisai R&D Management Co., Ltd. | Combined pharmaceutical composition as antifungal agent |
| CN101341160B | Dec 20, 2006 | Jan 25, 2012 | 卫材R&D管理有限公司 | Process for production of water-soluble azole prodrug |
| EP1345915A1 * | Oct 18, 2001 | Sep 24, 2003 | Bristol-Myers Squibb Company | Improved process for water soluble azole compounds |
| EP2291084A1 * | May 20, 2009 | Mar 9, 2011 | Neurogesx, Inc. | Carbonate prodrugs and methods of using the same |
| US7230023 | Aug 20, 2003 | Jun 12, 2007 | Sankyo Company, Limited | Triazole compound containing a phosphonate group |
| US8735376 | May 20, 2009 | May 27, 2014 | Acorda Therapeutics, Inc. | Carbonate prodrugs and methods of using the same |
some animations
![]()