
Home » Posts tagged 'bacterial infections'
Tag Archives: bacterial infections
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MEROPENEM

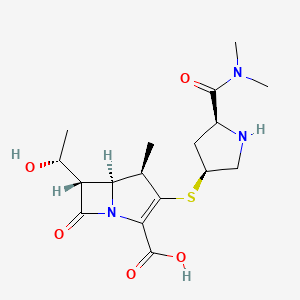
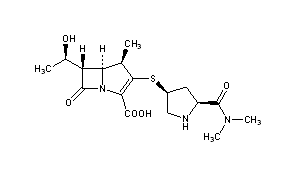
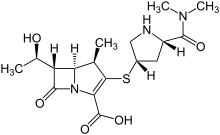
Meropenem
CAS number96036-03-2
IUPAC Name(4R,5S,6S)-3-{[(3S,5S)-5-(dimethylcarbamoyl)pyrrolidin-3-yl]sulfanyl}-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
WeightAverage: 383.463
Monoisotopic: 383.151491615
Chemical FormulaC17H25N3O5S
- Antibiotic SM 7338
- ICI 194660
- SM 7338
CAS Registry Number: 96036-03-2
CAS Name: (4R,5S,6S)-3-[[(3S,5S)-5-[(Dimethylamino)carbonyl]-3-pyrrolidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
Additional Names: (1R,5S,6S)-2-[(3S,5S)-5-(dimethylaminocarbonyl)pyrrolidin-3-ylthio]-6-[(R)-1-hydroxyethyl]-1-methylcarbapen-2-em-3-carboxylic acid
Molecular Formula: C17H25N3O5S
Molecular Weight: 383.46
Percent Composition: C 53.25%, H 6.57%, N 10.96%, O 20.86%, S 8.36%
Literature References: Carbapenem antibiotic. Prepn: M. Sunagawa et al.,EP126587; M. Sunagawa, US4943569 (1984, 1990 both to Sumitomo).
Structure-activity study: M. Sunagawa et al.,J. Antibiot.43, 519 (1990).Crystal structure: K. Yanagi et al.,Acta Crystallogr.C48, 1737 (1992).HPLC determn in serum and bronchial secretions: M. Ehrlich et al., J. Chromatogr. B751, 357 (2001). Pharmacokinetics: R. Wise et al.,Antimicrob. Agents Chemother.34, 1515 (1990).Series of articles on antimicrobial activity, metabolism: J. Antimicrob. Chemother.24, Suppl. A, 1-320 (1989); and clinical performance: ibid.36, Suppl. A, 1-223 (1995).Review of clinical experience in intensive care: M. Hurst, H. M. Lamb, Drugs59, 653-680 (2000).
Derivative Type: Trihydrate
CAS Registry Number: 119478-56-7
Manufacturers’ Codes: ICI-194660; SM-7338
Trademarks: Meronem (AstraZeneca); Meropen (Sumitomo); Merrem (AstraZeneca)
Properties: White to pale yellow crystalline powder. Sparingly sol in water; very slightly sol in hydrated ethanol. Practically insol in acetone, ether.
Therap-Cat: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Carbapenems.
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Meropenem sodium | Not Available | 211238-34-5 | UBQRNADYCUXRBD-NACOAMSHSA-N |
| Meropenem trihydrate | FV9J3JU8B1 | 119478-56-7 | CTUAQTBUVLKNDJ-OBZXMJSBSA-N |
International/Other BrandsAronem (ACI) / Aropen (Aristopharma) / Carbanem (Sanofi-Aventis) / Erope (Lincoln) / Fulspec (Acme) / I-penam (Incepta) / Merenz (Admac) / Merofit (FHC) / Meronem (AstraZeneca) / Meronis (Neiss) / Meropen (Swiss Parenterals) / Merotec (Zuventus) / Merrem I.V. (AstraZeneca) / Monan (AstraZeneca) / Ropenem (Drug International) / Zeropenem (Sanofi-Aventis)
Synthesis Reference
Yoon Seok Song, Sung Woo Park, Yeon Jung Yoon, Hee Kyoon Yoon, Seong Cheol Moon, Byung Goo Lee, Soo Jin Choi, Sun Ah Jun, “METHOD FOR PREPARING MEROPENEM USING ZINC POWDER.” U.S. Patent US20120065392, issued March 15, 2012.
SYN
Carbapenem antibiotic. Prepn: M. Sunagawa et al., EP 126587; M. Sunagawa, US 4943569 (1984, 1990 both to Sumitomo). Structure-activity study: M. Sunagawa et al., J. Antibiot. 43, 519 (1990).

SYN
https://patents.google.com/patent/WO2012062035A1/enCarbapenem, a type of β-lactam antibiotic, is known for its broad spectrum of antibacterial activity and strong antibacterial activity, such as meropenem (Me r0 p e nem), imine South (Imipenem) and Biabenem, etc., play an important role in the cure of severe infections.

Meropenem Imipenem For the synthetic methods of the Peinan type, the previous studies have mainly synthesized the corresponding Peinan side chain compound and the parent nucleus MAP, respectively, and then condensed and removed the protecting group to obtain the Peinan product. Such as US patentsUSP4933333, starting from 4-acetoxyazetidinone (4AA), obtained a matrix MAP after several steps of reaction. The mother nucleus is then condensed and deprotected from the side chain to obtain meropenem. However, this method is cumbersome, the synthesis step is long, and the total yield is low, and the noble metal catalyst is inevitably used in the synthesis of the compound (9).

MAP (10) Meropenem The Chinese invention patent document CN200810142137.5 has introduced a method for synthesizing meropenem.

(XII) (I)(TBD S = Si (CH 3 ) 2 C (CH 3) 3; PNB = p-N0 2 -C 6 H 4 CH 2; PNZ = 2 -C 6 H 4 CH 2 OCO N0 p-) This method of Scheme Short, easy to operate, easy to get raw materials, but there are some areas for improvement.

Example 11) (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,S, 4’R)- 1- (allyl Synthesis of oxycarbonylxiaodimethylaminocarbonylpyrrolidinothio]-2-azetidinone (II) In a 500 ml reaction flask, add 22.6 g (0.075 mol) of (3S,4S)-3-[( R) l-(tert-Butyldimethylsilyloxy)ethyl]-4-[(R)-1-carbonylethyl]-2-azetidinone (IV), 17.1 g (0.083 mol) Dicyclohexylcarbodiimide (DCC) in 100 ml of acetone and 0.76 g of 4-dimethylaminopyridine (DMAP), 20.3 g (0.078 mol) of (2S, 4R)-2-dimethylamine was added dropwise with stirring. A solution of carbonyl-4-mercapto (i-propoxycarbonyl)pyrrolidine (V) in 125 ml of acetone was reacted at room temperature for 14 hours. Filtration, collecting the filtrate, concentrating, adding 200 ml of toluene thereto, using 200 ml of a 5 % acetic acid solution, 200 ml of a saturated sodium hydrogencarbonate solution and 150 ml of saturation Washed with brine, dried over anhydrous magnesium sulfate and evaporated to dryness <mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj 4-[(2,8, 4, ) small (propoxycarbonyl dimethyl dimethylaminocarbonyl)pyrrolidinyl]-2-azetidinone (II), directly without further treatment Invest in the next step.1H-NMR (400 MHz, CDC 13): </ RTI> <RTIgt; m), 2.816-2.849 (lH, s), 2.935-2.953 (3H, m), 3.027-079 (3H, d), 3.378-3.401 (lH, m), 3.792-3.796 (1H, d), 3.807- 3.953 (lH, m), 4.042-4.160 (3H, m), 4.492-4.570 (2H, m), 4.670-4.739 (lH, m), 5.164-5.295 (1H, m), 5.807-5.921 (lH, m ), 6.214(1H, s). Example 22) (31,48)-3-[(1 )-1-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,8,4,1 )- 1- (allyl Synthesis of oxycarbonyl-1-dimethylaminocarbonylpyrrolidinothio]-1-(zincpropoxyl)-2-azetidinone (III) In a 1000 ml reaction flask, add 34.8 g (0.064) Mol) (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy)ethyl]-4-[(2,S, 4,R)-1-(allyl Oxycarbonyl-1-pyrimidinylcarbonyl)pyrrolidinylthio]-2-azetidinone (11), 15.0 ml of triethylamine and 350 ml of toluene, control temperature below -10 °C, add 18.9 g (0.128 mol) p-nitrobenzyl chloroacetate (VI), heated to 0 ° C (-20 ° 5 ° C can be) reaction l ~ 3h. Then slowly add 250 ml of ice water and stir for 10 min. The layers were static and the organic phase was washed three times with saturated sodium bicarbonate solution, 200 ml each time. Dry over anhydrous magnesium sulfate, filtered, and evaporated to dryness to give white crystals, 4,7g (0.0622mol, yield 97.3%) (3R, 4S)-3-[(R) small (tert-butyldimethylsilyloxy)ethyl ]-4-[(2,S, 4,R)-1-(allyloxycarbonyldimethyldimethylaminocarbonyl)pyrrolidinylsulfur]sodium (sweetoxypropanoyl)-2-azetidinone (III), the product was directly put into the next step without further purification.Mp: 33-34 °C1H-NMR (300 MHz, CDC 13):0.819(9H, s), 1.167(3H, d), 1.188(4H, d), 1.693(5H, s), 1.850-1.926(1H, m), 2.631-2.700(1H, m), 2.941-2.960( 3H,d), 3.029-3.080(3H,d), 3.357-3.433(lH, m), 3.506-3.545(2H, m), 3.918-3.968(1H, m), 4.054-4.123 (2H, m), 4.270-4.291(lH, m), 4.391(lH,s), 4.518-4.568(2H, m), 4.588-4.779(3H, m), 5.178-5.416(3H, m), 5.861-5.982(2H,m ). Example 33) (5R,6S,8R,2’S, 4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl) -1- dimethylaminocarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one- Synthesis of 2-carboxylate ![]() In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin.
In a 500 ml reaction flask, 40; 7 g (0.0622 mol) of (3R, 4S)-3-[(R)-l-(tert-butyldimethylsilyloxy) was added. Ethyl]-4-[(2,S,4,R)-1-(indolyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinylsulfate]small (sweetoxypropanoyl)-2-nitrogen Heterocyclic butanone (III) and 150 ml of toluene, 22 ml of trimethyl phosphite (furrowing lg of hydroquinone) were added under nitrogen. After reacting at 60 ° C for 16 hours, the solvent was evaporated under reduced pressure. It was recrystallized by adding 300 ml of ethyl acetate, and the solid was collected, and vacuum-dried at 40 ° C to obtain 32.8 g (0.0528 mol, yield: 85.0%) (5R, 6S, 8R, 2’S, 4,S)-[(R)- 1-(tert-Butyldimethylsilyloxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-ene Propoxycarbonyl ethoxy) small azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (oxime).1H-NMR (300 MHz, CDC 13):0.82(9H, s), 1.24(6H, d), 1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.69(1 H, m), 2.97-3.11(6H, m ), 3.15-3.74(4H, m), 4.35(2H,m), 4.37-4.67(5H, m), 5.24-5.28(4H, m), 5.84(1H, m). Example 44) (5R, 6S, 8R, 2, S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyrrolidinyl Synthesis of thio]-6-(1-allyloxycarbonylethoxy)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (Vffl) at room temperature , in a 2000ml reaction flask, add 32.8g (0.0528mol) (5R,6S,8R,2’S,4,S)-[(R)-1-(tert-butyldimethylsilyloxy)ethyl] 3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-indolyloxycarbonylethoxy)-1-azabicyclo[3.2.0 -Hept-2-ene-7-one-2-carboxylate (W), 27.4 ml of acetic acid, 41.3 g of fluorohydrogenamine and 1000 ml of dichloromethane, stirred at room temperature for 48 h. After completion of the reaction, 500 ml of a saturated aqueous solution of sodium hydrogencarbonate was added to the reaction mixture, and the mixture was stirred for 10 minutes, and the methylene chloride layer was separated and dried over anhydrous magnesium sulfate to give a white solid (26.2 g (0.0517 mol, yield 98.0). %) (5R, 6S, 8R, 2’S, 4’S)-[(R)小(hydroxy)ethyl]-3-[4-(1-allyloxycarbonylsuccinylcarbonyl)pyr Rhodium thio] -6-(l-allyloxycarbonylethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (ring The product was directly charged to the next step without further purification.1H-NMR (300 MHz, CDC 13):1.26(3H, s), 1.36(3H, s), 1.94(1H, m), 2.67(1H, m), 2.97-3.11(6H, m), 3.2-3.7(4H, m) ; 4.25(2H, m), 4.47-4.87 (5H, m), 5.15-5.50 (4H, m), 5.94 (2H, m). Example 55) (5R,6S,8R,2,S,4,S)-3-[4-dimethylaminocarbonyl)pyrrolidinyl]-6-(l-hydroxyethyl)-1-aza Synthesis of bicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) (5R, 6S, 8R, 2’S, 4’S) was added. – [(R)-l-(hydroxy)ethyl]-3-[4-(1-allyloxycarbonyl-1-dimethylaminocarbonyl)pyrrolidinyl] -6-(1-allyloxy Carbonyl ethoxy)-1-azabicyclo[3. 2. 0]-hept-2-en-7-one-2-carboxylate (VDI), 21.3 g (0.152 mol) dimethylcyclohexane The ketone and 550 ml of ethyl acetate were heated to 30 ° C, and a solution of 1.0 g (0.865 mmol) of tetratriphenylphosphine palladium in 150 ml of dichloromethane was added dropwise thereto, and the mixture was reacted at room temperature for 3 h under nitrogen atmosphere. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, the aqueous layer was washed with ethyl acetate, and then, 500 ml of tetrahydrofuran was added dropwise with stirring in an ice bath, and the crystals were stirred, and the crystals were collected and dried in vacuo to give pale yellow crystals of 13.4 g (0.0352 md, Yield 68.1%) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl) 1-Azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (I)-Meropectin.IR max KBr cm- 1 : 1755, 1627, 1393, 1252, 1130NMR (D20, 300Hz): 1.25 (3H, d), 1.81-1.96 (1H, m), 2.96 (3H, s), 3.03 (3H, s), 3.14-3.20 (3H, m), 3.31-3.41 (2H, m), 3.62- 3.72 (1H, m), 3.90-4.00 (1H, m), 4.14-4.26 (2H, m), 4.63 (1H, t). Example 6 6) (5R,6S,8R,2’S,4’S)-3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(l-hydroxyethyl)-1-azabicyclo[ Synthesis of 3.2.0]-hept-2-en-7-one-2-carboxylate (I)21.3 g (0.152 mol) of dimethylcyclohexanedione in Example 5 was replaced with 45.1 g (0.155 mol) of tributyltin hydride, and 0.125 g (0.108 mmol) of tetrakistriphenylphosphine palladium was added dropwise, and the other amount was added. And the same method, the obtained 16.2g (0.0426mol, 82.5%) (5R,6S,8R,2’S,4’S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl Sulfur]-6-(l-hydroxyethyl)-1-azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylic acid trihydrate (1) ~ meropenem. Example 7 7) (5R,6S,8R,2,S,4,S)-3-[4-(2-dimethylaminocarbonyl)pyrrolidinyl]-6-(1-hydroxyethyl)-1- Synthesis of azabicyclo[3.2.0]-hept-2-en-7-one-2-carboxylate (I) To the reaction flask, 26.2 g (0.0517 mol) of (5R, 6S, 8R, 2, S, 4’S)-[(R)-l-(hydroxy)ethyl]-3-[4-(1-allyl was added) Oxycarbonyl-1-ylaminocarbonylcarbonylpyrrolidinothio]-6-(1-allyloxycarbonylethoxy)azaabicyclo[3. 2.]-hept-2-ene-7- Ketone-2-carboxylate 01), 6.0 g (0.0387 mol) of N, N-dimethylbarbituric acid and 500 ml of dichloromethane, and 6.0 g (5.2 mmol) of tetratriphenylphosphine was added dropwise thereto. A solution of palladium in 100 ml of dichloromethane was reacted at room temperature for 5 h under nitrogen. After adding 300 ml of water to the reaction mixture, the aqueous layer was separated, and the aqueous layer was washed with ethyl acetate. THF was evaporated and evaporated, and the crystals were evaporated, and crystals were collected, and the crystals were dried in vacuo to give 15.7 g (0.0413 mol, yield: 80.1%). 5R, 6S, 8R, 2,S,4,S) – 3-[4-(2-Dimethylaminocarbonyl)pyrrolidinylthio]-6-(1-hydroxyethyl)-1-azabicyclo [3. 2. 0] -Hept-2-ene-7-keto-2-carboxylic acid trihydrate (I)-Meropectin.
ClaimsHide Dependent
Rights requesta synthetic method of meropenem, characterized in that the specific reaction route of the synthetic method

The reaction steps are as follows:1) The compound of the formula (IV) and the compound of the formula (V) are dissolved in an organic solvent and then subjected to a condensation reaction to obtain a compound of the formula (Π), the reaction time is 2 to 24 hours, and the reaction temperature is 0 to 40 ° C. ;2) The compound of the formula (Π) and the compound of the formula (VI) are dissolved in toluene, ethyl acetate or tetrahydrofuran and reacted with a base to form a compound of the formula (III), and the reaction time is ! ~ 3 hours, the reaction temperature is -20~5 °C;3) The compound of the formula (III) is dissolved in cyclohexanyl, n-glyoxime, n-octyl, toluene or xylene, and a Wittig ring-closing reaction is carried out under the action of an organophosphorus reagent to obtain a compound of the formula (VD), the organophosphorus reagent Is triphenylphosphine, tri-n-butylphosphine, triethyl phosphite or trimethyl phosphite;4) The compound of the formula (VII) is dissolved in methanol, tetrahydrofuran, acetone, n-pentane, n-hexane, diethyl ether, acetonitrile, dichloromethane, chloroform or ethyl acetate to hydrolyze the silyl ether bond under the action of an acid to obtain a formula (W). a compound; the acid is dilute hydrochloric acid, hydrofluoric acid, tetrabutylammonium fluoride, benzyltributylammonium fluoride, hydrofluoric hinge or vinegar The acid, the molar ratio of the acid to the compound of the formula ![]() is 5 to 15: 1; the temperature of the hydrolysis reaction is 0 to 40 ° C, and the reaction time is 8 to 24 hours;5) a compound of the formula
is 5 to 15: 1; the temperature of the hydrolysis reaction is 0 to 40 ° C, and the reaction time is 8 to 24 hours;5) a compound of the formula ![]() dissolved in one or more of methanol, ethanol, tert-butanol, isobutanol, isopropanol, tetrahydrofuran, dioxanthene, acetone, dichloromethane, chloroform and water After the solvent is formed, the allylic group is hydrogenated by a palladium catalyst to obtain the target product (1). The molar ratio of the palladium catalyst to the compound of the formula 1) is 0.0001 to 0.5:1; the reaction temperature is 0 to 40 ° C. , the reaction time is 2~24h.2. A method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (IV) to the compound of the formula (V) is 1.05 to 1.0: 1, the condensing agent and The molar ratio of the compound of the formula (IV) is 1.50 to 1.05:1.The method for synthesizing meropenem according to claim 1 or 2, wherein the condensing agent is a carbodiimide reagent or hydrazine, Ν’-carbonyldiimidazole; and the organic solvent is acetone. , acetonitrile, toluene, tetrahydrofuran, chloroform or dimethylformamide.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (VI) to the compound of the formula (VI) is from 1.5 to 2.5:1, the base and the The molar ratio of the compound of the formula (VI) is from 1.2 to 2:1.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the organophosphorus reagent to the compound of formula (III) in step 3) is 2-8: 1; The reaction temperature is 25 to 100 £ ^, and the reaction time is 10 to 24 hours.The method for synthesizing meropenem according to claim 3, wherein the carbodiimide reagent is dicyclohexylcarbodiimide, diisopropylcarbodiimide or 1-( 3-dimethylaminopropyl)-3-ethylcarbodiimide.7. A method for synthesizing meropenem according to claim 1, wherein the base in step 2) is an inorganic base or an organic base; when it is an inorganic base, it is sodium hydroxide, sodium carbonate or Sodium bicarbonate; when it is an organic base, it is pyridine, triethylamine, diisopropylethylamine or 2,6-lutidine.The method for synthesizing meropenem according to claim 1, wherein the palladium catalyst is palladium acetate, palladium chloride, palladium nitrate, bistriphenylphosphine palladium chloride or tetrakistriphenylphosphine. palladium.9. A method for synthesizing meropenem according to claim 1, wherein the protecting group acceptor in step 5) is morpholine, dimethylcyclohexanedione, tributyltin hydride, N, N-dimethylbarbituric acid, -ethylhexanoic acid or hexanoic acid.
dissolved in one or more of methanol, ethanol, tert-butanol, isobutanol, isopropanol, tetrahydrofuran, dioxanthene, acetone, dichloromethane, chloroform and water After the solvent is formed, the allylic group is hydrogenated by a palladium catalyst to obtain the target product (1). The molar ratio of the palladium catalyst to the compound of the formula 1) is 0.0001 to 0.5:1; the reaction temperature is 0 to 40 ° C. , the reaction time is 2~24h.2. A method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (IV) to the compound of the formula (V) is 1.05 to 1.0: 1, the condensing agent and The molar ratio of the compound of the formula (IV) is 1.50 to 1.05:1.The method for synthesizing meropenem according to claim 1 or 2, wherein the condensing agent is a carbodiimide reagent or hydrazine, Ν’-carbonyldiimidazole; and the organic solvent is acetone. , acetonitrile, toluene, tetrahydrofuran, chloroform or dimethylformamide.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the compound of the formula (VI) to the compound of the formula (VI) is from 1.5 to 2.5:1, the base and the The molar ratio of the compound of the formula (VI) is from 1.2 to 2:1.The method for synthesizing meropenem according to claim 1, wherein the molar ratio of the organophosphorus reagent to the compound of formula (III) in step 3) is 2-8: 1; The reaction temperature is 25 to 100 £ ^, and the reaction time is 10 to 24 hours.The method for synthesizing meropenem according to claim 3, wherein the carbodiimide reagent is dicyclohexylcarbodiimide, diisopropylcarbodiimide or 1-( 3-dimethylaminopropyl)-3-ethylcarbodiimide.7. A method for synthesizing meropenem according to claim 1, wherein the base in step 2) is an inorganic base or an organic base; when it is an inorganic base, it is sodium hydroxide, sodium carbonate or Sodium bicarbonate; when it is an organic base, it is pyridine, triethylamine, diisopropylethylamine or 2,6-lutidine.The method for synthesizing meropenem according to claim 1, wherein the palladium catalyst is palladium acetate, palladium chloride, palladium nitrate, bistriphenylphosphine palladium chloride or tetrakistriphenylphosphine. palladium.9. A method for synthesizing meropenem according to claim 1, wherein the protecting group acceptor in step 5) is morpholine, dimethylcyclohexanedione, tributyltin hydride, N, N-dimethylbarbituric acid, -ethylhexanoic acid or hexanoic acid.
SYN

Reference: Nadenik, Peter; Storm, Ole; Kremminger, Peter. Meropenem intermediate in crystalline form. WO 2005118586. (Assignee Sandoz AG, Switz)
SYN 2

Reference: Nishino, Keita; Koga, Teruyoshi. Improved process for producing carbapenem compound. WO 2007111328. (Assignee Kaneka Corporation, Japan)
SYN 3

Reference: Manca, Antonio; Monguzzi, Riccardo Ambrogio. Process for synthesizing carbapenem using Raney nickel. EP 2141167. (Assignee ACS Dobfar S.p.A., Italy)
SYN 4

Reference: Tseng, Wei-Hong; Chang, Wen-Hsin; Chang, Chia-Mao; Yeh, Chia-Wei; Kuo, Yuan-Liang. Improved process for the preparation of carbapenem using carbapenem intermediates and recovery of carbapenem. EP 2388261. (Assignee Savior Lifetec Corp., Taiwan)
STR5

Reference: Gnanaprakasam, Andrew; Ganapathy, Veeramani; Syed Ibrahim, Shahul Hameed; Karthikeyan, Murugesan; Sivasamy, Thangavel; Michael, Sekar Jeyaraj; Arulmoli, Thangavel; Das, Gautam Kumar. Preparation of meropenem trihydrate. WO 2012160576. (Assignee Sequent Anti Biotics Private Limited, India)
SYN 6
Reference: Gnanprakasam, Andrew; Ganapathy, Veeramani; Syed Ibrahim, Shahul Hameed; Karthikeyan, Murugesan; Sivasamy, Thangavel; Sekar, Jeyaraj; Arulmoli, Thangavel. Preparation of meropenem trihydrate. IN 2011CH01780. (Assignee Sequent Scientific Limited, India)
SYN7

Reference: Senthikumar, Udayampalayam Palanisamy; Sureshkumar, Kanagaraj; Babu, Kommoju Nagesh; Sudhan, Henry Syril; Kamaraj, Ponraj Pravin; Suresh, Thangaiyan. An improved process for the preparation of carbapenem antibiotic. WO 2013150550. (Assignee Orchid Chemicals & Pharmaceuticals Limited, India)
SYN 8

Reference: Ong, Winston Zapanta; Nowak, Pawel Wojciech; Kim, Jinsoo; Enlow, Elizabeth M.; Bourassa, James; Cu, Yen; Popov, Alexey; Chen, Hongming. Meropenem derivatives and uses thereof. WO 2014144285. (Assignee Kala Pharmaceuticals, Inc., USA)
SYN9

Reference: Cookson, James; McNair, Robert John; Satoskar, Deepak Vasant. Preparation of a carbapenem antibiotic by hydrogenation in the presence of a heterogeneous catalyst. WO 2015145161. (Assignee Johnson Matthey Public Limited Company, UK)
SYN 10

Reference: Gruenewald, Elena; Weidlich, Stephan; Jantke, Ralf. Process for the deprotection of a carbapenem by heterogeneous catalytic hydrogenation with hydrogen in the presence of an organic amine. WO 2018010974. (Assignee Evonik Degussa GmbH, Germany)
SYN 11

Some improvements in total synthesis of meropenem; Hu, Lai-Xing; Liu, Jun; Jin, Jie; Zhongguo Yiyao Gongye Zazhi; Volume 31; Issue 7; Pages 290-292; Journal; 2000
synhttps://www.researchgate.net/figure/Synthesis-of-MRPD-starting-from-meropenem_fig9_283306781

//////////////////////////

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Meropenem is an ultra-broad spectrum injectable antibiotic used to treat a wide variety of infections, including meningitis and pneumonia. It is a beta-lactam and belongs to the subgroup of carbapenem, similar to imipenem and ertapenem. Meropenem was originally developed by Sumitomo Pharmaceuticals. It is marketed outside Japan by AstraZeneca with the brand names Merrem and Meronem. Other brand names include Zwipen (India, Marketed by Nucleus) Mepem (Taiwan) Meropen (Japan, Korea) and Neopenem (NEOMED India) . It gained FDA approval in July 1996. It penetrates well into many tissues and body fluids including the cerebrospinal fluid, bile, heart valves, lung, and peritoneal fluid.
Meropenem, sold under the brandname Merrem among others, is an intravenous β-lactam antibiotic used to treat a variety of bacterial infections.[1] Some of these include meningitis, intra-abdominal infection, pneumonia, sepsis, and anthrax.[1]
Common side effects include nausea, diarrhea, constipation, headache, rash, and pain at the site of injection.[1] Serious side effects include Clostridium difficile infection, seizures, and allergic reactions including anaphylaxis.[1] Those who are allergic to other β-lactam antibiotics are more likely to be allergic to meropenem as well.[1] Use in pregnancy appears to be safe.[1] It is in the carbapenem family of medications.[1] Meropenem usually results in bacterial death through blocking their ability to make a cell wall.[1] It is more resistant to breakdown by β-lactamase producing bacteria.[1]
Meropenem was patented in 1983.[2] It was approved for medical use in the United States in 1996.[1] It is on the World Health Organization’s List of Essential Medicines.[3] The World Health Organization classifies meropenem as critically important for human medicine.[4]
Medical uses
The spectrum of action includes many Gram-positive and Gram-negative bacteria (including Pseudomonas) and anaerobic bacteria. The overall spectrum is similar to that of imipenem, although meropenem is more active against Enterobacteriaceae and less active against Gram-positive bacteria. It works against extended-spectrum β-lactamases, but may be more susceptible to metallo-β-lactamases.[5] Meropenem is frequently given in the treatment of febrile neutropenia. This condition frequently occurs in patients with hematological malignancies and cancer patients receiving anticancer drugs that suppress bone marrow formation. It is approved for complicated skin and skin structure infections, complicated intra-abdominal infections and bacterial meningitis.
In 2017 the FDA granted approval for the combination of meropenem and vaborbactam to treat adults with complicated urinary tract infections.[6]
Administration
Meropenem is administered intravenously as a white crystalline powder to be dissolved in 5% monobasic potassium phosphate solution. Dosing must be adjusted for altered kidney function and for haemofiltration.[7]
As with other ß-lactams antibiotics, the effectiveness of treatment depends on the amount of time during the dosing interval that the meropenem concentration is above the minimum inhibitory concentration for the bacteria causing the infection.[8] For ß-lactams, including meropenem, prolonged intravenous administration is associated with lower mortality than bolus intravenous infusion in persons with whose infections are severe, or caused by bacteria that are less sensitive to meropenem, such as Pseudomonas aeruginosa.[8][9]
Side effects
The most common adverse effects are diarrhea (4.8%), nausea and vomiting (3.6%), injection-site inflammation (2.4%), headache (2.3%), rash (1.9%) and thrombophlebitis (0.9%).[10] Many of these adverse effects were observed in severely ill individuals already taking many medications including vancomycin.[11][12] Meropenem has a reduced potential for seizures in comparison with imipenem. Several cases of severe hypokalemia have been reported.[13][14] Meropenem, like other carbapenems, is a potent inducer of multidrug resistance in bacteria.
Pharmacology
Mechanism of action
Meropenem is bactericidal except against Listeria monocytogenes, where it is bacteriostatic. It inhibits bacterial cell wall synthesis like other β-lactam antibiotics. In contrast to other beta-lactams, it is highly resistant to degradation by β-lactamases or cephalosporinases. In general, resistance arises due to mutations in penicillin-binding proteins, production of metallo-β-lactamases, or resistance to diffusion across the bacterial outer membrane.[10] Unlike imipenem, it is stable to dehydropeptidase-1, so can be given without cilastatin.
In 2016, a synthetic peptide-conjugated PMO (PPMO) was found to inhibit the expression of New Delhi metallo-beta-lactamase, an enzyme that many drug-resistant bacteria use to destroy carbapenems.[15][16]
Society and culture

Meropenem vial
Trade names
| Country | Name | Maker |
|---|---|---|
| India | Inzapenum | Dream India |
| Aurobindo Pharma | ||
| Penmer | Biocon | |
| Meronir | Nirlife | |
| Merowin | Strides Acrolab | |
| Aktimer | Aktimas Biopharmaceuticals | |
| Neopenem | Neomed | |
| Mexopen | Samarth life sciences | |
| Meropenia | SYZA Health Sciences LLP | |
| Ivpenem | Medicorp Pharmaceuticals | |
| Merofit | ||
| Lykapiper | Lyka Labs | |
| Winmero | Parabolic Drugs | |
| Bangladesh | ||
| Meroject | Eskayef Pharmaceuticals Ltd. | |
| Merocon | Beacon Pharmaceuticals | |
| Indonesia | Merofen | Kalbe |
| Brazil | Zylpen | Aspen Pharma |
| Japan, Korea | Meropen | |
| Australia | Merem | |
| Taiwan | Mepem | |
| Germany | Meronem | |
| Nigeria | Zironem | Lyn-Edge Pharmaceuticals |
| US | Meronem | AstraZeneca |
| … | Merosan | Sanbe Farma |
| Merobat | Interbat | |
| Zwipen | ||
| Carbonem | ||
| Ronem | Opsonin Pharma, BD | |
| Neopenem | ||
| Merocon | Continental | |
| Carnem | Laderly Biotech | |
| Penro | Bosch | |
| Meroza | German Remedies | |
| Merotrol | Lupin) | |
| Meromer | Orchid Chemicals | |
| Mepenox | BioChimico | |
| Meromax | Eurofarma | |
| Ropen | Macter | |
| mirage | adwic | |
| Meropex | Apex Pharma Ltd. | |
| Merostarkyl | Hefny Pharma Group[17] |
References
- ^ Jump up to:a b c d e f g h i j “Meropenem”. The American Society of Health-System Pharmacists. Retrieved 8 December 2017.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 497. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ World Health Organization (2019). Critically important antimicrobials for human medicine (6th revision ed.). Geneva: World Health Organization. hdl:10665/312266. ISBN 9789241515528.
- ^ AHFS Drug Information (2006 ed.). American Society of Health-System Pharmacists. 2006.
- ^ Commissioner, Office of the (24 March 2020). “Press Announcements – FDA approves new antibacterial drug”. http://www.fda.gov.
- ^ Bilgrami, I; Roberts, JA; Wallis, SC; Thomas, J; Davis, J; Fowler, S; Goldrick, PB; Lipman, J (July 2010). “Meropenem dosing in critically ill patients with sepsis receiving high-volume continuous venovenous hemofiltration” (PDF). Antimicrobial Agents and Chemotherapy. 54 (7): 2974–8. doi:10.1128/AAC.01582-09. PMC 2897321. PMID 20479205.
- ^ Jump up to:a b Yu Z, Pang X, Wu X, Shan C, Jiang S (2018). “Clinical outcomes of prolonged infusion (extended infusion or continuous infusion) versus intermittent bolus of meropenem in severe infection: A meta-analysis”. PLOS ONE. 13 (7): e0201667. Bibcode:2018PLoSO..1301667Y. doi:10.1371/journal.pone.0201667. PMC 6066326. PMID 30059536.
- ^ Vardakas KZ, Voulgaris GL, Maliaros A, Samonis G, Falagas ME (January 2018). “Prolonged versus short-term intravenous infusion of antipseudomonal β-lactams for patients with sepsis: a systematic review and meta-analysis of randomised trials”. Lancet Infect Dis. 18 (1): 108–120. doi:10.1016/S1473-3099(17)30615-1. PMID 29102324.
- ^ Jump up to:a b Mosby’s Drug Consult 2006 (16 ed.). Mosby, Inc. 2006.
- ^ Erden, M; Gulcan, E; Bilen, A; Bilen, Y; Uyanik, A; Keles, M (7 March 2013). “Pancytopenýa and Sepsýs due to Meropenem: A Case Report” (PDF). Tropical Journal of Pharmaceutical Research. 12 (1). doi:10.4314/tjpr.v12i1.21.
- ^ “Meropenem side effects – from FDA reports”. eHealthMe.
- ^ Margolin, L (2004). “Impaired rehabilitation secondary to muscle weakness induced by meropenem”. Clinical Drug Investigation. 24(1): 61–2. doi:10.2165/00044011-200424010-00008. PMID 17516692. S2CID 44484294.
- ^ Bharti, R; Gombar, S; Khanna, AK (2010). “Meropenem in critical care – uncovering the truths behind weaning failure”. Journal of Anaesthesiology Clinical Pharmacology. 26 (1): 99–101.
- ^ “New molecule knocks out superbugs’ immunity to antibiotics”. newatlas.com. 20 January 2017. Retrieved 2017-01-25.
- ^ K., Sully, Erin; L., Geller, Bruce; Lixin, Li; M., Moody, Christina; M., Bailey, Stacey; L., Moore, Amy; Michael, Wong; Patrice, Nordmann; M., Daly, Seth (2016). “Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo”. Journal of Antimicrobial Chemotherapy. 72 (3): 782–790. doi:10.1093/jac/dkw476. PMC 5890718. PMID 27999041.
- ^ “Hefny Pharma Group”. hefnypharmagroup.info. Retrieved 2018-05-22.
External links
- “Meropenem”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Merrem, others |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: B2 |
| Routes of administration | Intravenous |
| ATC code | J01DH02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | Approximately 2% |
| Elimination half-life | 1 hour |
| Excretion | Renal |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 119478-56-7 |
| PubChem CID | 441130 |
| DrugBank | DB00760 |
| ChemSpider | 389924 |
| UNII | FV9J3JU8B1 |
| KEGG | D02222 |
| ChEBI | CHEBI:43968 |
| ChEMBL | ChEMBL127 |
| PDB ligand | MEM (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID7045526 |
| ECHA InfoCard | 100.169.299 |
| Chemical and physical data | |
| Formula | C17H25N3O5S |
| Molar mass | 383.46 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4888344A *1986-07-301989-12-19Sumitomo Pharmaceuticals Company, LimitedCarbapenem compound in crystalline form, and its production and useCN101348486A *2008-08-292009-01-21深圳市海滨制药有限公司Preparation of meropenemCN101962383A *2010-11-122011-02-02上海巴迪生物医药科技有限公司Synthesis method of meropenemFamily To Family CitationsJPS6475488A *1987-09-171989-03-22Sumitomo PharmaProduction of beta-lactam compound* Cited by examiner, † Cited by third party
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsCN101962383A *2010-11-122011-02-02上海巴迪生物医药科技有限公司Synthesis method of meropenemCN102250096B *2011-09-052016-04-06江西华邦药业有限公司A kind of preparation method of meropenemCN104072523B *2014-07-142017-10-24上海上药新亚药业有限公司The preparation method of BiapenemCN108191869A *2018-01-222018-06-22重庆天地药业有限责任公司The purification process of Meropenem
PublicationPublication DateTitleEP0007973B11984-02-01Process for the preparation of thienamycin and intermediatesUS4631150A1986-12-23Process for the preparation of penemsWO2012062035A12012-05-18Synthesis method for meropenemWO2010022590A12010-03-04Method for preparation of meropenemUS4443373A1984-04-17Process for the production of antibiotic penemsWO2008035153A22008-03-27Process for the preparation of beta-lactam antibioticEP0167154B11990-01-03Process for preparing 4-acetoxy-3-hydroxyethylazetizin-2-one derivativesKR101059339B12011-08-24Method for preparing carbapenem compound for oral administrationKR100886347B12009-03-03Process for stereoselective preparation of 4-BMA using a chiral auxiliaryUS4841043A1989-06-20Stereoselective synthesis of 1-β-alkyl carbapenem antibiotic intermediatesUS4772683A1988-09-20High percentage beta-yield synthesis of carbapenem intermediatesJP2000344774A2000-12-12Production of carbapenem compoundAU745980B22002-04-11Titanium catalyzed preparation of carbapenem intermediatesUS5700930A1997-12-234-substituted azetidinones as precursors to 2-substituted-3-carboxy carbapenem antibiotics and a method of producing themJP2002338572A2002-11-27Method for producing carbapenemsJP3684339B22005-08-17Method for producing carbapenem compoundsEP0066301B11986-01-22Intermediates for the preparation of thienamycin and process for preparing the sameWO2001053305A12001-07-26Processes for the preparation of carbapenem derivativesAU737502B22001-08-23Preparation of beta-methyl carbapenem intermediatesJP3213734B22001-10-02New β-lactam compoundsJP2004107289A2004-04-08Method for producing vinyl sulfide compoundJPH085853B21996-01-24Lactam compound and its manufacturing methodJPH0827168A1996-01-30Carbapenem intermediate fieldEP0204440A11986-12-10Azetidine derivatives productionWO1994021638A11994-09-29Process for the preparation of condensed carbapeneme derivatives
ApplicationPriority dateFiling dateTitleCN 2010105416652010-11-122010-11-12Synthesis method of meropenemCN201010541665.52010-11-12
Nmrhttps://www.researchgate.net/figure/1HNMR-spectra-of-meropenem-hydrolysis-catalyzed-by-NDM-1-Ecoli-cells-Only-1H-signals-of_fig3_272515470


NMRNMR spectra monitoring meropenem hydrolysis catalyzed by NDM-1. a¹H NMR spectrum of hydrolyzed meropenem recorded before and 6 or 20 min after NDM-1 addition to the reaction system. b Part of a ROESY spectrum of the hydrolysis product. Diagonal and cross peaks are shown in blue and red, respectively. Proton signal assignments are labeled beside the peaks. The chemical shifts of H2, H1, H5, and H10 are highlighted by dashed linesSEEhttps://www.mdpi.com/1420-3049/23/11/2738/htm
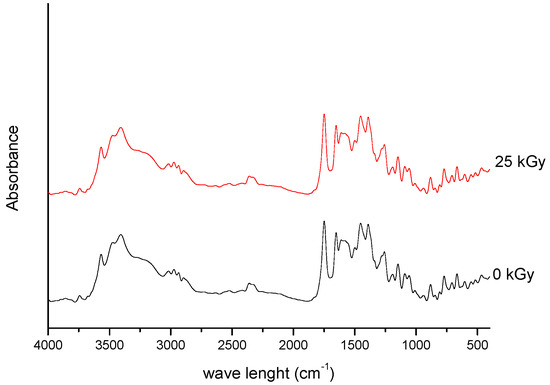
Figure 1. FT-IR spectra of unirradiated and irradiated (25 kGy) meropenem.
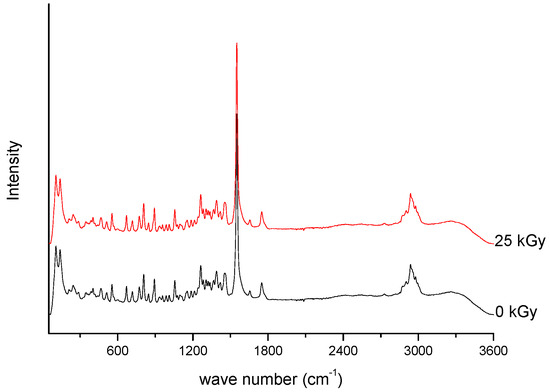
Figure 2. Raman spectra of unirradiated and irradiated (A-25 kGy) meropenem.
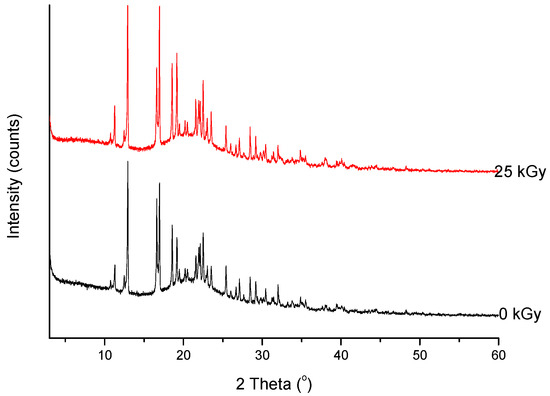
Figure 6. XRPD diffractograms of unirradiated and irradiated (25 kGy) meropenem.
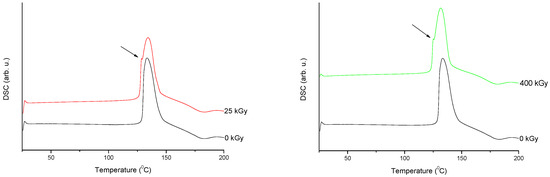
Figure 7. Differential scanning calorimetry (DSC) curves of non-irradiated and irradiated (A-25 kGy, B-400 kGy) meropenem. The arrows indicate the changes in the DSC spectrum after irradiation.
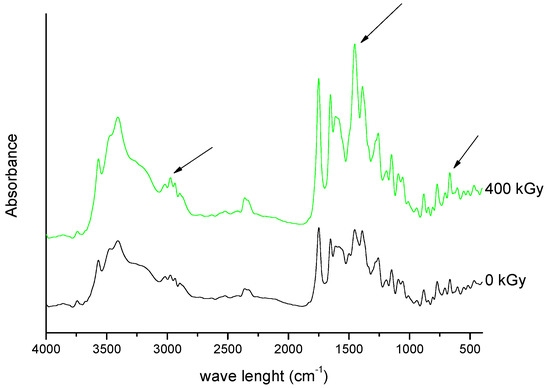
Figure 9. FT-IR spectra of unirradiated and irradiated (400 kGy) meropenem. The arrows indicate the changes in the FT-IR spectrum after irradiation.
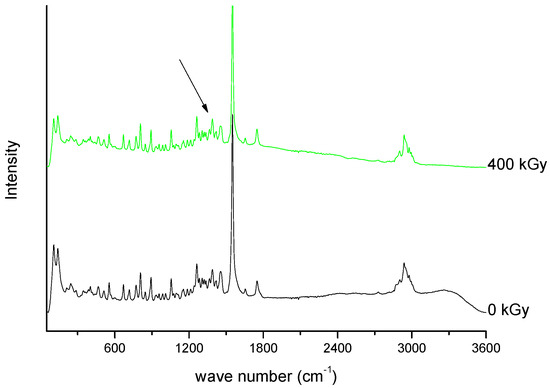
Figure 10. Raman spectra of unirradiated and irradiated (400 kGy) meropenem. The arrow indicates the change in the Raman spectrum after irradiation.
//////////////Meropenem, Merrem, intravenous β-lactam antibiotic, bacterial infections, meningitis, intra-abdominal infection, pneumonia, sepsis, anthrax, Antibiotic SM 7338, ICI 194660, SM 7338, ANTIBACTERIALS
[H][C@]1([C@@H](C)O)C(=O)N2C(C(O)=O)=C(S[C@@H]3CN[C@@H](C3)C(=O)N(C)C)[C@H](C)[C@]12[H]

NEW DRUG APPROVALS
ONE TIME
$10.00
VNRX-5133 from VENATORX PHARMACEUTICALS

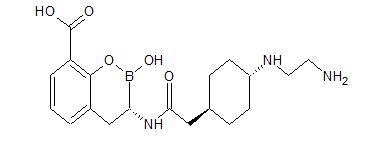
Chemical Formula: C19H28BN3O5
Molecular Weight: 389.26

- Originator VenatoRx Pharmaceuticals
- Developer National Institute of Allergy and Infectious Diseases; VenatoRx Pharmaceuticals
- Class Antibacterials; Cephalosporins; Small molecules
- Mechanism of Action Beta lactamase inhibitors; Cell wall inhibitors
Highest Development Phases
- Phase I Bacterial infections
Most Recent Events
- 19 Mar 2018 VenatoRx Pharmaceuticals plans phase III pivotal trials in mid-2018
- 03 Jan 2018 VNRX 5133 receives Fast Track designation for Bacterial infections (complicated urinary tract infections and complicated intra-abdominal infections) [IV-infusion] in USA
- 03 Jan 2018 VNRX 5133 receives Qualified Infectious Disease Product status for Intra-abdominal infections in USA
- clip
- https://cen.acs.org/articles/96/web/2018/03/Drug-structures-made-public-New-Orleans.html
 Credit: Tien Nguyen/C&EN
Credit: Tien Nguyen/C&EN
Presented by: Christopher J. Burns, president and chief executive officer of VenatoRx Pharmaceuticals
Target: β-lactamase enzymes, enzymes that inactivate β-lactam-based antibiotics enabling bacteria to resist their attacks
Disease: Gram-negative bacterial infections
Reporter’s notes: Another story with humble beginnings, this time with Burns and two colleagues sitting in a Panera Bread, with an idea. They wanted to offer a new compound in the class of β-lactam antibiotics, drugs which are “well-liked” by doctors, Burns said, and make up 60% of all antibiotic prescriptions. However, bacteria have developed defenses against these compounds in the form of β-lactamases, or as Burns dubbed them, “PAC-men.” These enzymes can chew up 1000 β-lactams per second, he said. VNRX-5133 was active against both serine-β-lactamases and metallo-β-lactamases in enzyme assays. It is being developed in combination with the antibiotic cefepime. VNRX-5133 fends off the PAC-men’s attacks, allowing cefepime to combat infection. The compound has gone through Phase I clinical trials and will be skipping ahead to Phase III later this year.
PATENT
WO 2014089365


| Applicants: | VENATORX PHARMACEUTICALS, INC [US/US]; 30 Spring Mill Drive Malvern, PA 19355 (US) |
| Inventors: | BURNS, Christopher, J.; (US). DAIGLE, Denis; (US). LIU, Bin; (US). MCGARRY, Daniel; (US). PEVEAR, Daniel C.; (US). TROUT, Robert E. Lee; (US) |
https://patents.google.com/patent/WO2014089365A1/en

Dr. Burns is Co-Founder, President and Chief Executive Officer of VenatoRx. He brings over 25 years of corporate and R&D experience within both major (RPR/Aventis) and specialty (ViroPharma, Protez…https://www.venatorx.com/leadership/
Antibiotics are the most effective drugs for curing bacteria-infectious diseases clinically. They have a wide market due to their advantages of good antibacterial effect with limited side effects. Among them, the beta-lactam class of antibiotics (for example, penicillins,
cephalosporins, and carbapenems) are widely used because they have a strong bactericidal effect and low toxicity.
[0004] To counter the efficacy of the various beta-lactams, bacteria have evolved to produce variants of beta-lactam deactivating enzymes called beta-lactamases, and in the ability to share this tool inter- and intra-species. These beta-lactamases are categorized as “serine” or “metallo” based, respectively, on presence of a key serine or zinc in the enzyme active site. The rapid spread of this mechanism of bacterial resistance can severely limit beta-lactam treatment options in the hospital and in the community.
EXAMPLE 15 : ( R)-3-( 2-( trans-4-( 2-aminoethylamino)cvclohexyl)acetamido)-2-hvdroxy-3-,4-dihydro-2H-benzo[el [l,21oxaborinine-8-carboxylic acid

Step 1 : Synthesis of (R)-3-(2-(trans-4-(2-(tert-butoxycarbonylamino)ethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e] [ 1 ,2]oxaborinine-8-carboxylic acid.
[00240] To (R)-3-(2-(trans-4-aminocyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid (Example 6, 15 mg) in MeOH (2 mL) was added tert-butyl 2-oxoethylcarbamate (20 mg). Pd/C (10% by weight, 10 mg) was added and the reaction mixture was stirred under ¾ balloon overnight. The reaction mixture was filtrated and the solvent was then removed under reduced pressure and the residue was carried on to the next step without further purification. ESI-MS m/z 490.1 (MH)+.
Step 2: Synthesis of (R)-3-(2-(trans-4-(2-aminoethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid.
[00241] To (R)-3-(2-(trans-4-(2-(tert-butoxycarbonylamino)ethylamino)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e][l,2]oxaborinine-8-carboxylic acid (20 mg) in a flask was added 1 mL 4N HC1 in dioxane. The resulting reaction mixture was stirred at RT for 2hr. The solvent was removed in vacuo and the residue was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 390 (MH)+.
Step 2: (R)-3-(2-(trans-4-((2-aminoethylamino)methyl)cyclohexyl)acetamido)-2-hydroxy-3,4-dihydro-2H-benzo[e] [ 1 ,2]oxaborinine-8-carboxylic acid
[00229] Prepared from 3-[2-(2-{4-[(2-tert-Butoxycarbonylamino-ethylamino)-methyl]-cyclohexyl}-acetylamino)-2-(2,9,9-trimethyl-3,5-dioxa-4-bora-tricyclo[6.1.1.02,6]dec-4-yl)-ethyl]-2-methoxy-benzoic acid tert-butyl ester and BC13 following the procedure described in Step 2 of Example 1. The crude product was purified by reverse phase preparative HPLC and dried using lyophilization. ESI-MS m/z 404 (MH)+.
/////////////////////////////VNRX-5133; VNRX5133; VNRX 5133, phase 1, VenatoRx Pharmaceuticals, BACTERIAL INFECTIONS, Christopher J. Burns
NCCN[C@@H]1CC[C@@H](CC(NC2B(O)OC(C(C(O)=O)=CC=C3)=C3C2)=O)CC1
RG 6080, Nacubactam
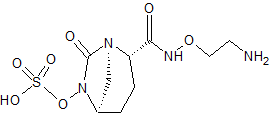

RG-6080
Sulfuric acid, mono[(1R,2S,5R)-2-[[(2-aminoethoxy)amino]carbonyl]-7-oxo-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
Phase I
A β-lactamase inhibitor potentially for the treatment of bacterial infections.
![]()
RG-6080; FPI-1459; OP-0595
CAS No. 1452458-86-4
| Molecular Formula | C9 H16 N4 O7 S |
| Formula Weight | 324.31 |
- Originator Fedora Pharmaceuticals
- Developer Meiji Seika Pharma
- Class Antibacterials; Azabicyclo compounds
- Mechanism of Action Beta lactamase inhibitors
- Phase IBacterial infections
Most Recent Events
- 13 Jan 2015 OP 0595 licensed to Roche worldwide, except Japan ,
- 30 Nov 2014 Meiji Seika Pharma completes a phase I trial in Healthy volunteers in Australia (NCT02134834)
- 01 May 2014 Phase-I clinical trials in Bacterial infections (in volunteers) in Australia (IV)
![]()
SYNTHESIS
WO 2015046207,
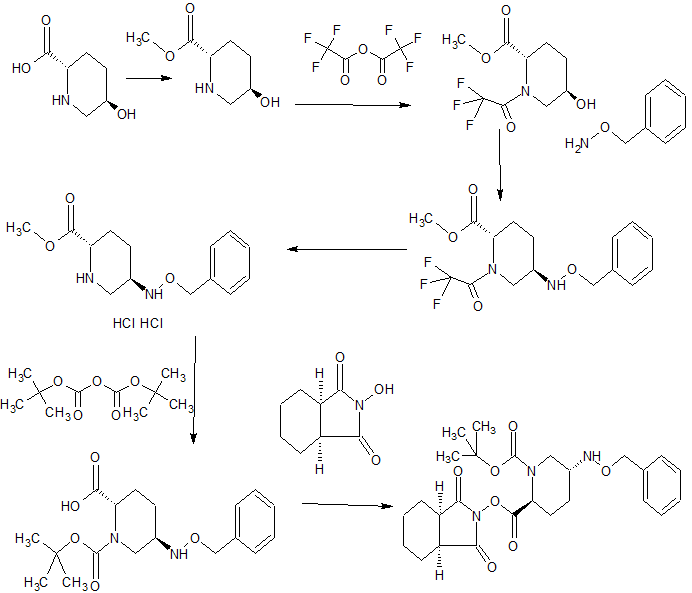
CONTD…………………..
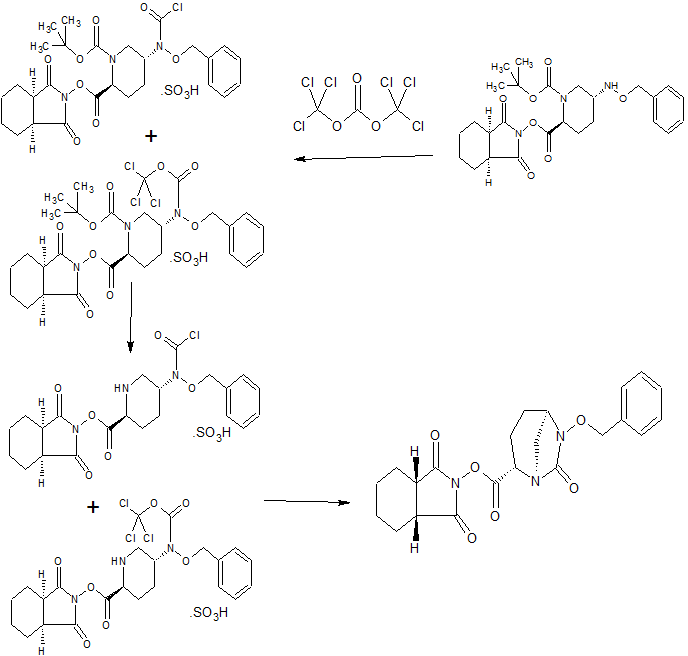
CONTD………………………………..
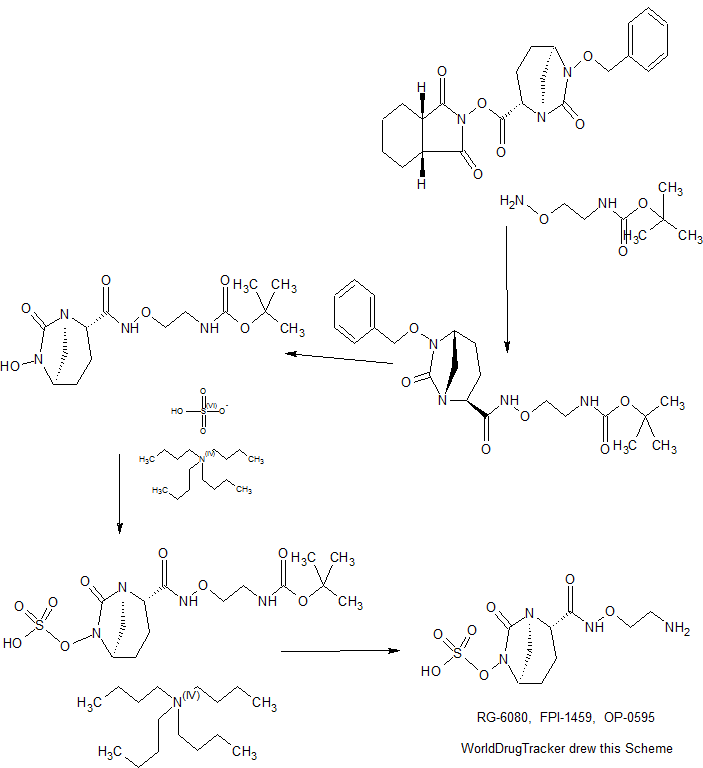
Patent
Patent Document 2: Japanese Patent Publication 2010-138206 Pat
Patent Document 3: Japanese patent publication 2010-539147 Pat
Patent Document 4: International Publication No. WO2011 / 042560 Patent
Patent Document 5: Japanese Patent No. 5038509 Pat
Patent Document 6: Japanese Patent Publication 2011-207900 Pat
Patent Document 7: International Publication No. WO2010 / 126820 Patent
Patent Document 8: Japanese Patent application 2012-122603 Pat.
5 of 5 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VII-1)
Formula 43]
(V-1) tert-butyl {2 – [({[( 2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl ] carbonyl} amino) oxy] ethyl} carbamate (3.91 g, to a methanol solution (80 mL) of 9.01mmol), 10% palladium on carbon catalyst (50% water, 803 mg) was added, under hydrogen atmosphere and stirred for 45 minutes . The reaction mixture was filtered through Celite, after concentrated under reduced pressure to give 3.11g of the title compound (quantitative).
HPLC: COSMOSIL 5C18 MS-II 4.6 × 150 mm, 33.3 mM phosphate buffer / MeCN = 75/25, 1.0 mL / min, UV 210 nm, Retweeted 3.9 from min; 1 H NMR (400 MHz, CD 3 OD) [delta] 1.44 (s, 9H) , 1.73-1.83 (m, 1H), 1.86-1.99 (m, 1H), 2.01-2.12 (m, 1H), 2.22 (br.dd., J = 15.0, 7.0 Hz, 1H), 3.03 (d, J= 12.0 Hz, 1H), 3.12 (br.d., J = 12.0 Hz, 1H), 3.25-3.35 (m, 2H), 3.68-3.71 (m, 1H), 3.82-3.91 (m, 3H); MS M / Z 345 [M Tasu H] Tasu .
Tetrabutylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl } amino) oxy] ethyl} carbamate
(VI-1) tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct 2-yl] carbonyl} amino) oxy] ethyl} carbamate (3.09g, in dichloromethane (80mL) solution of 8.97mmol), 2,6- lutidine (3.20mL), sulfur trioxide – pyridine complex (3 .58g) was added, and the mixture was stirred overnight at room temperature. The reaction mixture was poured into half-saturated aqueous sodium bicarbonate solution, washed the aqueous layer with chloroform, tetrabutylammonium hydrogen sulfate to the aqueous layer and (3.47 g) chloroform (30 mL) was added and stirred for 10 minutes. The aqueous layer was extracted with chloroform, drying the obtained organic layer with anhydrous sodium sulfate, filtered, and concentrated in vacuo to give the title compound 5.46g (91% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 80/20, 1.0ML / Min, UV210nm, RT 2.0 Min; 1 H NMR (400 MHz, CDCl 3 ) Deruta 1.01 (T, J = 7.4 Hz, 12H), 1.37-1.54 (m , 8H), 1.45 (s, 9H), 1.57-1.80 (m, 9H), 1.85-1.98 (m, 1H), 2.14-2.24 (m, 1H), 2.30- 2.39 (m, 1H), 2.83 (d, J = 11.6 Hz, 1H), 3.20-3.50 (m, 11H), 3.85-3.99 (m, 3H), 4.33-4.38 (m, 1H), 5.51 (br s , 1H), 9.44 (Br.S., 1H); MS yd / z 425 [M-Bu 4 N + 2H] + .
tetra butylammonium tert- butyl {2 – [({[( 2S, 5R) -7- oxo-6 (sulfooxy) 1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl} carbamate (5.20g, 7.82mmol) in dichloromethane (25mL) solution of ice-cold under trifluoroacetic acid (25mL), and the mixture was stirred for 1 hour at 0 ℃. The reaction mixture was concentrated under reduced pressure, washed the resulting residue with diethyl ether, adjusted to pH7 with aqueous sodium bicarbonate, subjected to an octadecyl silica gel column chromatography (water), after freeze drying, 1.44 g of the title compound obtained (57% yield).
HPLC: COSMOSIL 5C18 MS-II 4.6X150mm, 33.3MM Phosphate Buffer / MeCN = 99/1, 1.0ML / Min, UV210nm, RT 3.1 Min; 1 H NMR (400 MHz, D 2O) Deruta 1.66-1.76 (M, 1H), 1.76-1.88 (m, 1H ), 1.91-2.00 (m, 1H), 2.00-2.08 (m, 1H), 3.02 (d, J = 12.0 Hz, 1H), 3.15 (t, J = 5.0 Hz , 2H), 3.18 (br d , J = 12.0 Hz, 1H), 3.95 (dd, J = 7.8, 2.2 Hz, 1H), 4.04 (t, J = 5.0 Hz, 2H), 4.07 (dd, J = 6.4 3.2 Hz &, 1H); MS yd / z 325 [M + H] + .
PATENT
64 tert-butyl {2 – [({[( 2S, 5R) -6- hydroxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy ] ethyl} carbamate (V-1)
[of 124]
tert- butyl {2 – [({[(2S, 5R) -6- benzyloxy-7-oxo-1,6-diazabicyclo [3.2.1] oct-2-yl] carbonyl} amino) oxy] ethyl } carbamate (example 63q, net 156.42g, 360mmol) in methanol solution (2.4L) of 10% palladium carbon catalyst (50% water, 15.64g) was added, under an atmosphere of hydrogen, stirred for 1.5 hours did. The catalyst was filtered through celite, filtrate was concentrated under reduced pressure until 450mL, concentrated to 450mL by adding acetonitrile (1.5 L), the mixture was stirred ice-cooled for 30 minutes, collected by filtration the precipitated crystals, washing with acetonitrile, and vacuum dried to obtain 118.26g of the title compound (net 117.90g, 95% yield). Equipment data of the crystals were the same as those of the step 2 of Reference Example 3.
65 (2S, 5R)-N- (2-aminoethoxy) -7-oxo-6- (sulfooxy) 1,6-diazabicyclo [3.2.1] octane-2-carboxamide (VI-1)
[of 125]
PATENT
US 20140288051
WO 2014091268
WO 2013180197
US 20130225554
///////////RG-6080, 1452458-86-4, FPI-1459, OP-0595, Phase I , β-lactamase inhibitor, bacterial infections, Fedora parmaceuticals, Meiji Seika Pharma
WCK ? New molecules from Wochkardt to treat bacterial infections

(2S, 5R)-7-OXO-N-[(3S)-PYRROLIDIN-3-YLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO [3.2.1]OCTANE-2-CARBOXAMIDE
- (2S,5R)-7-Oxo-N-((3S)-pyrrolidin-3-yloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
- C11 H18 N4 O7 S, 350.35
- Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
CAS 1452458-72-8
KEEP WATCHING THIS POST
SODIUM SALT CAS 1629221-44-8
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(3S)-3-pyrrolidinyloxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester, sodium salt (1:1)
Patent
http://www.google.com/patents/WO2015110886A1?cl=en

Formula (II) Formula (III) Formula (IV)
Hydrogenolysis

Formula (I)
Scheme – 1

Formula (VII) Formula (VIII)
Hydrazine hydrate

Formula I
Scheme – 2
Example 1
Synthesis of tert-butyl (3S)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
Step 1; Preparation of 3-(R)-hydroxypyrrolidine hydrochloride (VIII):
To a stirred suspension of commercially available (25, 4i?)-4-hydroxy-2-pyrrolidinecarboxylic acid (L-hydroxyproline) (VII) (100 g, 0.762 mol) in anhydrous cyclohexanol (500 ml), was added 2-cyclohexen-l-one (5 ml). The resulting mixture was heated under reflux at about 154°C for about 48 hour. The obtained clear solution was allowed to cool to room temperature and then was cooled further to 10°C. To this, about 15 % solution of hydrochloric acid in ethanol (234 ml) was added and then stirred for 30 minutes. The separated solid was filtered under suction and washed with ethyl acetate (2 x 100 ml). The solid was dried under reduced pressure to obtain 47.5 g of 3-(R)-hydroxypyrrolidine hydrochloride (VIII) in 51 % yield. The solid was used without further purification in the next step.
Analysis:
Mass: 87.8 (M+l) as free base; for Molecular weight of 123.57 and Molecular Formula of C4Hi0ClNO; and
1H NMR (400MHz, DMSO): 5 9.58 – 9.32 (brd, 2H), 5.36 (brs, 1H), 4.36 – 3.39 (brs, 1H), 3.17 (brs, 2H), 3.11-2.96 (dd, 2H), 1.90 – 1.81 (m, 2H).
Step 2: Preparation of (3R)-l-(tert-butoxycarbonyl)-3-hydroxypyrrolidine (IX):
To a stirred suspension of 3-(i?)-hydroxypyrrolidine hydrochloride (VIII) (110 g, 0.9 mol) in dichloromethane (1100 ml), triethylamine (273 g, 2.7 mol) was added at 0-5°C. After 5 minute of stirring di-feri-butyldicarbonate [(Boc)20] (245 g, 1.125 mol) was added to the reaction mixture in small portions, followed by 4-dimethylaminopyridine (10.99 g, 0.09 mol). The reaction mixture was stirred for 2 hour and then poured in to water (1100 ml). The organic layer was separated and washed with saturated ammonium chloride solution (1×1100 ml) and water (1100 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residue was purified by silica gel (60-120 mesh) column chromatography using 1-5% mixtures of acetone: hexane as an eluent. The combined fractions were evaporated, to obtain the 118 g of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX), as a white solid, in 71 % yield.
Analysis:
Melting point: 55 – 58°C;
Mass: 188 (M+l); for Molecular Weight of 187.24 and Molecular Formula of C9H17N03; and
1H NMR (400MHz, CDC13): 54.428 – 4.424 (s, 1H), 3.46 – 3.43 (m, 2H), 3.37 -3.28 (m, 2H), 2.36 – 2.30 (d, 1H), 2.00 – 1.86 (m, 2H), 1.44 (s, 9H).
Step 3: Preparation of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-l-carbox lic acid tert- butyl ester (X):
To a stirred solution of di-isopropyl azodicarboxylate (97.17 g, 0.481 mol) in tetrahydrofuran (1200 ml), a solution triphenyl phosphine (125.9 g, 0.481 mol) in tetrahydrofuran (300 ml) was added at temperature below -10°C. The resulting reaction mixture was stirred for further 45 minute at the same condition and a solution of (3i?)-l-(ieri-butoxycarbonyl)-3-hydroxypyrrolidine (IX) (60 g, 0.3204 mol) in tetrahydrofuran (300 ml) was added over a period of 15 minute. After another 45 minute of stirring, N-hydroxy phthalimide (52.4 g, 0.3204mol) was added in one portion to the reaction mass. The reaction mixture was allowed to warm to room temperature and stirred for 16 hour.
The completion of the reaction was monitored by thin layer chromatography. After completion of reaction, the solvent was evaporated under reduced pressure. The residue thus obtained was stirred with di-isopropyl ether (600 ml). The precipitate formed was filtered under suction. The filtrate was concentrated under reduced pressure and the residual mass was purified by silica gel (60-120 mesh) column chromatography using 1-5 % mixtures of acetone: hexane as an eluent. The solvent from the combined fractions was evaporated to obtain 63 g of (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl)oxy]pyrrolidine-1-carboxylic acid tert-buty\ ester (X), as a white solid, in 59% yield.
Analysis:
Melting point: 112-115°C;
Mass: 333.2 (M+l); for Molecular Weight of 332.36 and Molecular Formula of ![]()
1H NMR (400 MHz, CDC13): 57.86-7.83 (m, 2H), 7.78-7.75 (m, 2H), 4.99 – 4.94 (d, 1H), 3.80 – 3.68 (m, 2H), 3.60 – 3.53 (m, 2H), 2.28-2.25 (m, 1H), 2.02 (m, 1H), 1.48 (s, 9H).
Step 4: Preparation of tert-butyl (35)-2-(aminooxy)pyrrolidine-l-carboxylate (III):
To a stirred suspension of the (5)-3-[(l,3-dihydro-l,3-dioxo-isoindol-2-yl) oxy]pyrrolidine-l-carboxylic acid tert-buty\ ester (X) (12.68 g, 0.0381 mol) in dichloromethane (200 ml) was added 99% hydrazine hydrate (3.81 g, 0.0762 mol) drop-wise over a period of 30 minutes, at 25°C. After 2 hour of stirring, the separated solid was filtered and washed with dichloromethane (2 x 50 ml). The filtrate and washings were combined and washed with water (2 x 65 ml) and finally with brine (1 x 65 ml). The organic layer was dried over anhydrous sodium sulphate and the solvent was evaporated under reduced pressure to obtain 7.71 g of tert-buty\ (3S)-2-(aminooxy pyrrolidine- 1-carboxylate (III) as pale yellow oil.
Analysis:
Mass: 203 (M+l); for Molecular Weight of 202.26 and Molecular Formula of C9H18N203.
Example 2
Synthesis of (25, 5R)-7-oxo-N-r(35)-pyrrolidin-3-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2. lloctane-2-carboxamide (I) :
Step 1: Preparation of fert-butyl-(35)-3-[({[25, 5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV):
To a clear, stirred solution of sodium (25, 5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II) (11.38 g, 0.0382 mol) in water (114 ml), was added EDC.HC1 (18.24 g, 0.0955 mol) at 15°C, in small portions. After 10 minutes, a solution of feri-butyl-(35)-3-(aminooxy) pyrrolidine- 1-carboxylate (III, 7.72 g, 0.0382 mol), prepared as per the literature procedure: US5233053, Chemistry Letters, 893-896, (1986) and depicted in scheme 2), in dimethylformamide (24 ml) was added drop wise, to the above stirred solution, at about 10°C. The reaction mass was allowed to warm to 25°C and HOBt (5.15 g, 0.0382 mol) was added in small portions over a period of 15 minutes and the reaction mixture was stirred further at room temperature for 16 hour. After completion of the reaction (monitored by thin layer chromatography using solvent system acetone: hexane (35:65)) the resulting mixture was filtered and the residue was washed with water (120 ml). The residual white solid was suspended in fresh water (120 ml) and the mixture stirred at 50°C, for 3 hour. The resulting suspension was filtered and the residual solid dried under reduced pressure to obtain 16.1 g of tert-buty\ (35)-3-[({ [25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino) oxy]pyrrolidine- 1-carboxylate (IV) as off white solid in 92% yield.
Analysis:
Mass: 461.3 (M+l); for Molecular weight of 460.53 and Molecular formula of ![]()
1H NMR (400MHz, CDC13): δ 9.08-9.03 (d, 1H), 7.43-7.36 (m, 5H), 5.06-4.88 (dd, 2H), 4.63-4.57 (d, 1H), 3.97-.396 (d, 1H), 3.64-3.53 (m, 2H), 3.47-3.37 (m, 2H), 3.31 (s, 1H), 3.02-2.99 (d, 1H), 2.75-2.73 (d, 1H), 2.29(m, 2H), 2.18-2.15 (m, 1H), 2.01-1.90 (m, 3H), 1.66 (m, 1H), 1.46 (s, 9H).
Step 2: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V):
ieri-Butyl-(35)-3-[({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (IV) (10 g, 0.02171 mol) was dissolved in a mixture of dimethylformamide and dichloromethane ( 1 : 1 , 50 ml : 50 ml) to obtain a clear solution. To this solution, was added 10% palladium on carbon (2.5 g, 50% wet) catalyst. The suspension was stirred for 4 hour, at 50 psi hydrogen atmosphere, at 25°C. After completion of the reaction (monitored by thin layer chromatography), the resulting mixture was filtered through a celite pad. The residue was washed with dichloromethane (50 ml). The solvent from the combined filtrate was evaporated under reduced pressure to obtain 8.04 g of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl
amino)oxy]pyrrolidine-l-carboxylate (V) as oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 371.2 (M+l); for Molecular Weight of 370.4 and Molecular Formula of
Step 3: Preparation of tert-butyl-(35)-3-[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate, tetrabutyl ammonium salt (VI):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-hydroxy-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate (V) (8.04 g, 0.0217 mol) in dimethylformamide (50 ml), was added sulfur trioxide dimethyl formamide complex (3.98 g, 0.0260 mol) in one portion, at about 10°C. The stirring was continued further for 30 minute and then the reaction mixture was allowed to warm to room temperature. After 2 hour, a solution of tetrabutylammonium acetate (7.83 g, 0.0260 mol) in water (25.8 ml) was added to the resulting reaction mass under stirring. After additional 2 hour of stirring, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2 x 20 ml) to obtain thick mass. This mass was partitioned between dichloromethane (100 ml) and water (100 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (50 ml). The combined organic extracts were washed with water (3 x 50 ml), dried over anhydrous sodium sulphate and the solvent evaporated under reduced pressure. The residual oily mass was triturated with ether (3 x 50 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 11.3 g of tert-butyl(3S)-3-[({ [2S,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy] pyrrolidine- 1-carboxylate, tetrabutylammonium salt (VI), as a white foam, in 75 % yield.
Analysis:
Mass: 449.3 (M-l, without TBA); for Molecular weight of 691.94 and Molecular formula of C32H61N5O9S; and
1H NMR (400MHz, CDC13): 59.14-9.10 (d, 1H), 4.63 (s, 1H), 4.35 (s, 1H), 3.94-3.92 (d, 1H), 3.66-3.35 (m, 5H), 3.29-3.27 (m, 8H), 2.83-2.80 (d, 1H), 2.35-2.17 (m, 3H), 1.98-1.87 (m, 2H), 1.73 (m, 1H), 1.70-1.62 (m, 8H), 1.49-1.40 (m, 17H), 1.02-0.99 (t, 12H).
Step 4: Preparation of (25,5R)-7-oxo-iV-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1]octane-2-carboxamide (I):
To a stirred solution of ieri-butyl(35)-3-[({ [25,5i?)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]pyrrolidine-l-carboxylate tetrabutyl ammonium salt (VI) (11 g, 0.0158 mol) in dichloromethane (55 ml), was added trifluoroacetic acid (55 ml) drop wise at about -10 °C over a period of 1 hour. After 1 hour of stirring, the resulting mixture was poured into hexane (550 ml), stirred well for 30 minute and the separated oily layer was collected. This procedure was repeated one more time and finally the combined oily layer was added to diethyl ether (110 ml) under vigorous stirring, at about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2 x 110 ml). The solid thus obtained was stirred with fresh dichloromethane (110 ml) for 30 minutes and filtered. The residual solid was dried at about 45 °C under reduced pressure to obtain 5.7 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)- l,6-diaza bicyclo[3.2.1] octane-2-carboxamide (I), as a white amorphous solid having XRPD as shown in Figure 1.
Analysis:
Mass: 349.2 (M-l); for Molecular Weight of 350.35 and Molecular Formula of ![]()
1H NMR (400MHz, DMSO-D6): δ 11.44 (brs, 1H), 8.80 (brs, 2H), 4.64-4.63 (m, 1H), 4.00 (s, 1H), 3.78-3.77 (d, 1H), 3.38-3.23 (m, 4H), 3.03-2.93 (dd, 2H), 2.48-2.11 (m, 1H), 2.00- 1.94 (m, 2H), 1.88- 1.86 (m, 1H), 1.71-1.65 (m, 2H).
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-jV-r(35)-pyrrolidin-2-yl-oxyl-6-(sulfooxy)-l,6-diaza bicyclor3.2.11 octane-2-carboxamide:
The solid (5 g) obtained in Step 4 of Example 2 was dissolved in water (30 ml) with stirring. To this solution, Isopropanol (210 ml) was slowly added at 25 °C and stirred for 12 hours. The separated solid was filtered and washed with additional isopropanol ( 10 ml) and dried under reduced pressure to obtain 3.9 g of (25,5i?)-7-oxo-N-[(35)-pyrrolidin-2-yl-oxy]-6-(sulfo-oxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I, having XRPD as shown in Figure 2, in 78 % yield.
Analysis:
Purity as determined by HPLC: 95.56 %; and
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 10.57 (± 0.2), 12.01 (± 0.2), 13.61 (± 0.2), 15.47 (± 0.2), 17.86 (± 0.2), 18.34 (± 0.2), 19.09 (± 0.2), 19.81 (± 0.2), 22.69 (± 0.2), 24.79 (± 0.2), 27.22 (± 0.2) and 33.41 (± 0.2)
//////
WCK ? , WCK Series by Wockhardt for treating the bacterial infection

(2S,5R)-7-0X0-N-[(2S)-PYRROLLIDIN-2-YL-METHYLOXY]-6-(SULFOOXY)-1,6-DIAZABICYCLO[3.2.1 ]OCTANE-2-CARBOXAMIDE
(2S,5R)-7-Oxo-N-((2S)-pyrrolidin-2-ylmethyloxy)-6-(sulfooxy)-1,6-diazabicyclo[3.2.1]octane-2-carboxamide
Sulfuric acid, mono[(1R,2S,5R)-7-oxo-2-[[[(2S)-2-pyrrolidinylmethoxy]amino]carbonyl]-1,6-diazabicyclo[3.2.1]oct-6-yl] ester
KEEP WATCHING THIS POST
MW 364.37, C12 H20 N4 O7 S
CAS 1452459-04-9 FREE FORM
CAS Na SALT 1572988-44-3
PATENTS, WO 2015079329, WO 2015079389 , WO 2015063714, US 20130225554
Emergence of bacterial resistance to known antibacterial agents is becoming a major challenge in treating bacterial infections. One way forward to treat bacterial infections, and especially those caused by resistant bacteria, is to develop newer antibacterial agents that can overcome the bacterial resistant. Coates et al. (Br. J. Pharmacol. 2007; 152(8), 1147-1154.) have reviewed novel approaches to developing new antibiotics. However, the development of new antibacterial agents is a challenging task. For example, Gwynn et al. (Annals of the New York Academy of Sciences, 2010, 1213: 5-19) have reviewed the challenges in discovery of antibacterial agents.
Several compounds have been described in the prior art for use in treatment of bacterial infections (for example, see Patent Application Nos. PCT/IB2012/054296, PCT/IB2012/054290, US20130225554, PCT/US2010/060923, PCT/EP2010/067647, PCT/US2010/052109, PCT/US2010/048109, PCT/GB2009/050609, PCT/EP2009/056178, PCT/US2009/041200, PCT/US2013/034562, PCT/US2013/034589, PCT/IB2013/053092 and PCT/IB2012054706). However, there remains a need for potent antibacterial agents for preventing and/or treating bacterial infections, including those caused by bacteria that are resistant to known antibacterial agents.

PATENT
https://encrypted.google.com/patents/WO2015079329A2?cl=en



Formula (I)
Scheme -1

Formula (VII) Formula (VIII)

Formula (III) Formula (X) Formula (IX)
Scheme 2
Example 1
Synthesis of fert-butyl (25)-2-r(aminooxy)methyllpyrrolidine-l-carboxylate
Step 1: Synthesis of l-(tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII):
To a stirred suspension of (2S)-pyrrolidine-2-carboxylic acid (L-proline) (200 g, 1.73 mol) in 1,4-dioxan and water mixture (1: 1, 1000 ml : 1000 ml) was added a solution of sodium hydroxide (138.97 g, 3.47 mol in 740 ml water) over a period of 20 minutes at 0 °C. Bi-feri-butyl dicarbonate (415.3 ml, 1.9 mol in 400 ml 1,4-dioxan) was added to the resulting clear solution over a period of 30 minutes, at temperature of about 0-5 °C. The reaction mixture was allowed to warm to room temperature and stirred for 16 hours. After completion of the reaction (monitored by thin layer chromatography), the reaction mixture was concentrated to 40 % of the initial volume under reduced pressure at 40-50 °C. The pH of the residual mixture was adjusted to 2 – 2.5 using 30 % aqueous potassium hydrogen sulphate at 15 °C under continuous stirring. The separated solid was filtered under suction and washed with water (2×400 ml) and dried under reduced pressure (4 mm Hg), to obtain 370 g of l-(ieri-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII) as white solid.
Analysis:
Mass: 216 (M+l), for Molecular Weight: 215.24 and Molecular Formula:
1H NMR (400 MHz, CDC13): δ 10.60 (s, 1H), 4.35-4.24 (dd, 1H), 3.54-.3.34 (M, 2H), 2.27-1.91 (unresolved, 4H), 1.47-1.41 (d, 9H);
Purity as determined by HPLC: 99.92 %.
Step 2: Synthesis of tert-iutyl-(25)-2-(hydroxymethyl)-pyrrolidine-l-carboxylate (IX):
N-Methylmorpholine (113 ml, 1.114 mol) was added to the suspension of \-{tert-butoxycarbonyl)-(25)-pyrrolidine-2-carboxylic acid (VII, 30 g, 139 mmol) in tetrahydrofuran (2000 ml) under stirring at temperature of about 0 °C. Ethyl chloroformate (106 ml, 1.114 mol) was added drop- wise to the above obtained clear solution over a period of 30 minutes. After stirring for 1 hour, the resulting suspension was filtered over celite and the residue was washed with tetrahydrofuran (2×200 ml). To the combined filtrate was added dropwise a solution of sodium borohydride (42.1 g, 1.114 mol) in 210 ml water, containing a catalytic amount of sodium hydroxide, at temperature of about -10 °C over a period of 1-2 hours under stirring. The reaction mixture was allowed to warm to room temperature and stirred further for an hour. The reaction mixture was filtered through celite bed and the filtrate concentrated under reduced pressure to yield 180 g of ieri-butyl(25)-2-(hydroxymethyl)-pyrrolidene-l-carboxylate (IX) as colorless oil.
Analysis:
Mass: 202 (M+l), for Molecular Weight: 201.2 and Molecular Formula: C10H19NO3;
1H NMR (400 MHz, CDC13): δ 3.94-.3.92 (m, 1H), 3.80 (board, 1H), 3.63-3.54 (m, 2H), 3.45-3.40 (m, 1H), 3.32-3.28 (m, 1H), 2.01-1.96 (m, 1H), 1.84-1.75 (m, 2H), 1.63 (m, 1H), 1.45 (s, 9H);
Purity as determined by HPLC: 87.7 %.
Step 3: Synthesis of fert-butyl (25)-2-[[(l,3-dihydro-l,3-dioxo-2H-isoindol-2-yl)oxy] methyl] -pyrrolidine-1 -carboxylate (X) :
Triphenylphosphine (328.4 g, 1.253 mol) in tetrahydrofuran (1260 ml) was added to solution of Diisopropyl azodicarboxylate (253.3 g, 1.253 mol) in tetrahydrofuran at temperature of -15 °C under stirring. After stirring for an hour, N-feri-butoxylcarbonyl-L-prolinol (IX) (180 g, 0.895 mol) in tetrahydrofuran (540 ml) was added to the resulting mixture over a period of 15 minutes. After stirring the mixture for 45 minutes, N-Hydroxy phthalimide (146 g, 0.895 mol) was added and the mixture was allowed to warm to room temperature and stirred further for 16 hours. The solvent was evaporated under reduced pressure and residual oil was dissolved in dichloromethane (5000 ml) and washed with an aqueous 5 % sodium hydrogen carbonate solution (2×300 ml). The organic layer was dried over anhydrous sodium sulfate and the solvent evaporated under reduced pressure to obtain viscous oil. Diisopropyl ether (720 ml) was added to the oil, the mixture was stirred well and separated solid was filtered under suction. The filtrate was concentrated under reduced pressure and the residue was further purified by chromatography over a silica gel column (60 -120 mesh) and eluted with mixtures of ethyl acetate and hexane. Upon concentration of the combined eluted fractions, 230 g of teri-butyl (25)-2-[[( l,3-dihydro- l,3-dioxo-2H-isoindol-2-yl)oxy]methyl]-pyrrolidine- l-carboxylate (X) was obtained as yellow oil.
Analysis:
Mass: 347.3 (M+l), for Molecular Weight: 346.39 and Molecular Formula: ![]()
1H NMR (400 MHz, CDCI3): δ 7.80-7.78 (m, 2H), 7.72-7.70 (m, 2H), 4.32 (brs, 1H), 4.05 (brs, 2H), 3.36-3.31 (m, 2H), 2.27-2.25 (m, 1H), 2.08(m, 1H), 1.88-1.87 (m, 2H), 1.43 (s, 9H).
Step 4: Synthesis of fert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (HI):
To a stirred solution of the compound of Formula (X) ( 100 g, 0.288 mol) in dichloromethane (2000 ml) was added 99 % hydrazine hydrate (28.9 g, 0.577 mol) drop-wise over a period of 30 minutes at temperature of about 25 °C. The stirring was continued further for a period of 3 hours. The separated solid was filtered and the solid washed with additional dichloromethane (2 x 500 ml). The combined organic layer was collected and washed with water (2 x 500 ml). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to obtain 62.4 g of tert-butyl (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III) as a colorless oil. This was used as such for the next reaction without further purification.
Analysis:
Mass: 215.1 (M- l), for Molecular Weight: 216.2 and Molecular Formula:
Example 2
Synthesis of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)-l,6- diazabicvclor3.2.11octane-2-carboxamide (I)
Step 1: Synthesis of tert-butyl (25)-2-{[({[25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate
(IV):
Sodium(25,5i?)-6-(benzyloxy)-7-oxo-l,6-diazabicyclo[3.2.1]octane-2-carboxylate (II, 77.4 g, 0.259 mol; prepared according to the procedure disclosed in Indian patent application No. 699/MUM/2013) was dissolved in water (774 ml) to obtain a clear solution. To the clear solution was added EDC.HC1 (120.8 g, 0.632 mol) at temperature of about 15°C and after 10 minutes a solution of tert-buty\ (25)-2-[(aminooxy)methyl]pyrrolidine-l-carboxylate (III, 62.4 g, 0.288 moles prepared as per the literature procedure depicted in scheme 2) in dimethylformamide (125 ml) was added drop wise under continuous stirring at temperature of about 10 °C. The reaction mass was allowed to warm to temperature of about 25°C and then HOBt (38.96 g, 0.288 mol) was added in small portions over a period of 15 minutes and the resulting mixture was further stirred at room temperature for 16 hours. The reaction progress was monitored using thin layer chromatography using mixture of acetone and hexane (35: 65) as solvent system. The resulting suspension was filtered and the residue was washed with water (200 ml). The residual white solid was suspended in water (200 ml) and the mixture stirred with heating at temperatyre of about 50 °C for 3 hours. The resulting suspension was filtered, the residue dried at atmospheric temperature and then dried under vacuum to obtain 105 g of ierr-Butyl(25)-2- { [( { [25,5R)-6-(benzyloxy)-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy]methyl}pyyrolidine-l-carboxylate (IV) as off white solid.
Analysis:
Mass: 475.4 (M+l), for Molecular Weight of 474.56 and Molecular Formula of ![]()
1H NMR (400 MHz, CDCI3): δ 10.16 (br s, 1H), 7.43-7.35 (m, 5H), 5.06-4.88 (dd, 2H), 4.12 (s, 1H), 3.94-.393 (d, 2H), 3.83 (unresolved s, 1H), 3.75-3.73 (m, 1H), 3.37-3.28 (dt, 2H), 3.02-2.86 (dd, 2H), 2.31-2.26 (m, 1H), 2.02-1.84 (m, 6H), 1.71-1.68 (m, 1H), 1.45 (s, 9H).
Step 2: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-hydroxy-7-oxo-l,6-diazabicylco
[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V):
tert-butyl(25)-2-{ [({ [25,5R)-6-(benzyloxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (IV, 85 g, 0.179 mol) was dissolved in a mixture of dimethylformamide and dichloro methane (1: 1, 425 ml : 425 ml) to obtain a clear solution. To this solution was added 10 % Pd-C (17 g, 50 % wet) catalyst. The suspension was stirred for 4 hours under 7 psi hydrogen atmosphere at temperature of about 25 °C. The resulting mixture was filtered through celite under suction. The residue was washed with dichloromethane (170 ml). The solvent from the filtrate was evaporated under reduced pressure to furnish 68.8 g of tert-buty\(2S)-2-{ [( { [25,5i?)-6-hydroxy-7-oxo- l,6-diazabicylco[3.2. l]oct-2-yl]carbonyl} amino)oxy] methyl}pyyrolidine-l-carboxylate (V) as oil. The obtained product was used as such for the next reaction without further purification.
Analysis:
Mass: 385.4 (M+l), for Molecular Weight of 384.4 and Molecular Formula of C17H28N406.
Step 3: Synthesis of tert-butyl(25)-2-{[({[25,5R)-6-(sulfooxy)-7-oxo-l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate, tetra butyl ammonium salt (VI):
To solution of ieri-butyl(25)-2-{ [({ [25,5i?)-6-hydroxy7-oxo-l,6-diazabicylco [3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine-l-carboxylate (V, 68.8 g, 0.178 mol) in dimethylformamide, (345 ml) was added sulfur trioxide dimethylformamide complex (30 g, 0.196 mol) under stirring at temperature of about 10 °C. The reaction mass was stirred at the same temperature for 30 minutes and then allowed to warm to room temperature. After 2 hours solution of tetra butyl ammonium acetate (59.09 g, 0.196 mol) in water (178 ml) was added to the reaction mixture under stirring. After 2 hours, the solvent from the reaction mixture was evaporated under reduced pressure to obtain an oily residue. The oily mass was co-evaporated with xylene (2×140 ml) to obtain thick mass. This mass was partitioned between dichloromethane (690 ml) and water (690 ml). The organic layer was separated and the aqueous layer re-extracted with dichloromethane (345 ml). The combined organic extracts were washed with water (3×345 ml) and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the resulting oily mass was triturated with ether (3×140 ml), each time the ether layer was decanted and finally the residue was concentrated under reduced pressure to obtain 102 g of ieri-butyl(25)-2- { [({ [25,5i?)-6-(sulfooxy)-7-oxo- l,6-diazabicylco[3.2.1]oct-2-yl]carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutyl ammonium salt (VI) as fluffy material.
Analysis:
Mass: 463.4 (M- l without TBA), for Molecular Weight of 705.96 and Molecular Formula of C33H63N5O9 S;
1H NMR (400 MHz, CDCI3): δ 10.2 (s, 1H), 4.35 (s, 1H), 4.14 (s, 1H), 3.91 -3.92 (d, 2H), 3.74 (m, 1H), 3.36-3.27 (m, 10H), 2.96-2.88 (dd, 2H), 2.31-2.26 (m, 2H), 2.19-1.98 (m, 2H), 1.95-1.70 (m, 4H), 1.68- 1.62 (p, 8H), 1.49- 1.40 (m, 17H), 1.02-0.98 (t, 12H).
Step 4: (25,5R)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diaza bicyclo [3.2.1]octane-2-carboxamide (I):
feri-butyl(25)-2-{ [( { [25,5i?)-6-(sulfooxy)-7-oxo-l ,6-diazabicylco[3.2.1]oct-2-yl] carbonyl}amino)oxy]methyl}pyyrolidine- l-carboxylate, tetrabutylammonium salt (VI) (50 g, 0.0708 mol) was dissolved in dichloromethane (250 ml) and to the clear solution was slowly added trifluoroacetic acid (250 ml) at temperature of about -10 °C over a period of 1 hour under stirring. After stirring for an hour, the resulting mixture was poured into hexane (2500 ml) and the oily layer was separated. This procedure was repeated one more time and finally the separated oily layer was added to diethyl ether (500 ml) under vigorous stirring at temperature of about 25 °C. The ether layer was removed by decantation from the precipitated solid. This procedure was repeated twice again with diethyl ether (2x500ml). The solid thus obtained was stirred with fresh dichloromethane (500 ml) for 30 minutes and filtered. The residual solid was dried at temperature of about 45 °C under reduced pressure to yield 25 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo[3.2.1]octane-2-carboxamide (I) in amorphous form. The XRD of the obtained amorphous form is shown in Figure 1.
Analysis:
Mass: 363.2 (M- l), for Molecular Weight: 364.37 and Molecular Formula: C12H2oN407S;
1H NMR (400 MHz, DMSO-D6): δ 1 1.73 (s, 1H), 8.62-8.83 (d, 2H), 3.88-4.00 (m, 3H), 3.74-3.81 (m, 2H), 3.19 (t, 2H), 2.94-3.04 (dd, 2H), 1.96-2.03 (m, 2H), 1.80-1.92 (m, 3H), 1.54- 1.73 (m, 3H);
Purity as determined by HPLC: 90.30 %.
Example 3
Preparation of Crystalline Form I of (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl- methyloxyl-6-(sulfooxy)-l,6-diaza bicvclor3.2.11octane-2-carboxamide
The amorphous solid obtained in the Step 4 of Example 2 was dissolved in water (75 ml) and to this solution isopropanol (200 ml) was slowly added at temperature of about 25 °C. The solution was further stirred for 12 hours. The separated solid thus obtained was filtered and washed with additional isopropanol (25 ml) and dried under reduced pressure to obtain 19 g of (25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l ,6-diazabicyclo[3.2.1]octane-2-carboxamide as crystalline Form I. The XRD of the obtained crystalline Form I is shown in Figure 2.
X-ray powder diffraction pattern comprising peak at (2 Theta Values): 8.08 (± 0.2), 1 1.45 (± 0.2), 16.26 (± 0.2), 17.89 (± 0.2), 18.15 (± 0.2), 19.66 (± 0.2), 21.15 (± 0.2), 23.55 (± 0.2), 24.23 (± 0.2), 24.94 (± 0.2), 25.66 (± 0.2) and 29.41 (± 0.2).
Typical X-ray analysis was performed as follows. Pass the test substance through sieve #100 BSS or gently grind it with a mortar and pestle. Place the test substance uniformly on a sample holder having cavity surface on one side, press the sample and cut into thin uniform film using a glass slide in such a way that the surface of the sample should be smooth and even. Record the X-ray diffractogram using the following instrument parameters:
Instrument : X-Ray Diffractometer
(PANalytical, Model X’Pert Pro
MPD)
Target source : CuK(a)
Antiscattering slit (Incident beam) : 1°
Programmable Divergent slit : 10 mm (fixed)
Anti- scattering slit (Diffracted beam) : 5.5 mm
Step width : 0.02°
Voltage : 40 kV
Current : 40 mA
Time per step : 30 seconds
Scan range : 3 to 40°
Example 4
Preparation of Pure (25,5R)-7-oxo-N-r(25)-pyrrolidin-2-yl-methyloxyl-6-(sulfooxy)- l,6-diazabicyclor3.2.11octane-2-carboxamide
(25,5i?)-7-Oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- l,6-diazabicyclo [3.2.1] octane-2-carboxamide (5 g) was slowly dissolved in water (50 ml) under stirring until clear solution appears. To this clear solution 350 ml of isopropanol was added drop wise under stirring over the period of 2 hours. Formation of fine white precipitates was observed after the completion of the addition of isopropanol. The resulted fine suspension was stirred at temperature of about 25 °C for 20 hours. The formed white precipitates were filtered and vacuum dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 4.4 g of (2S,5i?)-7-oxo-N-[(2S)-pyrroMin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo [3.2.1] octane-2-carboxamide.
The above obtained (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide (3.4 gm) was dissolved in 34 ml of water to get clear solution. To the obtained clear solution 170 ml of isopropanol was added drop wise over a period of 1 hour. Formation of fine oily globules was observed and allowed to stand still for 15 minutes. The upper clear water and isopropanol layer was decanted from the oily mass. The clear decanted solution was allowed to stand at temperature of about 25 °C for 48 hours. Formation of crystals was observed and were collected by filtration. The collected crystals were dried at temperature of about 30-40 °C, under reduced pressure (2 mm Hg) to get 2 g of (2S,5i?)-7-oxo-N-[(2S)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)- 1 ,6-diazabicyclo[3.2.1 ]octane-2-carboxamide which was analyzed for content of various components using HPLC and the results are described in Table 1.
The relative % content of (25,5i?)-7-oxo-N-[(25)-pyrrolidin-2-yl-methyloxy]-6-(sulfooxy)-l,6-diazabicyclo[3.2.1]octane-2-carboxamide with other substances (Table 1) was determined using HPLC (Agilent 1100 or equivalent). The HPLC column having 250 mm length and 4.6 mm ID packed with 5 μ particles of octa-decyl silane (ODS) was used. Mobile phase A used was a mixture of buffer (0.02 M potassium dihydrogen phosphate in HPLC grade water, pH adjusted to 2.5 with orthophosphoric acid and again readjusted to 7.0 with dilute ammonia), HPLC grade water and acetonitrile in a ratio of 40 : 60 : 0.2; v/v/v. Mobile phase B was mixture of buffer and acetonitrile in a ratio of 40 : 60; v/v. Mobile phase was run in gradient mode. Initially mobile phase A and B was run at 100 : 0 for 15 minutes, slowly ratio of mobile phase B was raised to 100 % in 10 minutes, held for 10 minutes at this concentration and brought back to initial condition in next 5 minutes and held for 10 minutes before next run. Flow rate of mobile phase was maintained at 1.0 ml/min. Column temperature was maintained at temperature of about 30°C. Detection was carried out using UV detector at wavelength 225 nm. Test solutions were prepared in mobile phase A. The method is capable of resolving diastereomers (Table 1, Sr. No. 1 and 2) with resolution of not less than 2.0.


{kind=link}
{kind=link}